
Page 167
5
Evaluation and Cost Coverage of Technologies
The United States is both the largest market for most new health care technologies and the largest producer of the research and development that generates them. The post-World War II era saw an ever-increasing investment in scientific research in both the public and the private sectors. Increased access to medical insurance coverage through employer-sponsored health benefits and government-sponsored programs such as Medicare and Medicaid made it possible for new medical technologies to be rapidly diffused throughout the medical care system and encouraged continued investment in their development. Initially, physicians essentially controlled the process of technology adoption because a test or procedure ordered by a doctor was deemed medically necessary and was thus eligible for coverage (reviewed by Braslow et al., 1998).
As the costs of health care escalated in the 1970s, the traditional model was questioned and concerns were raised about the lack of professional consensus in medical care as well as the wastefulness of medical interventions and procedures that were of unproven value. Concern was also raised as to whether expensive new technologies were deployed in an efficient manner. Most health economists believe that technology is the single largest driving force behind the long-term rise in health care spending in the United States (Fuchs, 1999; Rettig, 1997). Because the resources for health care are not infinite, there has been increasing pressure to make evidence-based decisions about the use of medical technology, and as a result, the need for technology assessment has risen.
In 1976, an amendment to the Federal Food, Drug, and Cosmetic Act, as discussed in the previous chapter, required approval by the Food and
Page 168
Drug Administration (FDA) for all medical devices. However, FDA approval does not guarantee that insurers will cover the cost of new technologies in health care. FDA approval permits legal marketing in interstate commerce but does not mandate or imply health plan approval or coverage, which are essential for successful technology diffusion. Such coverage decisions are now often based on critical assessments of the effects of technologies on patient outcomes, and the criteria for these assessments can be far more stringent than those of the FDA (Aubry, 1998). Organizations that assess technologies often ask two related questions: (1) Can the technology positively affect patient outcome, and (2) How does the technology compare with other technologies on the market? Some organizations may also ask whether the technology can achieve results similar to those of products already on the market, but in a more cost-effective manner. A recent report from the Milbank Foundation (2000)1 found that U.S. purchasers were most likely to use technology assessment data for new technologies that are costly and controversial.
Reimbursement decisions can be highly influential on the adoption of new technologies as well. Coverage decisions determine whether a particular service or product is eligible for reimbursement, whereas the actual rates of reimbursement (the methods and amounts of payment) for covered services and products may vary greatly depending on the specific case, location, insurance carrier, and so on.
As health care purchasers increasingly rely on scientific evidence of improved health outcomes to make coverage and reimbursement decisions, the demand for credible technology assessments will likely increase and the influence of such assessments on the coverage and diffusion of new technologies will grow. The challenge of evidence-based medicine is to develop a health care system that “rewards better outcomes, recognizes value, and encourages efficient use of limited resources” (Eisenberg, 1999, p. 1865).
As in many areas of health care, there will be increasing concern about when to introduce a promising new technology that costs more but that may be more effective. Methods that offer better value (accuracy, sensitivity, or reliability) at a lower cost are likely to be adopted quickly. Technologies that do something entirely new or that clearly offer major improvements are often readily adopted as well. The most difficult coverage decisions will come from technologies that are marginally better but that are also substantially more costly. Unfortunately, many technologies may fall into this category.
1Health care purchasers in the United Kingdom and the United States (including 13 public officials from 11 states, 4 private-sectors purchasers, 5 representatives of private purchasing coalitions, and 3 consultants) were interviewed by phone over a 3-month period in early 1999.
Page 169
OVERVIEW OF TECHNOLOGY ASSESSMENT ORGANIZATIONS AND THEIR ROLES
Early efforts to assess medical technologies so that they would not diffuse too rapidly were initiated by the National Health Planning and Resources Development Act (Public Law 93-641) of 1975. However, this legislation was later repealed because it was viewed as too limiting. From 1975 to 1995, the congressional Office of Technology Assessment (OTA) undertook studies of the methods for technology assessment and carried out several assessments of medical technologies. The National Center for Health Care Technology (NCHCT), established in the U.S. Department of Health and Human Services (DHHS) in 1978, did technology assessments to assist the Health Care Financing Administration (HCFA) with coverage decisions. However, despite its involvement in health technology assessment, the federal government has never really carried out the central technology assessment repository function that was originally envisioned (U.S. Congress, Office of Technology Assessment, 1994). This is in contrast to the situation in many European countries, in which there are either global budgets or some other form of real control by the government (Perry and Thamer, 1999).
Both federal organizations (NCHCT and OTA) have since been dissolved, and technology evaluation activities have increased greatly in the private sector (Perry and Thamer, 1999). Today, health technology assessment is undertaken by a variety of public and private organizations, including insurers and managed care organizations, professional medical societies, health technology companies, academic medical centers, and independent technology assessment institutions (U.S. Congress, Office of Technology Assessment, 1994). The various groups differ in their objectives and process, as is discussed briefly below for some of the major organizations.
Health Care Financing Administration
HCFA is the federal agency that administers Medicare, Medicaid (in collaboration with the states), and the State Children's Health Insurance Program. In addition to paying for health care, HCFA also performs a number of activities focused on quality, including regulation of laboratory testing (under the Clinical Laboratory Improvement Amendments), surveys and certification of health care facilities, development of coverage policies, and quality-of-care improvements. External advisory panels provide advice or make recommendations on a variety of issues relating to HCFA's responsibilities and activities. As the nation's largest health care provider, the Medicare program directly exerts significant influence on patient access to new medical technologies and also indirectly influ-
Page 170
ences coverage and reimbursement decisions in other sectors of the health care marketplace.
The Social Security Act of 1965 specified broad categories of services covered by Medicare, excluded services, and exceptions to the exclusions. Because the rationale for Medicare's creation was to pay for expensive hospitalization and later for medical care, preventive services were excluded from coverage. The Secretary of DHHS (and predecessor departments) was, however, given authority to add specific items for coverage under Medicare, and the U.S. Congress could always amend the Medicare legislation to include a specific benefit. Mammography, added as a benefit by congressional mandate,2 is one of only a handful of preventive services that have been added. Experimental drugs, devices, and procedures have traditionally not been covered because they have been defined as not meeting the basic Medicare criterion of being “reasonable and necessary” for diagnosis or treatment.
This changed somewhat in 1995 through an interagency agreement between HCFA and FDA, which established coverage and reimbursement for certain devices and related services in clinical trials carried out under FDA-approved investigational device exemptions (IDEs). Under this agreement, medical devices are categorized by FDA as either Category A (novel, not reimbursable) or Category B (“next-generation” devices, eligible for Medicare reimbursement) (Table 5-1). Category A devices are novel experimental Class III devices (see Chapter 4) that are excluded from Medicare reimbursement. Category B devices are those that FDA has approved for use for one indication but that have been technically altered or are being tested for a new use. They generally fall into Class I or II but may also include some Class III devices that are related to devices that have already been shown to be safe and effective. According to FDA, about 95 percent of device trials involve Category B devices and are thus eligible for reimbursement, although it is not always granted (Institute of Medicine, 2000).
Medicare coverage and reimbursement decisions can be made at the local or national level. Medicare contractors (private insurance companies that contract with Medicare to process claims from beneficiaries, providers, and suppliers) primarily make local decisions with input from advisory committees consisting of local specialists. Carriers and intermediar-
2Section 4163 of the Omnibus Budget Reconciliation Act of 1990 originally set forth payment limitations and conditions for coverage of screening mammography. A new law signed as part of the Balanced Budget Act of 1997 provides Medicare coverage for annual screening mammograms for all Medicare-eligible women age 40 and over and waives the Part B deductible for screening mammography.
Page 171
|
Category A: Experimental Subcategory |
|
|
1 |
Class III devicesa of a type for which no marketing application has been approved for any indication or use |
|
2 |
Class III devices that would otherwise be in Category B but that have undergone significant modification for a new indication or use |
|
Category B: Nonexperimental/Investigational Subcategory |
|
|
1 |
Devices, regardless of classification, under investigation to establish substantial equivalence to a predicate device (one that is or that could be legally marketed) |
|
2 |
Class III devices whose technological characteristics and indications are comparable to those of an approved device |
|
3 |
Class III devices with technological (“generational”) advances compared with an approved device |
|
4 |
Class III devices comparable to an approved device (no significant modifications) but under investigation for a new indication |
|
5 |
Class III devices on the market before the current regulatory requirements (1976) but now under investigation |
|
6 |
Devices not posing significant risks (Class I or II) for which an IDE is required |
NOTE: Some investigational devices may exhibit unique characteristics or raise safety concerns that make additional consideration necessary. For these devices, HCFA and FDA will agree on the additional criteria to be used. FDA will then use these criteria to assign the device to a category. As experience is gained in the categorization process, this attachment may be modified.
aDevices are classified by their inherent risks and benefits on the basis of the level of control necessary to ensure safety and effectiveness. Class I devices present minimal potential for harm to the user and are subject only to “general controls” (e.g., proper registration and labeling and good manufacturing practices). Class II devices are those for which general controls alone are insufficient to ensure safety and effectiveness, so they are also subject to special controls, which may include special labeling requirements, guidance documents, mandatory performance standards, and postmarketing surveillance. Class III is the most stringent regulatory category and includes devices for which safety and effectiveness cannot be ensured solely through general or special controls. Class III devices usually support or sustain human life, are of substantial importance in preventing impairment of human health, or present a potential, unreasonable risk of illness or injury. They require premarket approval, which may include evidence from clinical trials.
SOURCE: Health Care Financing Administration/Food and Drug Administration (1995).
Page 172
ies still make most coverage and reimbursement decisions,3 but in recent years, HCFA has attempted to move away from local decisions and more toward national coverage decisions and also toward more evidence-based medical decisions. At the national level, the agency has tried a number of different approaches to coverage policy and may seek input from entities such as the Agency for Healthcare Research and Quality (AHQR; see below); FDA; the U.S. Department of Veterans Affairs-the Civilian Health and Medical Program of the Uniformed Services; the American Medical Association; the National Institutes of Health (NIH); specialty-specific professional organizations; specialty advocacy groups; and health care providers, suppliers, and manufacturers. HCFA is also charged with tracking emerging technologies and patterns of care to determine the applicability of existing coverage policy and to assess the need for policy change, but the process for accomplishing this goal is not yet well established or standardized.
A recent reorganization of HCFA (Federal Register, 1999) separated the offices responsible for coverage decisions (i.e., the procedures or devices covered under Medicare) and cost decisions (i.e., the level of reimbursement for those procedures or devices). The Center for Health Plans and Providers determines reimbursement levels, and the Office of Clinical Standards and Quality decides on coverage issues. Requests for coverage review may come from industry, from patients or patient advocacy groups, or from within the agency itself. In making a decision for or against coverage, HCFA may solicit input from a Medicare Coverage Advisory Committee (MCAC) or an external technology assessment organization. The agency may also defer the decision back to the local level.
HCFA has recently proposed two new criteria for making national coverage decisions within the agency and has recommended that contractors use the same criteria to make local decisions (Federal Register, 2000). First, the item or service must demonstrate medical benefit, and second, it must demonstrate added value to the Medicare population. To consistently apply these criteria, the following sequential questions would be addressed:
1. Is there evidence that demonstrates that the service is medically beneficial for a defined population?
2. For that population, is a medically beneficial alternative already covered by Medicare?
3. If yes, is the new service substantially more beneficial than the current one that is covered?
4. If the service is equivalent in benefit, will it result in equal or lower costs for Medicare?
3 Decisions are always local unless national action is taken.
Page 173
The use of external advisory committees is also a new approach that is being undertaken in part to make the process more open and accountable. The mission of MCACs is to serve as an impartial panel that reviews and evaluates the evidence on the effectiveness of services that Medicare is considering for coverage. MCACs review the available data on a new technology, make a judgment on its effectiveness compared with that of the established standard of care, and consider the applicability of the evidence to Medicare patients. MCAC assigns the technologies to one of six categories:
1. breakthrough– (more effective than the current standard of care);
2. as effective as the current standard of care, but with advantages;
3. as effective as the current standard of care, but with no advantages;
4. less effective than the current standard of care, but with advantages;
5. less effective, but with no advantages; or
6. not effective.
The MCAC program is organized into six panels, each of which is composed of 15 voting members, a chair, one consumer representative, and one representative from industry. The six panels cover six broad areas: drugs, biologics, and therapeutics; laboratory and diagnostic services; medical and surgical procedures; diagnostic imaging; medical devices and prosthetics; and durable medical equipment. MCAC also has an executive committee that develops criteria for assessment of effectiveness, develops panel procedures, coordinates panels, develops an annual slate of technology assessments, and approves panel recommendations and then submits them to HCFA. MCAC's executive committee consists of the chair and vice-chair of each panel, as well as one consumer representative and one industry representative.
Two reports by The Lewin Group4
(1999, 2000) have recently examined the HCFA process for making coverage and reimbursement determinations and the effect of that process on the medical device industry. The group concluded that although the recently redesigned national coverage process is an improvement over the previous process, it can still be unpredictable and time-consuming, especially for novel or breakthrough tech-
4The Lewin Group was commissioned by the Advanced Medical Technology Association (AdvaMed) to conduct a study on the current situation of the U.S. medical device and diagnostic industry and produce a series of reports on the following topics: the state of the industry, the Medicare payment process and patient access to technology, technology assessment by public and private payers, and the impact of regulation and market dynamics on innovation.
Page 174
nologies. They further concluded that problems with the Medicare coverage and payment systems can influence provider behavior, impede access to health care technology, and affect the viability of small companies and the direction of innovation in both large and small companies.
Agency for Healthcare Research and Quality
AHRQ5 conducts research on health care outcomes, quality, cost, use, and access. AHRQ (formerly the Agency for Health Care Policy and Research) is a U.S. Public Health Service agency in DHHS. One of AHRQ's missions is to provide evidence-based information that can help health care decision makers—patients and clinicians, health system leaders, purchasers, and policy makers—make more informed decisions and improve the quality of health care services. Although the agency does not mandate practice guidelines or standards for the measurement of quality, AHRQ established the Center for Practice and Technology Assessment (CPTA) in 1997 to serve as a single contact for organizations and individuals searching for comprehensive evidence-based reviews of health conditions, treatments, and technologies. CPTA supports several major programmatic activities, including 12 evidence-based practice centers (EPCs)6 that develop scientific knowledge in health care. CPTA also supports and conducts research grants and evaluation projects that focus on two key areas: (1) methodologies used to conduct systematic, evidence-based reviews and syntheses, such as meta-analysis, cost and cost-effectiveness analyses, and decision analysis, and (2) approaches used to incorporate evidence-based clinical information and recommendations into the health care delivery system. Projects compare alternative strategies to facilitating change in provider behavior and investigate the effects of implementation efforts on health outcomes as well as patient knowledge, behavior, and satisfaction.
The U.S. Preventive Services Task Force (USPSTF) is an independent
5Authorized by the Healthcare Research and Quality Act of 1999 (see
http://www.AHRQ.gov/).
6The 12 EPCs are Blue Cross/Blue Shield Association Technology Evaluation Center, Chicago, Illinois; Duke University, Durham, North Carolina; ECRI, Plymouth Meeting, Pennsylvania; Johns Hopkins University, Baltimore, Maryland; McMaster University, Hamilton, Ontario, Canada; MetaWorks, Inc., Boston, Massachusetts; New England Medical Center, Boston, Massachusetts; Oregon Health Sciences University, Portland, Oregon; Southern California Evidence-Based Practice Center-RAND, Santa Monica, California; Research Triangle Institute and University of North Carolina at Chapel Hill, Chapel Hill, North Carolina; University of California, San Francisco, Stanford University, Stanford, California; and University of Texas Health Sciences Center, San Antonio, Texas.
Page 175
panel of preventive health experts convened by AHRQ who are charged with evaluating the scientific evidence for the effectiveness of clinical preventive services, including screening, and producing age- and risk factor-specific recommendations for these services. Currently, AHRQ provides the technical support for USPSTF through two of its EPCs and oversees implementation of USPSTF recommendations by providing tools for clinicians and health systems to improve the delivery of preventive care.
NIH Office of Medical Applications of Research
At the request of the U.S. Congress, NIH established the Office of Medical Applications of Research in 1977 (Office of Technology Assessment, 1994). Under the auspices of this office, the Consensus Development Program was established with the goal of reaching general agreement on whether a given medical technology is safe and effective. Panels consisting of physicians, consumers, scientists, and others are asked to weigh the available evidence and form a consensus recommendation on the use of a given technology. The first consensus development conference, held in 1977, was on breast cancer screening. Additional panels have since been convened to revisit the breast cancer screening issue, both in general and for specific age groups.
State Mandates
State legislatures often require public and private insurers to cover or at least offer coverage for specific medical interventions or procedures. Breast cancer screening is by far the most frequently mandated coverage among screening tests for cancer. Currently, 46 states mandate some form of coverage for screening mammography (Figure 5-1). In comparison, only 22 states mandate coverage for cervical cancer screening, 18 mandate coverage for prostate cancer screening, and 1 mandates coverage for colorectal cancer screening (Rathore et al., 2000). For most states, coverage is required for annual mammograms for women over age 50, but other details vary considerably among the states, especially with regard to the age and frequency of screening. This is due to differences in the selection and use of screening guidelines by each state (see Table 6-1). A recent study found that 23 states used American Cancer Society (ACS) guidelines only, 18 states used ACS as well as other guidelines, and 3 states used only non-ACS guidelines in determining coverage mandates. No state screening coverage mandate reflected the screening guidelines of USPSTF (Rathore et al., 2000).
Page 176
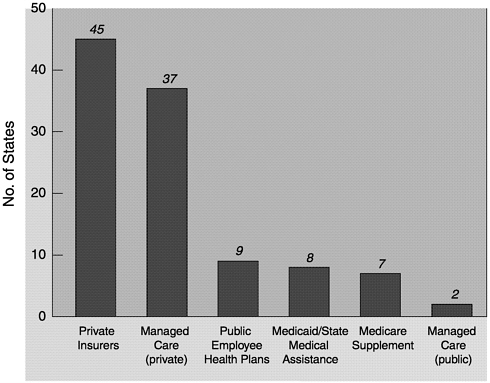
~ enlarge ~
FIGURE 5-1 States requiring specified insurer to provide coverage for screening mammograms (as of March 31, 2000).
Private Insurers and Managed Care
The evaluative science of technology assessment has been expanding in the private sector since the 1980s, and commercial health plans increasingly use these assessments, when they are available, to determine evidence-based coverage decisions (Aubry, 1998). In the past, health care insurers, purchasers, and providers relied on ad hoc opinion by experts in making coverage decisions, but virtually all now have formal technology assessment programs or more structured decision-making processes for determination of coverage (Perry and Thamer, 1999). However, different plans use different criteria to determine coverage for new technologies, so the results of the evaluations can vary significantly and thus coverage is far from uniform.
A number of large insurers have established their own staffed technology assessment divisions, including United HealthCare, CIGNA, Humana, and Aetna. One of the oldest and largest commercial health plan programs for assessment of new medical technologies is the Technology Evaluation Center (TEC) Program of Blue Cross/Blue Shield As-
Page 177
sociation (BCBSA). The TEC program, which relies heavily on scientific criteria and which is overseen by a multidisciplinary medical advisory panel, was initiated in 1985 and expanded in 1993 when BCBSA reached an agreement with Kaiser Permanente to collaborate on technology assessment. The program currently annually produces 35 to 40 formal assessments of both diagnostic and therapeutic technologies. The major focus of the evaluations is on health outcomes for patients, but the technologies are expected to meet five criteria (Aubry, 1998), as follows:
1. The technology must have final approval from the appropriate government regulatory bodies.
2. The scientific evidence must permit a conclusion to be made concerning the effect of the technology on health outcomes.
3. The technology must improve the net health outcome.
4. The technology must be as beneficial as any established alternatives.
5. The improvement must be attainable outside the investigational setting.
TEC and BCBSA do not themselves determine coverage. Rather, the scientific reports produced by TEC are available by subscription and are frequently used by Blue Cross and Blue Shield plans, as well as other health plans, as part of their coverage decision-making process. Once a coverage decision is made, the level of reimbursement may vary significantly. This is due to a multitude of factors, including network coverage, certificate language, and negotiated rates within a community.
Independent Technology Assessment Organizations
The increased interest in evidence-based medicine and technology assessment has also provided incentives to launch private firms that specialize in medical technology assessment. One of the oldest and largest private firms is ECRI, whose Health Technology Assessment Information Service has been designated an EPC by AHQR. Public and private payers, health systems, health care providers, purchasers, government agencies, and other health care constituencies in the United States as well as other countries use ECRI's services and resources, including a technology assessment clearinghouse funded by the World Health Organization. In evaluating emerging technologies, ECRI often uses decision modeling, which is one of the least-used tools for the assessment of new technologies but which is potentially useful when data from clinical trials are sparse.
Other technology assessment organizations include MetaWorks, Inc. (Medford, Massachusetts), which offers meta-analyses of clinical studies,
Page 178
and the Lewin Group (Fairfax, Virginia). In addition, medical professional organizations often develop clinical practice guidelines, and a number have published recommended guidelines for breast cancer screening (see Table 6-1).
CRITERIA BY WHICH NEW TECHNOLOGIES ARE JUDGED
Health care technology assessment entails the systematic evaluation of the properties and effects of medical technologies, both in absolute terms and in comparison with other competing technologies. The process may involve collection of new primary data as well as collection and pooled analysis of existing data, such as meta-analyses.7 To date, health outcomes have been measured primarily in terms of changes in mortality or morbidity, but the analysis may also include measures of clinical safety, efficacy or effectiveness, and cost and economic attributes or effects, as well as the social, legal, ethical, or political effects of a medical technology (Goodman, 1998). In the case of coverage decisions, the major focus is on efficacy and effectiveness. Efficacy is a measure of whether a device or procedure has utility among a group of patients in an ideal setting (e.g., a clinical trial). Effectiveness, in contrast, is a measure of the utility of a device or procedure for individual patients in a realistic clinical setting (e.g., community medical practice). Studies of effectiveness are necessary because new technologies may be associated with a complex learning curve for users and a limited range of applicability that make it difficult to duplicate, in the real world of the clinic, results produced with select populations in carefully controlled trial settings.
RELATIVE MERIT OF DATA FROM STUDIES WITH VARIOUS DESIGNS
Technology assessment can entail the examination of data from several studies with various designs that may differ in their strengths and the validity of their results from which investigators can draw conclusions. As a result, organizations that undertake such assessments have developed scales that can be used to rank the evidence from different studies and the strength of recommendations on the basis of the available data (Boxes 5-1 and 5-2).
7Meta-analysis refers to a group of statistical techniques that combine the results of multiple studies to obtain a quantitative estimate of the overall effect of a particular technology on a defined outcome. This combination may produce a stronger conclusion than that which can be provided by any individual study.
Page 179
BOX 5-1
|
Two basic types of study designs, experimental and observational studies, are used to evaluate technologies (Prorok et al., 1999). The randomized controlled trial is considered by many to be the “gold standard” of experimental designs and is often the method of choice. In a randomized clinical trial, study participants are randomly assigned to either (1) a group that receives the test or treatment in question or (2) a control group that receives a placebo or that does not undergo the procedure being tested. In observational studies, information is collected for groups of individuals who have chosen a particular course of medical intervention or who have a specific condition. They can be prospective, in which data about subsequent events are collected, or retrospective, in which information about past events is collected. Observational studies may include censuses, surveys,
case-control8
studies, or cohort studies.9
Observational
8In a case-control study, women who have a particular disease (cases) are compared with women who have similar characteristics but who do not have the disease (controls) by looking back in time to determine the frequency of a particular intervention (such as screening) in the two groups.
9In a cohort study, researchers identify a group of subjects of interest and follow them over time to see what happens.
Page 180
BOX 5-2
|
designs may be similar in many respects to experimental designs, but their results can be more difficult to interpret and analyze because the comparison groups are not established by randomization and thus the studies are subject to bias. They are often used when a randomized clinical trial is not possible, but the results of observational studies may also complement those of randomized clinical trials. A recent report in the New England Journal of Medicine even suggests that when they are well designed, the results of observation studies and experimental studies may be statistically indistinguishable (Concato et al., 2000).
If a screening test is already in common use, it can be very difficult to randomly assign test subjects to screened or unscreened test groups. This was the initial experience with prostate-specific antigen (PSA) screening for prostate cancer in men (Mandelson et al., 1995), although a random-
Page 181
ized clinical trial10 sponsored by NCI is under way to test the ability of PSA screening to reduce mortality from prostate cancer. A similar situation may be developing for lung cancer screening by spiral computed tomography, a relatively new form of computed tomography that has been shown to detect lung cancers at an earlier stage than would otherwise be possible but that has not yet been tested for its ability to reduce lung cancer mortality (Henschke et al., 1999; Newman, 2000). An alternative would be to examine the disease-specific mortality rate among a group of individuals who have chosen to undergo screening with that among a group of individuals who have foregone screening and who have been selected to match, as closely as possible, the screened population in terms of age, risk, and other characteristics (a case-control study). A second alternative would be to compare the disease-specific mortality rates in different geographic regions that have significantly different rates of voluntary screening (a cohort study). In both cases, there is a possibility that the individuals who volunteer for screening are at increased risk for cancer and may therefore be more likely to benefit from a screening program, which is a selection bias (see Chapter 1). In the case of mammography, for which a positive effect of screening has already been shown, it would be both difficult and unethical to randomize women to be screened by either mammography or a new modality. Rather, studies are likely to entail screening by both modalities, but such a design can make it difficult to assign the effects of screening to a single modality.
SPECIFIC CRITERIA FOR EVALUATION OF SCREENING AND DIAGNOSTIC TECHNOLOGIES
Medical technologies can be divided into two broad categories: therapeutic technologies that cure or prevent a disease and screening and diagnostic technologies that detect an abnormality so that therapy can subsequently be applied. In the latter case, the technology itself does not improve health outcomes, but it may produce a positive effect when combined with an effective therapeutic intervention. Because of this fundamental difference, models of evaluation are not equally applicable to medical technologies in both categories. Therapeutic technologies should be assessed differently than diagnostic and screening technologies, but definition of the assessment criteria and process for the latter category has lagged.
Several investigators have suggested criteria for the evaluation of new diagnostic tests (Lijmer et al., 1999; U.S. Preventive Services Task
10The Prostate, Lung, Colorectal, and Ovarian Cancer Screening Trial will study 148,000 volunteers.
Page 182
BOX 5-3
|
|||||||||||||||||||||||||||||||||||||||
|
1. |
How great is the burden of suffering caused by the condition in terms of: |
||
|
Death |
Discomfort |
||
|
Disease |
Dissatisfaction |
||
|
Disability |
Destitution |
||
|
2. |
How good is the screening test, if one is to be performed, in terms of: |
||
|
Sensitivity |
Cost |
||
|
Specificity |
Safety |
||
|
Simplicity |
Acceptability |
||
|
Labeling effects |
|||
|
3. |
a. For primary prevention, how effective is the intervention? or |
||
|
b. For secondary prevention, if the condition is found, how effective is the ensuing treatment in terms of: Efficacy Patient compliance Early treatment being more effective than later treatment |
|||
SOURCE: Fletcher et al., 1997.
Force, 1996; Fletcher et al., 1996) (Box 5-3). Key criteria include (1) the use of an appropriate spectrum of subjects (i.e., the subjects in the evaluation should resemble the kinds of people for whom the test might be used in practice); (2) the use of consecutively chosen subjects (to avoid any possibility of selection bias); (3) the use of both the new diagnostic test and the “reference standard” (i.e., the test against which the new test is compared) for all subjects and (4) blinded determination of test results (i.e., the results of the reference tests should not be known when the results of the diagnostic test under study are determined). When an appropriate evaluation of a diagnostic test is carried out in this way, the accuracy of the new test (specifically, its sensitivity and its specificity) can be calculated, as can the positive and negative predictive values (see Box 1-1). These are the key outcomes for diagnostic tests (not efficacy and effectiveness).
The criteria for an appropriate evaluation of a screening test include all the criteria for evaluation of a diagnostic test, but because of differences between screening and diagnosis, several features are unique. First, because most diseases are uncommon, studies that evaluate a new test to be used to screen for a disease such as breast cancer require much larger numbers of people than studies that evaluate a new diagnostic test. Sec-
Page 183
ond, the reference standard for a new screening test almost always must include a follow-up observation period to identify any false-negative test results. For example, in breast cancer screening, the reference standard is commonly a combination of mammography, pathological verification of breast cancer (to be certain that the breast cancer diagnoses are correct), and results from 1 year of follow-up to verify the results for subjects with true-negative results and to identify subjects with false-negative results (cancers that were missed on the original examination by both the test under study and the older test, usually mammography). Monitoring over time is an imperfect reference standard, but in screening, it is unrealistic and unethical to subject all participants to histological verification of a negative test result. Because of these two features, evaluation of a new screening test is more difficult than evaluation of a new diagnostic test. However, if a test is to be used for screening, it is critical to evaluate it for that purpose because many good diagnostic tests are poor screening tests.
Traditionally, diagnostic tests should be highly specific, with few false-positive results. For example, it is important that pathological diagnoses of breast cancer not include any false-positive results because such results could lead to unnecessary breast surgery. On the other hand, screening tests should be highly sensitive because they are looking for an uncommon event (for breast cancer, only a few women in 1,000 will have breast cancer at any given time). However, because screening tests are given to such large numbers of people, the numbers and percentages of people suffering false-positive test results is also important, because all these people must have further testing. In addition to added cost and inconvenience, false-positive test results can cause anxiety.
In calculating the sensitivities of screening tests, it is also important to consider the potential for overdiagnosis due to a technology's ability to diagnose early conditions such as ductal carcinoma in situ (DCIS), which may or may not progress to become a lethal cancer (see Chapter 1). If a new technology's sensitivity is calculated by counting all cases of DCIS detected as true-positive results, it will be judged to be superior to an older technology regardless of its effect on a patient's health. The new test will appear to be better because the numerator will include all cancers found, not just those with invasive potential. To deal with this possibility, a method for calculation of a test's sensitivity called the “incidence method” has been developed (Box 5-4).
A key issue in the evaluation of screening is the question of whether a test is efficacious and effective. As pointed out earlier in this chapter and in Chapter 4, neither a diagnostic test nor a screening test alone, in the absence of treatment, can be efficacious or effective. Thus, a particularly vexing challenge is the need to demonstrate that use of a test leads to improved health outcomes in combination with follow-up therapy. Measurement of a test's sensitivity, specificity, and positive and negative pre-
Page 184
BOX 5-4
|
dictive values provides end points that are only imperfect surrogate measures for the most critical measures of patient outcome: disease-specific morbidity and mortality. It is not sufficient to demonstrate that a screening test can be accurately and reliably performed. It must also be shown that testing leads to a change in treatment or management of a patient's condition that results in improved health outcomes or a net benefit to the patient (i.e., the benefits must outweigh the harms). Unfortunately, such trials are often slow to produce data and are expensive because of their size and duration. The first randomized controlled trial of screening mammography enrolled 62,000 women, and it took 8 years to publish the first results from the study showing a reduction in breast cancer mortality. Reaching the first consensus on recommendations for screening mammography required additional studies and demonstration projects that lasted many more years (Lerner, 2001).
Because multiple randomized controlled trials have demonstrated the effectiveness of mammography in combination with follow-up therapy, it may not be necessary to require that each new technology developed for breast cancer screening be evaluated in a new randomized clinical trial. Instead, the technology could be evaluated as outlined above, with the reference standard being a combination of mammography, pathological verification of cancer, and follow-up observation. A possible concern with this approach is that the new test may be detecting different types of breast cancer that respond differently to follow-up treatment and that would therefore lead to a different level of effectiveness if evaluated in combination with treatment.
Page 185
COST-EFFECTIVENESS
Cost-effectiveness analysis provides a framework for comparison of the economic efficiency of different therapies or programs that produce health. Although sometimes misconstrued as a method for assessing the “cost savings” from health interventions, cost-effectiveness analysis is in fact a measure of the relative value—the amount of health produced per dollar spent—of alternative therapies. In a cost-effectiveness ratio, two interventions are compared. The numerator of the ratio represents the difference in cost in dollars of the two alternatives, and the denominator represents the difference in health effects. The denominator is generally measured in years of life or the number of quality-adjusted life years (QALYs) gained (Table 5-2). The QALY represents a measure that combines survival and health-related quality of life. Because of the extensive gains made in the treatment of symptoms associated with chronic diseases such as diabetes and arthritis, a measure that captures both length of life and morbidity is an increasingly important gauge of the effectiveness of a health intervention. In addition, because all health care is not benign, that is, many diagnostic tests and treatments can initiate a series of untoward health outcomes, QALYs provide additional value in being able to capture the negative effects of health-related quality of life. For example, in the case where an antibiotic therapy were to “cure” an infectious disease but, as a side effect, induced permanent hearing loss, QALYs would record the increased life expectancy positively but could also capture the decrement in health-related quality of life associated with deafness. A major difficulty with this approach is objectively defining quality-
|
Strategy |
Treatment Costs |
Effectiveness (Life Expectancy) |
Utility (Quality of Life) |
Utility (Quality-Adjusted Life Expectancy) |
Benefits |
|
Treatment A |
$20,000 |
4.5 years |
0.80 |
3.6 QALYs* |
$4,000 |
|
Treatment B |
$10,000 |
3.5 years |
0.90 |
3.15 QALYs |
$2,000 |
|
Incremental cost-effectiveness ratio = ($20,000 − $10,000) ÷ (4.5 years − 3.5 years) = $10,000 per life-year gained. |
|||||
|
Incremental cost-utility ratio = ($20,000 − $10,000) ÷ (3.6 QALYs − 3.15 QALYs) = $22,222 per QALY gained. |
|||||
|
Incremental cost-benefit ratio = ($20,000 − $10,000) ÷ ($4000 − $2000) = 5 |
|||||
SOURCE: Detsky and Naglie (1990).
Page 186
of-life measures, particularly when comparing or ranking different aspects of life quality. The results of these analyses can be quite different depending on how quality is defined and measured.
Efficacy (performance of a therapy under ideal conditions) or effectiveness (performance of a therapy under real-life conditions) information for cost-effectiveness analyses is generally gathered from clinical trials or observational studies. The strongest measures of effectiveness come from studies in which the experimental design permits direct linkage of the intervention with changes in survival or quality of life. For screening tests, this linkage can be elusive. For example, although it may be possible to demonstrate that a particular screening test detects a disease earlier in the disease process than another test does, that information alone does not provide adequate information about the effectiveness of the test in either prolonging life or improving its quality. The identification and treatment of tumors at an earlier point in the progression of the disease by screening does not guarantee that the rate of tumor progression will be altered or that the ultimate outcome will be changed. (See the discussion of lead-time and length biases in Chapter 1.) Furthermore, a screening test may detect lesions that would not cause death but would precipitate potentially harmful (or costly) medical follow-up (overdiagnosis) (see Chapter 1).
Cost-effectiveness analyses of screening mammography for the detection of breast cancer have relied upon a relatively extensive literature that has demonstrated that early detection by mammography can extend survival for certain groups of women. Most studies have found that the cost per years of life gained for screening mammography falls within a generally accepted range ($50,000 or less/QALY) (Gold et al., 1996), although the cost-effectiveness varies depending on age, screening interval, and the assumed benefit of screening (percent decrease in breast cancer mortality) (Brown and Fintor, 1993; Rosenquist and Lindfors, 1998).
Recently, questions have been raised about the potential cost-effectiveness of full-field digital mammography (FFDM) compared with that of film-screen mammography (FSM). The cost of the new machines will be significantly greater (General Electric FFDM machines cost ∼$450,000 per unit, whereas FSM machines cost ∼$70,000 per unit), but to date, the sensitivity and specificity of FFDM have not been shown to be vastly improved over those of FSM. A simulation model for assessing the cost-effectiveness of breast cancer diagnosis in the United States suggests that an increased cost of $20 per digital mammogram could be cost-effective and could produce an overall cost savings, even if the positive predictive value (cases of breast cancer accurately detected) increased by as little as 2 percent, because of the reduced numbers of unnecessary follow-up biopsies (Nields and Galaty, 1998). The investigators noted, however, that
Page 187
the cost-effectiveness of digital mammography as a screening tool is more difficult to model and may require prospective, randomized trials.
A number of studies have also compared various imaging and biopsy techniques to determine the most cost-effective diagnostic modality for women whose mammograms suggest a suspicious lesion. For example, core-needle biopsy and magnetic resonance imaging (MRI) may be cost-effective diagnostic alternatives to open biopsy (Doyle et al., 1995; Hillner et al., 1996; Hrung et al., 1999). However, the model used in the MRI study was extremely sensitive to changes in estimates of the sensitivity and specificity of MRI, which can be quite variable depending on the patient population, the type of imaging technique used, and the diagnostic criteria used (Hrung et al., 1999b). For palpable lesions, fine-needle aspiration and ultrasonography may also be cost-effective alternatives to open biopsy for certain patients (Vetto et al., 1996; Rubin et al., 1997).
Data linking the newer breast cancer detection technologies to reduced breast cancer mortality rates for women are lacking. To conduct a cost-effectiveness analysis of any of the technologies examined in this report, investigators would need to estimate the level of effectiveness of a technology in extending the length of life or would need to estimate the improvements in health-related quality of life that would accompany use of the technology. Cost-effectiveness analyses that are done under speculative circumstances such as these are referred as “what-if” analyses (Siegel et al., 1996). For example, a recent computer modeling study suggests that MRI screening for young women at high risk (on the basis of germ-line mutations or a strong family history) may be cost-effective (Plevritis, 2000a). Computer models that simulate a variety of screening protocols and trial results could perhaps also be helpful in selecting the most promising trial design needed for evaluation of new detection technologies by inferring long-term outcomes from short-term end points (Plevritis, 2000b). This could potentially lead to a reduction in both the duration and the cost of screening trials. Although from a policy perspective such models may provide useful information about the potential efficacy of particular medical interventions, from a coverage perspective they are inadequate, since they are not based on a proven clinical effect from experimental trials.
Cost-effectiveness analysis is most straightforward when an alternative technology gives the same or a better result but at a lower cost. Unfortunately, this is rarely the case. Thus, the issue becomes a question of how much better the outcome is and how much one should spend to get that outcome. In the absence of societal agreement on how much of the gross national product should be spent on health care, it will be difficult to reach a consensus on the use of cost-effectiveness analysis in making coverage decisions. Because the United States does not have a global budget for medical care (in contrast to the situations in Canada and the
Page 188
United Kingdom), the pressure to decide what should be prioritized for payment with a defined pool of money is lacking, and there is an aversion on the part of policy makers, clinicians, and the public to initiate discussions on such topics.
Physicians, manufacturers, and patients have often opposed considerations of cost-effectiveness in technology assessment (Braslow et al., 1998), but their reasons may differ. Although physicians and patients are to some degree aligned in wanting “the best” no matter what the cost, manufacturers are more influenced by their need to sell their products in the marketplace. FDA and HCFA are not authorized to review the cost-effectiveness of medical devices, and at present, neither HCFA nor private insurers are explicitly using cost-effectiveness analyses in their coverage decisions. However, cost-effectiveness analyses have been performed for some technologies in recent years by private organizations such as the TEC program of BCBSA. This information is supplemental to rather than an integral part of the clinical TEC assessment.
As greater consensus is achieved regarding the best way to incorporate costs into the coverage decision-making process, cost-effectiveness analyses of competing imaging technologies may become increasingly important. This may be particularly true in diagnostic imaging, in which alternative tests may be available for a given condition. Cost-effectiveness is not just an issue of the absolute cost of a technology but is a measure of how that cost and all downstream costs compare to the current standard of care. If a new test is more costly than an established test but detects disease at an earlier stage, when treatment is more effective or treatment costs are substantially lower, the cost-effectiveness ratio of the diagnostic test will be more favorable. What appears initially to be an expensive technology may become a more attractive coverage option for insurers.
LIMITATIONS OF TECHNOLOGY ASSESSMENT PROCESS
The technology assessment process is complicated by the fact that technology development is often incremental and ongoing. If new technologies came out of the initial research and development phase in complete and final form, technology assessment might be relatively straightforward because new technologies could be evaluated one by one, with the good ones accepted and the poor ones rejected. However, most technologies that ultimately achieve widespread use go through a long period of development, variation, assessment of what they are good for and what they are not good for, and discovery of how to use them effectively (Gelijns and Rosenberg, 1999). During this process, there is a considerable amount of two-way interaction between research and development on the one side and actual experience with clinical use on the other. Physicians who are using the device in practice provide valuable feedback,
Page 189
including aspects that may not have been considered by the manufacturers. This information is different from the information about the efficacy of the device acquired earlier in the process for FDA approval. Thus, the process of technology development and diffusion can be caught in a sort of catch-22 that could potentially prevent technologies from reaching their full potential. Successful diffusion depends on whether a device is covered, but a device may need to be used and improved over time as it diffuses into everyday practice before it is deemed effective enough for general coverage.
Because of this conundrum, the concept of “conditional coverage” has been explored as a potential way to allow new medical technologies to enter the market before a final and definitive yes-or-no decision about coverage is made. Conditional coverage in either the public or private sector refers to limited, temporary coverage under specified conditions to allow collection of data that can be used to determine the value of a technology and to set a definitive coverage policy. Although neither the utility of this form of coverage nor the barriers to its implementation are fully understood, a recent report by the Medical Technology Leadership Forum11 identified the steps that have already been taken in the public and private sectors to facilitate coverage of experimental technologies and ascertained some remaining barriers to broad application of the concept.
Once a technology is adopted into practice, it can be difficult to restrict its use even if no evidence supports its effectiveness (unless direct harm is documented). It has been estimated that only 20 percent of medical technologies in current use have documented evidence of effectiveness (Braslow et al., 1998). However, vested economic interests, disputes over withdrawal of coverage, and political pressures make it difficult to restrict their use. Even the emergence of newer, better, or cheaper technologies does not necessarily lead to the elimination of older technologies from clinical use (Eisenberg, 1999).
In practice, there are few reassessments of “old technologies” that are already disseminated, despite arguments that this type of activity is greatly needed (Banta and Thacker, 1990). In theory, any technology, whether disseminated or not, could be subject to assessment and reassess-
11The Medical Technology Leadership Forum is a nonprofit, educational enterprise supported by members representing the broad range of leaders in the medical community. It has engaged in a series of explorations of issues relating to evidence of value for medical technologies, and in July 1999, it convened a summit on conditional coverage of new medical technologies. A panel of experts representing both the public and the private sectors included individuals from NIH, FDA, AHQR, HCFA, academia, the clinical research community, and those who provide and pay for medical services.
Page 190
ment based on the best available and current scientific data. However, current efforts in technology assessment have focused on new and emerging technologies before they have been disseminated. This is especially true of higher-cost technologies, which also tend to be more controversial and create more pressure for both an assessment and a formal coverage decision. Greater emphasis on technology assessment for both developing and disseminated technologies will require commitment and resources from both the public and the private sectors, but it could make the system more efficient and effective in the long run.
Some of the major challenges to developing and implementing a conditional coverage program include defining an evidence threshold at which new technologies would be considered eligible for conditional coverage, setting guidelines for the timeliness of conditional and subsequent definitive coverage decisions, and assigning financial responsibilities for the process. The criteria used to qualify technologies for definitive coverage decisions may serve as a model for the development of less stringent criteria for conditional coverage. Different decision paradigms would most likely need to be developed for different types of technologies (e.g., drugs versus devices) and would reflect the same issues discussed above with regard to definitive coverage decisions. For example, in the case of cancer screening, the evidence needed for a definitive coverage decision would ideally include reduced disease-specific mortality and morbidity rather than simply the indirect measures of sensitivity, specificity, and so forth, but it can take years to accumulate these data, so the time lines for conditional coverage may be longer.
Local Medicare coverage can be another way of allowing technologies to diffuse slowly, but this approach has its own complexities (Strongin, 1998). In this case, a national coverage decision could be made at a later date on the basis of an assessment of the results obtained in the areas where local coverage was previously approved. However, a negative decision at the national level could then be problematic in the areas in which the technology was formerly covered. Furthermore, because the local decision-making process is not standardized, it can be confusing and frustrating for both patients and providers. It could also raise the question of fairness by those not covered in the initial study area.
NCI BREAST CANCER SURVEILLANCE PROGRAM TO MEASURE EFFECTIVENESS
Technology assessments are perhaps most useful when they reflect everyday medical practice rather than just the experience of the technology developers in controlled environments. The benefit of medical technologies as predicted from controlled clinical trials may not be realized in general clinical practice because there are more variations in both the
Page 191
target population and the way in which the technology is used. Who should do or fund these effectiveness studies, however? What are the incentives? Once a technology has been approved for coverage, a company has very little incentive to carry out large, expensive surveillance studies to assess the effectiveness of disseminated products. In the case of cancer screening technologies, the National Cancer Institute (NCI) has made some effort to fill this gap, in part because of a congressional mandate.
The Applied Research Program of NCI supports research to examine the dissemination of cancer screening technologies, to understand factors that influence that dissemination, and to assess the accuracy of screening at the population level. In addition to extramural research activities, research programs have focused on the evaluation and development of methods and national database resources. The Breast Cancer Surveillance Consortium12 (BCSC) in particular was established to monitor the effectiveness and impact of breast cancer screening programs and to address issues that can be adequately examined only with a very large sample drawn from diverse geographic and practice settings.
NCI began pilot studies in 1990 to appraise the feasibility of creating a breast cancer surveillance system to determine if screening in community practice resulted in breast cancer mortality rate reductions comparable to those demonstrated in clinical trials. With a mandate from the Mammography Quality Standards Act (MQSA) to create such systems, NCI established BCSC in 1994 to evaluate population-based screening mammography in the United States.13
The three major objectives of the Surveillance Consortium are to:
-
the United States through an assessment of the accuracy, cost, and quality of screening programs and the relation of these practices to changes in breast cancer mortality or other shorter-term outcomes, such as stage at diagnosis or survival.
-
examine issues such as regional and health care system differences in the provision of screening services and subsequent diagnostic evaluations.
-
research that can improve understanding of breast cancer etiology and prognosis. The intent is to collect a core set of pathological data on established prognostic indicators and to provide the capability to examine the prognostic potential of other, more investigational indicators.
12http://www-dccps.ims.nci.nih.gov/ARP/BCSC.
13BCSC also works with the International Breast Cancer Screening Network.
Page 192
The consortium's database contains information on more than 3 million screening mammogram examinations and more than 24,000 breast cancer cases contributed by eight medical centers across the nation. The consortium has made an effort to collect data on women across a wide range of ages and from various racial or ethnic groups.14 The first major effort of the consortium was to create a standard set of carefully defined variables to facilitate pooling of data with sample sizes sufficient to examine various factors in subgroups for which the number of cancers in any one study is relatively low, such as younger women, women with a family history of breast cancer, or some ethnic or racial groups.
In addition to its intended purpose of evaluating population-based screening mammography in the United States, the database serves as a resource for future research. For example, BCSC studies are examining the hypothesis that the accuracy of screening mammography varies by biological characteristics, the stage of the breast tumor, and the rate of growth of the breast tumor. Furthermore, the BCSC database will provide information on demographic characteristics, risk factors, clinical characteristics, and treatments for women who subsequently develop breast cancer. It will also provide data on a large population-based sample of women at high risk for breast cancer, including those with a family history of breast cancer or benign breast disease. Therefore, this resource may be particularly useful for the identification of patients relevant for research into the population prevalence of genetic and other biological markers for breast cancer risk and prognosis and research into the potential associations of these markers with other known breast cancer risk factors. Data from BCSC will provide estimates of the prevalence of subsequent diagnostic follow-up and information about means of improving the communication of risks and benefits of screening. The mammography registry may also serve as a resource for intervention trials to study ways to improve compliance with recommendations for screening mammography.
REIMBURSEMENT
Reimbursement rates can vary greatly, depending on the location, health insurance carrier, and other factors, even if coverage decisions are
14The age distributions of women currently receiving mammography within the database are 8%, 31%, 26%, 19%, and 16% for women ages less than 40, 40 to 49, 50 to 59, 60 to 69, and 70 and older, respectively. The racial makeup of the study population is as follows: white, 67 percent; Hispanic, 7 percent; African-American, 4 percent; Asian and Pacific Islanders, 2 percent; and American Indians, 2 percent. The remaining 18 percent were categorized as “Other/Unknown.”
Page 193
relatively uniform. Reimbursement by Medicare for physician services is determined by three factors: practice expenses, physician work, and professional liability (reviewed by Farria and Feig [2000]). Each component is assigned a numerical value (a relative value unit [RVU]) that represents its relative contribution to the expense incurred in delivering that service. The practice expense component (∼41 percent of the total) was initially based on historical Medicare charge data, but a new method based on actual practice expense survey data (derived from 1995 to 1997) is being phased in.
The new system also differentiates the various facilities in which a service can be performed. (Under this new system, the RVU for diagnostic mammography is expected to decrease [Farria and Feig, 2000]). The physician work component (∼54 percent of the total) is determined by several factors, including the time required to perform the service, mental effort and judgment, the technical skill required, and stress due to the potential risk to the patient. The malpractice component is based on Medicare procedure charge data from 1991 and data from the American Medical Association, but in the future this component will also be based on actual resource information rather than historical data.
Screening mammography is unique in that it is not reimbursed using RVUs, but rather by a special statutory rule (Farria and Feig, 2000). The payment rate, which is updated annually, is split between a technical fee (68 percent) and a professional fee (32 percent).
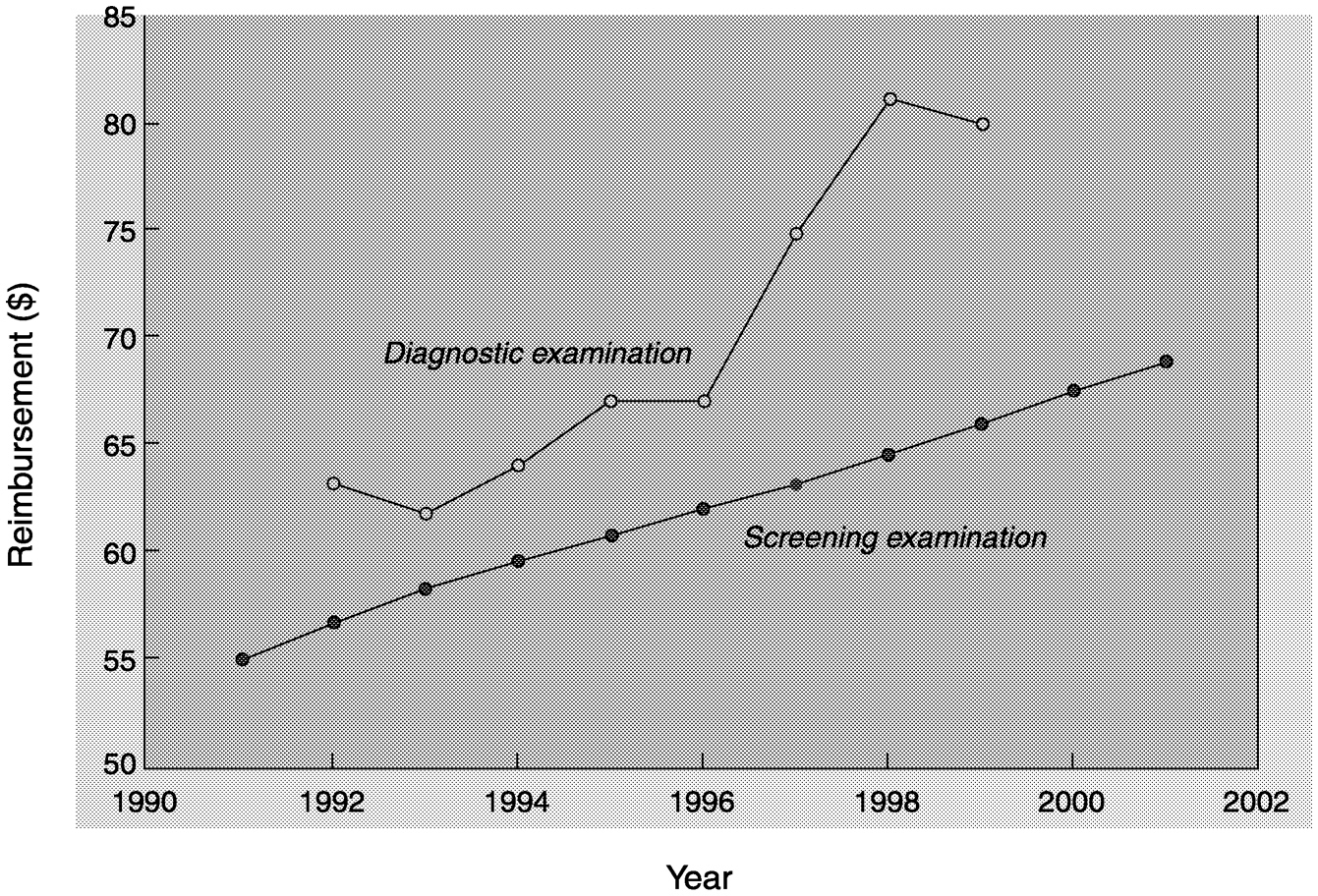
In the private sector the payment rate for mammography varies from $42 to $150, but it most commonly falls in the $60 to $70 range (Farria and Feig, 2000). For Medicare reimbursement, the 1999 cap was $66.22 for screening mammography (Figure 5-2). Radiologists have argued that the reimbursement for mammography is too low for the time, effort, and interpretive skill that it requires compared with that required for other imaging procedures (Farria and Feig, 2000; Feig, 2000a,b)15 (Table 5-3). The additional costs associated with new mammography technologies such as computer-assisted detection and digital detection are not included in the reimbursement rates.
MQSA may add additional financial pressures to mammography facilities. MQSA requires all mammography facilities to meet minimum quality standards for equipment, radiologists, physicists, and technologists. Regulations require extensive records of medical audits and outcome analyses, personnel qualifications, and medical reporting. Thus,
15In March 2000, the American College of Radiology formally requested that HCFA consider an increase in the physician work component of the RVU for diagnostic mammography for this reason.
Page 194
~ enlarge ~
FIGURE 5-2 Medicare reimbursement rates for mammography. Filled circles = screening examination; open circle = diagnostic examination. SOURCE: Farria and Feig (2000).
MQSA increases costs to facilities, but it does not mandate reimbursement levels to cover those costs. Inspections to ensure compliance with MQSA cost each facility $1,549 annually, and the average annual cost required to reach compliance with MQSA is $18,000 (Inman, 1998). These fees may be particularly burdensome for smaller, lower-volume centers (Inman, 1998; Eastern Research Group, 1996).
If the reimbursement rates for mammography are in fact artificially low, this could also have a negative effect on how new technologies are compared with the current standard of care with respect to cost-effectiveness. HCFA has recently proposed linking both coverage and reimbursement rates to patient outcomes (Federal Register, 2000). Since the commercial sphere tends to closely follow the actions of Medicare, if these criteria are adopted by HCFA, they may be used by health plans as well.
From a reimbursement perspective, new technologies that replicate current techniques or that make incremental improvements may have a particularly difficult time compared with those that are completely novel or that offer major improvements over technologies that are the current state of the art. A relevant model for this phenomenon is the ThinPrep Pap Test16 for cervical cancer screening. After several years of testing, the ThinPrep Pap Test was shown to have a small positive effect on patient
Page 195
|
Professional |
Technical |
|||
|
Procedure |
RVU |
Payment ($) |
RVU |
Payment ($) |
|
Screening mammographya |
NAa |
21.5 |
NAa |
45.8 |
|
Bilateral diagnostic mammography |
0.90 |
31.30 |
1.40 |
48.60 |
|
Unilateral diagnostic mammography |
0.74 |
25.70 |
1.14 |
39.60 |
|
Breast sonography |
0.71 |
24.70 |
1.13 |
39.20 |
|
MRI, unilateral breast |
2.15 |
74.70 |
19.10 |
663.80 |
|
Stereotactic core breast biopsy |
2.13 |
74.00 |
6.20 |
270.20 |
|
Wire needle localization |
0.73 |
25.40 |
1.40 |
48.60 |
|
Aortogram |
0.71 |
24.70 |
13.67 |
474.80 |
|
Chest radiograph |
0.29 |
10.10 |
0.67 |
23.30 |
|
Foot radiograph |
0.21 |
7.30 |
0.53 |
18.40 |
|
MRI of brain |
1.96 |
68.10 |
12.15 |
422.00 |
|
CT of abdomen with contrast |
1.68 |
58.30 |
7.42 |
257.70 |
|
CT of brain without contrast |
1.13 |
39.20 |
5.13 |
178.20 |
|
Three-phase bone scan |
1.35 |
49.60 |
5.18 |
179.90 |
|
Barium enema |
0.92 |
32.00 |
1.86 |
64.60 |
|
Transvaginal ultrasound |
0.92 |
32.00 |
1.66 |
57.70 |
aScreening mammography services for Medicare beneficiaries are not reimbursed by using RVUs but are reimbursed under a special statutory rule (Farria and Feig, 2000).
Page 196
outcomes compared with standard Pap smears. However, after health plans had agreed to cover it, the level of reimbursement was sometimes half of what the company was requesting. Meanwhile, several other related technologies, such as PapNet,17 were not able to demonstrate significantly better outcomes, and some are no longer on the market (Brown and Garber, 1999; Hutchinson et al., 2000).
SUMMARY
Developers of new technologies have major hurdles to clear in seeking coverage and reimbursement. Payers are increasingly looking for evidence of improved patient outcomes in making coverage decisions. That is, if improved outcomes cannot be demonstrated, then coverage may be denied or the level of reimbursement may be low. Given the immense and growing expenditures for medical care, more efforts to incorporate cost-effectiveness analysis as an aid to decision making may also be undertaken in the future. Many new technologies are only marginally better than the current standard of care, but they are also substantially more costly. These are the technologies for which coverage decisions will be the most difficult. In the absence of an ulimited budget for health care, decision makers in the health care industry may be unlikely to endorse spending on technical innovations unless a new device or test offers a real opportunity to lessen the disease burden by reducing morbidity and mortality.
The uncertainty of coverage and reimbursement decisions can have an indirect effect on technology development. Expectations that research firms and investors have about the market for new technologies affect the projects they choose to pursue. Uncertainty about the scientific outcome of research is inevitable, but uncertainty about the market profitability also results from the unpredictability of coverage decisions by public and private health insurers. HCFA coverage policy has been changing and evolving in recent years, adding to the high-risk atmosphere for device developers who face uncertainties about the specific coverage criteria that will be applied to devices. As in the case of FDA approval (discussed in
16With the ThinPrep Pap Test, the physician collects the cervical cell sample in the traditional manner, but rather than smearing it directly onto a glass slide, the collection device is rinsed in a vial of preservative solution. The cervical cell sample is then dispersed and filtered to reduce the levels of blood, mucus, and inflammation before applying a thin layer of the cervical cells to a glass slide. The slide is evaluated for cellular abnormalities by a cytologist, as usual.
17PapNet is a computer-assisted detection system for cervical cancer screening. It was developed to double check Pap smears that were deemed normal by a cytologist.
Page 197
Chapter 4), the dominant model for HCFA coverage decisions is based on drug development, which is not always applicable to the medical device industry.
Unfortunately, the process of technology development, evaluation, and adoption can also be quite slow and is generally iterative. That is, most technologies that ultimately achieve widespread use go through successive stages of development, variation, and appraisal of actual experience in the marketplace. For screening technologies in particular, proving a reduction in disease-specific mortality and morbidity due to screening is a long and difficult process, requiring large study populations and extended periods of time. It took more than 10 years from the start of the first randomized clinical trial before consensus was reached that mammography actually decreased breast cancer mortality as a result of early detection. For this reason, most new breast cancer detection technologies have been or are being evaluated by diagnostic studies rather than screening studies. However, once a technology has FDA approval for diagnostic use, manufacturers are often likely to advocate use of the technology for screening purposes.
One potential way to avoid this situation would be to use a conditional coverage policy that would provide a mechanism to bring new screening technologies into the clinic, but only in the context of clinical trials for assessment of the clinical outcome. When potential screening devices meet basic standards for safety and accuracy (sensitivity and specificity), FDA, NCI, HCFA, and private insurers should coordinate the oversight and support of clinical trials to assess patient outcomes through approval of an investigational device exemption from FDA and conditional coverage from the insurers. HCFA and private insurers would provide conditional coverage for use of the technologies in approved clinical trials, whereas the technology sponsors and NCI could cover additional costs attributable to the study design. Data review at appropriate intervals by all participants would determine whether the technology is sufficiently effective to obtain approval from FDA and to change the conditional coverage status to approved coverage for the general population (for those deemed sufficiently effective).
Such a mechanism would not only prevent new technologies approved for diagnostic use from being widely adopted as screening tools before their effectiveness for screening is proven, but it would also make it easier for the technology sponsors to conduct the clinical trials needed to gather the necessary data on outcomes. Data on diagnostic sensitivity and specificity are simply not adequate for assessment of the potential screening value of either newer technologies or technical improvements to established technologies. However, screening trials that could evaluate the effects of recently introduced breast cancer detection technologies on patient outcomes have not been designed thus far.
Page 198
When FSM was introduced, FDA approval was not required, and it represented a “void-filling” technology. As a result, new technologies face a much different level of assessment that will likely include comparison with mammography. The adoption process will be complicated in other ways as well. These issues will be revisited in Chapter 6.
































