
Page 241
5
Strategies for Future Research on Disease Mechanisms
NEUROBIOLOGY
Even though the histology of multiple sclerosis (MS) was described in early texts of neurology more than a century ago, the full repertoire of cellular players, and their roles in the disease process (which cells are actors, and which are victims?) are incompletely understood.
Oligodendrocytes, astrocytes, and neurons can, in a sense, all be regarded as the “victims” in multiple sclerosis. It is clear that oligodendrocytes and the myelin sheaths they form are damaged, astrocytes respond by forming a glial scar, and in some cases, axons (which are outgrowths of neurons) degenerate in MS. Although this much is known, additional questions remain, and their answers have important implications for therapy.
Understanding Injury of Neurons
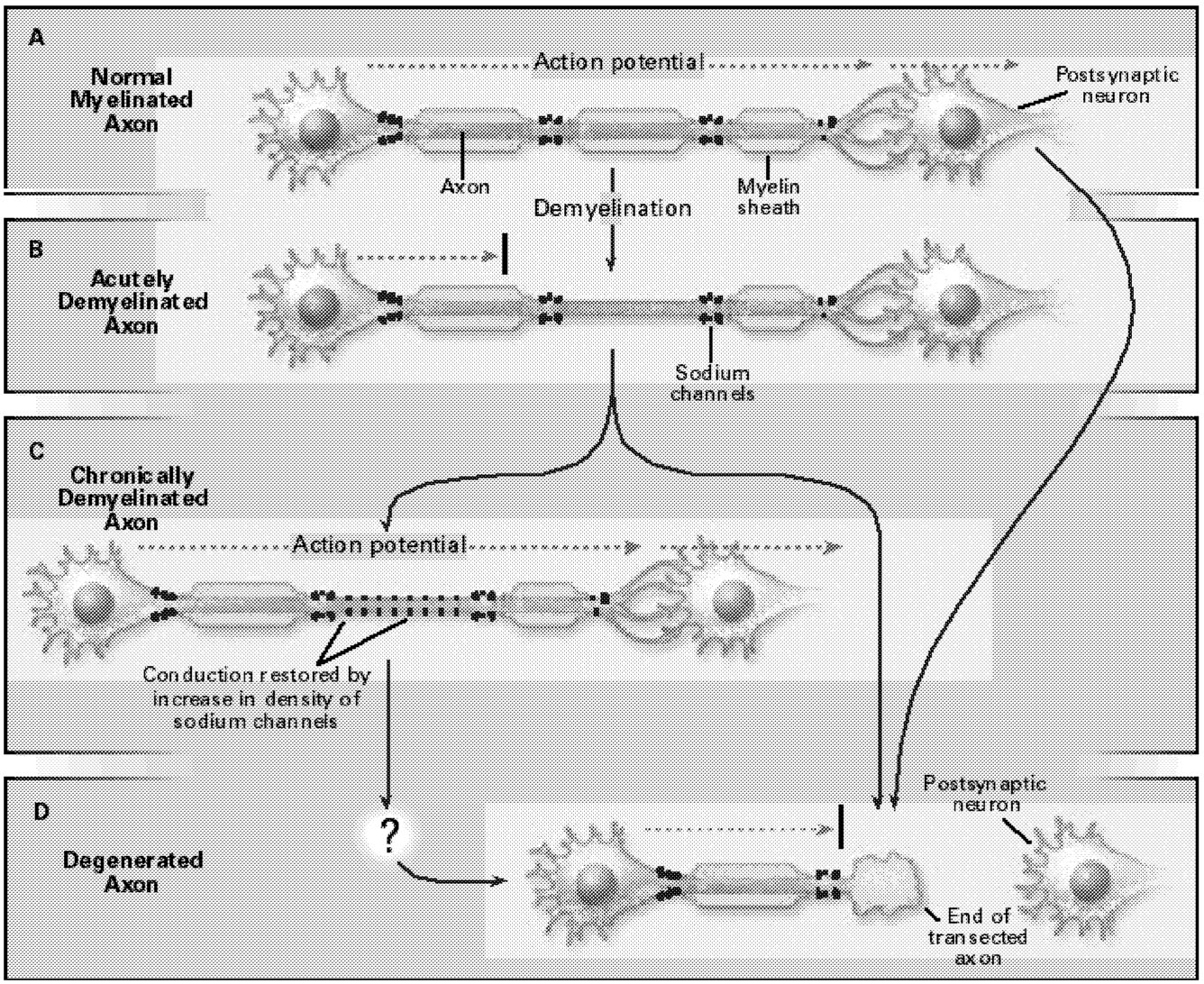
Even the earliest descriptions of multiple sclerosis mentioned axonal degeneration. Recent studies, using contemporary methods such as magnetic resonance imaging (MRI) and confocal microscopy, have more definitively demonstrated the degeneration of axons, both within plaques in multiple sclerosis and in normal-appearing white matter in this disorder (Figure 5.1). It has been speculated that axonal injury contributes to the development of irreversible neurologic deficits as in multiple sclerosis.23,60,66,93,99 If this turns out to be true, it will be important because the prevention of axonal loss might then prevent persistent
Page 242
~ enlarge ~
FIGURE 5.1 Demyelination and axonal degeneration in multiple sclerosis. (A) In a normal myelinated axon, the action potential (dashed arrow) travels, with high velocity and reliability, to the postsynaptic neuron. (B) In acutely demyelinated axons, conduction is blocked (black bar). (C) In some chronically demyelinated axons that acquire a higher-than-normal density of sodium channels, conduction is restored. (D) Axonal degeneration, by contrast, interrupts action potential propagation in a permanent manner. SOURCE: Waxman, SG, 1998.99 Copyright 1998 Massachusetts Medical Society. All rights reserved. Reprinted with permission.
disability. Synapses are not formed onto axons, so that the excitotoxic theory of neuronal death, which almost certainly applies to diseases of gray matter such as stroke, may not play an important role in multiple sclerosis.80
It is not yet known whether axonal injury is a consequence of demyelination, or the immune processes underlying demyelination, or is an independent process. There are some results suggesting that inflammation results in axonal damage in MS.60,93 This provides an important angle for further investigation. Cytokines are likely candidates as mediators of inflammatory-induced axonal damage, and their possible role in MS should be investigated. Another approach is suggested by demonstrations that ion channels and exchangers together form a “final common
Page 243
pathway” that can be modulated by neurotransmitters and that underlies axonal degeneration after various insults.13,25,26,91,92 Neuroprotective interventions can preserve axonal function and integrity after acute insult in two ways. Axons can be protected either by agents that block or modulate the various injurious ion fluxes that occur during this molecular death cascade or by agents that interfere with “downstream” degenerative events, such as activation of calpains and other destructive enzymes.24,32,92
It is well established that axonal transection can trigger dramatic changes in the neuronal cell body, but with a few exceptions, the effects of demyelination on the neuronal cell body have not been examined. The available evidence suggests that demyelination may produce significant molecular changes in the neuronal cell body, including changes in gene activation.9 Since these changes are likely to interfere with neuronal function, they should be studied.
Details of the molecular mechanisms underlying various pathological changes in neurons in MS remain to be elucidated. Rather than reflecting a pessimistic scenario, recognition of neuronal changes in the “demyelinating” diseases presents new therapeutic targets and opportunities. We know a lot about injured neurons, including injured axons, and about how to alter their behavior. Neuronal injury in demyelinating diseases is therefore not necessarily bad news. More information about neuronal dysfunction in MS and related disorders might provide inroads in the search for more effective therapies that will preserve function in people with MS.
How Oligodendrocytes and Myelin Are Injured
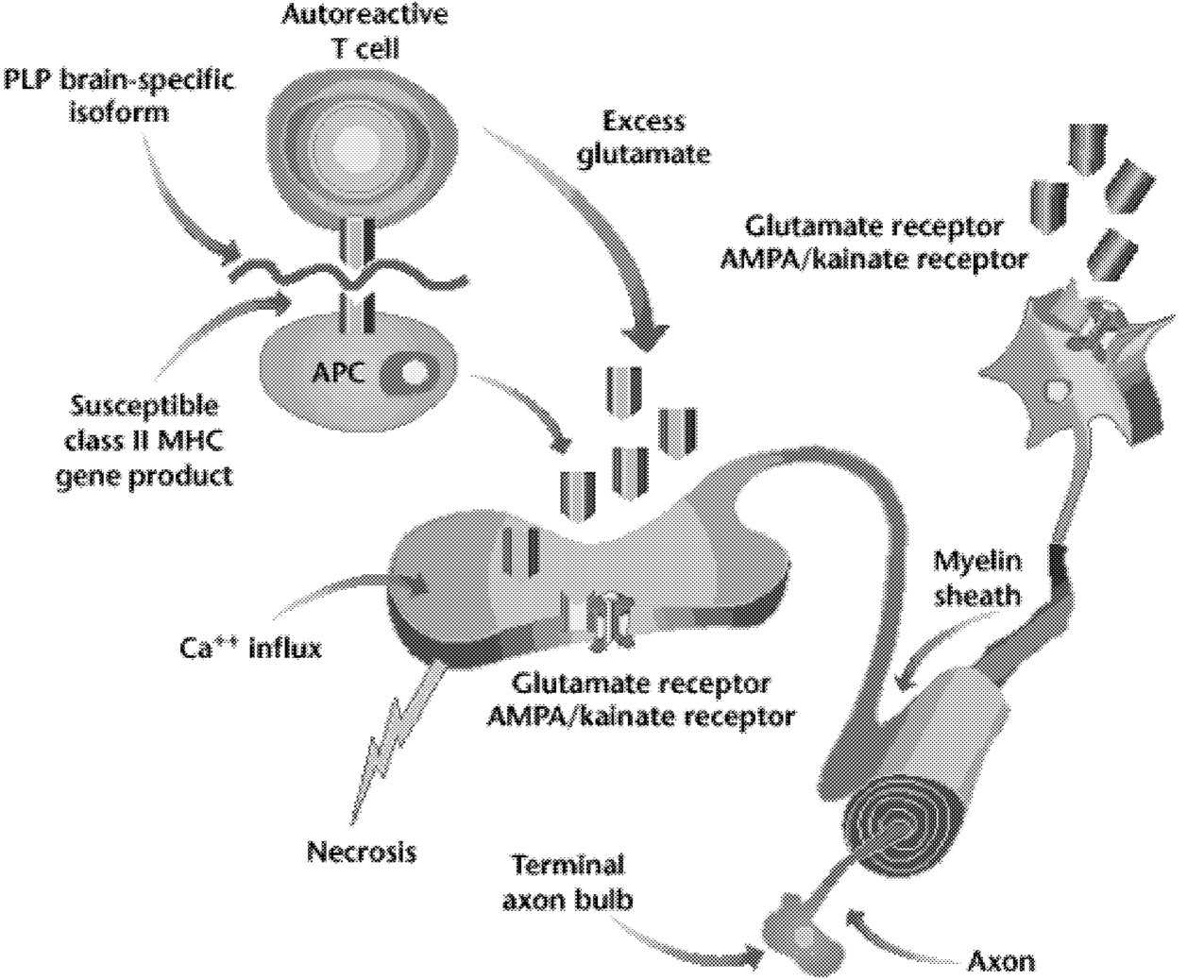
Demyelination is the hallmark of multiple sclerosis, and it is also known that oligodendrocytes degenerate in this disorder. Yet we still do not understand the primary target of MS. Is it the oligodendrocyte or the myelin sheath it forms? Much is known about the “death cascade” in neurons, which leads from initial insults, via a series of molecular steps, to the ultimate death of the cell. Less is known about the degenerative cascade in oligodendrocytes. Recent evidence suggests that excitotoxic mechanisms, possibly involving glutamate acting via AMPA/kainate receptors, may injure oligodendrocytes (Figure 5.2).47 (The AMPA/kainate receptor is one of several glutamate receptors in the brain; it also binds to kainic acid and AMPA.) If the details of mechanisms that injure oligodendrocytes and axons were better understood, it might be possible to protect oligodendrocytes, or their myelin sheaths, so that they are not injured in MS.
There is also the important question of whether oligodendrocyte progenitors (stemlike cells that can give rise to oligodendrocytes with the potential to form new myelin) are present within the adult brain. If they are, can these cells be awakened or activated, so that they will, in fact, form new myelin in multiple sclerosis? Evidence from animal models indicates that it is, in fact, possible to promote remyelination by endogenous cells by exposing oligodendrocytes to
Page 244
various factors. If this could be accomplished in humans, it might be possible to promote recovery of function in people with MS.
Astrocytes: Glial Cells Are More than “Glue”
Astrocytes are star-shaped glial cells that do not form myelin. They have traditionally been viewed only as “scarring” elements in multiple sclerosis. Yet there is evidence that these cells are much more complex than this. Astrocytes are capable of presenting antigens and promoting T-cell proliferation, which indicates that they might play a role in reactivation and regulation of inflammatory process in the brain.2,16,46 They may play an important role in the etiopathogen-
~ enlarge ~
FIGURE 5.2 Possible role of AMPA/kainate receptors on neurons and glia. Activation of T lymphocytes (in this case, a T lymphocyte reactive to the brain-specific loop of the myelin protein, PLP [proteolipid protein]), macrophages, and resident microglia at the site of inflammation in the white matter of the central nervous system causes release of glutamate. The increased extracellular glutamate binds to AMPA/kainate receptors on neurons and on oligodendrocytes. This leads to increased calcium fluxes and death of oligodendrocytes and neurons. NOTE: MHC = major histocompatibility complex. SOURCE: Steinman L, 2000.89 Reprinted with permission.
Page 245
esis of MS. More information is needed about the degree to which astrocytes can serve as antigen-presenting cells and, if so, about the role they play in triggering, driving, or amplifying the immune assault on myelin in MS.
The permeability of the blood-brain barrier reflects, to a major degree, the tightness of the junctions between endothelial cells. Astrocytes are the brain cells that are the nearest neighbors to these cells. Astrocytes send out “feet” that cover much of the surface of the endothelial barrier cells. While astrocytes play a role in brain permeability, this can be modified by many other factors, including a range of inflammatory mediators secreted by leukocytes.1,83 It is important to determine whether it is possible to modulate the leakiness of the blood vessels that occurs in multiple sclerosis by modifying the behavior of astrocytes.
It has also been suggested that astrocytes may play a role in axonal plasticity. Although astrocytes do not generate action potentials and have classically been considered “nonexcitable” cells, they produce sodium channels and insert them in their membranes.7,35 These astrocytic sodium channels have physiological and molecular properties very similar (if not identical) to those of neuronal sodium channels.4,87
The role of astrocytic sodium channels is not known. Interestingly, astrocytes extend finger-like processes that contact sodium channel-rich parts of the axon membrane, for example, at the node of Ranvier. It has been speculated that astrocytes might serve as subsidiary sites for the synthesis of sodium channels, which they donate to nearby axons, thereby helping to restore conduction.7 The last step in this hypothetical sequence, transfer of sodium channels from astrocytes to axons, has not been demonstrated however. Given the critical role that sodium channels play in restoration of conduction in demyelinated axons and the intimate spatial relationship that develops between astrocytes and demyelinated axons, there is a need to understand the role of astrocytic sodium channels, and thus of astrocytes, in MS.
Mechanisms of Recovery
The nervous system exhibits a remarkable degree of plasticity, and it seems likely that reorganization at several levels, ranging from the molecular to the circuit level, contributes to remissions.
Neuronal Plasticity at the Molecular Level
MS is unique among diseases of the brain and spinal cord in that many patients display remissions, in which lost functions (such as vision, ability to walk, tactile sensory function) are regained. Gaining a fuller understanding of the mechanisms underlying remissions is important for several reasons:
First, even if a totally effective immunotherapy were to be developed tomorrow, so that the immune assault on the myelin could be halted, 250,000 patients
Page 246
in the United States alone would be left with multiple sclerosis, and the disease would have left its footprints on their nervous systems, along with resultant neurologic deficits; if remissions could be induced in these patients, this might restore function.
Second, very few neurological diseases exhibit the degree of functional recovery that is seen during remissions in patients with MS. Thus, remissions might provide a unique model that could teach us important lessons about principles underlying recovery of function after various types of assaults on the nervous system. Although it has been demonstrated that, in principle, remyelination of denuded axons within the brain and spinal cord can promote restoration of conduction, this does not seem to provide a basis for remission in MS. There is, in fact, very little remyelination within the core of the plaques that characterize this disorder.
Molecular Plasticity of Demyelinated Axons
Neurons are remarkably dynamic cells, and it has recently become clear that even in the healthy nervous system, they are constantly rebuilding and retuning themselves, so as to meet functional needs. There is a constant background turnover, for example, of ion channels including sodium channels and potassium channels. Following injury to the nervous system, the magnitude and rate of changes in channel deployment appear to be even greater.
It is now well established that conduction block occurs in demyelinated axons, in part, because they do not possess an adequate number of sodium channels within the internodal (previously myelinated but denuded after demyelination) part of their membranes. Following loss of the overlying myelin insulation, electrical current is lost through this sodium channel-poor membrane, and the density of current through sodium channel-rich parts of the axon membrane falls. As a result, the conduction of action potentials is impaired and, in some cases, abolished.98 It has been well demonstrated in a number of animal models that in some chronically demyelinated axons, the denuded axon membrane, however, can acquire a higher-than-normal number of sodium channels, which is sufficient to support conduction.10,20,21,29,65 Experimental methods for demonstrating this sodium channel plasticity require access to demyelinated tissue, and to date, most demonstrations of this phenomenon have been confined to laboratory models of MS. It would be useful, in the future, to develop methods for “sodium channel scanning” that might permit the visualization of sodium channel plasticity in humans. Now that nearly a dozen different genes for sodium channels with distinct molecular structures have been cloned (eight of which are expressed in the nervous system), there is also the opportunity to study their promoter regions and to learn about the molecular control mechanisms that regulate their synthesis.
The edge of the demyelinated plaque is a critical zone in that events there can also contribute to remissions. Even if an adequate density of sodium channels is
Page 247
present in a demyelinated axon, conduction from the normally myelinated region to the demyelinated zone must traverse a critical transition region where the geometry of the nerve fiber changes as the myelin is lost. This produces a phenomenon called “impedance mismatch,” which is well-known to electrical engineers who study wires that have different properties at different points along their course. To overcome impedance mismatch, there must be a mechanism for “impedance matching.” This probably occurs, in some demyelinated fibers, as a result of the production of short myelin segments at the juncture between normally myelinated and demyelinated parts of the axon.97 This is an elegant example of impedance matching in the biological domain, but if it does not occur, conduction will be blocked. Thus, it is important for us to learn how the myelin-forming cell “knows” to form a myelin sheath of the appropriate length and to be able to control this process.
Neuroplasticity at the Cell and Systems Levels
The concept of neural plasticity is varied but usually means a change in the neural response. This change can include either increases or decreases in neural responsivity, due to some modification of the environment, or in the input or output of the neurons involved. After the occurrence of lesions in the central (CNS) or peripheral nervous system (PNS), neurons display plastic changes in response properties that can range from subtle to dramatic. Altered neural responses can occur quickly or progress over many years. For example, after a long-term deafferentation of the nerve supply to the upper limb (several years) or after amputation, neurons in the primary somatosensory cortex that normally responded to the missing arm discharge after stimulation to the face.74,77,78 and 79 This phenomenon reflects a substantial reorganization in the response properties of neocortical neurons distributed over many millimeters (a long distance in cortex) and is not seen after shorter periods of deafferentation. A quicker alteration in neural organization can be seen in patients with syndactyly.64 Before surgery to separate their fingers, maps of primary somatosensory cortex showed an abnormal organization, with the representation of all fingers focused in a restricted cortical site. After surgery, a rapid reorganization of the finger representation occurred so that the digits were more normally distributed across the somatosensory cortex. Whether the changes after a lesion occur rapidly or over long periods of time, neurons acquire the ability to respond to stimuli that were previously ineffective in producing a response.
Recent evidence suggests that neural reorganization might also contribute to recovery from MS relapses. An MRI study of a group of seven MS patients showed that their patterns of neural activation were altered after they had recovered from a single episode of optic neuritis.102 Visual stimulation of the recovered eye activated various areas of the brain that do not normally respond to visual
Page 248
stimulation (lateral temporal and posterior parietal cortices, thalamus, and insulaclaustrum), whereas stimulation of the unaffected eye showed normal activation patterns.
In relation to MS, the question is, How do neurons change their responses in the presence of lesions, and how can the potential for positive neuronal change be harnessed to promote restorative recovery of function? Although the lesions in MS are clearly related primarily to glial cells, it is now accepted that axonal damage accompanies the lesions, which will impact on neural function. Presented below is a synopsis of the more commonly accepted manifestations of neural plasticity and ideas about the mechanisms that may underlie their occurrence.
Changes in the Balance of Excitation and Inhibition. Neuroplastic changes can occur within minutes or can evolve over extended periods of time, from weeks to years. In many instances after an acute lesion, neurons at different levels of the CNS quickly alter their responses so that cells previously responding to a restricted region on one side of the body can expand their responsiveness dramatically to include input from widespread regions, including the contralateral side.13 One idea underlying the rapid changes is that they occur so quickly that only an immediate alteration in the balance of excitation and inhibition can account for the new level of activity. These changes are often referred to as an “unmasking” of responses, due to removal of a suppression or excitation, allowing a new type of activity to emerge. A delicate balance exists between excitatory and inhibitory inputs so that if a component is removed from the equation, a new response capability emerges rapidly. Several dramatic examples of these types of changes appear in reports dating back decades. One classic example is a study in which reversible blockade of the dorsal columns at a level in the spinal cord receiving input from the leg revealed neuronal responses to stimuli of remote parts of the body including the abdomen, which would not normally activate these neurons. After the blockade was removed, the neural responses returned to normal.63 The unmasking of latent responses suggests the existence of dormant or normally suppressed pathways that might be tapped for functional restoration. More recent studies report that subtle and experimental alterations in the balance of excitation and inhibition can produce changes in receptive field organization and in motor behavior. For example, small injections of GABAA (gamma-aminobutyric acid) agonists into motor cortex, which increase inhibition, result in severe incoordination and deficits in motor dexterity of primates, suggesting the necessity of proper inhibition for smooth and coordinated movements.84
Relatively longer-term changes after cortical lesions, which reflect continued alterations in the balance of excitation and inhibition, include hyperexcitability of neurons accompanied by enhancement of receptors known to mediate long term excitation of a subset of glutamate receptors (NMDA [N-methyl-D-aspartate] receptors), and downregulation of GABAA receptors.68 Numerous triggers
Page 249
for functional reorganizations of maps, primarily in the cerebral cortex, have also been reported over the past several decades. Maps can be altered after minor (practice at a skill) to major (removal of an input, such as blindness) changes in system inputs. Over time, the altered properties of neurons continually refine to result in neuronal representations that reflect numerous factors, including the ensuing experiences of the patient or subject.
Changes in Synaptic Strength. At its most elemental, the concept underlying altered synaptic strength is that a stimulus that previously elicited a relatively small neuronal response, now elicits a larger response, or an even smaller response. Several mechanisms for such a change have been proposed. One is activity dependence, in which the output of a given cell depends on both the quantity and the quality of the input. Much of this notion stems from the pioneering work of Hebb,37 who postulated in 1949 that synaptic strength can be increased when neurons receive inputs that are temporally or physically correlated (reviewed in 1998 by Buonomano and Merzenich12). Understanding the mechanisms underlying synaptic strength has been approached experimentally through the phenomena of long-term potentiation (LTP) and long-term depression (LTD). These processes were first identified in the hippocampus but have since been identified in many other loci of the CNS, including the cerebral cortex (reviewed in 1999 by Malenka and Nicoll56). Researchers initially observed that stimulation of afferent fibers in the hippocampus at a specific frequency resulted in a long-lasting enhancement of postsynaptic cell activity. Similarly, LTD can be elicited by a reversal of the neural mechanisms producing LTP. The mechanisms essential to LTP production are too numerous to consider in depth here. Several conditions are considered fundamental to the implementation of LTP; these include post-synaptic activation of NMDA receptors and subsequent inflow of calcium into the cell.
Although the experimental procedures that produce LTP may not be physiological, since the stimuli used are generally of a higher frequency than that normally active in the CNS, many researchers agree that a similar phenomenon may produce plastic changes in neural responses. For example, rats trained in a motor task subsequently display increased field potentials in the motor cortex, which are reminiscent of the electrophysiological enhancements occurring in LTP.81 In a different paradigm, cellular conditioning can be produced in visual and auditory cortex, by pairing a given cell's preferred stimulus with a decrease in activity and a nonpreferred stimulus with an increase in activity by manipulating current injected into the cell. This procedure also results in a change in synaptic strength because the conditioned cell often responds to the nonpreferred stimulus.12
Changes in Morphology of Neurons-Axonal Growth or Sprouting. Many types of CNS lesions result in morphological changes, particularly axonal growth
Page 250
or sprouting. Such changes have been observed after lesions in multiple regions and levels of the CNS, without further special treatment. The possibility of axonal regrowth obviously represents a potentially important mechanism for repair in MS. Although it has long been known that substantial morphologic reorganizations of major fiber tracts occur in young animals in response to central or peripheral manipulations, such dramatic patterns of regrowth do not occur in adults. For nearly half a century, however, evidence has accumulated reporting that various levels of the adult CNS attempt self-repair, which is difficult to correlate with changes in function (see, for example, Chambers et al.14). More recently, studies find that at specific levels of the nervous system, structural changes in response to specific deprivations or lesions occur in adults, which correspond more clearly to alterations in functional responses (see, for example, Kossut and Juliano44). For example, the ability to display morphologic reorganization appears impossible for thalamic axons terminating within the neocortex after a certain critical period, whereas structural reorganizations at the level of the neocortex itself continue to occur into adulthood.95 These observations encouraged researchers to search for properties present in young brains that allow axonal growth and for properties in adult brains that inhibit axonal reorganization. Several potential molecules and genes have been identified on both sides, and their capacity is beginning to be exploited. For example, growth factors are important molecules that appear to encourage axonal growth. The support of these molecules is withdrawn as the CNS develops. More detail about this family of neurotrophic and gliotrophic molecules is given in other sections. On the other side, proteoglycans such as chondroitin sulfate are molecules that appear to block axonal growth and may prevent successful attempts at reorganization.8,18,27
In summary, all the manifestations of neural plasticity described above hold the potential to be harnessed for recovery of function in diseases such as MS. More than likely, they are interdependent, so that the long-term changes, such as those occurring after amputation or deafferentation, may reflect sprouting or axonal growth, whereas the more quickly occurring reorganizations may reflect unmasking of responses due to a change in excitatory and inhibitory balance. Furthermore, structural alterations, such as local sprouting, may result from the changes in synaptic strength that occur during normal or repetitive stimulation. Another point to consider is that some manifestations of plasticity may be functionally maladaptive. For example, after long-term amputations the massive cortical reorganization that occurs may not be helpful to function. Other types of axonal sprouting have also been reported to result in impaired behavior.88 In relation to MS, it is possible that attempts at sprouting or axonal growth may contribute to a pattern of paresthesias or other dysesthesias that occur. On the other hand, it is likely that the redistribution of the finger representation that occurs after surgery to release individual digits in patients with syndactyly, will present as a positive functional change. It is important to distinguish the mechanisms responsible for alterations in functional responses and to harness their most
Page 251
useful components, whether axonal growth or modification of excitation and/or inhibition.
IMMUNOLOGY
MS appears to result from an autoimmune attack against myelin initiated by autoreactive T cells (see Chapter 2). However, there is no formal proof of an immunopathogenesis of MS. The classic experiment to reproduce the disease by transferring autoreactive T cells from affected to unaffected individuals is not ethically permissible in humans.
There are, however, reasons to suspect that MS is not a “pure” T-cell autoimmune disease. In animal models, T cells are able to cause autoimmune inflammation, but they are unable by themselves to create large-scale demyelination, the hallmark of the MS plaque.48 Demyelination, however, is produced by the addition of B-lymphocyte-derived autoantibodies, which then bind to the surface of myelin sheaths or myelin-forming oligodendrocytes. Both T cells and B cells are needed to produce demyelination in these animal models.
To determine how MS lesions are generated, it is essential for future research to sort out the precise roles of T cells, B cells, their target antigens, and other immune mechanisms. Greater understanding of immunopathology is pivotal for identifying new molecular and cellular targets for treatment. One key question is, Which cells are the actual pathogenic effector cells in the disease? Newly identified effector cells become candidates for immunospecific therapy.
Finally, the destruction of axons in MS lesions has gained attention, but how this destruction occurs is unknown. It remains unclear whether axons and the neuron cell bodies from which they extend are damaged by direct attack by CD8+ cytotoxic T cells, by other activated immune cells (microglia or macrophages), by soluble mediators of cytotoxicity, such as those contained in cytotoxic granules (for example, perform and granzymes), or by cytokines and neurotransmitters. Axons and neuronal cell bodies can be induced to express MHC class I and thus, in principle, could be recognized by cytotoxic T cells (CTLs). Neurons can also be damaged by perform released from cytotoxic T cells,57,61 but the potential target autoantigen and the mode of autoimmune attack remain completely unknown. Experimental systems for studying neuronal injury in MS will have to be developed further to answer these questions.
Identification of Pathogenic T Cells
Studies in healthy humans and primates have shown that peripheral blood lymphocytes normally contain numerous T-cell clones that are autoreactive to myelin antigens.58,70,72 More importantly, two studies showed that some, but not all, of these myelin autoreactive T cells isolated from healthy primates have the
Page 252
potential to induce experimental autoimmune encephalomyelitis (EAE) upon transfer.31,62
As a consequence, assays must be developed for the identification and enumeration of pathogenic—as distinguished from merely autoreactive—T cells in the blood of MS patients. One possible approach is the construction of “humanized” animal models to allow monitoring of the pathogenic potential of individual T-cell clones. These could be transgenic rodents with a suitable complement of human immune genes55 or immunodeficient mice carrying human tissue grafts serving as targets for potentially autoaggressive T cells. (Autoreactive T cells recognize an autoantigen but do not necessarily attack the autoantigen-presenting cells; an autoaggressive T cell is autoreactive, and, at the same time, will attack the autoantigen-presenting cells.) Alternatively, tissue culture systems could be developed to assess autoaggressive T-cell potential. Any technique that allows the identification of pathogenic T cells would provide a surrogate marker for assessing therapeutic efficacy.
Direct Visualization of Autoantigen-Specific T Cells
Myelin autoreactive T cells have been identified in the peripheral blood or the cerebrospinal fluid (CSF) of MS patients using microculture techniques.70,72 These techniques allowed the isolation and characterization of T-cell clones reactive against a variety of brain autoantigens. However, comparable clonal yields were found in the blood of MS patients and healthy donors. Unfortunately, cloning assays do not provide a reliable estimate of the absolute number of autoreactive T cells present within a particular blood sample, and they are of limited use in estimating fluctuations of brain-specific T-cell clones in MS patients over time.
Additional methods are required to correlate disease activity with the number of recirculating autoimmune T cells. These may include the use of recombinant MHC class II proteins complexed with autoantigenic peptide epitopes. These complexes resemble the MHC-peptide complexes formed on the surface of an antigen-presenting cell and therefore are bound to a specific T-cell receptor. Myelin basic protein (MBP) specific, class II restricted CD4 T cells, for example, bind recombinant complexes of appropriate MHC class II and peptide to their membrane receptors, and this binding can be visualized by fluorescence staining. The number of antigen-specific T cells can thus be quantified by cytofluorometry.
Recombinant MHC class I-peptide complexes have been used successfully to monitor the dynamics of T cells in microbial infection.3 Complexes of class II with suitable peptide are now available.15 To be of use in MS, complexes have to be tailored to contain both individual class II proteins (for example, those of the DR, DQ, or DP subclasses) and autoantigenic peptides suspected to have a role in the development of disease in a particular patient. Similar approaches, along with
Page 253
intracellular cytokine determination, ELISPOT (Enzyme-Linked ImmunoSPOT, an assay used to enumerate individual cytokine-secreting T cells), and T-cell receptor spectratyping technologies, will be of use in determining the number of T cells responding to altered peptide ligands and predicting their therapeutic effect in an individual.
Identification of Target Autoantigens
For many years, it was assumed that MBP was the dominant, if not the only, myelin autoantigen in EAE and perhaps in MS. Later it became clear that numerous other CNS proteins, myelin or nonmyelin, can serve as targets of T-cell-mediated EAE. The ever-growing list of candidate autoantigens includes proteolipid protein (PLP), heat shock proteins, myelin oligodendrocyte glycoprotein (MOG), myelin-associated oligodendrocyte basic protein (MOBP), and S100b—a calcium-binding protein produced by astrocytes.
However, recent evidence indicates that not all autoantigens are equal: the antigen specificity of T cells may mediate EAE syndromes of different lesion locations, and different degrees of inflammation and clinical severity.6 For example, T cells specific for MBP often mediate highly aggressive EAE in rodents, with lesions dominated by activated macrophages. In contrast, MOG- and S100b-specific T cells often induce huge infiltrates composed mainly of T cells, but with only marginal neurological deficits.6
It will be critically important, therefore, to identify all CNS autoantigens that can act as targets in MS and then to determine if there is a dominant autoantigen in individual patients and whether there are differences in clinical presentation with different principal autoantigens. Different autoantigens might predominate in different people because of individual variation in T-cell repertoire or in genetic makeup, or they might vary with different stages of disease. The significance of different CNS autoantigens is not yet known.
At present, the repertoire of strategies and technologies to determine potential target autoantigens is limited. Phage display technology refers to the insertion of a mutation at an appropriate location within the gene of a virus, resulting in the display of a mutation-encoded peptide on the surface of the virus.82 Large-scale growth of the viral particle results in a library of peptides that are physically linked to the encoded DNA, allowing for identification of a single peptide, whose presence has been increased with the help of viral replication. Phage display libraries have been used to directly test potentially binding target autoantigens of B-cell-derived autoantigens.17 In the case of T cells, which recognize peptide segments embedded in suitable MHC class I or class II products rather than native proteins, modified approaches must be developed. One example is the screening of large-scale libraries of synthetic peptide variants, which implies laborious determination of T-cell responses in multiple microcultures.38 In any
Page 254
event, identification of autoantigens is crucial for designing more powerful and specific immunomodulatory therapies.
Triggers of Autoimmune Attack
Studies of T-cell-mediated models of EAE have shown that brain-specific T cells are able to mediate EAE only when transferred directly after activation. However, the mechanisms that lead to the activation of an autoreactive T-cell population can be diverse. They can include presentation and recognition of the nominal antigen, as well as erroneous recognition of structurally similar microbial protein components (“mimicry”), microbial “superantigens,” or global T-cell mitogens (such as phytohemogglutinin [PHA] and concanavalin A [ConA]).100 T-cell mitogens activate lymphocytes by binding to carbohydrate components of T-cell receptors, which they do independently of antigens. They are thus “polyclonal” activators, because they activate many, if not all, T-cell clones at the same time.
Microbial factors can be critical in triggering an autoimmune attack. Indeed, in one model of transgenic mice with MBP-specific T-cell receptors, EAE developed only in animals exposed to a heavy microbial load, while unexposed mice remained healthy.34 However, the further course of the autoimmune disease involves regulatory controls inherent in the immune system. Evidence for self-regulatory T cells has been identified in several transgenic EAE models. An early study pointed to CD8+ T cells,43 while more recent work showed that CD4+ T cells could prevent spontaneous exacerbations of EAE.69 In future MS research it will be important to identify precisely the self-tolerogenic regulatory T cells (especially of human equivalents), and to determine their mode of action and the location of the regulatory interactions. Regulatory T cells, also known as “suppressor cells,” downregulate ongoing immune responses against self- or foreign antigens. Although the nature of these cells is still incompletely understood, they are important in fine-tuning the immune response. Certain regulatory cells might recognize peptide antigens, whereas others act independently of antigens. The entire field of regulation and suppression is still subject to debate.
Although it is likely that downregulation of autoimmune responses takes place in the immune tissues, the location of T-cell activation, for example by exposure to autoantigen, is less established. The development of local lesions in the CNS means of course that the autoantigen must be presented locally in the CNS. However, additional events, such as export of autoantigen via macrophages or dendritic cells warrant consideration. This possibility has recently received further attention because of the demonstration that dendritic cells preferentially engulf apoptotic parenchymal cells (such as oligodendrocytes), and then “home” to immune organs where they present their antigenic load to specific T cells.90 The expansion of target epitopes, or epitope spreading, observed in some (but not
Page 255
all) models of EAE might involve presentation of autoantigen by dendritic cells. Thus, one hypothesis is that dendritic cells engulf apoptotic brain cells, then migrate out of the CNS into the peripheral immune system (lymph nodes), where they present protein components of these engulfed cells as autoantigens to specific autoreactive T cells. Such spreading of an autoimmune response might cause a relapse or exacerbation of the clinical disease.
Clarifying the Role of B Cells and Antibodies
New emphasis is being placed on the role of B cells in MS. Upon transfer of T cells, most animal models display the inflammatory changes seen in the active MS plaque, but importantly, they do not exhibit large-scale demyelination. In rodent EAE, fully demyelinating, plaque-like lesions have been created by co-transferring autoimmune T cells along with autoantibodies bound to the surface of myelin, or oligodendrocytes, or both. These studies heralded MOG as a prominent target autoantigen for demyelinating autoantibodies (and B lymphocytes, implicitly).48 The pathogenic role of antibodies to MOG was spotlighted by a recent study that found MOG-specific antibodies bound to myelin debris in active lesions in people with MS.30
These findings, along with evidence of antigen-driven clonal expansion of B lymphocytes in the MS brain,76 urge new investigations into the physiological and pathological roles of B cells in inflammatory demyelination. Such investigations could include suitable transgenic experimental models with demyelinating B cells,50 the development of technologies allowing the establishment of antigen-specific B-cell lines, and B-cell receptor grafting techniques, which have been successfully used in studies of T lymphocytes.19 Further, these new approaches would be of help in learning more about the role of autoantigen-presenting B cells in inducing T cells toward a pro-inflammatory (Th1) or anti-inflammatory (Th2) cytokine-secreting phenotype.
Neurotoxic T Cells
As discussed earlier in this chapter, axonal damage is a common feature of the MS lesion and it appears to be responsible for brain atrophy and long-term, irreversible neurological deficits, although the mechanisms leading to neuronal damage are unknown. Two recent reports described a protective effect of glutamate receptor antagonists in preventing destruction of myelin and axons in mouse models of EAE.36,73 These observations are possibly related to the earlier discovery that neuroreactive T cells profoundly interfere with the electrical conductance of axons in optic nerve explant cultures, a process requiring histocompatibility between lymphocytes and CNS tissue.104 In addition, cytotoxic CD8+ T cells are able to kill neurons by release of perform, possibly also a process involving glutamate cytotoxicity.57
Page 256
Clearly, the new field of T-cell neurotoxicity will require much attention. The conditions of antigen presentation by neurons must be better understood, and there are important deficits in our knowledge of the mechanisms by which T cells mediate damage to neurons.
Importantly, it now appears that neurons are by no means just passive targets of immune destruction. It has become apparent that neurons have an essential role in regulating immune reactivity within the CNS. Intact neurons efficiently suppress the induction of immunologic molecules (MHC products, as well as humoral mediators), not only in surrounding glia cells but also in the neurons themselves. Nerve growth factor seems to be one mediator involved in this regulation.67 Conversely, areas with failing neuronal function preferentially attract immune cells and favor local inflammatory responses, as in Alzheimer's disease and other cases of neurodegeneration.
Research into the interactions between neurons and immune cells thus may shed light on the generation of MS lesions and generate new therapeutic targets.
Gene Expression in MS Lesions: Microarrays and Proteomics
Thus far, most explorations of autoimmune interactions have relied either on global, morphological descriptions of CNS lesions or on the study of isolated cell cultures. Both approaches have quite narrow technical limits. More recently, techniques are emerging that allow direct and detailed investigation of the cellular microenvironment in situ. They combine the advantages of in vitro and in vivo approaches.
One example is the isolation of single cells from tissue sections followed by exploration of the expressed genome using microarrays.54 This new technology, also known as gene chip technology, can profile gene expression patterns for thousands of genes simultaneously. Microarrays, especially if combined with biological markers,28 may lead to better understanding of cell-to-cell interactions in MS CNS lesions. Microarrays are useful to identify and quantify genes involved in a response—but the actual function of these activated genes must be verified via functional tests.
Another emerging technology to be used in determining disease-related changes in the CNS and immune organs comes from proteomics. In contrast to a global genetic approach, proteomics uses microtechniques to first separate protein units of a particular tissue (or cell populations) and then identify these molecules by microsequencing methods. Proteomics yields information about protein concentrations and posttranscriptional chemical modifications, neither of which can necessarily be predicted on the basis of mRNA expression via microarrays. Thus, proteomic approaches are the ultimate tool to characterize the expressed gene repertoire.
Page 257
Therapeutic Perspectives
Initial strategies to design immunotherapies were aimed at physically eliminating, or functionally blocking, pathogenic T cells. One such strategy, vaccination, was tested as a possible approach to activate immunoregulatory mechanisms that could suppress the autoimmune T-cell response. Vaccinations with inactivated, “mitigated” autoreactive T-cell lines5 or peptide segments representing their antigen receptors were tested.94 There have also been attempts to “blind-fold” pathogenic T cells, for example, by injecting either peptide antagonists that bind to restricting MHC loci or soluble MHC class II proteins complexed with autoantigenic peptide.
More recently, researchers have attempted to modify the pathogenic potential of autoreactive T cells by exposing them to altered peptide ligands. Under optimal conditions, they may reeducate the T cells to produce anti-inflammatory cytokines instead of their natural pro-inflammatory products. However, it is not clear whether these T-cell modulatory therapies will work. Although such treatments may benefit some patients, they might aggravate the disease in others. As pointed out earlier, diagnostic tests allowing the exact monitoring of immune status will be indispensable to safe application of these approaches.
A more radical therapy uses brain-specific T cells merely as vehicles to transport therapeutically effective transgenes through the endothelial blood-tissue barrier into the brain. For example, recombinant plasmid DNA and retroviruses have been used to express genes for neurotrophins or anti-inflammatory cytokines.45,59,85 This approach must be tailored for each individual patient because it requires the isolation and engineering of T-cell clones from that patient.
Cytokines in MS
Cytokines are intercellular signaling proteins produced by cells of the immune system and CNS. They are involved in various aspects of disease processes, including altering the permeability of blood-brain barrier, recruitment of inflammatory cells, cytotoxicity, but also stimulating repair processes.
Blood-Brain Barrier Permeability
MS is characterized by significant infiltration of leukocytes into the CNS. Normally, the CNS is protected from leukocyte infiltration by the blood-brain barrier. This barrier consists of a layer of specialized endothelial cells, that are interconnected by tight junctions and foot-like projections of astrocytes, and it is responsible for the brain's “immunoprivileged” status. Only some highly activated lymphocytes pass through the resting blood-brain barrier. Among these are the freshly activated MBP-specific T cells that initiate the pathological processes culminating in EAE.101
Page 258
After T cells reach and recognize their CNS targets, they become activated, thereby triggering a complex series of inflammatory events that enhance the permeability of the blood-brain barrier. The mediators of this complex process are pro-inflammatory cytokines (for example, interleukin-1 [IL-1] and tumor necrosis factor-α [TNF-α]) and chemokines produced and released locally within the CNS by activated glia cells (also by endothelial cells).39 As a consequence of their release, endothelial cells acquire a new set of cell adhesion molecules, to which recirculating lymphocytes and monocytes adhere in order to gain access to the CNS. One adhesion molecule is VCAM-1, which binds to the receptor, VLA-4, that is expressed on activated lymphocytes and serves as a signal to recruit activated lymphocytes into the brain. In EAE, the interaction between endothelial cells and lymphocytes leads to a massive invasion of inflammatory cells into the CNS, a process that results in a general permeabilization of the blood-brain barrier. Leukocyte enzymes, including matrix metalloproteinases and glycosidases, digest the extracellular blood-brain barrier matrix, including the basement membrane, thereby loosening the inter-endothelial junctions.53 The critical importance of adhesion molecules and chemokines has been demonstrated in animal models in which immunization with antibodies against adhesion molecules and chemokines prevented the onset of EAE.41 Other cytokines, such as transforming growth factor-β (TGF-β) and IL-10, might be involved in reversing the process of blood-brain barrier permeability.
It is well established that the blood-brain barrier becomes more permeable during inflammatory attacks in MS, yet the cytokines, chemokines, and other molecular mechanisms underlying enhanced permeability are just beginning to be defined.33 Greater understanding of key events that initiate blood-brain barrier permeability and trafficking of immune cells into the CNS may lead to means for preventing the development of MS lesions or controlling the degree of inflammation once an MS attack is in progress. Fresh experimental and clinical studies are required to identify the best therapeutic targets.
Cell-Cell Interactions in Inflammation
In the active MS lesion, cytokines appear to be responsible for recruitment of inflammatory cells, astrocytes, and microglia into the inflammatory process by causing their migration, proliferation, and activation. Production of TNF-α, IL-1, and interferon-γ (IFN-γ) for instance, has been implicated in the production of EAE in rodents and in the progression of MS. Other cytokines that have been demonstrated to be elevated in MS patients also possess anti-inflammatory properties. These include TGF-β, IL-6, IL-4 and IL-10. In EAE models, administration of IL-4, IL-10, TGF-β, or IL-13 can ameliorate clinical and histological disease by downregulating aspects of the inflammatory response.
Page 259
Cytotoxicity
Cytokine signaling contributes to cell dysfunction and death by inducing antibody and complement production, direct activation of cell death signaling pathways, and induction of antigen-presenting class II MHC molecules on both glial and inflammatory cells. Cytokine signaling also contributes to the generation of toxic intermediaries such as hydrogen peroxide and nitric oxide in macrophages, glia, and astrocytes. Direct toxicity of cytokines to oligodendroglial cells has not been convincingly demonstrated.
Induction of Restorative Processes
Paradoxically, several cytokines can also induce what can be considered protective, restorative, or reparative processes in the brain. Several cytokines, including IL-6, TGF-β, and ciliary neurotrophic factor (CNTF), both protect and support the proliferation of oligodendroglia. Pro-inflammatory cytokines, such as IL-1, perpetuate lesion production, yet they also induce the production of counter-regulatory cytokines that dampen the immune response and stimulates the production of neurotrophic factors. For example, TGF-β is a potent chemotactic factor for inflammatory cells, yet it also inhibits aspects of immune function. It is also an important differentiation factor for oligodendrocytes. It is thus vital to understand the precise temporal patterning of cytokine release, interactions, and responses, as well as the nature and localization of cytokine receptors and the specificity of intracellular signaling pathways that they command. The precise roles of various cytokine and cytokine-related molecules in dampening the inflammatory process and initiating restorative mechanisms in the MS plaque is a fertile area for future research.
Cytokine Production as a Systemic Marker of Disease Activity
Cytokine levels and the production of cytokines by circulating peripheral blood cells have reportedly been increased in MS patient sera and CSF. Increased TNF levels have been observed in the prodromal period before an attack, but they do not correlate with disease activity as shown by MRI. Careful, prospective longitudinal profiling of cytokine activity in the periphery may give clues to predict the pattern of disease activity in patients.
Cytokines and Cytokine Antagonists as Therapeutic Agents
The demonstrated utility of beta-interferon in the clinical management of MS is presumably due to its ability to downregulate inflammatory cytokine activity and perhaps to enhance the activity of anti-inflammatory cytokines such as IL-
Page 260
10. Other anticytokine approaches have not yet proven their utility. Agents with promising results in animal models (for example, IFN-γ and anti-TNF, which are effective in EAE models) have not been successful when tested in humans with MS. Many cytokine-based strategies are currently under development. There are currently no cytokine-based strategies other than the use of corticosteroids, which appear to alter the course of individual MS attacks. In the absence of completely effective disease course-modifying agents, the availability of anticytokine therapies with the ability to interrupt the acute disease process would be desirable.
IDENTIFICATION OF INFECTIOUS AGENTS IN MS
Standard and conventional approaches to the isolation of pathogens in MS have failed to find any consistent and convincing result. For this reason, it seems appropriate to pursue new directions with respect to the identification of an etiologic agent. These approaches should pursue cultivation-independent methods primarily involving the identification of genomic information relevant to the pathogen. The methods include polymerase chain reaction (PCR), differential sequence analysis based on subtraction or display, and sequence screening using the host immune response. These new directions are especially attractive since the powerful methods that will be employed in these studies have not yet been applied to investigations of MS tissues in any concerted and organized way.
One method of differential sequence analysis, representational difference analysis, has recently been used successfully to identify a new pathogenic herpesvirus in Kaposi's sarcoma. Representational difference analysis employs PCR to amplify DNA fragments that are present in the diseased tissue, but not in healthy tissue from the same patient. The identification of even a small amount of sequence information may be sufficient to characterize the agent.
In addition, it might be valuable to probe tissues for transcription expression profiles that are associated with particular pathogens. This will involve microchip array approaches.
The use of phage display libraries as a screen for the antigenic target of oligoclonal immunoglobulin G (IgG) is a valuable method, however, there may be problems interpreting these studies. The antigenic targets of the oligoclonal IgG may be merely a result of immunodysregulation rather than relevant from an etiological point of view.
Methods similar to those used to probe the antigenic target of oligoclonal IgG can be used to identify the antigenic targets of T cells that are present in the MS brain. The results are more difficult to interpret in the case of T cells than B cells, since the antigenic targets that are identified in the case of the T-cell target epitopes might have little sequence identity with the actual sequence of the antigen within the MS brain.103 Isolation of peptides from human leukocyte antigen (HLA) class I molecules followed by purification and Edman sequencing or mass
Page 261
spectrometry should also be valuable as a means of identifying cytolytic T-cell epitopes. In some situations, however, the application of these techniques can be time-consuming and problematic because of the complexity and low abundance of the epitopes presented.
IDENTIFICATION OF DISEASE SUBTYPES
Identification of specific biologic or surrogate markers that characterize each of the different disease subtypes will help determine whether the different disease subtypes represent distinct entities or are, instead, a spectrum of single pathogenic disorders. If the latter is true, it will be important to determine what modifies disease expression. The identification of markers of disease phenotype would also permit one to identify these forms prior to full clinical expression and to determine the effectiveness of therapy.
Improved identification of different disease subtypes could be derived from clinical observations, neuroimaging, and genetic or acquired biological markers. The biological markers to be considered would include those related to products of the immune system (immunological) and products of the nervous system (neurobiological). The latter could reflect tissue injury or repair.
Clinical Data Collections
Organized clinics focused on MS have been in existence for more than 25 years. Most have attempted to adopt standardized measures to document neurological status, clinical disease course, and occurrence of clinical relapses. National and international clinical databases have been and continue to be developed (EDMUS in Europe, Costar in Canada). No one uniform database has been adopted, although measures of neurological disability have become standardized. Emerging data from these databases are providing important information on the relation between early clinical patterns of disease and subsequent disease course. The overall scope of existing databases in different regions of the world remains to be documented. Comparative studies would help confirm observations already reported and identify regional differences (see also discussion of data registries in Chapter 6).
Neuroimaging
In the context of disease heterogeneity, one would wish to know whether lesion frequency and topography as defined by current MRI techniques correlate with specific clinical or pathologically defined disease phenotypes. MRI studies indicate that single clinical episodes have markedly different prognoses with regard to recurrent disease, depending on the presence or absence of multifocal
Page 262
lesions. Further data are needed to define the imaging correlates of either human cases of MS or animal demyelinating models with distinct pathologies. Neuroimaging could facilitate the classification of patients with regard to their prognosis and the identification of subgroups of patients that would respond best to different treatment strategies. If patterns of neuropathology revealed by neuroimaging could be linked to biochemical, immunological, or genetic markers, it might become possible to understand the variation among patients in the clinical course of their disease. In general, neuroimaging should play a critical role in bridging the gap between understanding the underlying biological mechanisms of the disease and the resulting damage to the brain and spinal cord.
New methods in neuroimaging are described in a later section.
Biological Markers
One would like to develop markers that reflect or serve as surrogates for the actual biology of the disease process. We do not yet know what the critical biological mediators of the disease process are. One would have to establish whether such markers also serve as surrogates for the clinical disease course.
The interpretation of measures of biological markers during clinical trials can be complex. One could theoretically derive the following during a clinical trial with a new agent (Table 5.1).
Genetic Markers
Specific genetic markers are covered in greater depth in Chapter 2. Genes related to immune function are candidate markers of disease susceptibility or progression. Precedent exists in animals for the importance of genes regulating
|
TABLE 5.1 Hypothetical Results of a Clinical Trial |
||
|
Clinical Response |
Biological Marker |
Implications |
|
Positive |
Correlates with clinical response |
One would still have to determine whether there is a true cause-effect relation, but such a result would encourage seeking more selective agents that act on this marker |
|
Positive |
Does not correlate with clinical response |
No conclusion is possible |
|
Negative |
Altered by therapy response |
Biological marker is not central to the disease |
|
Negative |
No change |
No conclusion |
Page 263
blood-brain barrier permeability and antigen presentation within the CNS. In the EAE model, disease susceptibility and disease course are highly dependent on which mouse strain is studied.
Acquired Markers
One has to consider both what to measure and from which body compartment one can obtain the necessary samples to perform the measure. The site of injury in MS, namely the CNS tissue, is not routinely available for clinical correlative-type studies. The CSF is considered to more closely reflect events that occur in the CNS than does the peripheral blood compartment, but it is also not readily available, especially for serial analyses.
Immunologic Markers
The most accepted postulate regarding MS pathogenesis is that the disease process is initiated by autoreactive T cells that migrate from the systemic compartment into the CNS. The status of the overall immune environment may be a critical factor in determining the extent and persistence of the initial response. As the disease evolves, this initial immune response may expand to involve additional antigens (determinant spreading). The actual effectors of tissue injury could involve a wide array of immune mediators derived from the adaptive (αβ T cells, antibody) and innate (macrophages, NK [natural killer] cells, γδ T cells) immune systems.
Immunologic markers would be relevant in MS with regard to defining a disease-related antigen, that is, identifying the presence of a T-cell or B-cell population in MS patients that reacts with a candidate disease-relevant antigen, and demonstrating ongoing immune activation or altered immune regulation that contributes to autoimmunity.
T-Cell-Related Markers
The molecular events associated with antigen presentation, cell activation, and migration have increasingly been defined. Studies of disease-specific immune responses—the measurement of antigen reactive T cells—are dependent on having a candidate disease-relevant antigen. Most studies continue to focus on myelin antigens, although the same assay techniques could be applied to any such candidate. These assays are considered in terms of whether they require in vitro manipulation of the cells or whether they are based on ex vivo analysis of harvested cells.
In Vitro Manipulation Required. Repeated cycles of stimulation with the selected antigen is the classic technique used to generate and determine the
Page 264
frequency of antigen-specific T cells. At issue is whether the properties of the derived cells, such as their T-cell receptor (TCR) phenotype and cytokine production profiles, are representative of the in vivo situation. Memory T-cell stimulation assays can be done in which memory cells are enriched either by addition of IL-7 or by preselecting cells that have undergone somatic mutation (HPRT mutant T cells).51
Analysis of Harvested Ex Vivo Cells. These techniques remain to be established and standardized. Single-cell PCR techniques are used to sequence the TCR of individual T cells and compare them to the sequence of myelin-reactive T-cell clones derived by in vitro cell culture (as above).
Tetramer technology allows one to assess the direct binding of labeled peptide-MHC constructs to T cells. The MHC tetramer technology is a general, robust and extremely powerful method for the precise and rapid measurement of T cell responses to a broad range of antigens. (MHC tetramers consist of four complexes of an antigenic peptide, beta-2 microglobulin and a biotinylated MHC class I molecule conjugated to streptavidin linked to a fluorescent marker, hence the name tetramer.) The ability to link multiple MHC class I/peptide complexes together allows for identification of specific receptor-bearing T cells by flow cytometry. In addition, investigators can use tetramers to quickly and efficiently purify a specific T-cell population using fluorescent activated cell sorting (FACS).
Other T-cell related markers include assays for antigen presentation, T-cell activation, and T-cell migration.
Antigen Presentation. T-cell activation by antigen is dependent on antigen presentation in context of MHC molecules in the presence of costimulatory molecules (CD40/CD40L; CD80:86/B27:CTLA-4) expressed on the antigen-presenting cell (monocytes, dendritic cells, B cells). Levels of expression of many of these molecules are themselves subject to regulation by cytokines. MHC and costimulatory molecules can be monitored by antibody fluorescence-activated cell sorter (FACS) analysis techniques.
T-Cell Activation. When activated, T cells upregulate an array of surface molecules that contribute to their capacity to proliferate (IL-2 receptor) and migrate (adhesion molecules, chemokine receptors). These cell surface molecules can be assayed by immunostaining and FACS analysis as described in the previous section.
Activated T cells increase the production of soluble molecules (cytokines) that promote communication between cells of the immune system, between immune cells and cells that form the vascular barriers, and between immune cells and cells within specific body compartments. Families of cytokines are identified that promote (pro-inflammatory or Th1—IFNγ and TNF) or inhibit (anti-inflammatory or Th2—IL-4, 5, and 13) the inflammatory response. Antigen-presenting
Page 265
cells also produce cytokines that can regulate immune function (TNF, IL-10, TGF, IL-12, IL-1).
Cytokines can be assayed in terms of either protein or mRNA by a number of methods. The former includes the techniques of intracellular cytokine staining (ICS) and ELISPOT. The latter involve PCR or RNA protection assays. DNA microarray techniques, focused on the immune system, are beginning to be used as a means to survey for a wide array of known and unknown genes. The ideal would be to analyze immune cells directly ex vivo, but the low level of cytokines has often required the use of in vitro activation techniques to amplify the signal. In vitro, one can apply enzyme-linked immunosorbent assay (ELISA) measures to the supernatant in which cells are cultured. Measures of serum cytokine levels do not permit one to determine what cell is the source of the molecule being measured.
Measures of cytokine levels in the CSF have been technically difficult to perform due to the paucity of cells present in MS cases and the low levels that can be detected in the fluid.
T-Cell Migration. This property can be assessed either in functional assays or in terms of participating molecules. In the context of migration of lymphocytes into the CNS, in vitro models of blood-brain barrier have been used. These include assessment of lymphocyte adherence to isolated microvessels or migration through barriers comprised of extracellular matrix proteins or endothelial cells. The molecular families involved in the migration process include chemokines and chemokine receptors, adhesion molecules (such as integrins and selectins), and proteases such as the matrix metalloproteinases that digest the extracellular matrix.
There is an extensive literature indicating that the release of immunoregulatory molecules described above correlates with clinically or MRI-defined disease activity in MS. There are significant differences related to disease phase (for example, IL-12 and IFN-γ levels are higher in mononuclear blood cells from primary progressive MS patients than in those from relapsing-remitting MS patients) and disease phenotype (for example, lymphocyte migration rate and IFN-γ production are increased in primary progressive patients with high lesion volume compared to primary progressive patients with low lesion volume).75
B-Cell-Related Markers
In terms of disease-specific immune responses, the direct approach has been to look for the presence of antibodies in the serum or CSF that may directly participate in the MS disease process. The search has focused largely on myelin-directed antibodies. These can be assayed directly from serum or CSF. An alternative approach has been to determine whether stimulated blood or CSF-derived cells produce such antibodies in vitro.
Page 266
There has long been recognition of the oligoclonality of the immunoglobulin fraction of the CSF in MS (oligoclonal bands). Molecular biologic techniques have been used to sequence the hypervariable regions of the immunoglobulin genes expressed by B cells recovered from CSF or CNS tissues of MS cases, usually of recent onset.76,86 These studies have shown significant restriction of the B-cell repertoire. The search now focuses on what antigen is being recognized by these specific immunoglobulin molecules.
More indirect evidence of antibody participation in the immune response in MS is provided by reports of increased presence of autoantibody-producing B-cell populations (CD5+) and by detection of immune products such as those of the complement cascade and immune complexes that participate in antibody-mediated immune responses.
Neurobiological Markers
These can be considered in terms of molecules produced by resident cells of the CNS that reflect interaction with the constituents of the immune system and in terms of molecules that reflect the tissue injury or repair process. Again, here one faces the dilemma that CNS tissue cannot be readily sampled; thus, one is dependent on measures of molecules that are shed from the tissues and reach either the CSF or a systemic compartment.
CNS Immune Interaction-Related Markers. For such studies, the cell source of the markers being measured is not always certain since, in most cases; the molecules can be produced by both the immune system and the resident CNS cells. The CSF would for the most part seem preferable to blood for conducting such assays. Molecules to be measured are those with which lymphocytes interact during the migration process (chemokines, adhesion molecules), those that are expressed by resident CNS cells in their role as antigen-presenting cells, and those that may serve as targets of immune effector mechanisms (fas, TNF receptor).
CNS Injury and Repair Markers. With the evidence that oligodendrocytes and myelin are injured as part of the MS disease process, the CSF (± blood) can be assayed for the presence of products that would reflect such injury. The presence of myelin debris and MBP can be demonstrated in the CSF when there is active tissue destruction in MS cases. Although MBP-like material was found in serum and urine, this material has now been identified as p-cresol sulfate. Since myelin regeneration is now also a recognized feature of MS, the search for molecules that are upregulated during the repair process should also be undertaken.
With the recent emphasis on the contribution of axonal injury to neurologic disability in MS, there is a search for axonal products that would be released
Page 267
consequent to such injury. These would be the same candidates as those claimed to be released in other tissue-destructive CNS disorders (for example, neurofilament proteins).
The marked response of astrocytes in the MS disease process (the basis of “sclerosis”) suggests that astrocyte-derived molecules could also be detected.
TECHNOLOGIES AND RESEARCH STRATEGIES
New Methods in MRI
Magnetic resonance imaging is a nonradioactive, noninvasive way to produce images of internal structures such as the brain. MRI has been an important tool for studying the characteristic white matter lesions of MS.
The varying amounts of water in different structures within the brain produce the light and dark regions of an MRI image.71 The viscosity, temperature, and general molecular structure of a brain region also affect the MRI signal.40 Like a photographic flash bulb, contrast agents increase this signal. Older methods used contrast agents as general image enhancers. Now researchers can use contrast agents to selectively enhance visualization of specific tissues or cell types. With this technique, immune cells have been visualized as they traveled to an inflammation site105 or rejection site of a transplanted organ in the rat (Chien Ho, personal communication). Implanted stem cells or genetically altered cells might be tracked this way, as could the immune cellular involvement in demyelination and remyelination. In the EAE mouse model, this technique could allow researchers to watch immune cells migrate to the CNS long before any obvious damage occurs. With improved methods of targeting, they might be able to determine which immune cells lead the attack on myelin and the remyelination process.
Improvements in MRI equipment and methodology have increased the level of resolution of the images as well as the MRI signal-to-noise ratio. In particular, new ways to use contrast agents have great potential for investigating the early stages of MS.
Another new application for contrast agents uses one of two different methods to “turn on” a contrasting agent only in certain cells. Both rely on a contrast agent, such as gadolinium, that is injected in a caged (chemically neutralized) form into animals. Caged gadolinium cannot do its job as a contrast agent until it is released from its chemical enclosure. In the first method, the cage is made with a sort of hinged door that opens in response to cellular signaling molecules, such as calcium. This method has been used in frog embryos to see cell groups with active calcium signaling (Scott Fraser, personal communication). In the EAE mouse, this technique might highlight cell signaling and biochemical changes that occur in the CNS or in immune cells long before lesions are apparent.
Page 268
In the second method, an enzyme cuts open the cage and releases gadolinium. Researchers have made frog embryos that produce the necessary enzyme only in certain cells. When the caged gadolinium and the enzyme are in the same cell, gadolinium is released and these cells light up on the MRI image.52 Researchers can use this technique to probe when and where specific proteins are made.
For each protein there is a piece of DNA, a gene, that is the molecular blueprint for that protein. This blueprint includes the information that tells a cell when it should make the protein. This information is called the promoter. Researchers can take the promoter from one gene, such as the TNF-α promoter, and hook it up to another gene, in this case the gene for the uncaging enzyme. This engineered DNA is then put into a mouse, so that whenever a cell in that mouse makes the TNF-α protein, it will also make the uncaging enzyme. In combination with the caged contrast agent, MRI images from this mouse will show which cells make TNF-α. In an EAE mouse, this technique might provide information about which proteins are made early in diseased animals and which might have a direct effect on lesion formation.
These new MRI techniques hold great promise for increasing our understanding of animal models of MS and, perhaps eventually, the disease itself. Because they are applicable to large fields of research, such as developmental biology and immunology, our knowledge of their advantages and limitations should grow rapidly.
Genes and Genomics
Available data support the hypothesis that inherited susceptibility to MS involves the interaction of different susceptibility genes, each of which individually contributes a small amount to the overall risk. Whole genome screens confirm the importance of the major histocompatibility complex region in chromosome 6p21 in conferring susceptibility to MS. Susceptibility is likely to be mediated by the MHC class II genes themselves (DR, DQ, or both) and is most likely related to the known function of these molecules in the normal immune response, antigen-binding, and T-cell repertoire determination. The data also show that although the MHC region carries significant susceptibility, much of the genetic effect in MS remains to be explained. By analogy to emerging data on the genetic basis of experimental autoimmune demyelination, it will be of particular interest to identify whether some gene loci are involved in the initial pathogenic events while others influence the development and progression of the disease.
The following genetic approaches should be pursued:
1. Groups and consortia with the appropriate experimental, clinical, and financial resources should be supported to continue the analysis of the
Page 269
MS genome with larger DNA data sets and dense and informative genetic markers, for example, single nucleotide polymorphisms (SNPs). In all likelihood, the use of phenotypic and demographic variables will assume increasing importance as stratifying elements for genetic studies of MS and in addressing the fundamental question of genotype-phenotype correlation in autoimmune demyelination. These studies will necessarily be linked to the development of novel mathematical formulations designed to identify modest genetic effects, as well as epistatic interactions between multiple genes and interactions between genetic, clinical, and environmental factors.
2. With the advances in deciphering the human genome code and sequences readily available in the public domain, analysis will focus on the detailed analysis of candidate genes, particularly genes located in chromosomal segments linked to MS susceptibility. The problematic “case-control” population-based studies with limited statistical power will be replaced by the analysis of large collections of nuclear or singleton families (the patient and the biological parents or the patient and healthy siblings) using transmission-disequilibrium test (TDT) and Sib-TDT tests of association. For complex disorders such as MS, genomic analysis of multiple candidate genes must be performed on an extremely large group of individuals if small genetic effects are to be detected. Hence, key to the success of the proposed studies will be the availability of rapid, reliable, non-labor-intensive methods for high-throughput polymorphism screening. The inclusion of non-Caucasian patient populations, both in their native environment and after migration, will provide important new insights and clues about MS genetic and clinical heterogeneity.
3. The critical importance of identifying rare families that might have a monogenic variant of MS cannot be overstated; this approach has been extraordinarily fruitful in neurodegenerative diseases such as Alzheimer's disease and Parkinson's disease.
4. Continued analysis of the genes responsible for different forms of demyelinating disease in experimental models such as EAE and Theiler's virus infection is likely to identify syntenic regions in humans that may prove fruitful in MS.
5. Studies with DNA microarrays (DNA chips) to look at the coordinated expression, in both the CNS and the periphery, of ensembles of critical genes encoding cytokines, adhesion molecules, metalloproteinases, molecules involved in apoptosis, and molecules participating in myelin destruction and repair will contribute to our understanding of how these genes influence susceptibility and pathogenesis in MS. As gene chip methodologies mature, there is also the opportunity to perform wider
Page 270
“whole-genome” analyses of gene expression unbiased by the selection of known candidate genes (see
Box 5.1).
BOX 5.1Tracking Gene Activity in Disease: Microarray TechnologyScientists use genes to figure out what is going on in a cell. Genes are the molecular master switches. The 100,000 or so human genes control everything from how a cell grows to how it responds in times of stress and disease. In the past, researchers have been able to study only a handful of genes at a time. (Imagine trying to navigate in Boston with a road map that shows only two streets.) With microarray technology, scientists can simultaneously keep tabs on thousands of genes. For the first time, researchers can generate detailed genetic profiles of cells. These profiles can give us an integrated picture of gene regulation that can help explain susceptibility to MS and the mechanisms behind myelin destruction and disease progression and even suggest new directions for therapy. Genes are the functional units in genetic material, or DNA. Each gene is the molecular blueprint for a particular protein. When a gene is turned on (or expressed), it is used as a template to make RNA. This RNA, in turn, serves as the working copy of the blueprint, instructing the cell to build that protein. Genes are turned on and off in different situations, and their expression patterns provide important clues about what is going on in a cell, tissue, or organism. For example, in the central nervous system of mice with experimental autoimmune encephalomyelitis (EAE), a model for MS, there is increased expression of genes for chemokines.42 These are small proteins that regulate immune responses. Chemokine expression in the CNS is just one of the many pieces of evidence demonstrating that the immune system plays a role in EAE (and also MS). Microarrays measure the expression patterns of many genes simultaneously. A microarray, as the name suggests, is a microscopic array of DNA targets. Each target corresponds to a region of a gene. Each of the DNA targets on the array will specifically bind (or hybridize) only to DNA with a complementary sequence. A robot precisely spots the DNA target onto the substrate (for example, a glass microscope slide or membrane). Arrays can contain as many as 300,000 targets on a 1.28-cm2 surface.49 Researchers commonly use microarray technology to compare gene expression between two samples, for example, RNA from a brain region in an EAE mouse that is in remission versus a mouse that has relapsed. In this case, researchers use each RNA sample as a template to make |
Page 271
|
complementary DNA (cDNA). Incorporated into the cDNA is a fluorescent dye. Using a different dye for each cDNA sample (for example, a red dye for the remission sample and a green dye for the relapsed sample), researchers can visualize the two populations simultaneously. The combined, fluorescently labeled cDNAs bind, or hybridize, to the target DNA on the array. Following the removal of unbound DNA, the microarray will contain a representation of the relative amounts of RNA from the two samples. Using a laser to excite the fluorescent dyes and a confocal microscope to read the emitted light, researchers can measure the fluorescence at a given DNA target. The ratio of the two fluorescent signals (for example, the amount of red versus green fluorescence) corresponds to the relative amounts of RNA for that target in the two samples. (Some researchers hybridize with one sample, remove the sample, and then hybridize with the second sample, or they compare side-by-side hybridizations on two separate arrays.) It is then up to computers and ever-improving data analysis programs to process these huge amounts of data into a format that is both meaningful and easy to understand.22,106 As with any method, it is important to understand both the strengths and the limitations of using microarrays. This technology has given researchers a means to monitor thousands of genes simultaneously. Yet when a scientist contemplates a list of the hundred or so genes that are turned on or off in the relapsing EAE mouse, it is difficult to determine which of these may be important. Also, because sequencing projects have leapt far ahead of the actual annotation of the genome, the function of many of these genes is unknown. Finding the truly significant results will become easier as data analysis improves and as researchers generate and make available more experimental data about gene expression in MS. Reproducibility is also a concern in microarray studies. RNA is notoriously unstable. If not handled properly, duplicate samples (even those taken from the same batch of tissue) can vary greatly. Well-planned experiments incorporate various controls within the hybridization. (This also allows researchers to compare data from one experiment to the next.) Researchers also routinely confirm microarray results with older methods that look at expression changes in one gene at a time. Collecting the sample is itself an important issue, especially when, as in the case of MS, researchers are analyzing human RNA. Microarrays use relatively large amounts of RNA, and increased risk of RNA degradation is greater in postmortem samples than in samples collected from living tissue.11 However, ethical considerations limit the use of fresh human tissue. Sample homogeneity is another consideration. The brain is made up of many cell types, each of which express different genes. For the sake of accurate comparisons (both within and across experiments), researchers must collect precisely defined samples (for example, through the use of microdissection).11 Finally, it is important to consider what microarrays measure. These techniques measure only relative levels of gene expression. Moreover, turning a gene on is just the beginning of the story. The real cellular actors are the proteins. Many proteins, once they are built, are not active until they undergo further processing in the cell, or their actions can change under different cellular conditions. Although microarrays have greatly expanded our view of life in a cell, they still reveal only part of the picture. We will have to wait for such advances as complete genome annotation and protein arrays to capture the entire panorama. |
Page 272
REFERENCES
1. . 2000. Inflammatory mediators and modulation of blood-brain barrier permeability. Cell Mol Neurobiol.; 20: 131-47.
2. , , , . 1998. Microglia are more efficient than astrocytes in antigen processing and in Th1 but not Th2 cell activation. J Immunol.; 160: 4671-80.
3. , , , . 1996. Phenotypic analysis of antigen-specific T lymphocytes. Science.; 274: 94-6.
4. , , . 1989. Glial and neuronal forms of the voltage-dependent sodium channel: characteristics and cell-type distribution. Neuron.; 2: 1375-88.
5. , , . 1981. Vaccination against autoimmune encephalomyelitis with T-lymphocyte line cells reactive against myelin basic protein. Nature.; 292: 60-1.
6. , , , , , . 1997. Experimental autoimmune encephalomyelitis: the antigen specificity of T lymphocytes determines the topography of lesions in the central and peripheral nervous system. Lab Invest.; 76: 355-64.
7. , , , . 1985. The presence of voltage-gated sodium, potassium and chloride channels in rat cultured astrocytes. Proc R Soc Lond B Biol Sci. ; 225: 299-313.
8. , , , . 1994. Thalamocortical axons extend along a chondroitin sulfate proteoglycan-enriched pathway coincident with the neocortical subplate and distinct from the efferent path. J Neurosci.; 14: 3500-10.
9. , , , , , . 2000. Sensory neuron specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc Natl Acad Sci U S A.; 97: 11598-11602.
10. , , . 1987. Macromolecular structure of axonal membrane during acute experimental allergic encephalomyelitis in rat and guinea pig spinal cord. J Neuropathol Exp Neurol.; 46: 167-84.
11. . 1999. Options available—from start to finish—for obtaining expression data by microarray. Nat Genet.; 21: 25-32.
12. , . 1998. Cortical plasticity: from synapses to maps. Annu Rev Neurosci.; 21: 149-86.
13. , . 1991. Immediate expansion of receptive fields of neurons in area 3b of macaque monkeys after digit denervation. Somatosens Mot Res.; 8: 249-60.
14. , , . 1973. Anatomical and physiological correlates of plasticity in the central nervous system. Brain Behav Evol.; 8: 5-26.
15. , , . 2000. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity.; 12: 241-50.
16. , , , . 2000. Role of astrocytes in antigen presentation and naive T-cell activation. J Neuroimmunol.; 106: 69-77.
Page 273
17. , , , , , . 1996. Identification of peptides specific for cerebrospinal fluid antibodies in multiple sclerosis by using phage libraries. Proc Natl Acad Sci U S A.; 93: 11063-7.
18. , , , , , . 1997. Regeneration of adult axons in white matter tracts of the central nervous system. Nature.; 390: 680-3.
19. , , , . 1986. Transfer of specificity by murine alpha and beta T-cell receptor genes. Nature.; 320: 232-8.
20. , , , . 1995. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci.; 15: 492-503.
21. , , , . 1990. Changed distribution of sodium channels along demyelinated axons. Proc Natl Acad Sci U S A.; 87: 6777-80.
22. , . 2000. Microarray technology—enhanced versatility, persistent challenge. Curr Opin Biotechnol.; 11: 36-41.
23. , , , . 1997. Axonal damage in acute multiple sclerosis lesions. Brain.; 120: 393-9.
24. , , , . 1993. Pharmacological protection of CNS white matter during anoxia: actions of phenytoin, carbamazepine and diazepam. J Pharmacol Exp Ther.; 266: 1549-55.
25. , , . 1995. Voltage-gated calcium channels in CNS white matter: role in anoxic injury. J Neurophysiol.; 74: 369-77.
26. , , . 1995. Endogenous GABA attenuates CNS white matter dysfunction following anoxia. J Neurosci.; 15: 699-708.
27. , . 1997. Glial cell extracellular matrix: boundaries for axon growth in development and regeneration. Cell Tissue Res.; 290: 379-84.
28. , , , . 1999. Gene transfer into CD4+ T lymphocytes: green fluorescent protein-engineered, encephalitogenic T cells illuminate brain autoimmune responses. Nat Med.; 5: 843-7.
29. , , . 1980. Reorganization of the axon membrane in demyelinated peripheral nerve fibers: morphological evidence. Science.; 210: 661-3.
30. , , , . 1999. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med.; 5: 170-5.
31. , , , . 1994. In healthy primates, circulating autoreactive T cells mediate autoimmune disease. J Clin Invest.; 94: 1339-45.
32. , , . 1995. Axotomy-induced axonal degeneration is mediated by calcium influx through ion-specific channels. J Neurosci.; 15: 6445-52.
33. , . 1999. Sentries at the gate: chemokines and the blood-brain barrier. J Neurovirol.; 5: 623-34.
34. . 1999. Tolerance and autoimmunity in TCR transgenic mice specific for myelin basic protein. Immunol Rev.; 169.
35. , . 1985. Ion channels in Schwann and glial cells. Trends Neurosci.; 8: 411-15.
36. , , . 2000. The age-range of risk of developing multiple sclerosis: evidence from a migrant population in Australia. Brain.; 123: 968-74.
37. . The Organization of Behavior: a Neuropsychological Theory. New York, NY: Wiley; 1949.
38. , , , , . 1998. Probing degeneracy in T-cell recognition using peptide combinatorial libraries. Immunol Today.; 19: 163-8.
39. , . 1999. Chemokine and chemokine receptor expression in the central nervous system. J Neurovirol.; 5: 13-26.
40. , , , . 1999. Looking deeper into vertebrate development. Trends Cell Biol.; 9: 73-6.
Page 274
41. , , , , , . 1995. An important role for the chemokine macrophage inflammatory protein-1 alpha in the pathogenesis of the T cell-mediated autoimmune disease, experimental autoimmune encephalomyelitis. J Immunol.; 155: 5003-10.
42. , . 1999. Role of chemokines in the regulation of Th1/Th2 and autoimmune encephalomyelitis. J Clin Immunol.; 19: 273-9.
43. , , , , , . 1992. Less mortality butmore relapses in experimental allergic encephalomyelitis in CD8-/- mice. Science.; 256: 1210-3.
44. , . 1999. Anatomical correlates of representational map reorganization induced by partial vibrissectomy in the barrel cortex of adult mice. Neuroscience.; 92: 807-17.
45. , , , , , . 1995. Gene transfer through the blood-nerve barrier: NGF-engineered neuritogenic T lymphocytes attenuate experimental autoimmune neuritis. Nat Med.; 1: 1162-6.
46. , , , . 2000. Visualization of myelin basic protein (MBP) T cell epitopes in multiple sclerosis lesions using a monoclonal antibody specific for the human histocompatibility leukocyte antigen (HLA)-DR2-MBP 85-99 complex. J Exp Med. ; 191: 1395-412.
47. , . 2000. Mechanisms of ionotropic glutamate receptor-mediated excitotoxicity in isolated spinal cord white matter. J Neurosci.; 20: 1190-8.
48. , , , , . 1988. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol.; 130: 443-54.
49. , , , . 1999. High density synthetic oligonucleotide arrays. Nat Genet.; 21: 20-4.
50. , , , . 1998. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med.; 188: 169-80.
51. , , . 1996. Frequency of MBP and MBP peptide-reactive T cells in the HPRT mutant T-cell population of MS patients. Neurology.; 46: 1410-5.
52. , , , . 2000. In vivo visualization of gene expression using magnetic resonance imaging. Nat Biotechnol.; 18: 321-5.
53. , , , . 1999. Extracellular matrix degradation by metalloproteinases and central nervous system diseases. Mol Neurobiol.; 19: 267-84.
54. , , , . 1999. Gene expression profiles of laser-captured adjacent neuronal subtypes. Nat Med.; 5: 117-22.
55. , , , . 1999. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet.; 23: 343-7.
56. , . 1999. Long-term potentiation—a decade of progress? Science.; 285: 1870-4.
57. , , , , , . 1999. Involvement of the N-methyl-D-aspartate receptor in neuronal cell death induced by cytotoxic T cell-derived secretory granules. Eur J Immunol.; 29: 3053-62.
58. , , , . 1990. Fine specificity and HLA restriction of myelin basic protein-specific cytotoxic T cell lines from multiple sclerosis patients and healthy individuals. J Immunol.; 145: 540-8.
59. , , , , . 1997. Treatment of experimental autoimmune encephalomyelitis with genetically modified memory T cells. J Exp Med.; 186: 159-64.
60. , , . 1992. The pathological evolution of multiple sclerosis. Neuropathol Appl Neurobiol.; 18: 319-34.
Page 275
61. , , , , , . 2000. MHC class I-restricted killing of neurons by virus specific CD8+ T lymphocytes is effected through the Fas/FasL, but not the perform pathway. Eur.J.Immunol.; 30: 3623-3633.
62. , , , . 1997. Encephalitogenic potential of myelin basic protein-specific T cells isolated from normal rhesus macaques. Am J Pathol.; 150: 445-53.
63. , . . Selective inhibition of distant afferent input to lamina 4 and 5 cells in cat dorsal spinal cord. J Physiol (Lond).; 278: 51P.
64. , , , . 1993. Somatosensory cortical plasticity in adult humans revealed by magnetoencephalography. Proc Natl Acad Sci U S A.; 90: 3593-7.
65. , , , . 1991. Increase of sodium channels in demyelinated lesions of multiple sclerosis. Brain Res.; 556: 311-6.
66. , , , . 1997. Imaging of axonal damage in multiple sclerosis: spatial distribution of magnetic resonance imaging lesions. Ann Neurol.; 41: 385-91.
67. , . 1998. Neuronal control of the immune response in the central nervous system: linking brain immunity to neurodegeneration. J Neuropathol Exp Neurol.; 57: 1-9.
68. . 1999. Recovery after damage to motor cortical areas. Curr Opin Neurobiol.; 9: 740-7.
69. , , . 1998. Regulatory CD4(+) T cells expressing endogenous T cell receptor chains protect myelin basic protein-specific transgenic mice from spontaneous autoimmune encephalomyelitis. J Exp Med.; 188: 1883-94.
70. , , , , , . 1990. T-cell recognition of an immunodominant myelin basic protein epitope in multiple sclerosis. Nature.; 346: 183-7.
71. , . Magnetic Resonance Imaging Changes as Living Pathology in Multiple Sclerosis. Paty DW, Ebers GC, eds. Multiple Sclerosis. Philadelphia, PA: F.A. Davis Company; 1998: 328-369.
72. , , , . 1990. Myelin basic protein-specific T lymphocyte lines from MS patients and healthy individuals. Neurology.; 40: 1770-6.
73. , , . 2000. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med.; 6: 67-70.
74. , , , , , . 1991. Massive cortical reorganization after sensory deafferentation in adult macaques [see comments]. Science.; 252: 1857-60.
75. , , , , . 2000. Heterogeneity of T-lymphocyte function in primary progressive multiple sclerosis: relation to magnetic resonance imaging lesion volume. Ann Neurol.; 47: 234-7.
76. , , , , , . 1998. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest.; 102: 1045-50.
77. , . 2000. Phantom limbs and neural plasticity. Arch Neurol.; 57: 317-20.
78. , , . 1992. Perceptual correlates of massive cortical reorganization. Science.; 258: 1159-60.
79. , , . 1992. Perceptual correlates of massive cortical reorganization. Neuroreport.; 3: 583-6.
80. , , . 1990. Anoxic injury of CNS white matter: protective effect of ketamine. Neurology.; 40: 1399-403.
81. , , , . 1998. Strengthening of horizontal cortical connections following skill learning. Nat Neurosci.; 1: 230-4.
82. , . 1999. Phage-display technology—finding a needle in a vast molecular haystack. Curr Opin Biotechnol; 10: 87-93.
83. , . 1999. The cell biology of the blood-brain barrier. Annu Rev Neurosci.; 22: 11-28.
Page 276
84. , . 1998. Partial inactivation of the primary motor cortex hand area: effects on individuated finger movements. J Neurosci.; 18: 9038-54.
85. , , , . 1997. Local delivery of interleukin 4 by retrovirus-transduced T lymphocytes ameliorates experimental autoimmune encephalomyelitis. J Exp Med.; 185: 1711-4.
86. , , , , , . 2000. Comparison of immunoglobulin G heavy-chain sequences in MS and SSPE brains reveals an antigen-driven response. Neurology.; 54: 1227-32.
87. , . 1992. Ion channels in spinal cord astrocytes in vitro. II. Biophysical and pharmacological analysis of two Na+ current types. J Neurophysiol.; 68: 1001-11.
88. . Brain injury and theories of recovery. Goldstein LB, ed. Restorative Neurology: Advances in Pharmacotherapy for Recovery After Stroke. Armonk, NY: Futura Publishing Co., Inc.; 1998.
89. . 2000. Multiple approaches to multiple sclerosis. Nat Med.; 6: 15-6.
90. , , , . 2000. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med.; 191: 411-6.
91. , , , . 1993. Noninactivating, tetrodotoxin-sensitive Na+ conductance in rat optic nerve axons. Proc Natl Acad Sci U S A.; 90: 6976-80.
92. , , . 1992. Ionic mechanisms of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na(+)-Ca2+ exchanger. J Neurosci.; 12: 430-9.
93. , , , , , . 1998. Axonal transection in the lesions of multiple sclerosis. New Engl J Med.; 338: 278-285.
94. , , . 1989. Immunization with a synthetic T-cell receptor V-region peptide protects against experimental autoimmune encephalomyelitis. Nature.; 341: 541-4.
95. , . Organization and development of the forepaw representation in forepaw barrel subfield. Jones EG, Diamond IT, eds. Cerebral Cortex. New York: Plenum Press; 1995; 11.
96. , . 2000. Methodological problems in evaluating efficacy of a treatment in multiple sclerosis. Pathol Biol (Paris).; 48: 104-13.
97. . 1978. Prerequisites for conduction in demyelinated fibers. Neurology.; 28: 27-33.
98. . 1982. Membranes, myelin, and the pathophysiology of multiple sclerosis. N Engl J Med.; 306: 1529-33.
99. . 1998. Demyelinating diseases—new pathological insights, new therapeutic targets. N Engl J Med.; 338: 323-5.
100. . 1992. Myelin specific, autoaggressive T cell clones in the normal immune repertoire: their nature and their regulation. Int Rev Immunol.; 9: 231-41.
101. , , , . 1986. Cellular immune reactivity within the CNS. Trends Neurosci.; 9: 271.
102. , , , . 2000. Recovery from optic neuritis is associated with a change in the distribution of cerebral response to visual stimulation: a functional magnetic resonance imaging study. J Neurol Neurosurg Psychiatry.; 68: 441-9.
103. , , , . 1999. Immunogenicity. I. Use of peptide libraries to identify epitopes that activate clonotypic CD4+ T cells and induce T cell responses to native peptide ligands. J Immunol.; 163: 6424-34.
104. , , , , , . 1983. Immunospecific inhibition of nerve conduction by T lymphocytes reactive to basic protein of myelin. Nature.; 303: 246-7.
105. , , , . 1995. In vivo dynamic MRI tracking of rat T-cells labeled with superparamagnetic iron-oxide particles. Magn Reson Med.; 33: 200-8.
106. . 1999. Knowledge discovery in gene-expression-microarray data: mining the information output of the genome. Trends Biotechnol.; 17: 429-436.




































