
Page 29
2
Clinical and Biological Features
Multiple sclerosis (MS) literally means “many scars,” which refers to the lesions that accumulate in the brain and spinal cord throughout the course of the disease. These scars, or lesions, consist mostly of dead nerve cells, whose axons have been denuded of the myelin sheaths that normally protect them and permit the conduction of nerve impulses. MS is a chronic, degenerative disease that usually begins in young adulthood and most visibly destroys muscular control, although many other brain functions are affected. Most people will live with MS for decades after their diagnosis. MS reduces life expectancy after onset (as measured by current diagnostic criteria) by only about 10-15 years, and about half of the patients survive 30 years or more from onset.110
THE CLINICAL PICTURE: SYMPTOMS, DISEASE COURSE, VARIATION, AND DIAGNOSIS
Disease Activity and Progression
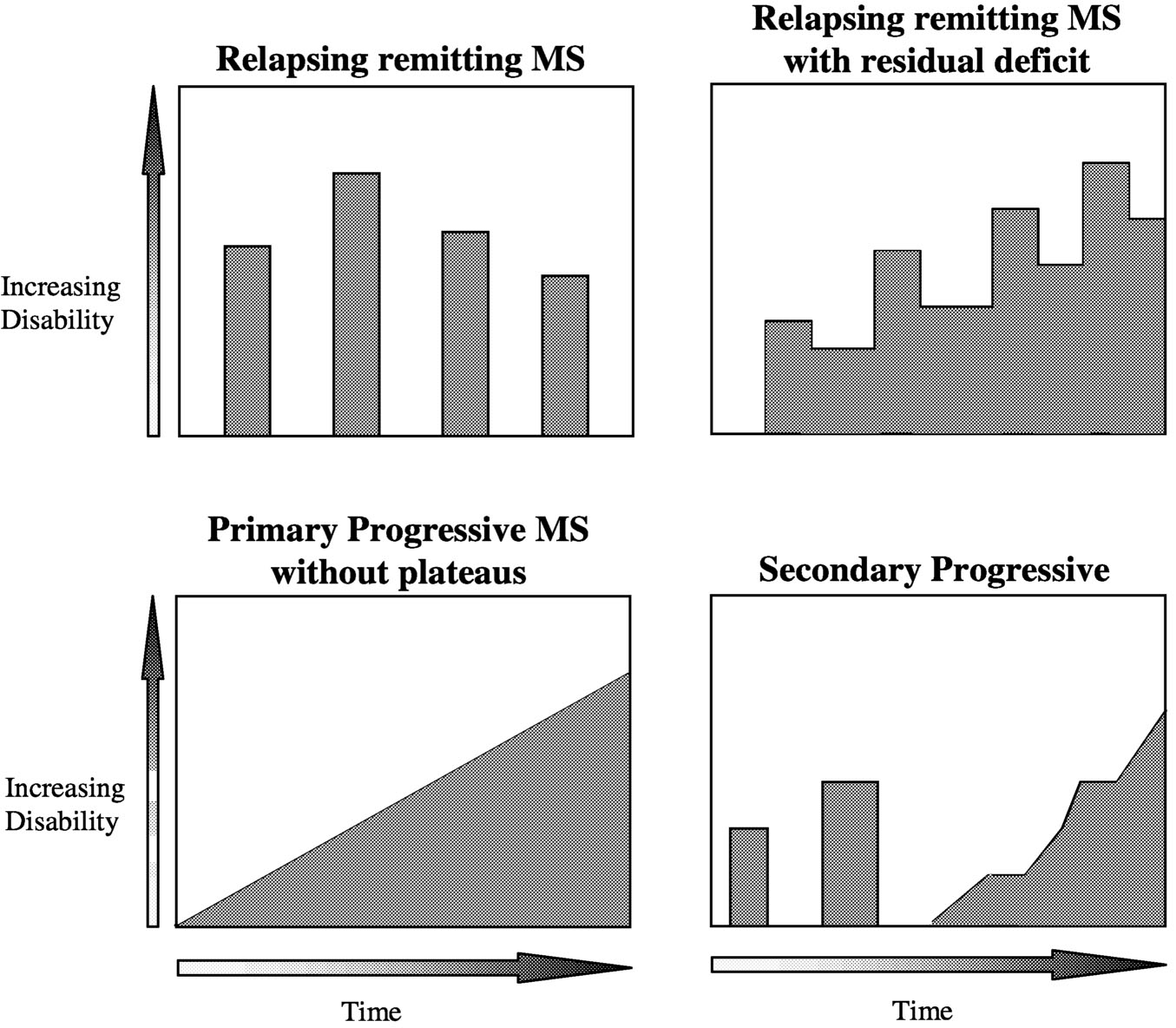
MS, as defined by ongoing central nervous system (CNS) lesion formation and increasing cumulative damage, is now recognized as a disease that is active in most patients most of the time. Disease activity has reversible and irreversible sequelae; irreversible sequelae ultimately lead to progressive impairment and disability in most patients. MS takes a variety of forms, distinguished by the clinical pattern of disease activity (Table 2.1, Figure 2.1). Accumulated deficit can produce sustained worsening in both relapsing and progressive MS. In re-
Page 30
|
Asymptomatic MS |
Autopsy studies indicate there are individuals without any known clinical history who have neuropathologic changes typical of MS. It is difficult to get an accurate estimate of subclinical disease, but one recent review suggested asymptomatic MS might account for up to 25% of all cases. |
|
Relapsing-remitting MS |
This is the major MS subtype. Approximately 85% of patients with a diagnosis of MS start out with relapsing MS. Overall, this subtype accounts for 55% of MS. Relapsing MS patients show a high rate of inflammatory lesion activity (gadolinium-enhancing lesions). |
|
Benign relapsing MS |
This category represents a subset of relapsing patients who have few attacks and make an excellent recovery. They show minimal impairment and disability, even after 20-30 years. The proportion of MS patients with benign disease is controversial. Reasonable studies suggest 10-20% of people with MS fit into this category. |
|
Primary progressive MS |
This subtype accounts for 10% of MS. Patients show gradual worsening from onset, without disease attacks. These patients tend to be older and often present with a spinal cord dysfunction without obvious brain involvement. This subtype is the least likely to show inflammatory lesion activity on MRI (gadolinium-enhancing). Unlike the other subtypes of MS, men are as likely as women to develop primary progressive MS. |
|
Progressive relapsing MS |
This subtype accounts for 5% of MS. Patients show slow worsening from onset, with superimposed attacks. Recent studies suggest these patients are similar to primary progressive patients. |
|
Secondary progressive MS |
This is the major progressive subtype and accounts for approximately 30% of MS. Relapsing MS patients usually transition to secondary progressive disease. They show gradual worsening, with or without superimposed relapses. Natural history studies of untreated relapsing MS indicate 50% of patients will be secondary progressive at 10 years and almost 90% by 25 years. This form of MS shows a lower rate of inflammatory lesion activity than relapsing MS, yet the total burden of disease continues to increase. This most likely reflects ongoing axonal loss. |
|
Acute MS |
Also referred to as Marburg variant MS, this is the most severe form of MS. Significant disability develops much more rapidly than usual, over weeks to months. Pathologic changes are widespread and destructive. These cases are rare and generally occur in young people. |
|
Clinically isolated syndromes |
This refers to patients who present with an isolated CNS syndrome (optic neuritis, incomplete transverse myelitis, brainstem or cerebellar lesion), which is often the first MS attack. Clinical, MRI, and CSF studies indicate that such patients with normal brain MRI and CSF have a low risk of developing MS. In contrast, those with abnormal MRI have a high risk of developing MS. |
NOTE: CSF = cerebrosinal fluid; MRI = magnetic resonance imaging
Page 31
~ enlarge ~
FIGURE 2.1 Spectrum of disease course (refer to Table 2.1 for definitions). SOURCE: Adapted from Lublin and Reingold, 1996.125
lapsing MS, worsening occurs in most patients during acute attacks with incomplete recovery. In progressive MS, the dominant pattern is a gradual accumulation of neurologic deficits, with slow clinical worsening.
Disease activity and progression have both clinical and subclinical components. Clinical disease activity and progression are judged by observation and neurologic examination. Subclinical components refer to pathological changes that are not observable in a clinical examination but are observed using a variety of laboratory tests, predominantly neuroimaging parameters.
Clinical Activity
Relapses. Relapses are variously referred to as acute attacks, exacerbations, or disease flare-ups. They involve the acute, or sudden onset, of focal neurologi-
Page 32
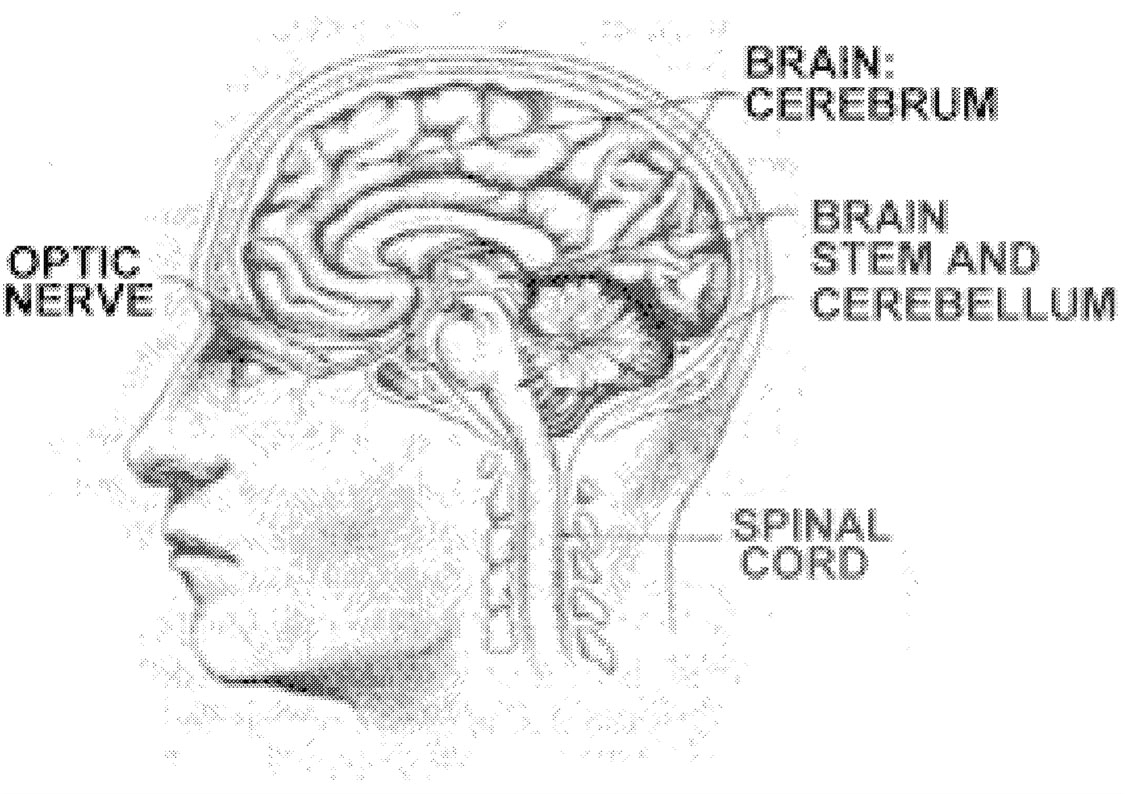
cal disturbances. Examples of typical MS relapses include blurring of vision in one eye (optic neuritis), persistent numbness or tingling of a body part (sensory system relapse), weakness of a body part (motor system relapse), or loss of coordination (cerebellar system relapse). Early in the MS disease process, relapses are likely to involve sensory, motor, cerebellar, or visual system abnormalities ( Figure 2.2, Table 2.2). Later in the disease process, relapses are likely to involve bladder, bowel, cognitive, and sexual function abnormalities. Acute disease attacks are a characteristic feature of the relapsing-remitting MS subtype. Relapses also occur in patients with progressive relapsing disease and in a number of patients with secondary progressive disease. The only clinical disease subtype in which relapses never occur is primary progressive MS.
~ enlarge ~
FIGURE 2.2 Areas of the CNS often affected by MS. Reprinted with permission from University of Delaware.
|
COMMON |
UNCOMMON |
|
|
Page 33
Relapses generally consist of three phases. There is a period of worsening, with onset of new deficits or increasing severity of old deficits. This is followed by a period of stability, with no change in deficits. The final phase is the period of recovery, with variable degrees of improvement in deficits. Most patients recover within six weeks, although for some, improvements can continue over months. Recovery can be complete return to baseline status, partial return, or no improvement. However, some degree of improvement is typical, particularly early in the disease. Relapsing patients then remain clinically stable until the next disease attack.
To be considered a relapse, deficits must persist for a minimum of 24 hours. This avoids confusion with deficits lasting only minutes to hours, which are believed to be a consequence of impaired nerve conduction through old lesion areas rather than the formation of a new lesion. Alternatively, new abnormalities that last seconds to minutes, such as Lhermitte's sign (a tingling sensation radiating down the arms, neck, or back on neck flexion), or paroxysmal attacks (stereotypic neurologic deficits occurring multiple times a day that last less than a minute) are also considered relapses if they occur repeatedly over several weeks. Sequential relapses are considered distinct only when they occur at least 30 days apart with a month of clinical stability in between. Although clinical relapses always produce changes in a patient's condition, they are not always associated with changes on neurologic examination. Maximal deficit in an MS relapse typically develops over several days but in some cases can develop much faster, over hours or even minutes, or much more slowly, over a period as long as several weeks.
Physiologic factors such as temperature, pH, or electrolyte balance can temporarily disrupt nerve conduction and produce neurologic abnormality. A relapse must be distinguished from a pseudoexacerbation, which is a neurologic deterioration associated with a physiologic change such as infection or fever. This condition can last for days, mimicking a true relapse. Pseudoexacerbation deficits disappear once the precipitating factor has been corrected. They reflect a temporary disruption in nerve conduction, rather than the formation of a new lesion.
Approximately 85 percent of MS patients begin with relapsing-remitting disease.222 MS relapses can involve a single neural system, as in optic neuritis, or several anatomically distinct systems at the same time, for example, combined motor and sensory problems. Attacks involving single neural systems are somewhat more common in the first MS relapse.
Most patients experience their second attack within two to three years of the first, but 5 percent of patients remain free of relapses for 15 years or more. In most cases, there is substantial recovery from the first relapse; only 4 percent of patients show no improvement. The average relapse rate is one to two attacks a year, but this rate normally declines over time. The longer a person has MS, the less likely it is that relapses will be followed by complete recovery and the more likely it is that relapses will be associated with residual deficits and increasing disability.
Page 34
|
Feature |
Favorable Prognosis |
Unfavorable Prognosis |
|
Relapse rate in first 2 years |
<5 relapses |
≥5 relapses |
|
Relapse rate after 5 years |
No increase |
Increasing |
|
Duration between relapses |
Long |
Short |
|
Number of neural systems involved |
One |
Multiple |
|
Relapse recovery |
Complete |
Incomplete |
|
Type of systems involved |
Visual, sensory, brainstem |
Motor, cerebellar, bowel or bladder |
Relapse features have prognostic significance (Table 2.3). In the first few years after disease onset, the number and type of relapses, as well as the degree of recovery, help predict future disease course.8 Relapses that involve visual, sensory, or brainstem systems have a better prognosis than those that involve cerebellar, motor, or sphincter systems. In the first two years of disease, a low relapse rate with excellent recovery indicates a better prognosis than a high relapse rate with poor recovery. Relapses restricted to single neural systems are prognostically better than those involving multiple systems. The relapse rate also has prognostic significance in the later stages of MS. With a disease duration of five or more years, an increasing relapse rate, polyregional relapses that involved multiple systems, and incomplete recovery from relapses indicate a worse prognosis.8
Progression. The relapsing form of MS is characterized by acute disease exacerbations. In contrast, progressive MS is characterized by slow deterioration and increasing neurological deficits. There are three forms of progressive MS. Approximately 15 percent of MS patients show slow deterioration from onset. In the second form, 10 percent have either primary progressive MS and never experience acute disease attacks or progressive relapsing MS (5 percent), and have occasional subsequent attacks. The third form, secondary progressive MS, is the major progressive subtype. These are relapsing patients who begin to slowly worsen 5 to 15 years after the first relapse. Once relapsing patients enter a progressive phase, they either stop having relapses or continue to experience exacerbations superimposed on slow worsening.
Documentation of a progressive course requires at least six months of observation. Observation over a year or two is often necessary to be confident of progression, since deficits can accumulate at a very gradual rate. The major defining feature of progressive MS is slow deterioration that occurs independently of acute disease relapses and does not reflect residual deficits from acute disease attacks. An analysis of the disease course among 1,844 patients indicated that the presence or absence of relapses during the progressive phase does not significantly affect the progression of irreversible disability45 (4 percent of
Page 35
patients in this study had been treated for up to one year with beta-interferon, but this did not affect the study results). Progressive MS patients can be clinically stable for up to several years at a time and can even show slight improvement for a period of time. Ultimately, however, all progressive MS patients develop disability with limited ability to walk. Progressive MS is a more severe form than benign or relapsing-remitting MS and has a worse prognosis.
Subclinical Disease Activity and Progression
Clinical parameters such as relapses and progression underestimate the actual damage to tissue that occurs in MS. When macroscopically normal-appearing brain tissue is looked at under the microscope, one can detect inflammation, gliosis (scarring), and myelin damage. Chemical studies of normal-appearing brain tissue often reveal changes in organelles such as lysosomes, in enzymes, and in myelin constituents. In addition, a number of the new research neuro-imaging techniques can detect changes in brain and spinal cord areas that appear free of lesions on conventional magnetic resonance imaging (MRI). Some of these abnormalities are detectable several months to years before they can be seen with conventional MRI. Changes in normal-appearing brain tissue are generally pronounced in MS patients with severe impairment. As a group, secondary progressive MS patients show more abnormalities in normal white matter and brain tissue than relapsing patients. (White matter corresponds to brain regions where axons are ensheathed in myelin; gray matter corresponds to brain regions that are rich in cell bodies.) Primary progressive patients often show subtle but diffuse changes in normal-appearing brain areas.
Even conventional MRI indicates that most new lesion formation is clinically silent, meaning that clinical exam does not reveal any corresponding symptoms. Approximately 80 to 90 percent of new brain lesions do not produce identifiable relapses. They might, however, be associated with subtle cognitive changes or other neuropsychological changes that are not detected in clinical examination. The total lesion burden increases in MS patients, on average, 5 to 10 percent per year, reflecting in large part the development of clinically silent lesions. (This does not apply to patients on the disease-modifying therapies discussed later in this section.) Atrophy of both brain and spinal cord can be detected even in patients with minimal symptoms. Atrophy can progress without obvious lesion formation, most likely reflecting loss of axons. MS patients show an accelerated rate of age-related brain and spinal cord atrophy that is three- to tenfold higher than the rate in control populations.76
Spinal cord lesions are generally similar to those in the brain except for the absence of “black holes” (see discussion in Box 2.1 of T1-weighted lesions).69 Spinal MS lesions rarely cover more than half of the cross-sectional area of the cord or exceed two vertebral segments in length. They are found more often in the cervical spinal cord (neck region) than thoracic region (midback) and are most
Page 36
BOX 2.1Basic Technical Principles of MRIMRI involves application of a magnetic field to the body that causes nuclei with odd numbers of protons, such as hydrogen nuclei, to behave like tiny magnets. These protons align themselves either parallel or antiparallel to the applied external magnetic field. The net magnetization induces an electric current that forms the basic MR signal. An MR image is formed by determining the spatial distribution of the signal and reconstructing the data into detailed images. The signals are picked up by a very sensitive antenna and forwarded to a computer for processing. Two time constants, T1 and T2 relaxation times, are important in determining the appearance of MR images. T1, or the longitudinal relaxation time, is the time constant when 63 percent of the original longitudinal magnetization is regained as the nuclei return to alignment with the external magnetic field. T2 or the transverse relaxation time, is the time constant when the transverse magnetization decreases to 37 percent of its original value as the nuclei lose alignment with each other following the initial application of an external magnetic field (a radio-frequency pulse). By altering the imaging parameters and pulse sequences used, differences between tissues with intrinsically different proton densities and T1 and T2 relaxation times can be highlighted or obscured. Image contrast can be either T1 weighted or T2 weighted in order to emphasize the differences between normal and pathological tissues. For example, cerebrospinal fluid (CSF) is dark on T1-weighted images and bright on T2-weighted images. White matter is bright on T1-weighted images, whereas a matter is dark but not as dark as CSF. |
common in the midcervical region. Disease activity is much less frequent in the spine than in the brain.
In summary, the clinical manifestations of MS possibly represent only the “tip of the iceberg,” with most of the CNS damage occurring much earlier and being detectable only when the accumulated damage overwhelms the ability of the CNS to compensate. The mechanisms through which CNS tissue is damaged or destroyed are discussed in greater detail later in the chapter.
Disease Markers
At the present time, neuroimaging provides the best assessment of disease activity in MS (Box 2.1, Figure 2.3).
Neuroimaging Abnormalities
A number of neuroimaging techniques can measure distinct pathologic changes and thereby provide markers for different aspects of the MS disease
Page 37
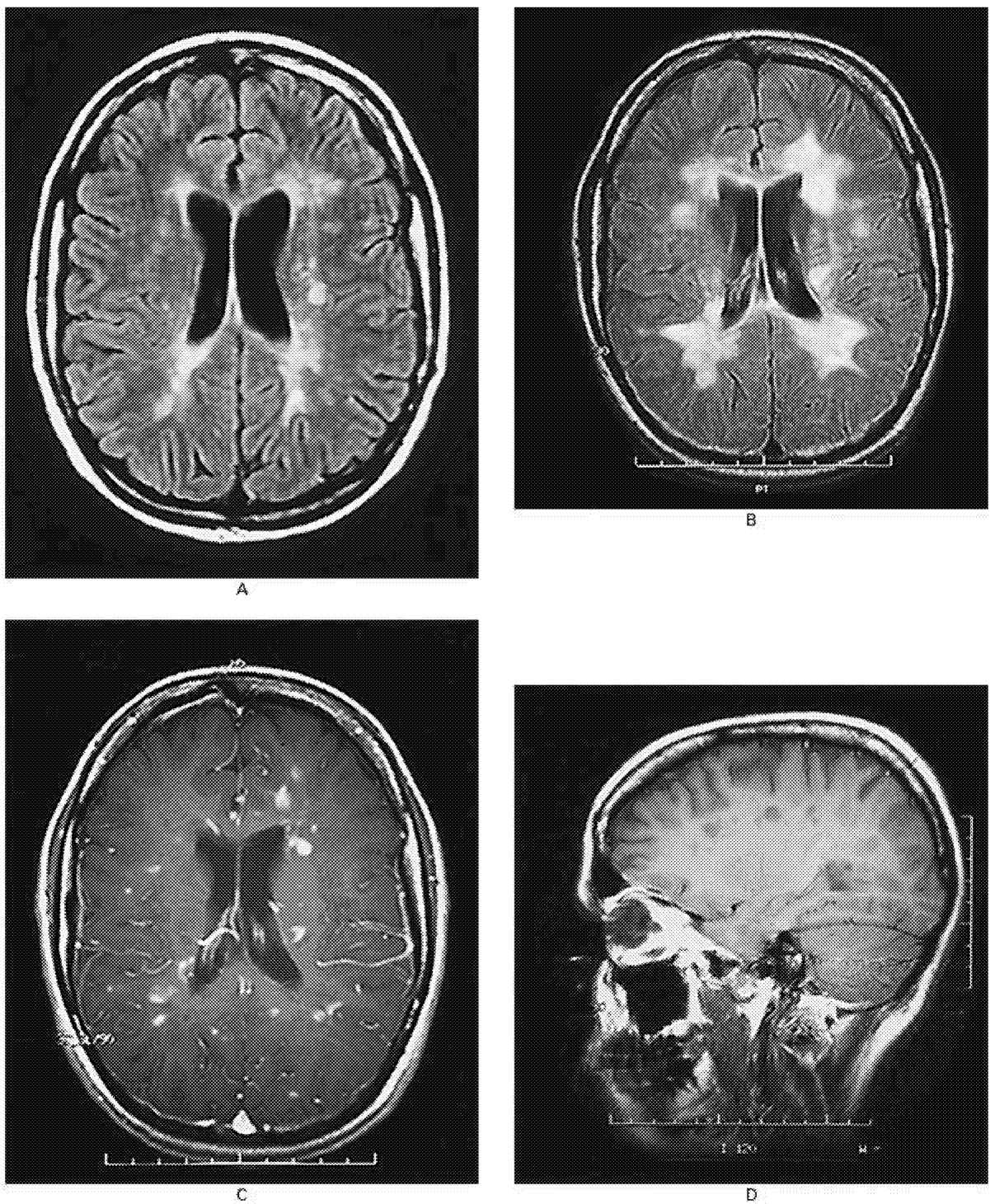
~ enlarge ~
FIGURE 2.3 MRI scans of the brain of a 25-year-old woman with relapsing-remitting multiple sclerosis.
(A) An MRI image shows multiple ovoid and confluent hyperintense lesions in the white matter surrounding the ventricles (the ventricles appear in the center of this image as a dark butterfly shape; they are the spaces through which cerebrospinal fluid [CSF] flows). (B) Nine months later, the number and size of the lesions have increased substantially. (C) After the administration of gadolinium, many of the lesions demonstrate ring or peripheral enhancement, indicating the breakdown of the blood-brain barrier. (D) A parasagittal T1-weighted MRI scan shows multiple regions in which the signal is diminished (referred to as “black holes”) in the periventricular white matter and corpus callosum. These regions correspond to the chronic lesions of multiple sclerosis. SOURCE: Reprinted with permission from Noseworthy et al.154 Copyright 2000 Massachusetts Medical Society. All rights reserved.
Page 38
process (Table 2.4). Magnetic resonance imaging is a technique that creates cross-sectional images of the brain using a magnetic field and radio waves (Box 2.1). It is a versatile, powerful, and sensitive tool for measuring abnormalities in the brain. This is especially valuable with MS, because so much of the pathological activity of the disease is neurologically asymptomatic.
Indeed, until neuroimaging results proved otherwise, the disease appeared to be quiescent during remissions. Neuroimaging has revealed a previously unsuspected level of activity and pathology throughout the course of disease.
Contrast-Enhanced Lesions. Contrast agents are used in MRI in cases where contrast between two tissues is poor. The contrasting agent, gadolinium, is normally excluded from the brain by the blood-brain barrier. Its presence in the
|
TABLE 2.4 Information Provided by Neuroimaging |
|
|
Observation or Method |
What it Reveals |
|
T1 gadolinium-enhancing lesions |
Detects blood-brain barrier leakage, inflammatory disturbances, and recent (≤6 weeks) activity, with lesion formation. |
|
T2 hyperintense lesions |
Provides total burden of disease measure, including reversible and irreversible pathologies. Most predictive of disease course in early MS. |
|
T1 hypointense lesions (black holes) |
Reflects more severe tissue pathology, including axon loss, and correlates with disability. |
|
Atrophy |
Reflects axon loss, as well as other tissue component loss. Correlates with disability. Atrophy is detectable in both brain and spinal cord of MS patients. CNS atrophy is ongoing and accelerated compared to normal age-related changes. |
|
MR spectroscopy measure of N-acetyl aspartate levels |
Decreased NAA levels reflect axon damage. Often shows abnormalities in normal brain tissue. Can be measured in whole brain (NAA) or in region of interest. |
|
Magnetization transfer imaging and magnetization transfer ratio |
Indicates more severe lesions, with tissue destruction. Abnormalities noted within both lesions and normal-appearing CNS tissue. Marker for disability. Can be measured in whole brain or in region of interest. |
|
Diffusion-weighted MRI |
Detects abnormalities in both lesions and normal-appearing CNS tissue. Detects white matter changes. |
|
High field MRI |
Increased sensitivity for MS lesions. Can be used in conjunction with MS spectroscopy or magnetization transfer imaging. |
|
Functional MRI |
Measures critical circuitry involved in response to injury, activation, loss of function, and recovery of function. |
Page 39
brain, therefore, indicates a breakdown of the blood-brain barrier. Gadolinium-enhancing activity on MRI correlates with clinical relapses and predicts increased risk or further disease activity. However, since most new brain MRI lesions are clinically silent, gadolinium-enhanced lesions are seen more often than clinical relapses.
T2-Weighted Hyperintense Lesions. In T2-weighted images, MS lesions appear as very bright white areas against a gray or more neutral background and are the most readily visualized MS lesions by MRI. They reflect lesions with different pathology and of various ages, and reversible as well as irreversible abnormalities. T2-weighted hyperintense lesions can be used to measure the total lesion volume (burden-of-disease). The variable pathology, which is not distinguished in T2 burden-of-disease measures, is probably a determinant of associated disability. Only a modest relationship has been observed between T2 burden of disease and clinical disability in relapsing and secondary progressive MS. However, in patients with clinically isolated syndromes who are in the early stages of MS, T2 burden-of-disease has been correlated with the development of MS, as well as the clinical subtype of MS and disability 10 years later. The magnitude of T2 burden-of-disease changes very early in the disease process and may be valuable for predicting subsequent course.
Atrophy. Atrophy of both brain and spinal cord can be detected in MS patients, including relapsing patients with minimal neurologic deficits.200 Both axon and myelin loss contribute to tissue atrophy. Recent studies suggest that CNS atrophy may be the best neuroimaging correlate for clinical disability (reviewed in 1999 by Trapp et al.213). A number of different methodologies are used to measure atrophy. Current advances involve measurement of the whole brain and improved automation, but the optimal technique has not been decided.
MR Spectroscopy. Axonal injury can be measured on proton MR spectroscopy by estimating N-acetyl aspartate (NAA) levels in brain tissue. NAA is a molecule that is virtually confined to axons and neurons. Levels of NAA can fluctuate, suggesting that they can be used to measure reversible as well as irreversible damage. Persistent reduction of NAA on MR Spectroscopy correlates with axon loss, damage, or dysfunction. Reduced NAA is found not only within MS lesions but also in the normal-appearing white matter of relapsing-remitting, secondary progressive, and primary progressive MS patients. The reduction in NAA is more severe in secondary progressive MS than in relapsing MS. In addition, NAA decrease in cerebellar white matter has been correlated with clinical ataxia.51 NAA can be measured in a discrete region of interest within the brain. Recently, whole-brain NAA has been measured in MS. This appears to be a more meaningful neuroimaging marker to evaluate axon damage. MR spectroscopy can also be used to measure lipid changes within both lesions and normal-
Page 40
appearing brain tissue, but these studies are very preliminary and NAA measurements are the major focus of current MR spectroscopy studies in MS.
T1-Weighted Hypointense Lesions. Also referred to as black holes, T1-weighted hypointense lesions have lower signal intensity than the surrounding white matter. T1 hypointense lesions are most common in the supratentorial region (cerebral hemispheres). They are much less common in the infratentorial (brainstem and cerebellum) region and are not reported in the spinal cord. Compared to T2-weighted lesions, they represent more severe tissue pathology, with axon loss, demyelination, and extracellular edema. 24 In postmortem studies of progressive MS, T1-weighted hypointense lesions correlate strongly with axon density measurements. T1-weighted hypointense lesions show a stronger correlation with disability than T2-weighted hyperintense lesions.
Magnetization Transfer Imaging. Magnetization transfer imaging (MTI) can be used to study global brain function or to measure changes within a local region of interest. 71 , 198 Populations of bound and soluble protons produce different signals in response to the external magnetic field. The magnetization transfer ratio (MTR) is the ratio of the different signals produced by these two populations. It is reduced in MS and is believed to reflect both demyelination and axon loss, thereby producing an index of tissue destruction. MTR measurements are correlated with MS disability, as measured by the Expanded Disability Status Scale (EDSS; see Appendix D), as well as cognitive measures. MTI shows great promise as a disease marker. Lower MTR values occur with disease worsening in relapsing, secondary progressive, and primary progressive MS patients and even in patients with clinically isolated MS syndromes. In primary progressive patients who have a relatively small T2 burden of disease, MTR is significantly reduced, suggesting that axon damage is significantly greater in this clinical subtype.
Differences in MTRs are associated with different lesion pathology. Lesions that are more destructive (as indicated by T1 hypointensity) have reduced MTR values. Lesions that remain hypointense show a persistent reduction in MTR, whereas lesions that become isointense recover in MTR. New lesions in secondary progressive patients have a lower MTR than those in relapsing patients. The decline in MTR over three years is significantly greater in secondary progressive MS than in relapsing MS, supporting a relationship between MTR changes and disease progression. MTR measures allow more significant lesions to be detected and may provide a better potential correlate with clinical disability. Reduction in MTR can precede the development of new lesions on conventional MRI.
Diffusion-Weighted MRI. Diffusion-weighted MRI is sensitive to the diffusion, or random motion, of water molecules in tissue. It can detect subtle pathological changes that are not seen on conventional MRI. This technique
Page 41
might allow detection of pathological change in white matter tracts, including demyelination and loss of axons,223 by quantifying anisotropy through a measure of diffusion tensor imaging (DTI). DTI can identify significantly altered diffusion properties in normal-appearing white matter. Lesions with the highest diffusion are the more destructive black holes, while the greatest change in anisotropy is seen in acute inflammatory lesions. Coincident with new lesion formation, diffusion-weighted imaging has shown changes in contralateral normal-appearing white matter.
High-Field-Strength MRI. High-field-strength magnets, which are 4 tesla (T) or higher, increase the signal-to-noise ratio (conventional imaging machines are 1.5 T). They allow enhanced detection of small (less than 5 mm) MS lesions, particularly those aligned along blood vessels. Both MR spectroscopy and MTI can be conducted on high-field machines with enhanced sensitivity.
Functional MRI. Functional magnetic resonance imaging, or fMRI, is a technique for determining which parts of the brain are activated by different types of sensation such as sight or sound, by different types of tasks such as moving one's fingers or legs, or by different mental tasks such as adding sums, reading, or memorizing. This “brain mapping” is achieved by using an MRI scanner to measure changes in blood flow to different areas of the brain. When a particular brain region is activated, blood flow into the region increases. The incoming arterial blood is rich in oxygenated hemoglobin, and there is a corresponding decrease in local deoxygenated hemoglobin. Changes in the MRI signal are derived from regional changes in the concentration of deoxygenated hemoglobin, which is a paramagnetic molecule (reviewed in Hirsch et al.91). The fMRI signal is, thus, determined by the balance between oxygenated and deoxygenated hemoglobin.
FMRI can provide second-by-second images of changes in response to different stimuli and during performance of mental tasks.183 It provides a unique tool for assessment of neural circuits involved in loss and recovery of function, as well as for measuring the circuits underlying symptoms that are as difficult to study as cognitive changes, fatigue, pain, and sensory disturbances.
Cerebrospinal Fluid
Cerebrospinal fluid (CSF) is the fluid that circulates around and within the brain and spinal cord. CSF provides a vehicle for removing waste products of cellular metabolism from the nervous system and is believed to be nutritive for both neurons and glial cells and to function as a transport system for biologically active substances such as releasing factors, hormones, neurotransmitters, and metabolites. Sampling this fluid thus provides an index to substances active in the CNS and possibly those involved in MS pathology.
Page 42
A number of potential CSF disease markers have been reported in MS, including markers that are proposed as distinguishing between different types of MS (Table 2.5). For the most part, these are markers of tissue damage or immune disturbance. None are currently used in routine clinical practice, since they have not proved useful enough to justify serial lumbar punctures.
There has been particular interest in the specificity of oligoclonal bands in MS. Oligoclonal bands are produced by the overrepresentation of particular anti-bodies that can be visualized when CSF proteins are separated by gel electrophoresis where they appear as separate bands of protein on a gel matrix. Each of the bands contains a single type of antibody produced by a single clone of B cells. Oligoclonal bands are typical for the CSF of MS patients, but they are not exclusive to MS patients. For example, they are also found in the CSF of patients with other inflammatory status, such as viral brain infections. In MS, however, the particular antigens that elicit each antibody band are unknown.210 Investigators have recently used molecular approaches such as phage display libraries to probe MS oligoclonal immunoglobulin G (IgG) bands for sequence information related to their antigenic target.30,48 These are powerful methods that should allow for the identification of antigenic targets for the oligoclonal IgG. A main question, however, is whether the oligoclonal IgG bands represent an immune response directed against the etiologic agent of MS or merely constitute a by-product of immune system activity. In other words, upregulation of the antibody response and the heterogeneous distribution of antibodies into oligoclonal IgG bands could be a result of B-cell hyperactivity rather than an immune response to a specific etiologic pathogen.
Other Studies
A variety of blood, urine, and mucosal fluid disease markers have been studied in MS, but none of them have provided a reliable disease marker. Again they are either markers of tissue damage (such as S-100) or immune activation (such as neopterin). Blood markers have included matrix metalloproteinases and their tissue inhibitors, circulating adhesion molecules, levels of various cytokines and their receptors, different subpopulations of cells, a variety of antibodies including antiviral and autoreactive antibodies, S-100 levels, and neopterin levels. Urine disease markers have included myelin basic protein-like material, free light chains, neopterin, gliotoxin, and neuron-specific enolase. Mucosal fluid cells and immunoglobulins have also been studied.
Diagnosis
The diagnosis of MS is based on both clinical parameters, such as medical history and neurological examination, and paraclinical parameters such as MRI, CSF oligoclonal banding, and evoked potentials. There is no MS-specific diag-
Page 43
nostic test, and the intermittent nature of the disease and high variability in presenting symptoms make diagnosis difficult (listed in Table 2.2). 163 The presentation of MS can be monosymptomatic or have multifocal signs and symptoms, and many neurodegenerative disorders are similar to MS in their presentation. 184
The general diagnostic criteria, established in 1965 by a committee sponsored by the National MS Society (the MS Society), state that a diagnosis of “clinically definite” MS (CDMS) requires clinical evidence of two or more white matter lesions on at least two occasions.195 In 1983, these criteria were expanded by Poser et al. to include the use of paraclinical parameters, and they have since become the standard MS diagnostic criteria (Table 2.6).168 In July 2000, an international committee met to further revise these criteria, in particular to make MRI information a more integral component and to incorporate diagnostic criteria for primary progressive MS. The results of that meeting, however, were not available at the time of this writing.
The failure of the Poser criteria to incorporate primary progressive MS has recently been addressed by revised criteria that define definite, probable, and possible levels of diagnostic certainty.209 These criteria are based on clinical findings, CSF abnormalities, brain and spinal cord MRI abnormalities, and evoked potentials. Using these criteria, at least one year of clinical progression must be documented before a diagnosis of primary progressive MS can be made.209
MRI reveals neuropathological damage in 70 to 95 percent of people with MS and, because of its sensitivity, is the most helpful paraclinical diagnostic test. However, the use of MRI in MS diagnosis has led to concern that its high sensitivity combined with limited MS specificity leads to misdiagnosis, since other conditions including myelopathy and disseminated encephalomyelitis can cause MRI lesions similar to MS lesions.69,167 Thus, it is important that imaging be used in combination with clinical data for the diagnosis of MS. Recently, several sets of criteria for the definition of “MRI-definite” MS have been suggested (Table 2.7).14,69,70,164 Patients with clinically isolated syndromes are particularly difficult to diagnose, and Barkhof et al.14 and Fazekas69 have identified criteria that are relevant to such cases.
Although assessment of spinal cord damage using MRI remains behind the development of brain methodology, it can be useful in diagnosing patients suspected of MS, particularly in cases with equivocal or negative brain MRI results.34,85,128,186 Spinal cord imaging increases the diagnostic sensitivity of MRI and might also enable earlier diagnosis.128,199
Evoked Potentials
When demyelination or sclerosis (scarring) occurs, the conduction of nerve impulses along axons is slowed or interrupted. Impaired conductance is reflected in an increased latency of evoked potentials or an increase in the amount of time that elapses between the presentation of a sensory stimulus and the resulting
Page 44
change in the brain's electrical field. Evoked potentials are measured by placing small electrodes on the head in the region corresponding to the stimuli presented ( Table 2.8).
Abnormal evoked responses to different types of stimuli provide clues to the location of plaques or lesions and are useful in detecting “clinically silent” lesions that do not produce easily observable symptoms. However, abnormal evoked responses are not unique to MS. For example, although abnormal visual
|
TABLE 2.5 Proposed CSF Disease Markers in MS |
|
|
Marker |
Description |
|
IMMUNE MARKERS |
|
|
Free light chains |
IgG antibodies are composed of light and heavy polypeptide chains. Free light chains are found in patients with chronic infections or inflammatory diseases such as rheumatoid arthritis, but also in healthy individuals particularly following strenuous exercise. |
|
Cytokines and cytokine receptors |
Cytokines are intercellular signaling proteins produced by cells of the immune system and CNS. They are involved in various aspects of disease processes, particularly inflammatory responses. |
|
Oligoclonal bands |
Oligoclonal bands are produced by the overrepresentation of particular antibodies. They are typical of the CSF of MS patients, but not exclusive to it. |
|
Antiviral antibodies |
Antiviral antibodies are produced by B cells in direct response to an antigen's presence, and certain antiviral antibodies are increased in the CSF of some patients with MS. 194 |
|
Intrathecal immunoglobulin production (IgG, IgM) |
Immunoglobulins are produced by plasma cells and are integral in adaptive immune responses. Polyclonal increases of IgG occur in chronic infection and inflammation. |
|
T cells |
White blood cells responsible for cell-mediated immune responses to antigens, including viral infections. |
|
Adhesion, costimulatory, and other surface of MS. molecules |
Upregulated adhesion molecules in blood and CSF indicate sustained potential for inflammation in the CNS throughout the clinical spectrum |
|
CNS TISSUE MARKERS |
|
|
Myelin basic protein (MBP) |
A major component of myelin, MBP is increased in the CSF of some, but not all, MS patients following a demyelinating episode. |
|
continued |
|
Page 45
evoked potentials are common in MS, they also occur in compressive lesions of the visual pathway and spinocerebellar degeneration.184
Evoked potentials can aid in the localization of lesions, confirm clinically ambiguous lesions, and confirm the organic basis of symptoms.84 In addition, changes in evoked potentials can be used to measure disease progression and the effectiveness of therapeutic treatment, including treatments designed to improve conduction.61,155
|
Marker |
Description |
|
S-100 |
S-100 protein is an astroglial-specific protein that binds calcium. When a brain lesion occurs, S-100 is released into both the CSF and the blood. 142 |
|
Neuron specific enolase (NSE) |
As a marker of brain damage, NSE reflects the severity of disease in patients with intracerebral hematoma, subarachnoid hemorrhage, head injury, and certain tumors.165 |
|
Glial fibrillary acidic protein (GFAP) |
A major constituent of glial filaments in differentiated CNS astrocytes, GFAP has been used for the diagnosis of astrocytic tumors, the study of astrocytic gliosis, and CNS regeneration and transplantation.62 |
|
Neurofilaments |
Neurofilaments are important for axonal structure, transport, and regeneration. Accumulation of neurofilaments in motor neurons can trigger a neurodegenerative process and may be a key intermediate in the pathway of pathogenesis leading to neuronal loss. 185 , 230 |
|
Neural cell adhesion molecules |
A modulator of axon outgrowth and cell adhesion that adapts its structure to requirements during development by alternative splicing and posttranslational modification. 60 |
|
Ciliary neurotrophic factor (CNTF) |
CNTF appears to promote remyelination, as well as formation of oligodendroctyes.132 |
|
INFLAMMATORY AND OTHER MARKERS |
|
|
Gliotoxin |
Highly cytotoxic for astrocytes and oligodendrocytes, gliotoxin may represent an initial pathogenic factor leading to the neuropathological features of MS, such as blood-brain barrier involvement and demyelination.139 |
|
Neopterin |
A marker of immune activation, neopterin is increased in CSF of relapsing-remitting patients and correlates with a decrease of L-tryptophan, reflecting interferon-gamma-mediated activation of macrophages.201 |
|
Matrix metalloproteinases |
Matrix metalloproteinases are enzymes that can dissolve the extracellular matrix of the blood-brain barrier.196 |
Page 46
|
TABLE 2.6 Poser Diagnostic Criteria for MSa |
||||
|
Category |
Attacks |
Clinical Evidence |
Paraclinical Evidence |
CSF OB/ IgG |
|
Clinical Diagnosis |
||||
|
Definite |
2 |
2 |
||
|
2 |
1 |
and |
1 |
|
|
Probable |
2 |
1 |
||
|
1 |
2 |
|||
|
1 |
1 |
and |
1 |
|
|
Laboratory-Supported Diagnosis |
||||
|
Definite |
2 |
1 |
or |
1 + |
|
1 |
2 |
+ |
||
|
1 |
1 |
and |
1 + |
|
|
Probable |
2 |
+ |
||
aCombinations of various types of evidence are used to diagnose MS under the Poser criteria. More than one combination of clinical and paraclinical evidence can support a diagnosis within a single category. Laboratory-supported diagnosis requires one of two possible immune disturbances in CSF: IgG oligoclonal bands or intrathecal IgG production.
NOTES: CSF, Cerebrospinal fluid. OB, Oligoclonal bands. IgG, Immunoglobulin G. Clinical evidence refers to symptoms recorded in medical history or signs observed in neurological examination. Paraclinical evidence might include neuroimaging, evoked potentials, CSF oligoclonal banding, or IgG levels.
|
Paty et al.163
|
|
Barkhof et al.14
|
|
Fazekas et al.69,70
|
Page 47
|
TABLE 2.8 Evoked Potentials as a Diagnostic Tool in MS |
|||||
|
Frequency of Abnormal Responses (%) |
|||||
|
Evoked Response |
Primary Purpose of Test |
Stimulus Presented |
Location of Recording Electrodes |
People with Definite MS |
People with Probable MS |
|
Visual evoked responses |
Evaluation of optic nerve function |
Strobe light flash or reversible checkerboard pattern flash on a computer screen |
On the scalp along the vertex and cortex lobes |
85-90 |
58 |
|
Brainstem auditory evoked potentials |
Evaluation of hearing pathways in the brainstem |
Series of clicking noises or tone bursts played into earphone |
On the scalp along the vertex and on each earlobe |
67 |
41 |
|
Somatosen-sory evoked responses |
Evaluation of sensory nerve tracts in spinal cord, thalamus, and sensory cortex |
Mild electrical stimulus via electrodes on wrists or knees |
On the scalp, each wrist (medial nerve), and the knees (peroneal nerve) |
77 |
67 |
The use of evoked potentials as a diagnostic tool has greatly declined since the advent of the MRI, which provides a more comprehensive picture of disease activity. In at least some cases of progressive MS, visual evoked potentials show changes over time where none are detected in MRI scans.192 In May 2000, the Quality Standards Subcommittee of the American Academy of Neurology concluded that although visual evoked potentials are probably useful to identify patients at increased risk of developing clinically definite MS, somatosensory evoked responses are only possibly useful for that purpose, and there is insufficient evidence to recommend brainstem auditory evoked potentials as a diagnostic tool.84
Disease Variants: Is MS One Disease or Many?
Although MS is postulated to have an underlying immune-mediated pathogenesis, there is as yet no biologic marker that is disease specific and can be used for diagnostic purposes. Similarly, there is insufficient evidence to allow detection of any putative disease-related infectious agent as a basis for defining the disease. Thus, MS continues to be defined by sets of criteria that have been
Page 48
developed based on clinical and pathologic observations. MS may well be heterogeneous when viewed from the perspective of genetics, pathogenic mechanisms, clinical phenotypes, and immunopathology. To be considered a truly distinctive variant of MS, any putative distinct disease subtypes defined in one of these categories would have to be correlated with the distinctive features identified in each of the other categories.
Cases classified as MS are recognized where disease distribution is mainly in the spinal cord, hind brain (cerebellum or brainstem), or cerebrum. Different animal models have distinct topographic distributions, some of which seem to have distinct immunopathologies. For example, Theiler's murine encephalomyelitis virus, the demyelinating disease that afflicts mice, is mainly a spinal cord disease. There is an apparent overrepresentation of specific phenotypes in certain geographic regions. For example, MS that is relatively restricted to the optic nerve and spinal cord is more common in Japan than in other countries.
The Devic's pattern of MS features a predominance of spinal cord and optic nerve involvement. The pathology is considered more destructive than classical MS, and the prognosis is worse. At issue is whether these differences reflect different immunopathogenic mechanisms in a given individual, even when the disease trigger, or initiating event, is not distinct among such individuals. Even in identical twins with MS, the disease course can be markedly different.
Disease-Modifying Therapies
A number of immunomodulatory agents have been shown in double-blind, placebo-controlled, multicenter Phase III trials to benefit patients with relapsing MS ( Table 2.9; see also 1999 review by Rudick 187). These agents help clinical disease features (they decrease the number of attacks, the severity of attacks, and sustained worsening on neurologic examination) as well as MRI disease features (they decrease the formation of new lesions, the number of contrast-enhancing lesions, the total burden of disease, and brain atrophy). Although all of these drugs have side effects, they are manageable in most patients. The benefit of treatment is sustained for at least several years. It is not yet known whether these agents prevent, reduce, or delay transition from relapsing to progressive MS, but preliminary data suggest that this may be the case. Throughout this report, the term “disease-modifying therapy” is used to distinguish these agents from other medications used to relieve the symptoms of MS that do not alter either the frequency of relapses or the rate of progression.
Beta-interferon (IFN-β) is an anti-inflammatory regulatory cytokine with antiviral, antineoplastic, and immunomodulatory activity. It has a number of effects on the immune system that would be beneficial in MS. For example, it decreases cell migration into the CNS, inhibits T-cell proliferation and expression of cell activation markers, inhibits inducible nitric oxide synthase (the enzyme that produces nitric oxide, a potentially damaging substance), and enhances
Page 49
|
Therapy |
Dosing |
Major Side Effects |
|
CYTOKINE THERAPIES |
||
|
Interferon-β1b (Betaseron) |
250 μg s.c. alternate days |
Flu-like symptoms, injection site reactions, menstrual irregularities, decreased white blood cells, elevated liver enzymes |
|
Interferon-β1a (Avonex) |
30 μg i.m. once a week |
Flu-like symptoms, pain from intramuscular injection |
|
Interferon-β1a (Rebif) |
22 μg and 44 μg s.c. three times a week |
Flu-like symptoms, injection site reactions, decreased white blood cells, elevated liver enzymes |
|
T-CELL THERAPIES |
||
|
Glatiramer acetate (Copaxone) |
20 mg s.c. daily |
Injection site reactions (mild), Immediate postinjection reaction |
|
IMMUNOSUPPRESSIVE THERAPIES |
||
|
Mitoxantrone (Novantrone) |
12 mg/m2 i.v. once every 3 months |
Nausea, hair thinning, menstrual irregularities, infertility, decreased white blood cells, transient discoloration of urine and sclera |
NOTE: i.m. = intramuscular; i.v. = intravenous; s.c. = subcutaneous.
production of the anti-inflammatory cytokine interleukin-10 and of nerve growth factor (which might enhance remyelination and axon repair) (reviewed in 1999 by Rudick187). There are two types of recombinant (artificially made) beta-interferon. Beta-interferon-1a (Avonex, Rebif) is a duplicate of human beta-interferon. * Beta-interferon-1b (Betaseron) has three molecular differences from human beta-interferon: it is not glycosylated, there is an amino acid substitution at position 17, and there is no “N-terminal” methionine.6 The three available beta-interferon therapies are given in different amounts and dosing schedules (Table 2.9). There are well-recognized side effects (most commonly flu-like reactions), which occur maximally during the first weeks or months of therapy. Flu-like reactions can be minimized by initiating therapy with a dose escalation schedule and consistent use of anti-inflammatory premedication during the first few weeks of therapy.
*Rebif has been approved for the treatment of relapsing-remitting MS by the European Commission but has not been approved by the Food and Drug Administration in the United States because of the Orphan Drug Act. If tentative approval is received, Rebif could enter the U.S. market in 2003, when the exclusivity periods for Avonex and Betaseron end.
Page 50
Glatiramer acetate (Copaxone) consists of random polymers of four amino acids, designed to mimic myelin basic protein, an important component of CNS myelin. Glatiramer acetate is believed to work by activating antiinflammatory regulatory T cells, which then migrate into the CNS to inhibit local immune reactions. Glatiramer acetate has an excellent side effect profile. Patients may experience injection site reactions, but they tend to be quite minor. Some 10 to 15 percent of patients experience at least one immediate postinjection reaction characterized by chest tightness, palpitations, flushing, and anxiety within a few minutes of injection. The reaction lasts only minutes and is not dangerous.
Mitoxantrone (Novantrone) is a cytotoxic agent that interferes with DNA synthesis and repair, and suppresses a variety of immune system cells. It also enhances suppressor cell activity. It is given as an intravenous infusion over 5 to 15 minutes, every three months. Mitoxantrone is fairly well tolerated at low doses. In the recent Phase III trial, both the low (5 mg/m2) and high (12 mg/m2) doses showed efficacy, but the high dose gave the best overall results.111 Mitoxantrone should not be given at a cumulative dose of 140 mg/m2 or higher because of concerns about cardiotoxicity, which also means that this drug can be used for only a few years.
Currently available treatments are highly effective in preventing the type of MS damage that can be visualized using MRI. They are moderately effective in preventing and reducing the severity of relapses, but they are generally disappointing in preventing long-term disability—the most important goal of treatment. This might reflect the timing of treatment, and there has been a recent emphasis on starting therapy at the time MS is first diagnosed. This type of early therapy is likely most effective in delaying or preventing long-term disability, although this effect has not yet been clearly demonstrated through empirical research studies (Richard Rudick, personal communication). Clearly, much remains to be done in the development of therapies for people who suffer from MS.
Two recent studies, the interferon beta-1a (Avonex) prevention study (CHAMPS, Controlled High Risk Subjects Avonex Multiple Sclerosis Prevention) and the interferon beta-1a (Rebif) early treatment (ETOMS, Early Treatment of Multiple Sclerosis) trial, have compared the use of disease-modifying therapy with placebo in patients after their first attack who also have an abnormal brain MRI. These are patients at high risk for MS, but who do not meet current criteria for a definite diagnosis. In both studies, early treatment with a disease-modifying agent significantly delayed onset of a second clinical attack over the two-year study period. Patients who received treatment also showed significantly less MRI disease activity over the next two years. These two trials have led to a reassessment of when disease-modifying therapy should be started. The National MS Society consensus statement endorsed treatment of patients as soon as a definite diagnosis of relapsing MS is made. If new diagnostic criteria are formulated for an MRI-based diagnosis at the time of a first attack, it is likely that the use of disease-modifying therapy will expand to include these early patients.
Page 51
Beta-interferon and glatiramer acetate have been tested mainly in relapsing MS. It is more controversial whether they benefit progressive MS. Several Phase III trials have examined beta-interferon therapy in patients with secondary progressive MS, with conflicting results. The European Secondary Progressive Study on beta-interferon-1b (Betaseron) showed a significant effect on slowing progression. In contrast, the North American Secondary Progressive beta-interferon-1b study and the European SPECTRIMS beta-interferon-1a (Rebif) study showed no significant effect on progression. These trials did, however, show positive results on secondary outcomes such as relapse rate and MRI disease parameters. The European study, which showed a treatment effect on progression in contrast to the two negative studies, included secondary progressive patients who had a shorter disease duration, were still experiencing relapses, and had contrast-enhancing brain MRI lesions. Considered as a whole, these studies suggest that in the earlier stages of MS, when there is still a significant inflammatory component (reflected in clinical relapses and gadolinium-enhancing lesion activity), beta-interferon may have a positive effect on clinical progression. In the later, nonrelapsing, progressive stages of MS, where there appears to be ongoing atrophy relatively independent of contrast-enhancing lesion activity, beta-interferon does not seem to slow progression. The European Phase III trial of Mitoxantrone enrolled both relapsing and secondary progressive MS patients.111 The drug had a positive effect on progression as indicated by a decrease in the EDSS impairment scale at the end of the study (change of -0.13 compared to +0.23 in the placebo group, p = .038) (for EDSS scale, see Appendix D). An ongoing trial (IMPACT Study) in secondary progressive MS is testing double-dose interferon beta-1a (Avonex) once a week. There have been no major treatment trials in primary progressive and progressive relapsing MS. There currently is an ongoing three-year Phase III trial of glatiramer acetate in primary progressive MS (the Promise trial).
Treatment Failures
Each of the currently available disease-modifying treatments has shortcomings, including partial efficacy for patients as a group and potential adverse effects. There are four reasons why treatments fail—nonadherence on the patient's part, adverse side effects, production of neutralizing antibodies, and nonresponsive disease (reviewed by Cohen at al., 199943).
Patient adherence is a factor in the efficacy of any medication, but particularly so when a patient's hopes exceed the outcome—which is particularly salient for therapies that are preventive, but not restorative. The primary principles to increase adherence are appropriate selection of patients for treatment, availability of adequate medical support throughout treatment, and perhaps most importantly, patient education before and during treatment. MS patients need to be fully informed that the therapy can prevent relapses and the accumulation of disability,
Page 52
but that it will not improve preexisting manifestations. In addition, some patients are averse to self-injection and will not be persuaded to inject themselves with any medication (unless, perhaps, they are convinced of a substantial and certain benefit, which many MS patients are not).
Health care financing policies vary widely among different countries and even within countries such as the United States and Canada. This will also influence patient adherence. In countries such as France where the costs of MS therapies are fully covered by the state, cost is unlikely to influence adherence, but in countries where patients must assume the full costs of MS disease-modifying therapies themselves, many of them will decide that they cannot afford to pay more than $10,000 (U.S.) annual expense for the modest health benefit they might gain.
Adverse effects are a common reason for discontinuing treatment, but they are generally not serious health threats. The most common side effect of glatiramer acetate is irritation at the injection site, although it is typically mild. The most common side effects of beta-interferon are flu-like symptoms, and these usually resolve after three to six months. (Depression is also a possible side effect and is discussed in greater detail in Chapter 3.) Although only 4 percent of actively treated patients withdrew from beta-interferon clinical trials because of side effects, in clinical practice, 11 percent discontinue within four months of initiating treatment (reviewed in 1999 by Mohr et al.144). (The percentage of patients who discontinue treatment because of side effects might, however, decline as physicians become more experienced in managing these side effects.)
Defining nonresponsive disease in individual patients is difficult, because unfortunately patients can continue to experience relapses after initiation of disease-modifying therapy with beta-interferon or glatiramer acetate. Comparison of pre- and post-treatment relapse rates is fraught with problems, and it is often difficult to identify whether an individual patient is responsive to therapy. At a minimum, a patient's level of neurological impairment and disability should remain stable on therapy. Persistent gadolinium-enhancing or T2 lesion accrual should be considered a worrisome feature, even in the absence of clinical evidence of activity or worsening.
Finally, neutralizing antibodies, which can interfere with the effects of interferons, appear in up to 20 percent of patients after two years of beta-interferon treatment. They do not appear to be an issue with glatiramer acetate. Increased neutralizing antibodies appear to be associated with reduced clinical benefit, although there is still some controversy about this point. For example, it is still not known what levels of neutralizing antibodies are clinically significant, how often they persist, and what is the most reliable method of measuring them. More research is needed on testing for neutralizing antibodies in MS patients treated with beta-interferons and how to best use the results to properly manage patient treatment.
Page 53
Cost-Effectiveness
Disease-modifying therapies are expensive, costing roughly $8,000 to $10,000 (U.S.) per year.187 Several studies have indicated that their costs outweigh their benefits, but these analyses have been heavily criticized.29,177 Forbes and colleagues73 argued that it is not cost-effective to treat progressive MS patients with interferon beta-1b in Britain and that the money spent on interferon beta-1b would be better spent on other services, such as supportive care and simple interventions to reduce the burden of patients' symptoms. Analyses of cost-effectiveness of medical treatment often use quality-adjusted life-years (QALYs) to measure health benefits (see Box 2.2). The estimated costs of beta-interferon treatment per gain in QALY for relapsing-remitting MS range from 809,000 British pounds ($1,140,000 U.S.)162 to 2,038,400 British pounds ($2,870,000 U.S.).152 The estimates are considerably lower in Canada (406,000 to 490,000 Canadian dollars, or $270,000-$330,000 U.S.).159 For secondary progressive MS, the estimated costs of beta-interferon treatment per gain in QALY range from 874,600 British pounds ($1,230,000 U.S.)152 to 1,024,000 British pounds ($1,440,000 U.S.).73 While disease-related expenditures are relatively easy to calculate, the benefits of these expenditures are not so easy to calculate, particu-
BOX 2.2What Are Quality-Adjusted Life-Years?Quality-adjusted life-years are a health status measure that includes both quantity and quality of life in a single outcome measure.21,98 Multiplying life expectancy by a quality-of-life adjustment fraction, or utility, results in a QALY value. Utilities represent the level of quality or value associated with a particular health state, and range in value from 0, representing death, to 1, representing optimal health. Utilities are determined in two ways: by researchers through literature searches for previously conducted utility studies or by direct measurement of utility values based on the assessment of people's values for different levels of health. The use of QALYs for resource allocation has been widely criticized as discriminatory against the elderly21 and the disabled98 by placing less value on extending their lives due to the lower potential for health status improvement. The determination of utility values that accurately reflect the many ways quality of life can be affected is difficult in part because one of the underlying assumptions of QALYs— that the severity of a disease state or disability and the corresponding value represent a fixed quantity—is unlikely to be true. The effect of health status on quality of life depends in large part on an individual's unique perspective.59 Although the use of QALYs is one of the current standards for such evaluations, it is a crude attempt to express a qualitative, subjective reality in quantitative terms and has been criticized as being of limited value in this respect.64 |
Page 54
larly in long-term diseases where treatments might slow the progress of disease, but neither cure it nor restore impaired functions.
In addition to the limitations inherent in the use of QALYs, there are other problems with the conclusion that disease-modifying therapies are not cost-effective. The studies done to date have been criticized for using poor economic methodology in interpreting the data.64,177 In a year 2000 review of immunomodulatory drugs used to treat MS, the British National Health Service committee on health technology assessment concluded that the cost-effectiveness of these drugs is simply not known.29 The committee cited a lack of quality clinical trials for each drug, including methodological limitations, poor reporting of data, small sample sizes, short duration, inconsistent treatment regimes and outcome measures, and uncertainty about the clinical significance of reported benefits.29 The latter criticism might, however, change. New data about these drugs are emerging at such a rapid pace that conclusions about their benefits should be reconsidered as new data from clinical trials become available.
In general, cost-effectiveness analysis should be considered skeptically. Cost-effectiveness is a highly politicized issue in which economic principles are often misapplied.64 Indeed, the United States health care system often favors economically inefficient delivery of some products—for example, liver transplants—in that health care providers are willing to underwrite additional costs to gain market share. Further, cross-national comparisons have little merit because of differences in national health care systems, as well as other economic and social factors. Finally, although economic analysis reveals important financial trade-offs, all societies hold certain social values that outweigh economic considerations. For many people, the health and well-being of their loved ones and themselves is among the deepest of these values.
UNDERLYING DISEASE MECHANISMS
Ultimately, the pathogenic mechanisms underlying MS will have to be better understood to design rational therapies.
Physiology of Myelin and Axons: Normal Function, Demyelination, and Repair
The integrative activity of the nervous system, which underlies motor, sensory, cognitive, and psychological behavior, depends on electrical signaling between neurons. Each neuron encodes its message in the form of action potentials (small all-or-none electrical impulses) that are carried to other neurons via axons, the cable-like fibers that extend from neuron cell bodies. Many axons within the brain and spinal cord are myelinated.
Page 55
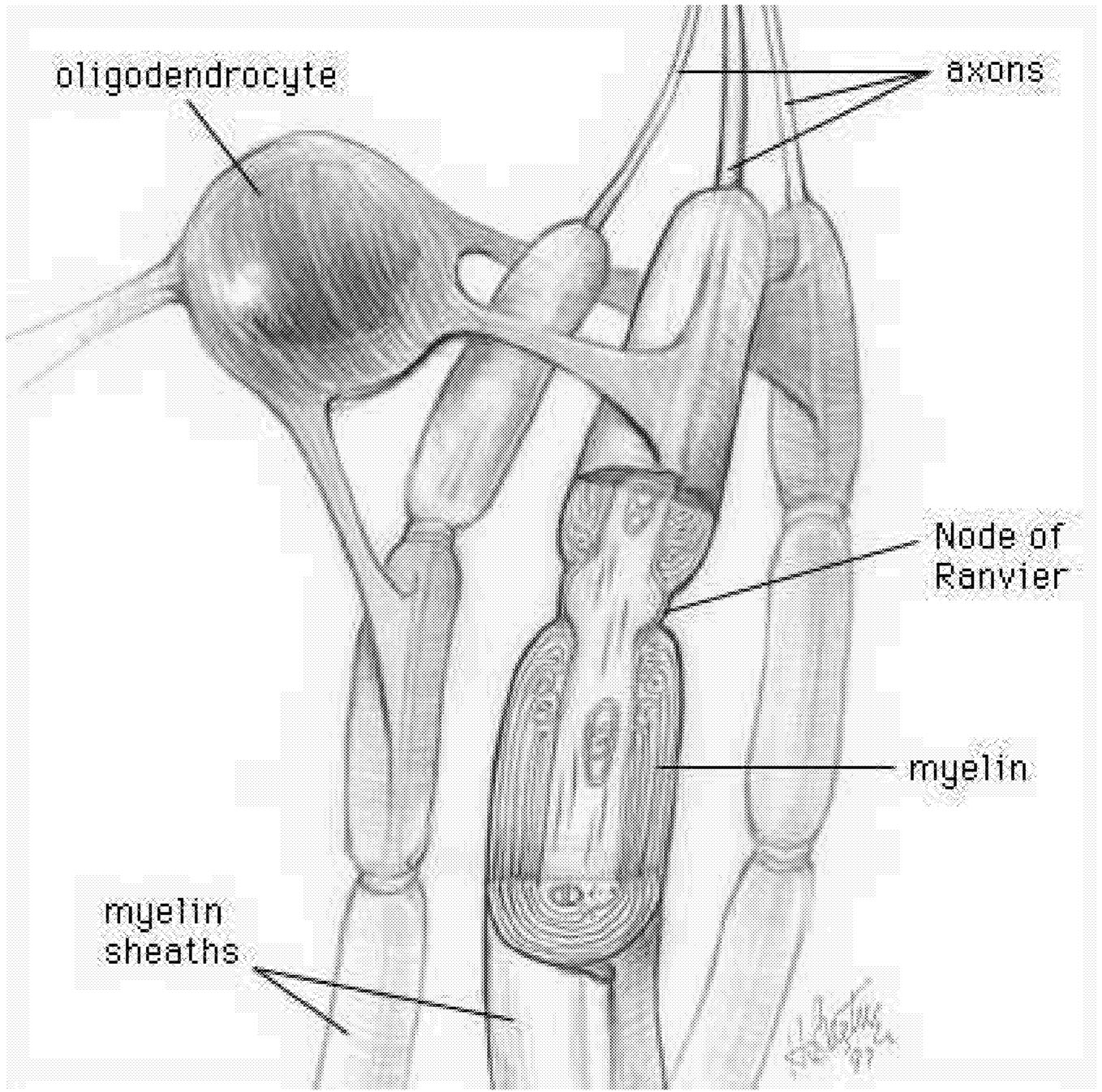
Myelin Acts as an Insulator
The myelin sheath provides a high-resistance, low-capacitance insulator that increases the reliability and speed of action potentials conducted along axon fibers. Myelin is what makes the white matter of brain white. It is a multilayered sheath formed by the oligodendroglial cells, or oligodendrocytes, that insulate axons ( Figure 2.4).169,173 Each segment of the myelin sheath surrounds the axon in a segmented fashion, with segments (called internodes) periodically interrupted at nodes of Ranvier. The internodal axon is normally surrounded by myelin sheaths whose thicknesses are related to the caliber of the ensheathed axon.219 In normal
~ enlarge ~
FIGURE 2.4 Oligodendrocyte making myelin. The processes of a given oligodendrocyte wrap themselves around portions of the surrounding axons. As each process wraps itself around, it forms layers of myelin. Each process thus becomes a segment of the axon's myelin sheath. SOURCE: National Institutes of Health Office of Science Education.
Page 56
myelinated fibers, the action potential does not travel in a continuous manner. On the contrary, it jumps from one node of Ranvier to the next, in a manner known as “saltatory” (derived from the Latin word for “jumping”). Saltatory conduction is a rapid process, with the impulse taking only 20 one-thousandths of a second to jump from one node of Ranvier to the next; as a result, myelinated fibers conduct impulses with a high velocity. Significantly, voltage-dependent sodium channels are clustered at the nodes of Ranvier but are relatively more scarce in the internodal axonal membrane. In contrast, potassium channels, which exist in low density at nodes, are more abundant in the internodal and paranodal axonal membrane, under the myelin sheath.219 Therefore, loss of the myelin sheath exposes relatively inexcitable axonal membranes; the consequence is that nerve impulses are conducted more slowly or not at all.
Axonal Conduction Is Impaired in Demyelinated Axons
Following damage to the myelin, conduction velocity is reduced, and conduction slows along the demyelinated axon ( Figure 2.5). Studies using evoked potentials to examine human subjects with MS have demonstrated that this slowing of conduction does not, in itself, necessarily produce clinical deficits.88,96,134 In addition, however, conduction failure can occur in demyelinated axons. When conduction failure occurs, the axon potential is not propagated from one end of the fiber to the other, and information is lost. This produces a clinical deficit. Conduction failure in demyelinated axons is now known to result not only from loss of the insulating myelin, but also from the molecular organization of the axon membrane. Following damage to the myelin, internodal parts of the axon membrane (which had previously been covered by myelin) are uncovered.
Myelinated Axons Exhibit Complex Molecular Architecture
Prior to the last decade, axonal dysfunction in demyelinating diseases was considered to be due entirely to the loss of the myelin insulation. According to this schema, after the myelin is damaged, there is a “short circuit,” and impulse conduction is slowed or fails. It is now known that although the schema described above is partially correct, it is not the whole story. The axon itself exhibits an elegant molecular architecture, and following damage to the myelin, this architecture is disrupted. The molecular architecture of the axon is manifest by the placement of specialized protein molecules, called ion channels, within the membrane of the axon. Sodium channels act as tiny molecular batteries, which produce the depolarization that is necessary for the generation of action potentials. In contrast, potassium channels act as molecular brakes, damping electrical activity. Within myelinated axons, these two types of ion channels have a complementary structure. Sodium channels are clustered in high density in the axon membrane at small gaps in the myelin called nodes of Ranvier, where they support the produc-
Page 57
tion of action potentials; they are sparse, however, in the “internodal” parts of the axon membrane beneath the myelin. Their numbers there are too low to support secure conduction, which contributes to conduction failure. Potassium channels, on the other hand, tend to be located in the internodal parts of the axon membrane, beneath the myelin sheath; as a result of this, they are masked by the
~ enlarge ~
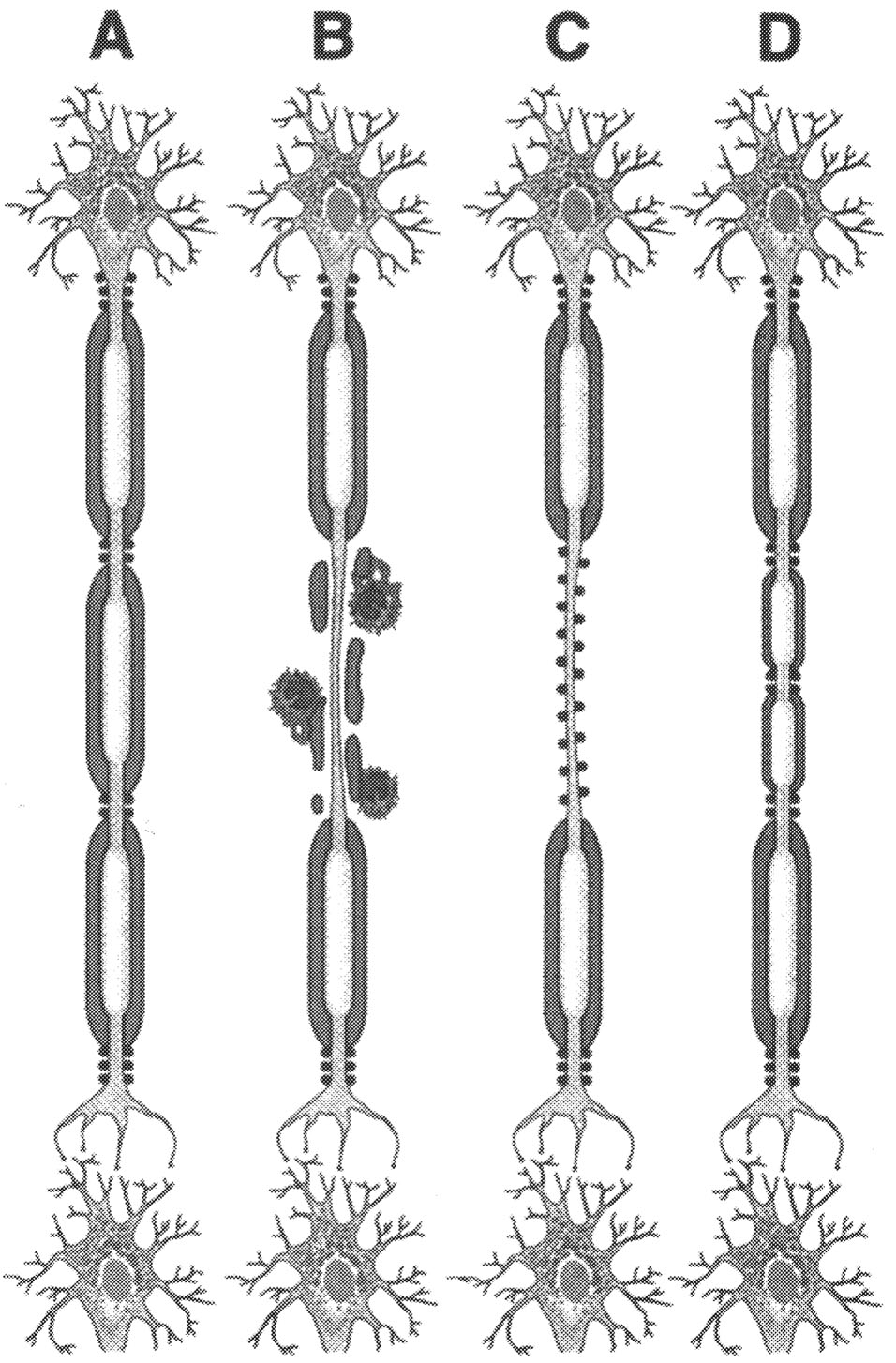
FIGURE 2.5 Pathogenesis. Current concept of pathogenesis of neurological dysfunction associated with acute multiple sclerosis lesion in relapsing-remitting MS patient. Normal myelinated fibers (A) are demyelinated by inflammatory process (B), which causes conduction block. Na+ channel redistribution (C) and remyelination (D) restore conduction and contribute to clinical remission. SOURCE: Trapp et al.213 Reprinted with permission of Sage Publications, Inc.
Page 58
overlying myelin in normal myelinated fibers.177,181,218 The unmasking of potassium channels by demyelination thus introduces another factor that tends to interfere with the conduction of action potentials.
Molecular Plasticity in Demyelinated Axons Underlies Restoration of Impulse Conduction
Given that impulse conduction fails in demyelinated axons and that this contributes to clinical deficits, how do remissions occur? It is now clear that demyelinated axons possess a remarkable capability to rebuild themselves at the molecular level. In the weeks following demyelination, demyelinated axons acquire, within regions where myelin has been lost, a density of sodium channels that is high enough to support action potential conduction even in the absence of insulating myelin. The demyelinated nerve fibers insert additional amplifiers (sodium channels) in their membranes so that they are able to conduct action potentials reliably even though there is a short circuit.23,55,63,74,145
This is a striking example of neuronal plasticity, in this case at the molecular level. Although this molecular plasticity has been clearly demonstrated in laboratory experiments, a number of important questions remain: How do neurons “know” that their axons have been demyelinated and that there is a need to activate the machinery for synthesis of sodium channels? How do neurons control the synthesis and deployment of sodium channels? What turns on the genes for sodium channels and ensures that the correct types of sodium channels (there are nearly a dozen different types, which are like different types of batteries) are produced following demyelination? Also, how are sodium channels transported and inserted into the correct parts of the axon membrane so that they can function normally? These questions have important therapeutic implications and are currently under study.
Axonal Degeneration Also Occurs in MS
The presence of axonal degeneration in MS was recognized even in the early descriptions of this disease,36 but its presence has recently been reemphasized (Figure 2.6).135,212,219 Axonal transection might be the structural basis for acquisition of permanent (nonremitting) neurological deficits, making it an especially important part of the pathology of MS.51,119,120 A corollary of this proposition would be that neuroprotective interventions that limit axonal injury should prevent, or at least lessen, the acquisition of new, permanent signs and symptoms in MS. As a step toward the development of neuroprotective strategies in MS, it will be important to delineate the mechanisms that underlie axonal injury in this disorder. Is it a consequence of demyelination? Alternatively, is the axonal damage a by-product of the inflammatory or immune processes involved in triggering demyelination? What is the nature of the “injury cascade” that leads from the
Page 59
initial insult to ultimate degeneration of axons in MS? Understanding the pathogenesis of the process might lead to the development of new therapeutic targets for MS. These questions are being approached in models of other neurological diseases such as trauma and cerebrovascular disease, including stroke, and should be actively pursued in MS research as well.
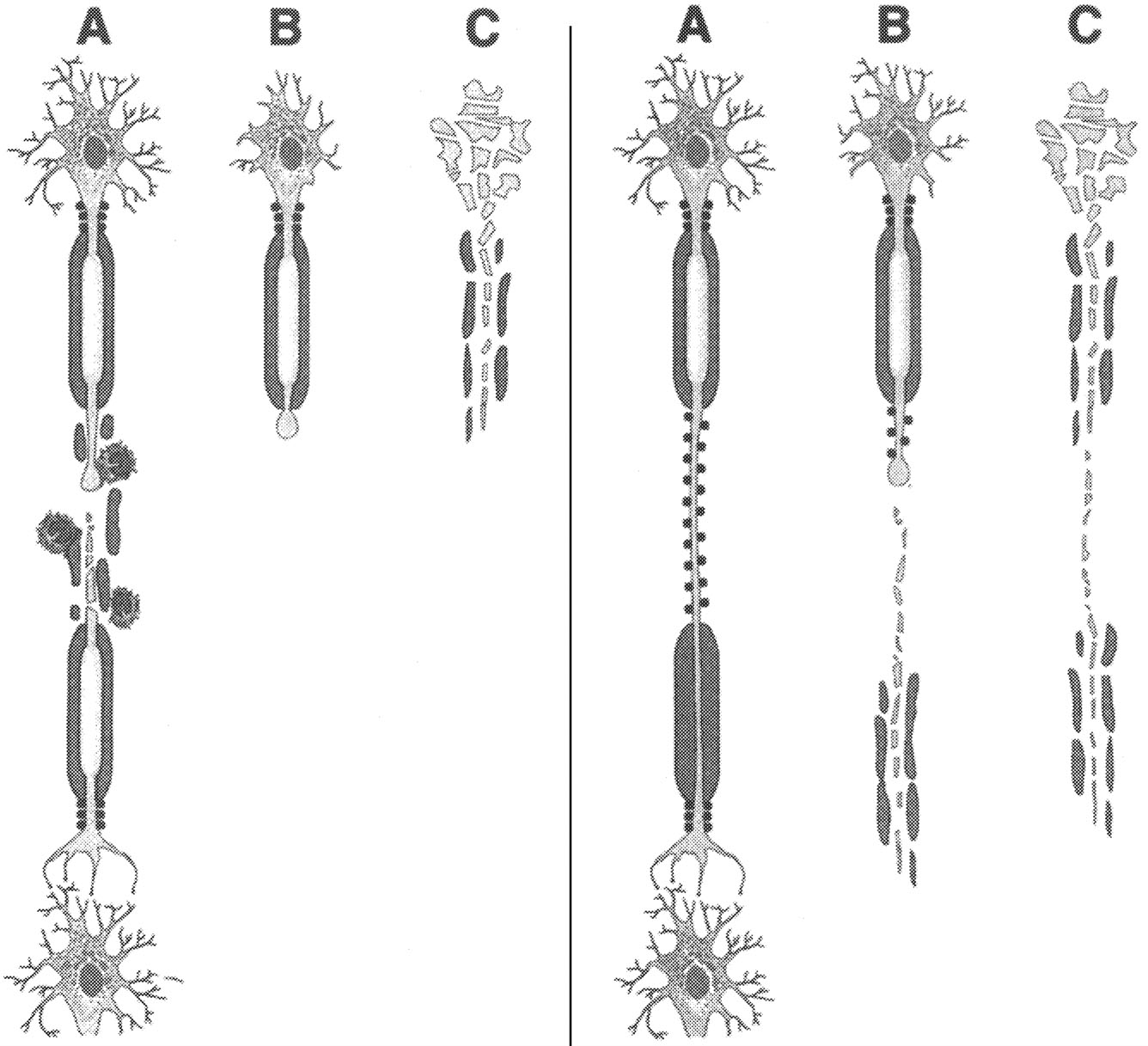
~ enlarge ~
|
Axonal transection during inflammatory demyelination. According to this schema, axonal transection during (A) is a consistent feature of inflammatory demyelinating lesions. This results in degeneration of the distal axonal segment (B) and irreversible loss of neuronal function. During the relapsing-remitting course of multiple sclerosis (RR-MS), the CNS compensates for axonal destruction. |
Axonal degeneration as a result of chronic demyelination. This model posits that axonal viability depends upon oligodendrocyte-derived trophic effect. Chronically demyelinated axons (A) may undergo nerve transection (B) or wallerian degeneration (C), which are caused by lack of myelin trophic support. |
FIGURE 2.6 Axonal transection and degeneration. SOURCE: Trapp et al.213 Reprinted with permission of Sage Publications, Inc.
Page 60
Demyelination and Clinical Signs
MS is defined as a demyelinating disease because the myelin sheaths and their parent cells, the oligodendroglia, are major targets of immune-mediated damage (Figure 2.7).46,153 The classical lesions are discrete plaques of demyelination, which, depending on disease stage, are associated with varying evidence of inflammation. The clinical signs, which are episodic and are not clinically predictable, are presumably related to the location of the lesions, although clinically “silent” demyelinating lesions also occur. These predominantly white matter lesions occur in multiple brain regions and appear at different times throughout the disease. Common syndromes correlated with lesions in specific areas include visual deficits, weakness and spasticity, eye movement abnormalities, and ataxia (Table 2.10). Lesions are often described as active or chronic, depending on whether there are signs of active inflammation, usually associated with ongoing demyelination, or whether the lesion is stable and does not show signs of inflammatory activity.
Lesions
Active Lesions. Disruption of the blood-brain barrier that normally insulates the brain from pathogenic blood-borne substances is an early event in the development of MS lesions (Figure 2.8).136 Antigen-specific T cells enter the nervous system, and when they encounter and recognize their specific antigen, a cascade of cytokine expression begins that contributes to the damage of the blood-brain barrier. This can be detected on contrast-enhanced MRI.136 Examination of active demyelinating lesions in autopsy cases of MS reveals structural and immunopathological abnormalities related to demyelination and abnormalities of oligodendrocytes.126 The inflammatory response is dominated by lymphocytes and macrophages, but the data on the relative numbers of CD4+ and CD8+ cells are still not settled. A plaque is characterized by loss of myelin sheath and infiltration by macrophages (which show myelin basic protein and myelin-associated glycoprotein immunoreactivities). As the
Page 61
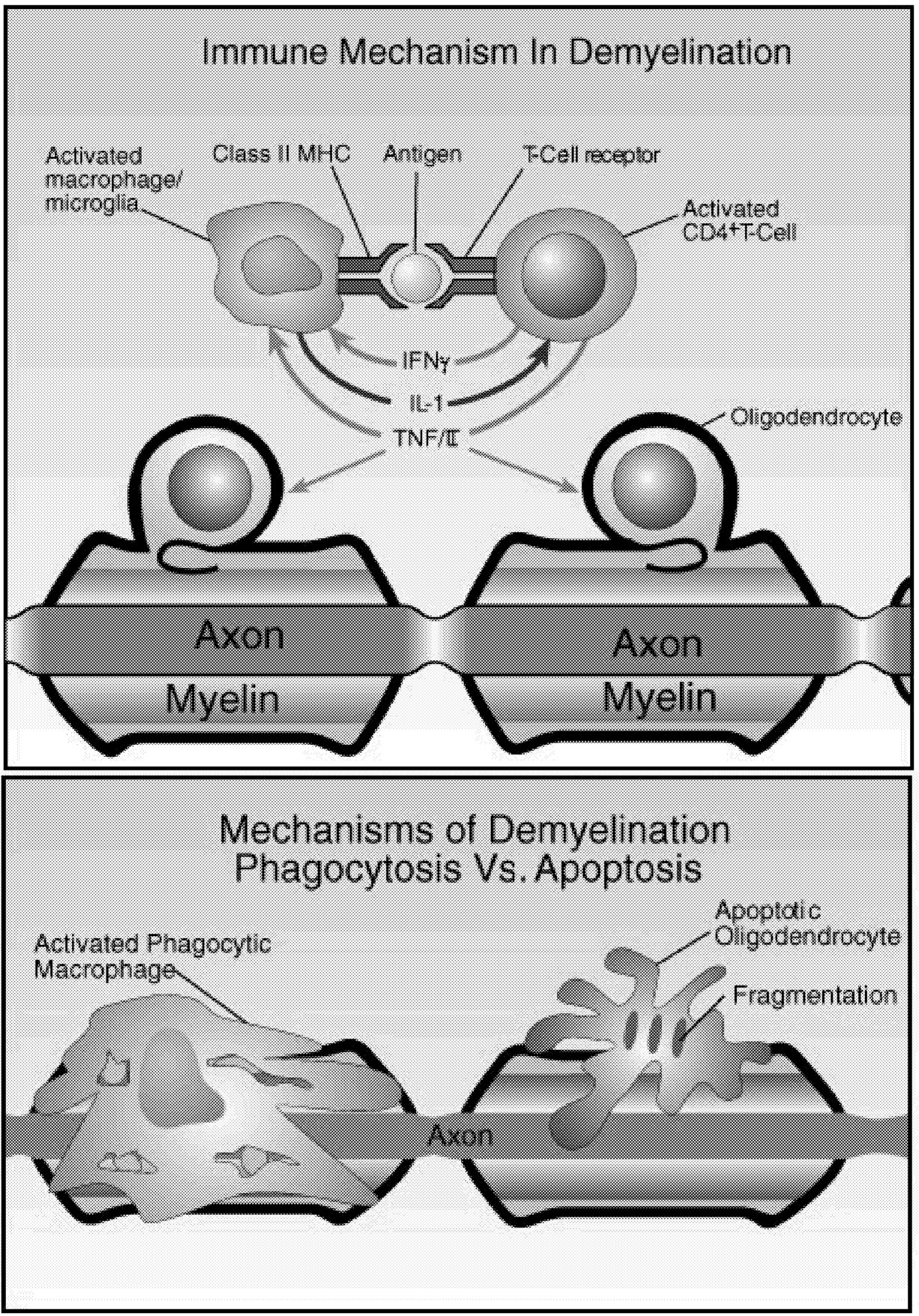
~ enlarge ~
FIGURE 2.7 Possible mechanisms of demyelination. The mechanisms causing myelin damage are not completely known. Possible mechanisms include a direct toxic effect of tumor necrosis factor (TNF) on myelin (upper panel) or macrophage-mediated damage through either phagocytosis, in which the cell is engulfed and destroyed, or apoptosis, in which cells are induced to self-destruct (lower panel).136 NOTE: IL = interleukin; MHC = major histocompatibility antigen. SOURCE: Adapted from New Directions in the Management of Multiple Sclerosis. 1994. Berlex Laboratories. Courtesy of John Rose and the Knowledge Weavers, University of Utah.
Page 62
|
Visual deficits |
Often related to the involvement of the optic nerve as occurs in optic neuritis |
|
Eye movement abnormalities |
Frequently associated with plaques involving connections between the brainstem nuclei subserving eye movements |
|
Weakness and spasticity |
One of the consequences of lesions involving the spinal cord or descending motor tracts in the white matter of internal capsule or brainstem |
|
Ataxia |
Usually the result of lesions in cerebellum or its input-output pathways |
inflammatory responses amplify, macrophages are filled with lipids, myelin damage occurs, and there is apparently some collateral damage to axons. Scattered B cells and plasma cells are also sometimes associated with these lesions.
Immunocytochemical analysis has suggested that there may be leakage of immunoglobulins and complement from vessels at the margins of active plaques. There is evidence for upregulation of a variety of cytokines within MS plaques including various interleukins (IL-1, 2, 4, 6, 10, and 12); beta-interferon; tumor necrosis factor (TNF); and transforming growth factor (TGF). Cytokines are intercellular signaling proteins produced by cells of the immune system and CNS. Their involvement in any disease is complex. They can initiate, sustain, or terminate various aspects of disease processes and are particularly involved in inflammatory responses. Specific chemokine receptors are expressed by infiltrating cells in demyelinating MS brain lesions and in CSF. These results imply pathogenic roles for specific chemokine-chemokine receptor interactions in MS and suggest new molecular targets for therapeutic intervention.163,202
Some investigators have emphasized that the pattern of pathology suggests a dying-back oligodendrogliopathy (those parts of the cell, such as the most distal process, farthest from the cell body are the most vulnerable).126 During the active stages of disease, the number of oligodendrocytes can be reduced near the demyelinating foci. Subsequently, there might be partial recruitment of oligodendrocyte precursors that may, in part, repopulate the margins of the plaque and contribute to remyelination.
Chronic Lesions. Trapp and colleagues212 have emphasized that the relapsing-remitting course is intimately related to the inflammatory demyelination and classical plaques, while more chronic progressive forms of the disease are linked to transection, or severing, of axons at sites of inflammation and demyelination. They have shown that severed axons were a consistent component in the lesions of individuals with MS and suggest that the number of these injured axons is correlated to the magnitude of the inflammation within the lesion. Axonal damage might be a pathological correlate of irreversible neurological deficits that occur in patients with progressive MS.51,212,213,220 In individuals with chronic MS, plaques are often sharply demarcated with scattered lipid-containing macrophages and little evidence of ongoing myelin destruction. Demyelination can be incomplete. The density of axons may be significantly reduced. There is usually astrogliosis within these lesions. It is not clear exactly how the axonal damage that occurs at later stages plays into this complex evolving pathology, but the extent of axonal damage appears to be a critical determinant of whether a person recovers from an attack or not. Lack of recovery from attacks and disease progression are more likely when axons are more severely damaged and repair mechanisms fail.
Page 63
~ enlarge ~
FIGURE 2.8 The blood-brain barrier. The tight seal of the cells lining the blood vessels forms a blood-brain barrier that keeps many substances out of the brain. Leaky blood vessels in the body allow many molecules to cross through to other tissues, but the tight construction of the vessels in the head guards against entry of most molecules into the brain. Normally, only certain molecules, for example, blood gases such as oxygen and small nutritional molecules, can cross the blood-brain barrier, but this barrier breaks down when the brain is injured or in certain diseases, such as MS. SOURCE: Reprinted with permission by Lydia Kibiuk/Society for Neuroscience.
Page 64
A correlation between demyelination and axonal degeneration observed at autopsy is supported by imaging studies, including those that compare the concentrations of N-acetyl aspartate.119 For example, one study measured the levels of NAA, an amino acid found only in neurons that serves as a biological marker for integrity of the axon and neuronal cell body. The brains of MS patients showed significantly greater side-to-side differences in levels of NAA, indicating decreased neuronal integrity on the side of the brain with lower NAA levels. There was a correlation between this asymmetry in motor function and the asymmetry of NAA concentrations in the internal capsule.119 Insights into processes that lead to demyelination and axon damage can be obtained by analyzing T1-weighted image hypointensities, proton spectroscopy, MTI, and DTI and by correlating these measures with clinical and pathological findings.
Gliosis. Gliosis is a prominent feature of the MS lesion, but it is best regarded as a secondary phenomenon.146 Whenever the CNS is damaged, it undergoes an injury response, usually called reactive gliosis or glial scarring. The response is broadly the same whatever the source of injury, although the details vary somewhat with different types of pathology.67 The glial reaction to injury includes recruitment of oligodendrocyte precursor cells, stem cells, microglia, and astrocytes. Formation of the glial scar after CNS injury generally occurs over a period of weeks. Microglia are typically the first cell types to enter the lesion. In the normal brain, they are quiescent with short, branched processes. Following injury, they exhibit various changes, including activation, cell division, and migration to the injury site.67 During activation, microglia display conspicuous functional plasticity, which involves changes in cell morphology, cell surface receptor expression, and production of growth factors and cytokines, and they become, in general, more macrophage like.208 Microglia can be either neurotoxic or neurotrophic.
The final glial scar is made up mainly of a meshwork of tightly interwoven astrocyte processes, attached to one another by tight junctions and gap junctions and surrounded by extracellular matrix (reviewed in 1999 by Fawcett and Asher67). (Astrocytes are irregularly star-shaped, background structural cells of the nervous system.) Gliosis is usually restricted to the area of demyelination, but it sometimes extends beyond that area. There is no specific way to identify the presence and extent of gliosis in MS lesions through MRI, although the T1 signal might be more sensitive to gliosis than the T2 signal.24
The role of astrocytes in gliosis is not completely known.146 Since there is evidence that glial scars can inhibit both axon growth and myelination, it is clearly important to know what causes them to form, what cells are involved, why they are inhibitory, and how to manipulate them. Finally, although gliosis is generally considered harmful, there is also evidence that the gliotic ensheathment of demyelinated axons might favor the restoration of nerve conduction.221
Page 65
New Directions for Research on Axons and Myelin
The relapsing-remitting form of MS appears to be related to the demyelination of axons during relapses, followed by the remodeling and remyelination with consequent restoration of conduction that underlies remission. Remyelination is carried out by surviving oligodendrocytes or by the proliferation of progenitor cells that are then stimulated to become oligodendrocytes. Molecular remodeling of demyelinated axons, in terms of their redistributing their sodium channels along the axon, might act as a form of adaptive plasticity; this process may represent a target for future therapies. In contrast, the progressive form of the disease, which appears clinically as an unremitting accumulation of deficits, might reflect the superimposition of axonal injury or degeneration on multiple chronic foci of demyelinated axons. The finding that axonal damage can occur frequently in MS and the suggestion that these lesions contribute to persistent neurological deficits are important issues in MS research.51,135,212,213,220 Thus, it will be important to understand the mechanisms whereby axons are damaged and to define ways of protecting axons, which may be vulnerable to degeneration, in part due to their proximity to inflammatory and demyelinating foci. It will also be important to search for molecules that promote the regrowth of injured axons to their appropriate targets. Some of the lessons from studies of the repair of spinal cord injury are likely to be relevant here.81,133 Moreover, because abnormal patterns of ion channels have been found within neurons whose axons are undergoing demyelination,22 it will be important to understand the factors that influence the regulation of these channels.219 These lines of research, involving strategies can restore conduction in demyelinated axons represent opportunities to restore that protect axons from degeneration or promote repair,81,133 and those agents that functions and could have significant implications for therapy.219 Similarly, future therapeutic approaches might involve replacement of oligodendroglia with pluripotent stem cells (discussed further in Chapter 6). Progenitor cells, those either already present in an individual or provided from another source by injection, can produce myelin in demyelinated foci in experimental animals.28 Future experimental therapeutics will involve approaches directed at restoring oligodendrocytes to ensheath axonal processes and to induce physiological modeling of axons to restore conduction.
Immunopathology
The most striking pathology in MS is the immune system's attack and destruction of the body's own myelin sheath, which is why it is believed to be an autoimmune disease, although this has not yet been definitively proven (Box 2.3). The pathogenic trigger that first causes the immune system to attack myelin is unknown, but the immunopathology, or pathological activity of the immune system, that ensues after that initial attack is becoming clearer.
Page 66
BOX 2.3Autoimmunity and DiseaseThe immune system defends the body against foreign invaders such as bacteria and viruses. It does so by recognizing that foreign invaders have special markers distinguishing them as “non-self,” compared to the body's own tissue (or “self”) (Figure 2.9). Normally, the immune system reacts only to non-self invaders, not to its own tissues. Unfortunately, this process is not foolproof. Autoimmunity is an immune response mounted against antigens that are naturally produced within the body, or self-antigens, to cause lasting tissue damage. In the strictest sense, an autoimmune disease must meet several criteria. First, the disease must be reproduced by transfer of autoantibodies or autoreactive T lymphocytes (T cells) from affected to unaffected individuals. Second, the self-antigen that elicits the immune attack must be identified. Third, this antigen (or a closely related one) should cause a similar disease in an animal model. (Scientifically, it would be best to show that the antigen caused the disease in humans, but it would be unethical to intentionally infect humans, hence, the compromise for evidence in animals). It is now feasible to transfer human genes into animals in an attempt to satisfy these criteria. One of the first so-called transgenic studies that introduced certain human genes into an animal without MS led to the development of a disease resembling MS.129 Among the newly added genes were those for a specific type of histocompatibility antigen and for a particular type of T-cell receptor that binds to a fragment of a myelin protein (myelin basic protein). This type of study begins to confirm the autoimmune nature of MS. Human autoimmune diseases, however, are generally classified as such without meeting these three stringent criteria. Circumstantial evidence often is marshaled for classification. For example, patients are classified as having autoimmune disease if they have high levels of autoantibody or autoreactive T cells, or because there is a correlation between the level of immune activity and disease severity. Some examples of autoimmune diseases are listed in Table 2.11. Why does the immune system have autoreactive lymphocytes? During development, the immune system randomly builds a vast repertoire of cells that respond to a multitude of foreign antigens. At the same time, the immune system must weed out those cells that react to self-antigens. Most self-reactive B cells and T cells are removed early during development. Other regulatory mechanisms exist to keep self-reactive lymphocytes unresponsive later on. These are among the normal regulatory mechanisms resulting in immunological tolerance to most self-antigens. Even though there are usually small numbers of autoreactive lymphocytes in a normal adult, most do not cause disease, and some might even serve a (currently unknown) beneficial purpose.182 With autoimmune disease, however, there are large numbers of autoreactive immune cells. Autoimmune disease thus can be thought of as a failure of normal regulatory mechanisms that guard against autoimmunity. |
Page 67
|
What causes a pathological autoimmune response? The causes of autoimmune response in MS and most other autoimmune diseases are unknown but likely include a combination of genetic susceptibility and exposure to environmental agents. For most autoimmune diseases, the actual genes and environmental agents are unknown. Gender also plays a role because women are disproportionately affected by autoimmune disease. The reasons for the gender difference are also unknown but appear to relate to distinct immune environments in women and How can genes and environment trigger autoimmune pathology? Genes control many properties of the immune system. Theoretically, autoimmune disease can occur if any of the genes controlling the immune system's ability to distinguish self from non-self are defective. The genes often suspected of predisposing to autoimmune disease encode proteins, such as histocompatibility antigens, that participate in this complex process of self versus non-self recognition. Environmental or infectious agents can stimulate pathological autoimmune reactions through at least two possible mechanisms: molecular mimicry, superantigens, and bystander damage (Figure 2.10). Molecular mimicry occurs when a bacterial or viral epitope—the fragment of an antigen that elicits an immune response—is very similar to a self-epitope. This can occur following an infection where an immune cell targeting an epitope on a bacterium subsequently cross-reacts with a self-antigen.5,46,157,229 Superantigens are proteins from bacteria or retroviruses that lead to widespread activation of T cells. Unlike most antigens, which activate only a specific T cell, superantigens activate approximately one out of every ten T cells.92 Because of their generic ability to activate so many types of T cells, superantigens may inadvertently activate autoreactive T cells, which then attack self-tissues. In bystander damage, a virus upregulates a nonspecific immune response that then leads to pathology; for example, proinflammatory cytokines might activate Th1 cells or macrophages that contribute to the immunopathology. Once damage occurs, new cellular epitopes become exposed and trigger an immune response in a process called epitope spreading, which can also lead to autoimmune pathology. During an immune response against a particular epitope, the number of lymphocytes that recognize the epitope normally multiplies. Yet, in epitope spreading, the immune response escalates to target other epitopes on the same antigen or on related antigens.54 In this way, an initially nonpathological autoimmune response expands to produce a pathological response. The underlying basis for epitope spreading in autoimmune diseases is poorly understood. Another way to produce a pathological autoimmune response is for autoreactive lymphocytes to gain access to a target antigen from which they are ordinarily separated. The brain is one example of an anatomical sanctuary site, because it is protected by the blood-brain barrier of the central nervous system. Normally, T cells that might react against myelin do not pass across this barrier into the central nervous system. In MS, however, T cells become activated, which enables them to penetrate the blood-brain barrier and reach their targets—myelin antigens—thereby generating an autoimmune response. |
Page 68
One of the first pathological processes leading up to MS attacks is thought to be activation of autoreactive T lymphocytes, or T cells, and their migration into the central nervous system.25,93,116 However, T cells and the inflammatory molecules they secrete are not the only players. Many cells and molecules of the immune system—likely unleashed by T-cell activation—participate in demyeli-
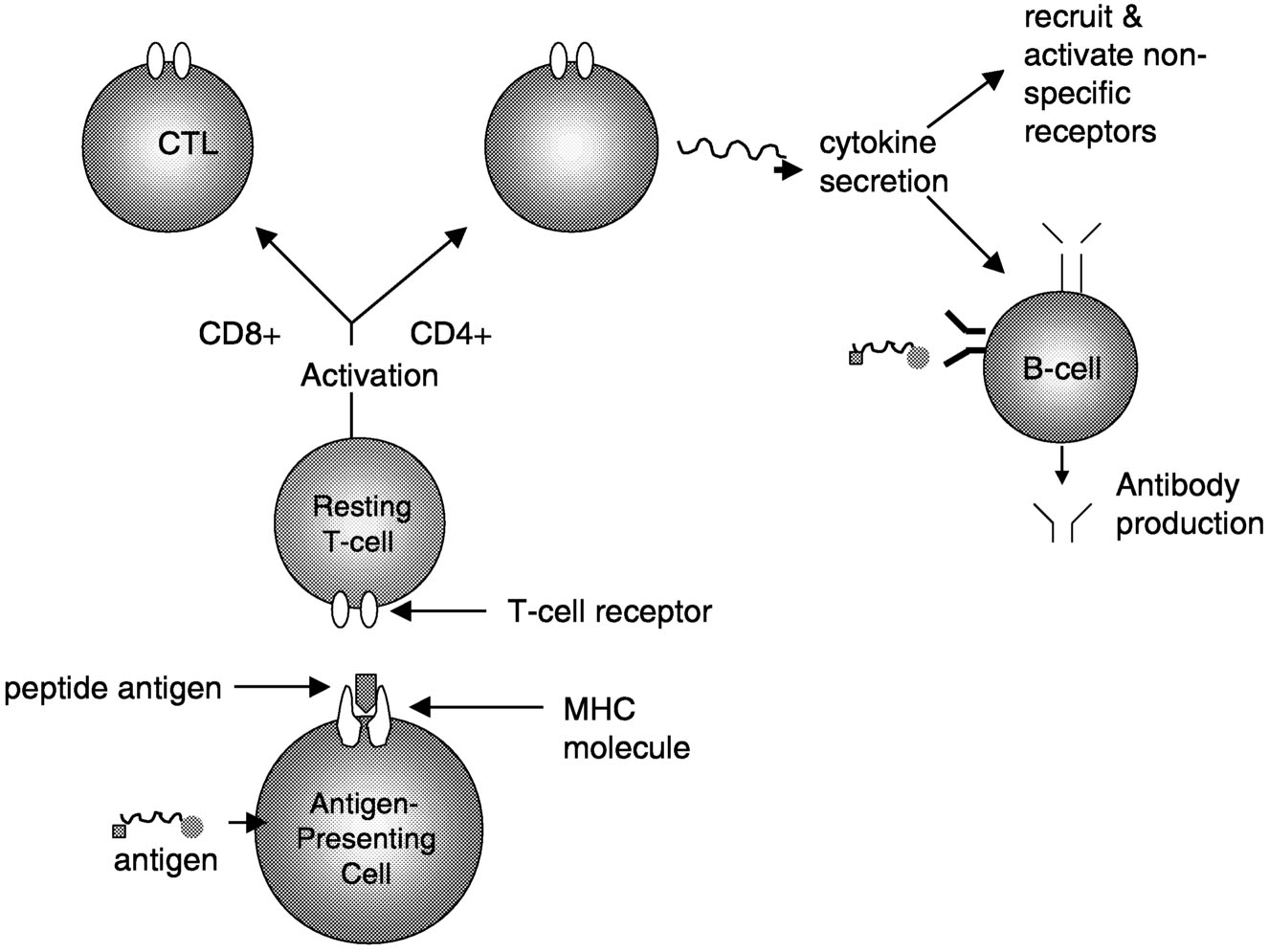
~ enlarge ~
FIGURE 2.9 Interactions between major cell components of the immune system. This simplified outline shows interactions that occur in response to a foreign antigen. The antigen could be an epitope from a virus particle, bacterium, or other foreign agent. The antigen-presenting cell (APC) ingests the antigen (or in the case of an infecting virus, it may already be within the cell) and processes it into peptide fragments. The major histocompatibility complex (MHC, the cell surface structure characteristic of each individual) presents this target to a resting T cell. Binding of the peptide-MHC complex by the T-cell receptor activates the T cell. In the case of a CD8+ cell, it becomes a cytotoxic T lymphocyte (CTL), which will destroy any cells that have the same peptide-MHC complex on their surfaces. In the case of a CD4+ cell, the activated cell produces and secretes various cytokines that recruit and activate nonspecific effector cells (such as macrophages) or stimulate antigen-bound B cells to produce antibodies. When T and B lymphocytes are activated by a specific antigen, they undergo proliferation, producing more cells with their same antigen specificity. This serves to amplify the immune response against the foreign antigen.
Page 69
|
TABLE 2.11 Selected Diseases That Are Believed to Be Autoimmune Based |
||
|
Disease |
Common Symptoms |
Proposed Mechanism |
|
Graves' disease |
Hyperthyroidism |
Antibodies against the thyrotropin receptor stimulate thyroid function |
|
Insulindependent diabetes mellitus (Type I diabetes) |
Hypoinsulinemia and hyperglycemia |
Destruction of insulin-producing cells in pancreas |
|
Pemphigoid (various diseases) |
Blister formation |
Antibodies block adhesion of epidermis to dermis |
|
Rheumatoid arthritis |
Joint pain and loss of mobility |
T-cell-mediated inflammation in the joints |
|
Systemic lupus erythematosus |
Arthritis, rash, CNS dysfunction, kidney damage |
Immune response to numerous cellular antigens, especially DNA |
|
Systemic sclerosis (scleroderma) |
Thickening of skin; kidney, lung, and gastrointestinal damage |
Immune response against topoisomerase I leads to increased formation of collagen in the skin and internal organs |
|
Thyroiditis |
Hypothyroidism |
Destruction of thyroid cells |
|
Chronic inflammatory demyelinating polyneuropathy |
Weakness and sensory loss |
Demyelination of peripheral nerve fibers |
|
Guillan-Barré syndrome |
Paralysis and loss of reflexes |
Demyelination and/or axonal degeneration of peripheral nerve fibers |
|
Lambert-Eaton myasthenic syndrome |
Muscle weakness |
Antibodies against presynaptic calcium channel of the neuromuscular junction (NMJ) disrupt function |
|
Myasthenia gravis |
Muscle weakness |
Antibodies against postsynaptic acetylcholine receptor of the NMJ disrupt function |
|
Neuromytonia (Isaac's syndrome) |
Muscular twitching, cramps, stiffness, and weakness |
Antibodies against potassium channel at the NMJ cause increased muscle activity |
|
Rasmussen's encephalitis |
Epileptic seizures and neurological dysfunction |
Antibodies against subunit of ionotropic glutamate receptor lead to degeneration of one cerebral hemisphere |
|
Stiff man syndrome |
Axial and limb rigidity; spasms |
Antibodies block production of GABA (γ-aminobutyric acid), an inhibitory neurotransmitter |
Page 70
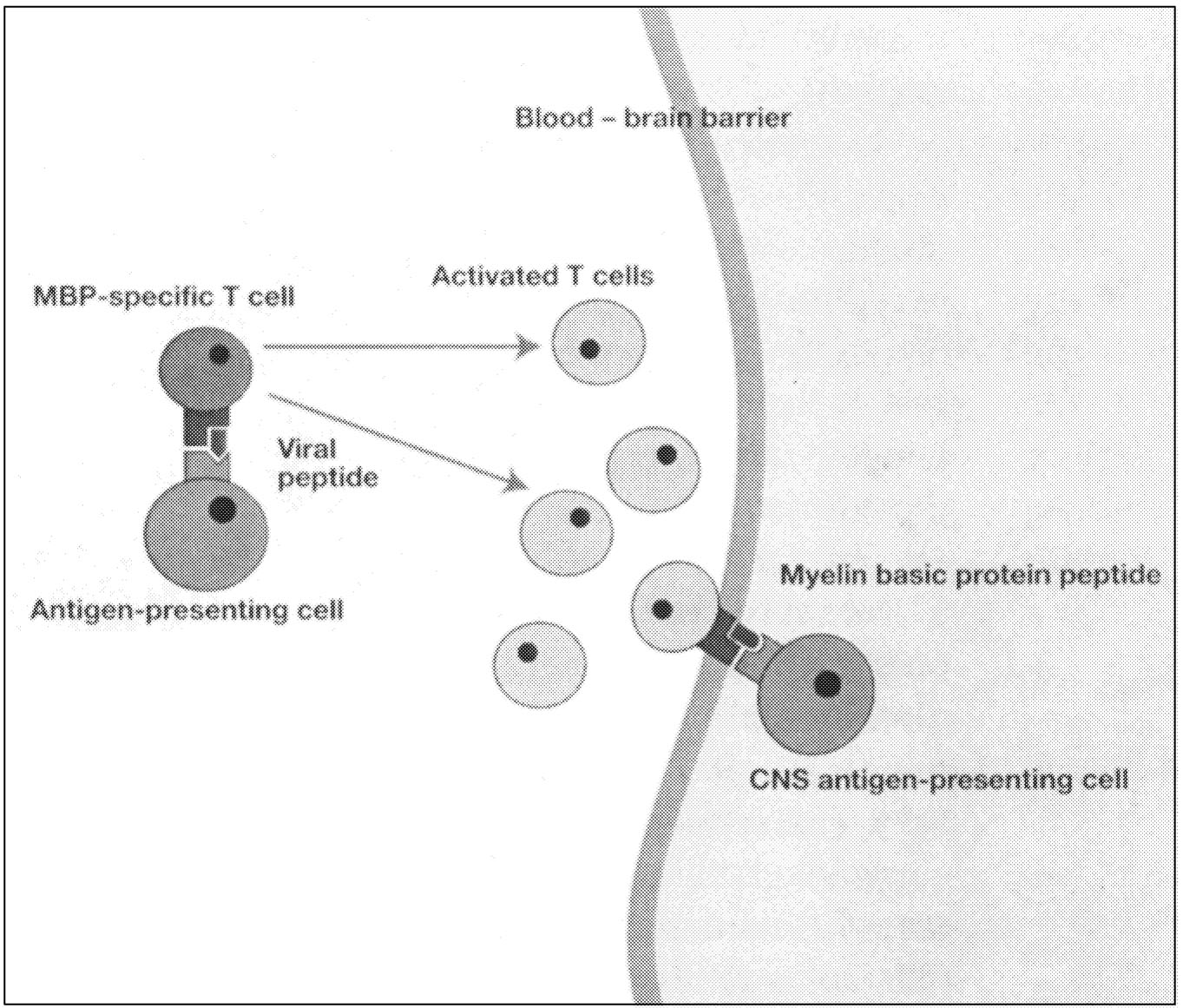
~ enlarge ~
FIGURE 2.10 Possible mechanism of viral etiology. Although no virus has yet been shown to contribute to the etiology of MS, there are several ways in which this could occur, and one of these is shown in this figure. Activated T cells cross the blood-brain barrier following activation by a microbe with a structural similarity to a component of the myelin sheath. Once inside the brain, these cells attack self-antigens, such as the various myelin proteins that are attacked in MS. NOTE: MBP = myelin basic protein. SOURCE: Steinman and Oldstone, 1997.206 Reprinted with permission. Adapted from Wucherpfennig and Strominger, 1995.229
nation. The entire cascade of immune system events eventually culminates in myelin destruction. The key features of this cascade are not fully understood, including the precise ordering of events, the antigens targeted by T cells, and the contributions of B lymphocytes, or B cells, and other cells of the immune system. Yet, as this section explains, much insight has been gained into the immunopathology of MS. This knowledge has been—and continues to be—fundamental for devising therapies targeted to the immunopathology of MS (Chapter 5).
Page 71
T Cells in MS
As much as a century ago, researchers observed that T cells were particularly abundant in MS lesions.116 Over time, using modern immunologic techniques, they managed to isolate and characterize a particular type of T cell, the autoreactive T cell, from the blood and CSF of MS patients.225
These and related findings gave credence to the hypothesis that autoreactive T cells played a dominant role in MS. After all, immunologists have long known that T cells are capable of orchestrating a multifaceted autoimmune attack. However, this was not enough to explain MS pathology. First, elevations in certain types of autoreactive T cells were not unique to MS patients. Second, and more critically, T cells were necessary but not sufficient to cause demyelinating disease in animal models. The transfer of myelin-specific T cells into normal animals initiated only inflammation, not demyelination.117 This suggested that other immune cells, particularly antibody-producing B cells (Table 2.12) and macrophages, might also play key roles, even if autoreactive T cells launched the process.
|
MYELIN PROTEINS |
|
|
Myelin basic protein (MBP) |
An important component of the myelin sheath, MBP is located on the cytoplasmic face of the myelin membrane, and constitutes 30-40% of myelin protein by weight. |
|
Myelin oligodendrocyte glycoprotein (MOG) |
Surface protein on the myelin sheath. |
|
Myelin-associated oligodendrocytic basic protein (MOBP) |
Structurally similar to MBP, but expressed exclusively in the CNS myelin. Possibly involved in myelin compaction. |
|
Proteolipid protein (PLP) |
PLP constitutes approximately 50% by weight of myelin protein. PLP spans the myelin membrane, providing increased stability. |
|
Myelin-associated glycoprotein (MAG) |
The major mediator of axonal-glial contacts essential for the initiation of myelination. |
|
OTHER PROTEINS |
|
|
S-100b |
Calcium-binding protein associated with astrocytes. |
|
Glial fibrillary acidic protein (GFAP) |
Major constituent of glial filaments in astrocytes, providing structural stability. Rapidly synthesized in response to CNS trauma or disease. |
|
Heat shock proteins |
Broad class of stress-responsive proteins that are normal components of the myelin sheath. |
|
αB-crystallin |
A stress protein that is an immunodominant antigen of CNS myelin in MS patients. |
Page 72
The prime autoantigen that elicits the autoimmune response in MS is not known. While there are many candidate autoantigens, as yet none is preeminent.
Much MS research focused on myelin basic protein (MBP), located at the inner surface of the myelin membrane. Clinical studies were shaped by research on experimental allergic encephalomyelitis (EAE), the classic animal model of demyelinating disease. In many species, MBP acts as a classical encephalitogenic autoantigen (an antigen capable of serving as the focus of an inflammatory attack in the brain). EAE research further established that in some strains of mice and rats, the autoimmune response to MBP displays two unique features. First, in the initial stages of the immune response in EAE, T cells react only to very small regions of the MBP molecule (“epitope dominance”), even though they later react to many more regions of the molecule (“epitope spreading”) (see Box 2.3). Second, the encephalitogenic T cells in EAE use an unusually narrow repertoire of genes for their antigen receptors.205 (T cells recognize their target antigen by its capacity to bind to specific receptors on their surface membrane, called T-cell receptors [TCRs]). These two features raised hopes for developing immune-based therapies because a more limited range of therapies might succeed in combating MS in early stages.
Unfortunately, these features turned out to be much less prominent in humans with MS. First, human T cells respond to a broader set of MBP epitopes. While there might be a relatively dominant epitope in the central portion of the MBP molecule, there are clearly many other target epitopes along the full sequence of this large polypeptide.131 Second, MBP-specific T cells use multiple genes for their TCRs, which allows for large variability among individuals.86 Generally, it appears that the T-cell response against myelin proteins differs greatly between individual patients, a property that suggests the potential need for individually designed immune therapies.
Several research teams have attempted to identify MBP-specific T cells in MS patients.216,228 Unfortunately, these attempts did not show significant increases in MBP-specific T-cell counts in MS patients compared to healthy blood donors. However, when more definitive assay systems were used, increased frequencies of activated MBP-specific T cells were found in MS patients.7,20,232 These studies relied on complex methods and were influenced by numerous factors that complicate the interpretation of results. More direct assays, such as those that use binding of oligomeric class II-peptide complexes to specific T cells,41 might resolve the problem.
To add a further degree of complexity, MBP does not appear to be the only autoantigen in MS: there are a number of additional myelin and nonmyelin proteins that are potential autoantigens in MS. Earlier hypotheses had implicated MBP on the basis of two lines of research. First, studies of EAE had suggested that MBP was indeed the most important, if not the only, encephalitogenic myelin autoantigen. Second, due to its particularly convenient molecular properties, MBP was the only myelin protein available both at high purity and in large
Page 73
quantities.118 However, thanks to modern biotechnology, even minor myelin proteins are now available in large quantities and can be studied for their encephalitogenic capacity.
Later studies established that many, if not all, myelin proteins are potentially encephalitogenic. Especially interesting among these newly recognized autoantigens is myelin oligodendrocyte glycoprotein (MOG). In contrast to MBP, which lies on the inside of the myelin sheath, MOG lies on the surface. As one of the few myelin proteins accessible to humoral autoantibodies, MOG is a target for demyelinating immunoglobulins (see next section). In addition, MOG is a very effective autoantigen in rodents and in primates for encephalitogenic T cells.17 Some studies even demonstrated increased frequencies of MOG-reactive T cells in MS patients.108,217 These findings clearly warrant general confirmation. It is also important to investigate whether different subtypes of MS, which are distinguished by divergent clinical, genetic, and morphological features, are associated with enhanced T-cell responses against different target autoantigens.127
Thus far, research has spotlighted one class of T cells—those with a TCR termed αβ. * αβ T cells are the major class of immune cells centrally involved in adaptive immune responses against infections and tumors. Much less attention has been given to other T-cell classes (γδ T cells and NK1 T cells), which may function both as effector cells in autoimmune attacks and as suppressor cells that dampen autoimmune responses.90,161 There are reports linking the presence of γδ T cells in the brain (presumably) with the induction of heat shock proteins in MS lesions.25 Certain members of this broad class of proteins are a normal component of the myelin sheath,46 yet they are also found outside the CNS as well as in pathogens. The role of heat shock proteins in the development of the MS lesion is unknown, but there are various possibilities. Some of these proteins might act as a target autoantigen, as has been shown in autoimmune diabetes. They might also reflect inflammatory stress inflicted on local CNS cells, or they might be determinants of beneficial anti-inflammatory control mechanisms.25,42
Even though the precise pathological roles of T cells and their autoantigens are unresolved, this line of research has generated many approved or emerging therapies. These include vaccination strategies, which use either attenuated myelinspecific T cells20733 or peptides representing myelin-specific T-cell receptors9 as vaccines to strengthen the body's own regulatory responses against pathogenic T cells (reviewed in Zhang et. al., 1998).233 Also under development are “altered peptide” therapies that use peptide analogues of myelin protein segments to induce autoreactive T cells to produce protective, rather than pathogenic, cytokine mediators.205 Cytokines, as discussed later, are a functionally diverse set of signaling molecules produced by T cells.
*Each (α and β) refers to a polypeptide chain that forms the T-cell receptor.
Page 74
B Cells in MS
B cells (also known as B lymphocytes) have been detected in MS lesions for many years, although less consistently than T cells.26 While most MS research naturally focused on T cells, evidence also has accumulated for participation by autoreactive B cells. It now appears that both types of lymphocytes actively contribute to MS immunopathology. Autoreactive T cells are thought to launch inflammation and, through their release of cytokines, to stimulate B cells to secrete antibodies that cause demyelination.93 This is consistent with animal studies finding that EAE can be produced only by injecting myelin-specific T cells in combination with myelin-specific antibodies.78,123 Without injecting the antibodies, demyelination does not occur.
In MS patients, levels in cerebrospinal fluid of the type of protein known to consist of antibodies (immunoglobulin) are often higher than in healthy people. The increased immunoglobulin is due to production by only a few different clones of B cells that have been induced to proliferate. B cells from the CSF of MS patients have been reported to contain mutations in the DNA sequences that encode antibodies, which is consistent with the notion of an antigen-driven selection of antibodies with high-affinity antigen-binding sites.170 Such events are commonly observed in immune responses against foreign antigens such as bacterial infections, as well as in humoral autoimmune responses.171 However, no foreign antigen or autoantigen responsible for the generation of oligoclonal bands in MS has yet been identified.
One recent study using sophisticated immunocytochemistry furnished direct evidence of a pathologic role for autoantibodies. For the first time, MOG-specific antibodies were demonstrated to be bound to myelin debris in active MS lesions.77 If confirmed and extended to a larger group of patients, this finding would suggest that B-cell-derived autoantibodies might induce myelin destruction. On the other hand, although anti-MOG antibodies are found in the CSF of MS patients, they are also found in CSF of patients with other neurological diseases that are not demyelinating.103 Thus, despite tantalizing leads, the role of anti-MOG antibodies in MS patients is still unclear.
Cytokines
Cytokines are soluble proteins produced and released by T cells, macrophages, and certain other cell types. Interferons (IFN- α, β, and γ), interleukins (including IL- 1, 4, 6, 10, 12, and 13), TGF-b, and the neuropoietic cytokines * (such as ciliary neurotrophic factor and leukemia inhibitory factor) are all different types of cytokines. They generally act as intercellular signaling molecules that regulate
*Neuropoietic cytokines are the family of neural growth factors that act on both the nervous and the hematopoietic or immune systems.
Page 75
and carry out immune functions, but their repertoire is complex. Some function as pro-inflammatory, others as anti-inflammatory agents. Some even have divergent functions during different phases of disease.32 Cytokines, including the subset known as chemokines (for chemotactic cytokines), also alter the permeability of the blood-brain barrier105 or act on neural cells.175 Thus, particular cytokines can initiate, sustain, or terminate inflammatory disease processes. Proinflammatory cytokines and other secretory products of immune cells are proposed in several neurological diseases—including MS—to be toxic to neurons and oligodendrocytes if they are secreted in sufficiently high concentrations over a sustained period of time.49,82
In MS, the initial entry of autoreactive T cells into the CNS is thought to trigger the local production of cytokines and chemokines, which in turn begins the inflammatory process and enhances the permeability of the blood-brain barrier.93 A more permeable blood-brain barrier allows infiltration into the CNS of more immune cells which in turn contribute to the ongoing inflammation. Thus, understanding the roles of cytokines and their temporal sequence of activation is crucial to modifying the course of MS. Much of our present understanding of cytokine action in demyelinating disease comes from studies of animal models, including EAE.
A large body of research is being compiled on the expression and possible function of cytokines as pro- and anti-inflammatory mediators in MS. Some studies have used in situ immunocytochemistry and in situ hybridization to visualize gene expression in lesions, whereas others have relied on the activation of inflammatory cells (T cells, B cells, macrophages) in vitro. Much of the research is comparing material from MS patients with or without treatment with immunomodulatory agents, especially glatiramer acetate and beta-interferon.93
Pro-inflammatory cytokines within active MS lesions have been localized to both infiltrating immune cells and glia.172 Longitudinal studies have linked clinical exacerbation and remission to high and low expression, respectively, of proinflammatory cytokines, especially TNF-γ.16 For example, secretion of TNF-γ and cell adhesion molecules by inflammatory cells is upregulated immediately prior to relapse.40,179,197 Yet, not all findings could be verified in all patient groups studied.31,80,178
Understanding of cytokines and their diverse roles throughout the course of disease, although still incomplete, has nevertheless spawned new treatments. One explanation for the success of glatiramer acetate and beta-interferon relates to their control over cytokine expression: they can induce T cells to switch from a pro-inflammatory phenotype (Th1) to an anti-inflammatory phenotype (Th2).93 T cells that are switched to an anti-inflammatory phenotype release a variety of cytokines, such as IL-10, that reduce inflammation.
Immunologically Special Features of the CNS. CNS tissues were traditionally thought to be exempt from active immune reactivity, but it is now known
Page 76
that the brain is subject to immune surveillance and can be the site of immune responses. Only activated and not resting T cells can cross the blood-brain barrier and interact with local CNS cells. Local glial cells can be stimulated by proinflammatory cytokines to express immunologically active molecules, such as the major histocompatability complex (MHC) products, cytokines, and chemokines required for local immune responses. Neurons are capable of suppressing immune responses within the CNS. Thus, immune responses are more likely to occur in areas of neuronal degeneration than in intact CNS tissues. Immune reactivity within the CNS must hence be viewed as the balance between the proinflammatory signals contributed by activated T cells and other inflammatory cells entering the brain and the anti-inflammatory signals from functional neurons.
Epidemiology
Epidemiologists define what causes a disease and what puts an individual at risk of getting this disease. They look for correlations such as whether Caucasians are more likely to have MS than Asians or whether residents of one county are more likely to get MS than residents of another. These correlations lead to hypotheses or working models as to which factors actually cause MS and which are only associated with it. These hypotheses or models can then be tested experimentally. Epidemiological studies have limitations, but for a complex disease like MS, they can rule out some factors and highlight others. They are the first step toward finding a biological mechanism for MS, or possibly a cure.
Most epidemiological studies of MS have been observational and retrospective; researchers collected the information (for example, ethnicity, age of onset) from an individual (or from records) that had been diagnosed with MS. Such studies rely on an individual's ability to accurately recall information from years ago, for example, the infections that she or he had before the age of five. Other sources of data, such as death certificates, can contain incorrect information. Despite these shortcomings, a few factors consistently correlate with MS prevalence. Determining how these factors lead to an increased risk of MS has proven more difficult.
The risk of MS increases with increasing distance from the equator. In the United States, this is seen as a gradient of risk, with higher risk in northern regions and a lower risk in the south.56,114 In Australia, the prevalence of MS increases similarly from lower, subequatorial latitudes to the more southerly latitudes.89 Studies on migrating populations, although inconclusive, offer tentative support for the hypothesis that environment influences MS risk. Individuals who have moved from a region with one risk level to a region with a higher or lower risk, in general, adopt the risk level of their new home.56,113 This is especially true for individuals moving from a low-risk to a high-risk area.
Studies carried out in the 1960s and 1970s suggested that this geographical effect had a defined susceptibility period before age 15. Although these data still
Page 77
appear in the MS literature and have led to several hypotheses about the effects of puberty or early dietary effects on MS risk, the sample numbers are too small to support such a tight cutoff.56,150 A later and larger study assayed the effects of migration by comparing location at birth with location at approximately 24 years of age.113,114 Although individuals did adopt the risk of their new home, the age values in that study are not precise enough to add to the argument for or against a cutoff. Most recently, a study comparing the prevalence of MS among native-born Australians and Australian immigrants from the United Kingdom (thereby providing a rough control for genetic background) suggests that rather than being established around age 15, environmental risk factors operate over many years and into early adulthood.89
Although the migrational studies suggest an environmental correlation with MS, the root cause of this latitude effect is unknown. Differences in diet150 or sunlight1,97,121 have been proposed, but neither has been supported by rigorous studies. Alternatively, the geographical distribution of MS could result from the migration either of a viral agent113 or of individuals (perhaps originally Scandinavians) who carried a pool of susceptible genes.193
Ethnicity is another definite risk factor. MS is more prevalent among Caucasians than other groups. In a study of 5,305 U.S. veterans with MS, Caucasian males had twice the risk of MS as African-American males.114 Other factors can modulate the effect of ethnicity on risk of MS. MS is almost absent among black Africans. African Americans, however, show a low risk of MS; this might be due to genetic mixing with Caucasian Americans or to an environmental effect.56 MS occurrence among Asians is also quite low. Again, Asian Americans show an intermediate risk for MS between Asians and Caucasian Americans.137 A discussion of the role of genetics in MS can be found in the next section.
When applied to a complex disorder such as MS, conclusions derived from epidemiologic data of this type must be interpreted cautiously because inapparent explanations may be present. For example, a higher-than-expected incidence of MS observed in South Vietnamese immigrants (a low-risk group) residing in France (a moderate-risk area) superficially suggest a modifying role of the environment on MS. However, this immigrant population was in fact racially mixed and contained substantial numbers of individuals with mixed French and Asian ancestry. Thus, the higher-than-expected MS incidence in these immigrants could have been due to either genetic or environmental factors. Neither factor can be ruled out. A similar argument could explain the incidence of MS in West Indian immigrants residing in Great Britain. On the other hand, the increased incidence of MS observed in Japanese-Americans compared to individuals residing in Japan is not easily explained by racial admixture and does support a role of the environment on MS risk.
As in other autoimmune diseases, women are much more likely to get MS than men, suggesting that hormonal or genetic factors are involved. The ratio of women to men with MS is about 2:1.57 Among both sexes, the age of onset for
Page 78
MS ranges between 10 and 59 years,56 with the highest incidence occurring among individuals in their mid-20s to early 30s, depending on the population examined.112 Males tend to have a slightly later mean age of onset than females. This results, at least in part, from an increase in the percentage of males with the primary progressive variant. This form of MS has a later onset than other types and affects approximately 15 percent of patients. The ratio of males to females with primary progressive MS is approximately 1:1.
Epidemiological studies have provided conflicting data as to whether an infectious agent (viral or bacterial) either causes or triggers MS. The occurrence of MS epidemics has suggested that an infectious agent might be at work. The two MS epidemics cited most often, one in the Faroe Islands and one in Iceland, were directly preceded by the influx of foreign troops during World War II. Both, however, are open to multiple interpretations. Coincident with the stationing of foreign soldiers, the population of the Faroe Islands received increased medical services56 and changed its diet.150 The Icelandic epidemic, on the other hand, began shortly after the arrival of that island's first neurologist.56 In both cases, the increased prevalence of MS might have resulted from an increased level of detection or a combination of other factors, such as nonspecific immune stimulation resulting from the introduction of a variety of infectious agents into previously unexposed populations. No infectious agent has been associated with either of these MS clusters.
Another area of investigation has explored whether environmental factors contribute to the onset of MS or the probability of MS attacks. Some studies suggest that MS attacks are more likely to occur in the spring and fall than in the winter or summer. Such a finding, if true, suggests that a relationship exists between some viral infections and the risk of exacerbations. Many patients with MS are also at heightened risk for urinary tract, pulmonary, or skin infections, yet the relationship between these potentially preventable infections and the course of MS has never been adequately studied. Additional research in this area is needed.
Perhaps the most clear-cut epidemiologic link to MS attacks is the effect of pregnancy and the postpartum period. Pregnancy is associated with a decrease in the risk for MS attacks, particularly during the third trimester. The postpartum period is, conversely, associated with a significant increase in risk. An immunosuppressive state in the pregnant mother is created by increased numbers of regulatory T cells (Th2 cells) which, presumably, dampen the autoimmune reaction that produces attacks of MS. The explanation for the increase in attack risk during the postpartum period is less clear but might involve immunostimulation by prolactin, the hormone responsible for milk production.
The absence of supporting evidence does not prove that a virus is not connected with the disease. Unrelated individuals (in the case of the adoptee and conjugal studies) may differ in their susceptibilities to infectious agents. Researchers have isolated a variety of viruses from individual MS patients, but to
Page 79
date, no one virus has been isolated from all MS patients examined.18 It might, however, be the case either that MS is not one single disease or that there are multiple ways to trigger it. Researchers continue to look for causative infectious agents.18,83 (See section below on infectious causes of MS for further discussion.)
The aggregation of MS in some geographic areas, ethnic populations, or families could be explained by a common environmental exposure, a shared genetic background, or a combination of both environmental and genetic susceptibility. It is likely that in MS, as in other complex disorders, both factors contribute. It is also possible that the relative contributions of environment versus genetics might vary in different situations, depending on the degree to which an individual is genetically susceptible and the specific environmental context. The role of genetic factors in MS is discussed in the next section.
MS Susceptibility Genes
MS is not considered a genetic disease in the classic sense because it usually occurs sporadically. However, population and family studies are consistent with a principal pathogenic role for genetic risk factors in MS etiology. This genetic component is indicated primarily by the increased relative risk to siblings of affected individuals compared with the general population. Familial aggregation (λs) is measured by estimating the ratio of the prevalence (frequency) in siblings versus the population prevalence of the disease. A λs of 1 represents no familial clustering of the trait. For MS, the λs is estimated to be between 20 (0.02/0.001) and 40 (0.04/0.001).180 Half-sibling189 and adoption58 studies confirm that genetic, and not environmental, factors are responsible for familial aggregation. In addition, twin studies from different populations consistently indicate that a monozygotic twin of an MS patient is at higher risk (25 to 30 percent concordance) for MS than a dizygotic twin (2 to 5 percent),148,188 providing additional evidence for a significant but complex genetic etiology. Finally, the frequent occurrence of MS in some ethnic populations (particularly those of northern European origin) compared to others (African and Asian groups), irrespective of geographic location, also provides evidence for a complex genetic etiology.44,156,166 Overall, adoption and family studies suggest that being related to a person with MS is a greater risk factor than living with someone with MS.
A simple genetic model for the inheritance of MS is unlikely to be valid. Such a single-gene hypothesis is at odds with concordance data in twin and family studies and with the observed nonlinear decrease in disease risk as the genetic distance from the relative with MS is increased. It is likely that susceptibility is determined by multiple independent genetic loci (polygenic inheritance), each with a relatively modest contribution to overall risk. It is also possible that there are different genetic causes of susceptibility to MS (genetic heterogeneity). Finally, the genes that contribute to MS susceptibility are likely to be normal, common variants (or alleles) of genes rather than obviously defective mutations.
Page 80
Most individuals who carry such susceptibility genes would have no obvious deleterious consequences. For example, the DR2 gene (described below) is the most important genetic contributor to MS susceptibility identified to date. Approximately half of patients with MS have this gene, but so do 15 to 20 percent of healthy Caucasians. Thus, only approximately 1 in 250 people who have DR2 develop MS.
The cumulative action of several susceptibility genes, each with weak effects and limited penetrance, is thought to underlie genetic susceptibility to MS. (Penetrance refers to the likelihood that a person carrying an allele will develop specific manifestations caused by that gene.) The effects of individual susceptibility genes may also be influenced by interactions with other genes and by specific environmental exposures. Locus heterogeneity is also likely, meaning that there are different susceptibility genes in different MS patients. The possibility that MS is a heterogeneous disease with different causes or pathological processes adds an additional level of complexity to the analysis. In addition to MS, similar issues are present in other autoimmune diseases such as diabetes mellitus that are genetically complex, and common research tools will be needed to decipher specific disease genes in these different conditions.
Major Histocompatibility Complex. The genetic region most clearly associated with MS susceptibility is the major histocompatibility complex (MHC, or HLA [human leukocyte antigen] in humans) locus on the short arm of chromosome 6 (6p21) (Box 2.4). This association has been seen in different population studies that have relied primarily on sporadic patients.156 Formal genetic linkage to 6p has also been found in several recent whole-genome scans of multiple affected MS families. Many of the MHC genes are extraordinarily variable or polymorphic, reflecting the importance of genetic variation of these critical antigen-presenting molecules in the maintenance of a heterozygous advantage and the need to effectively present a diverse array of antigens if immune homeostasis is to be maintained. Immune homeostasis refers to the capacity of the immune system to respond appropriately to a diverse number of infectious pathogens and tumors without initiating unhealthy responses against self-constituents (auto-
BOX 2.4MHC, HLA, and DR2The major histocompatibility complex is a chromosomal region that contains more than 100 genes,214 many of which make proteins involved in the immune system. It is named for the role it plays in rejecting tissue transplants (histo- means tissue). In humans, this region resides on chromosome 6 and is called the human leukocyte antigen gene complex. The terminology is slightly confusing because |
Page 81
MHC and HLA also refer to the proteins that some of these genes make. In this report, the term “MHC molecules” is used to indicate the proteins.
|
The role of the immune system is to differentiate between self and non-self. This allows it to tell the difference between, for example, muscle tissue (self) and an invading virus (non-self) and to respond appropriately. To do this, the immune system relies on several different proteins that specifically bind antigens. This process is analogous to a lock and key. The “lock” is an antibody, a T-cell receptor, or an MHC molecule. The “key” is an antigen, which can be a carbohydrate, lipid, nucleic acid, or protein—anything that binds specifically to a component of the immune system.15 This discussion focuses on T-cell receptors and MHC molecules. T-cell receptors sit on the surface of T cells and bind to antigens outside the cell. (In the case of T-cell receptors and MHC molecules, antigens are small protein fragments.) Binding to an antigen signals the T cell either to die, to do nothing, or to become active. The signal context, such as whether the antigen is self or non-self, determines which of these signals is relayed. However the T cell cannot bind the antigen alone. It needs the help of an MHC molecule. MHC molecules sit on the surface of other cells. The MHC molecule, like two outstretched arms, holds onto an antigen and presents it to a T-cell receptor. There are two major classes of MHC molecules. MHC I molecules, which are expressed on most cell types, present antigens to CD8+ T cells. MHC II molecules, which are expressed on special antigen presenting cells (APCs), present antigens to CD4+ T cells. In humans, there are three main class I molecules, HLA-A, HLA-B, and HLA-C, and three main class II molecules, HLA-DP, HLA-DQ, HLA-DR. Each arm of the MHC molecule is made up of a separate protein. In the case of MHC I molecules, there is an α-chain (A, B, or C) that pairs with the protein b2-microglobulin. An MHC II molecule is made up of an α-chain and a β-chain (for example, DP-α and DP-β), but the story becomes more complex. Each MHC molecule can bind many but not all of the thousands of antigens that confront the immune system. The immune system relies on diversity among the MHC proteins to help stack the odds in its favor. The gene for each MHC protein chain comes in different varieties, or alleles. One of the MHC β-chains has more than 150 alleles.10 Each of these alleles produces a slightly different protein. The proteins vary just enough that although they all function as MHC proteins, they can bind different antigens. Any one person will have two alleles, at most, for a particular MHC protein (there are two copies of each gene in human cells), but across a population of individuals, this variety becomes more important. For example, in Gambia, West Africa, 25 percent of HLA-B genes are the HLA-B53 allele, compared to less than 1 percent in Europe. HLA-B53 is very effective at presenting an antigen from the parasite that causes malaria to CD8+ T cells. Researchers hypothesize that because HLA-B53 can protect people from the most severe forms of malaria, it is more prevalent in Gambia.138 Having a particular MHC allele can also be a disadvantage, as researchers have shown in the case of HLA-DR2 and MS. (HLA-DR2 actually designates both a specific DR-a chain allele and a DR-b chain allele.) Scientists still do not understand how HLA-DR2 predisposes individuals to MS, but this might have to do with the MHC molecule's ability to present specific antigens (for example, a fragment of myelin basic protein) to T cells.129 |
Page 82
immune responses). In Caucasian MS populations of northern European descent, the critical MS-associated genetic region is thought to reside near the class II locus and is comprised of a group of genes with specific polymorphisms (alleles) that tend to occur in certain fixed combinations, termed haplotypes. In molecular terms, the “DR2” haplotype is designated as HLA-DRB1*1501, DQA1*0102, DQB1*0602. DR molecules are comprised of alpha and beta chains (encoded by A and B genes, respectively), and the polymorphisms are predominantly present in the beta chain. Of the more than 100 beta-chain sequence variations identified in humans, only one (1501, also designated as DR2) is associated with MS.
How can the DR2 association with MS be explained? The DR2 molecule itself may have a propensity to bind peptide antigens of myelin and stimulate disease-inducing T cells. DR2 is known to bind with high affinity to a region of MBP (spanning amino acids 89-98) thought to be “immunodominant” in humans. X-ray crystallography of the DR2-MBP peptide complex revealed that the DR2 molecule contains a distinctive hydrophobic pocket in its antigen-binding region, created by a unique alanine residue at the B71 position into which glutamic acid at position 93 of MBP is tightly bound, anchoring the MBP-DR2 complex.75 In a larger sense, the structure of the antigen-binding domain of DR2 molecules likely facilitates binding of many peptides containing certain amino acid residues, specifically aromatic amino acids. Glatiramer acetate (copolymer 1), a currently available disease-modifying therapy for MS, is a random synthetic protein composed of four amino acids, including tyrosine. The tyrosine residues of processed copolymer peptides likely also bind to the hydrophobic pocket of DR2, perhaps interfering with presentation of this key MBP peptide to encephalitogenic T cells. It is surprising that no data exist on the interaction of DR2 and the response to glatiramer acetate in MS. Another possibility is that the DR2 molecule does not itself predispose to MS but that another nearby gene (perhaps another HLA gene such as DQ) is responsible. DR2 is also linked to other diseases. Besides MS, narcolepsy is the disease most strongly linked to DR2.
Other MS Susceptibility or Modifier Genes. In various studies, the HLA region has been estimated to confer somewhere between 10 percent and half of the inheritability of MS. To date, no other genes of major effect have been identified in genomic screens. Several studies appear to demonstrate that a deletion mutation in the CCR5 chemokine receptor gene (a coreceptor for HIV) on chromosome 3 confers a later age of onset or a more benign course of MS; this mutation is also associated with protection against HIV. This is particularly important because the expression of CCR5, which is increased in MS brain lesions, is thought to attract inflammatory cells into tissue. Another locus on chromosome 19q22 near apolipoprotein C1 has been linked to MS in several genomic screens, but the estimated λs is only 1.4. A polymorphism near the gene for myelin basic protein on chromosome 21 was reported to be linked to MS in a family from Finland, but not in other populations. Some studies have suggested linkages or
Page 83
associations with the TCR beta-chain locus on chromosome 7, the immunoglobulin heavy-chain locus on chromosome 14, and a region on chromosome 5, but others have not found similar linkages. The inability to confirm some genetic regions as containing MS susceptibility genes might reflect the small genetic contribution of these putative genes or genetic heterogeneity; alternatively, the original claim might have been spurious.
As noted above, it is likely that an additive model consisting of multiple independent genes, each with small effects, explains the non-MHC genetic contributions to MS. It should be emphasized, however, that the identification of specific genes that have even very minor genetic effects on MS can have an enormous payoff, both in terms of helping to decipher the underlying biology of MS and in pointing to new potential treatments. For example, the genetic studies discussed earlier that identified a role for the CCR5 chemokine receptor suggest that therapies aimed at this receptor could be investigated in people with MS.
Genetic Heterogeneity in MS
Perhaps the strongest indication that MS is a heterogeneous disorder comes from HLA studies showing an absence of DR2 association in particular ethnic groups or perhaps in some clinical variants. In Japanese patients, one form of MS (“Western type”) is characterized by disseminated CNS involvement and is associated with the DR2 haplotype. In contrast, a more restricted form of MS in which optic nerve and/or spinal cord involvement predominate (“Asian MS”) is not associated with DR2. Lesions in the non-DR2-associated condition are frequently more severe and necrotizing than in the disseminated form.109 In one report, the Asian MS form was genetically associated with an HLA gene named DP (the DPB1*0501 allele).231 Another area of uncertainty is the strength of the association between primary progressive MS (PPMS) and DR2. A number of (relatively) small studies failed to show any association between PPMS and DR2, although a recent larger study from northern Ireland appeared to show an association; it is possible that PPMS represents more than one underlying disorder.
Evidence for genetic heterogeneity is not limited to case-control HLA association studies but is also derived from formal linkage studies. Analysis of the MHC locus in an American multiple affected member MS data set confirmed the significant genetic linkage to this region (lod score of 4.60), *and the specific association with the DR2 allele; however 25 percent of the families that were DR2 negative showed no linkage to the HLA region on 6p21. This indicated most likely the presence of locus heterogeneity in familial MS in Caucasians. A related
*Lod scores are based on the logarithm of the odds of linkage between two genes. A score of 3 or more is considered evidence of a genetic linkage between a known gene (or gene marker) and another unknown gene that underlies a trait (such as MS). That information thus indicates that the trait has a genetic basis and localizes the gene to a specific chromosomal region.
Page 84
observation in MS-prone Caucasian families is that the phenotypic expression of MS aggregates within families in some cases, suggesting that some clinical manifestations of MS are influenced by an individual's genetic background.
The extent to which distinct clinical forms of MS are associated with different susceptibility genes, as may be the case in EAE (see discussion of animal models), is not known. Also unknown is whether specific genes interact with certain causative agents or triggers. Genetic studies have the potential to answer these questions, particularly when the information is analyzed in combination with epidemiologic, clinical, and neuroimaging data.
Other Demyelinating Diseases
There are several human and animal diseases of known etiology or pathogenesis that resemble either the clinical or the pathological features of MS (Table 2.13). Animal diseases that resemble MS are discussed under animal models. CNS demyelinating diseases include those mediated by immune responses, infection, and toxins, as well as inherited disorders. Infectious agents can induce direct injury of oligodendrocytes and their myelin membranes, as well as indirect injury via the immune system.
A variety of toxins, such as diphtheria, lysolecithin, cuprizone, and ethidium bromide, have been associated with demyelinating lesions. Many of these toxins induce lysis of the oligodendrocyte, with demyelination as a secondary effect. In addition, nutritional deprivation can be associated with demyelination in the central and peripheral nervous system.
Immune-Mediated and Virus-Induced CNS Demyelinating Diseases
ADEM as a Consequence of Vaccination. Acute disseminated encephalomyelitis (ADEM), also known as post vaccination encephalomyelitis, occurs as a consequence of vaccination with neural antigens. EAE, the most widely used animal model of MS, is the animal counterpart of this human disease. ADEM is characterized pathologically by widespread perivenular inflammation and demyelination. The uniformity of lesions differs from the multi-age lesions found in even the most acute case of MS. Post vaccination immune-mediated damage can also affect the peripheral nervous system.
Since there is no standard laboratory-based test to diagnose the human disorder, the most reliable descriptions of the clinical spectrum of the disease are derived from collections of cases in which epidemiologic and statistical studies support the association of a triggering stimulus and disease. Such criteria are best met by cases associated with immunization with CNS tissue containing vaccines, for example, original Pasteur rabies vaccine. This vaccine complication can occur at all ages. A clinical hallmark of the rabies vaccine-associated form of ADEM is its uniphasic course evolving over days to several weeks.
Page 85
|
TABLE 2.13 CNS Demyelinating Diseases That Resemble MS |
|
|
Disease Type |
Disease Characteristics |
|
Immune-Mediated Diseases |
|
|
Acute disseminated encephalomyelitis (ADEM) |
ADEM is characterized pathologically by widespread perivenular inflammation and demyelination. |
|
Systemic inflammatory or autoimmune diseases |
Multifocal CNS lesions can occur as a component of an array of systemic collagen vascular disorders including systemic lupus eryhemtosus and polyarteritis nodosa. The CNS manifestations may be the presenting feature. The peripheral nervous system is also frequently involved. |
|
Infection-Mediated Disease |
|
|
Progressive multifocal leukoencephalitis (PML) |
PML is caused by the JC virus, which infects and destroys oligodendrocytes with minimal associated immune response. It typically occurs in immunosuppressed or immunocompromised individuals and is common in AIDS. The disease can feature a subacute progressive or relapsing clinical course. The imaging and pathologic features can be distinguished from classical MS. |
|
HTLV-1 myelopathy |
HTLV-1 (human T-cell lymphotropic virus type I) infection is sometimes associated with a neurological syndrome called HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Patients with HAM/TSP have a progressive myelopathy, usually with spastic paraparesis, sensory disturbance, bladder dysfunction, and occasionally, optic neuritis. |
|
Inherited Disorders |
|
|
Dysmyelinating disorders (leukodystrophies) |
These inherited disorders are characterized by specific gene defects that result in either inadequate formation or excess breakdown of myelin. There may be a prominent inflammatory response in the region of myelin breakdown, but this is considered to be secondary to tissue breakdown. To date, therapeutic attempts with immunomodulatory agents in the leukodystrophies, specifically, adrenoleukodystrophy, have been ineffective. |
|
Toxic Disorders |
|
|
Toxic optic neuropathy, subacute myelo-optic neuropathy (SMON) |
Outbreaks of toxic optic neuropathy and SMON have been described in Cuba and in Japan.11,12,99 Sporadic cases of toxic optic neuropathy likely account for the disorder tobacco-alcohol amblyopia. Conversely, one need consider whether deficiency syndromes could underlie development of demyelinating syndromes; deficiency of vitamin B12, a cofactor in myelin formation, has also been implicated. |
Page 86
ADEM as a Consequence of Infection. ADEM, or postinfectious encephalomyelitis, has been implicated as a consequence of a wide array of viral infections, although for many, the epidemiologic data are not strong enough to support a causal link. Measles virus epidemics have, however, been convincingly linked to ADEM. Postinfectious encephalomyelitis (PIE) is thought to be autoimmune in nature rather than secondary to a direct virus injury for the following reasons: (1) encephalitis appears after the rash clears; (2) there is generally little or no evidence of infectious virus or viral genome in the CNS at the time of the demyelinating disease;143 and (3) there is evidence of increased reactivity against myelin antigens during the demyelinating disease.102 Postinfectious encephalomyelitis is generally uniphasic.
Immune system disturbances, which are known to occur following measles virus infection,104 might underlie the immunopathological response of post-infectious encephalomyelitis. A new direction for studies of PIE involves the possibility of establishing a model for this disease in transgenic mice that express the CD46 measles virus receptor.158
Molecular cross-reactivity, or molecular mimicry, has been demonstrated between myelin antigens and an array of viruses. Homologous sequences from viruses have been used to induce EAE (for example, hepatitis virus antigens). The extent of T-cell receptor degeneracy (meaning that the same receptor can respond to a wide sequence of peptides) and T-cell receptor heterogeneity in humans suggests that a wide array of exogenous agents could induce such a disease mechanism. As in MS, the putative infectious trigger could be different in individual MS patients depending on their immunogenetic makeup and the status of their immune system at the time of infection. A remaining challenge in MS is to determine whether any infectious agents detected in the CNS of such cases also persist in the CNS without causing harm or whether they are responsible for generating a pathogenic immune response.
Recurrent ADEM. ADEM cases are usually sporadic, and it is sometimes difficult to identify the initiating factor. Relapsing cases of ADEM have been described, especially in younger-age patients. For some cases, characteristic pathologic material has been available. Similarly, the EAE model can be manipulated to produce a relapsing chronic disorder by selecting animals with specific immunogenetic backgrounds and timing their immunizations so that the underlying systemic immune response is amplified or the blood-brain barrier is altered. Such variables could also determine the nature of the response of humans when they are exposed to potential disease-inducing antigens.
Progressive Multifocal Leukoencephalopathy (PML). JC virus,* a member of the papovavirus family, is a common pathogen in humans, although the
*JC refers to the initials of the individual from whom the virus was isolated in 1970.
Page 87
primary disease caused by this virus is not characterized. Common symptoms include hemiparesis, aphasia, focal seizures, and visual disturbances. JC virus is thought to reactivate in immunosuppressed hosts, especially individuals with AIDS, producing the opportunistic infection PML. The disease involves a progressive CNS syndrome with symptoms and signs that suggest white matter involvement. Demyelination is usually most prominent in the occipital lobes of the cerebral hemispheres. The histopathology is characterized by the presence of oligodendrocytes with intranuclear inclusion bodies filled with papovavirus infectious particles, indicating that this disease involves a direct, lytic infection of the oligodendrocyte; that is, the oligodendrocyte is broken apart after being infected with the virus. Enlarged astrocytes are also seen, suggesting that the JC virus can transform these cells. Demyelination is a result of the direct oligodendrocyte infection by JC virus. Thus, in PML the immune response is considered to be protective rather than pathogenic.
Human T-Lymphotropic Virus-1 (HTLV-1). This retrovirus is common in the tropics as well as Japan, and is usually associated with asymptomatic disease. HTLV-1 infection is infrequently associated with a T-cell leukemia or lymphoma or a neurological syndrome called HTLV-1-associated myelopathy/tropical spastic paraparesis (HAM/TSP). Patients with HAM/TSP have a progressive myelopathy usually with spastic paraparesis, sensory disturbance, bladder dysfunction, and occasionally, optic neuritis. The CSF generally shows a lymphocytic pleocytosis with elevated IgG and oligoclonal IgG bands directed against HTLV-1. Necrotizing lesions with inflammation in the white matter are present in the spinal cord.
HAM/TSP can resemble MS clinically. In fact, a case of HAM/TSP might be diagnosed as MS if it were not for the presence of HTLV-1 antibody and the observation that the HTLV-1 genome can be detected in the CNS.
The pathogenesis of HAM/TSP remains unclear. There are numerous CD8+ T cells that recognize the virus, suggesting that this immune response might foster white matter disease.20 Infected glial cells are a possible source of inflammatory pathogenic cytokines and might also be the target for these cytolytic T cells.140 In addition, there is some evidence that molecular mimicry plays a role in disease pathogenesis. Host genetic factors, for example, HLA type, might also be important in determining susceptibility to HAM/TSP after infection.
Infectious Causes of MS
It has not been proven that MS is caused by an infectious agent, but various data, including the inflammatory nature of the disease, epidemiological studies, and a heightened immune response against several pathogens, suggest an infectious etiology (reviewed in 1998 by Kastrukoff et al.107). Demyelinating diseases that clinically and pathologically resemble MS can be caused by viral agents or
Page 88
|
TABLE 2.14 Koch’s Postulate on Causation of Disease by a Pathogen |
|
|
Postulate |
Limitations |
|
The pathogen is always present in pathologically affected tissue. |
It would be rare to consistently isolate a pathogen from all cases of the disease that it causes because of shortcomings in isolation procedures. |
|
The pathogen is not present in tissues from controls. |
Pathogens can have a variety of clinical manifestations, from asymptomatic disease to varied diseases. |
|
The pathogen can experimentally induce disease. |
Host restriction may prevent experimental transmission. |
immunopathological responses (Table 2.14). Infectious agents might be able to cause demyelination either directly, as a result of oligodendrocyte lysis, or indirectly, by means of an immunopathological response. Demyelination mediated by an immunopathological response can occur by a number of mechanisms. For example, the infectious agent can induce a pathogenic cross-reactive immune response (molecular mimicry), or the release of myelin antigens can stimulate an immune response that is directed against white matter antigens and becomes more broad over time (epitope spreading). (See Box 2.3 for a summary of autoimmunity and disease.)
Identification of MS Pathogens
There are clear limitations to using classical criteria to implicate a pathogen isolated in MS as a causal agent for the disease, 101 as shown in Table 2.14. These issues make it especially difficult to establish the significance of a positive isolation from tissues of a patient with MS.
However, all cases in which an infectious agent causes a disease will clearly not fit standard criteria, and individualization of the requirements is sometimes necessary. Because of these limitations, it is prudent to consider criteria that may be more appropriate and realistic. One could consider the following additional guidelines in analyzing disease causation by a pathogen: consistent transmission or isolation of the pathogen; cure or effective treatment of the disease following elimination of the pathogen; absence of the disease in geographic regions where the pathogen is not present. It may also be appropriate to consider particular molecular signatures related either to the genes of the pathogen or to the transcription expression profile associated with a particular pathogen. Many pathogens have been implicated over the years as etiological agents in MS (Table 2.15).
Page 89
|
TABLE 2.15 Agents Isolated or Implicated in the Etiology of MS |
|
|
Spirochete |
Simian cytomegalovirus |
|
Rabies virus |
Epstein-Barr virus |
|
Scrapie-like agent |
Measles virus |
|
Parainfluenza virus |
HTLV-1 |
|
“Carp” agent |
MS-associated retrovirus |
|
Coronavirus |
Human herpesvirus-6 |
|
Canine distemper virus |
Chlamydia |
|
Herpes simplex-1 |
|
Some of these claims have been doubted for the following reasons: contamination (for example, spirochete), artifacts of isolation methods (for example, simian cytomegalovirus), and normal flora (for example, herpes simplex-1 [HSV-1]). Although some of these agents are clearly capable of inducing a CNS demyelinating syndrome that resembles MS, it remains uncertain whether particular cases in which a pathogen is isolated represent a rare MS-like case or whether the pathogen may actually be a common cause of MS.
There are various possible explanations of why so many pathogens have been isolated in MS but no single pathogen has been consistently observed. First, there might be a variety of pathogens that can independently cause MS (i.e., the disease is multifactorial), perhaps inducing heterogeneous forms of the disease. Second, MS might not be an infectious disease. The isolations of different pathogens might all be artifactual or related to rare events associated with a particular pathogen. Third, the relationship of the pathogen to MS might be a relatively minor one, possibly through interactions with genetic factors. Finally, the true pathogen might not yet have been identified. Thus far, there is inadequate evidence to either accept or reject suggestions that any particular pathogen is causally related to MS.
A variety of members of the herpes group of viruses (for example, simian cytomegalovirus, Epstein-Barr virus, HSV-1, and human herpesvirus-6 [HHV-6]) have been implicated in the etiology of MS. Members of this group of viruses remain attractive candidates as etiologic agents in MS since they are common pathogens that are known to persist and reactivate from a latent stage (and therefore could trigger the attacks and remissions seen in MS) and in some cases can induce focal demyelination in animals (see discussion of animal models of virus-induced demyelination).
The most recent herpesvirus candidate to generate attention is HHV-6. HHV-6, a common pathogen, is the cause of the childhood disease, roseola (exanthem subitem). This virus is associated with febrile seizures in children,87 can invade the CNS, and can persist in peripheral blood mononuclear cells and the spinal fluid. In some cases, HHV-6 induces an MS-like disease, which raises the issue
Page 90
of its broader involvement in MS. Some recent studies of HHV-6 have found no130 or rare72 evidence of HHV-6 genome in MS CSF (as well as little, if any, evidence of genome from several other members of the herpesvirus group). One recent study reported that there was an increased incidence of HHV-6 (as well as HSV and varicella-zoster virus) genome in CNS tissue from MS cases compared to tissues from controls; however, the differences were not statistically significant (see Sanders190). Some investigators have argued that the cell type that is infected by HHV-6 differs in MS CNS compared to controls and, therefore, that the virus plays a role in the pathogenesis of MS.35 The interpretation of HHV-6 studies may be complicated because HHV-6 infection and the localization of the virus may be altered secondary to inflammation associated with MS or to immunosuppressive treatment of MS patients; therefore, a change in localization of HHV-6 in MS CNS compared to control CNS may be unrelated to any pathogenic role of HHV-6 in MS. Although HHV-6 may not induce the white matter lesions of MS, it remains a possibility that HHV-6 contributes to the demyelination seen in some cases of MS. The recent identification of CD46 as a cellular receptor for HHV-6191 and the availability of transgenic mice that carry CD46158 provide an opportunity to develop an experimental model of HHV-6-induced CNS disease pathogenesis.
A recent study found that CSF from MS patients was culture-positive for Chlamydia pneumoniae and PCR (polymerase chain reaction)-positive for this agent more commonly than CSF from patients with other neurological disease.203 In addition, there was evidence of increased antibody against chlamydia in MS CSF, as well as evidence that MS CSF oligoclonal IgG bands were absorbed by chlamydia antigens. This work clearly needs confirmation.
ANIMAL MODELS OF MS
Members of the public awaiting cures for specific diseases often express impatience with the fact that most research on the biological basis of disease is based on animal studies, complaining that the time and money spent on animal studies would be better invested in clinical trials. However, animal studies are not simply interchangeable with clinical studies. Ultimately, every biologically active substance exerts its effects at the cellular and molecular levels, and the evidence has shown that this is remarkably consistent among mammals, even those as different in body and mind as rats and humans. Thus, animals can serve as models for basic biological processes in humans and can provide information about how drugs work that would not be obtainable in clinical studies.
Animal models in which both spontaneous and induced disease occur have contributed greatly to our knowledge of the pathogenesis of diseases, and in the future, they will be increasingly used to aid in the assessment of various treatment modalities. A variety of animal models have been used to study the pathogenesis and experimental treatment of diseases that share features with MS (Table 2.16).174
Page 91
|
TABLE 2.16 Animal Models of MS |
|
|
Type of Model |
Description |
|
EAE |
Immunization of mice, rats, or primates with myelin proteins (MBP, MOG, PLP) or other autoantigens (PLP). CREAE is a chronic, relapsing type of EAE |
|
B-cell models |
In contrast to T cells, there is no technology available to routinely clone autoantigen specific B cells. As an alternative approach, B-cell “monoclonal” mice have recently been generated by gene replacement transgenesis |
|
Humanized models |
Genetic engineering is used to produce animals that express particular human genes hypothesized to be involved in MS |
|
Virus-induced demyelinating disease |
A variety of viruses can induce CNS demyelination, including Theiler's murine encephalomyelitis virus, mouse hepatitis virus, and herpes simplex virus |
Most of our present knowledge of myelin-specific autoimmunity and, more generally, of immune reactivity within the CNS emanates from experimental animal models. It should, however, be noted that there is a diversity of distinct models, defined by the animal species, the target autoantigen, and the mode of induction. Three basic types of animal models have been developed to understand the disease mechanisms underlying MS: EAE, virus-induced demyelination, and genetically modified animals.
Experimental Autoimmune Encephalomyelitis (EAE)
EAE models have served, in many respects, as the prototype for current thinking on the pathogenesis of MS. This paralytic disease is characterized by the presence of inflammation and demyelination in the CNS. It is an autoimmune syndrome induced in different susceptible strains and species, generally by intradermal immunization with myelin antigens (natural or synthetic) or by adoptive transfer of T lymphocytes reactive against myelin proteins. The antigens capable of inducing EAE vary depending upon the strain and species of animal, the adjuvants employed, and perhaps also the history of environmental exposures experienced by the animal. EAE should be considered not as a single model but rather as a heterogeneous family of related disorders. Each of the EAE variants reflects different aspects of human MS, and conversely, there is no one EAE model that represents the entire complexity of MS (Table 2.17).
As is the case in MS, susceptibilities to EAE are determined as complex genetic traits. In both disorders, the most evident susceptibility locus resides within the HLA locus. Also in both disorders, locus heterogeneity is extensive (i.e., different loci and genes). Different genes can act at specific stages of the
Page 92
|
TABLE 2.17 Comparison Between Multiple Sclerosis and EAE |
||
|
MS |
EAE |
|
|
Clinical Presentation |
||
|
Relapses and remissions |
Present |
Present |
|
Paralysis |
Present |
Present |
|
Ataxia |
Present |
Present |
|
Visual impairment |
Present |
Present |
|
Genetics |
||
|
MHC-linked susceptibility |
Yes |
Yes |
|
Females more susceptible |
Yes |
Yes |
|
Pathology |
||
|
Demyelination |
Present |
Present |
|
Axonal damage |
Present |
Present |
|
T cells reactive to myelin |
Present |
Present |
|
Antibodies to myelin |
Present |
Present |
|
α4-integrin, complement |
Present |
Present |
|
TFN-α, γ-IFN |
Present |
Present |
|
Therapy |
||
|
γ-IFN, systemic |
Worsens |
Cures |
|
Anti-TNF-α, systemic |
Worsens |
Cures |
|
IL-4 transduced T cells |
Not done |
Cures |
|
TNF-α transduced T cells |
Not done |
Worsens |
|
Copaxone (glatiramer acetate) |
Improves |
Cures |
|
Beta-interferon |
Improves |
Improves |
SOURCE: Larry Steinman, presentation to the committee, November 17, 1999.
EAE or Theiler's murine encephalomyelitis virus (TMEV) disease process, influencing severity, recovery, susceptibility to relapse, remyelination, and other elements of the phenotype. Knowledge of the extensive heterogeneity in disease susceptibility and modifier genes in these MS models should provide targets for study in human MS.
Even within genetically identical littermates, the immune response following immunization with whole myelin is heterogeneous, with different antigenic targets dominating in different individuals. When groups of genetically identical animals are housed in different facilities, the resulting clinical syndromes can be markedly different, presumably reflecting the effects of different individual microenvironments.
Studies of EAE established that the pathogenic agents of the disease are CD4+ T cells, which produce cytokines of the proinflammatory Th1 pattern (IFN-γ and TNF-α, but no IL-4) upon stimulation. The precursors of these T cells are contained within the normal immune repertoire, but they unfold their pathogenic potential only on activation, by either specific antigens, microbial superantigens, or mitogens.
In EAE induced in the Lewis rat strain (and H-2u mice) by immunization
Page 93
with MBP, the encephalitogenic T cells recognize almost exclusively single, circumscript epitope segments of the MBP in the molecular context of MHC class II (clonal dominance). This dominance can be gradually lost over time, a phenomenon referred to as determinant spreading. Furthermore, in these models, the T-cell receptor for most autoreactive T cells is based on a highly simplified repertoire of structural genes, such as the Vβ8.2 gene for the TCR β-chain. These unusual features of antigen recognition have raised hopes with regard to immunospecific therapies. Unfortunately, however, they seem to be limited to only a few experimental models. Most EAE models, as well as the human myelin-specific T cells, do not show either striking epitope dominance or T-cell receptor biases.
To date, attempts to identify possible functions of CNS-specific T-cell classes distinct from CD4+ T cells have not led to consistent pictures. Evidence from T-cell transfer models and TCR transgenic mice suggests that CD8+ T cells might help control and limit an ongoing CD4+ dependent EAE process.
B Cells in EAE. Most acute EAE models show profuse inflammatory CNS reactions with a conspicuous absence of large-scale demyelination, implying that MS-like demyelination is not caused directly by myelin-specific T cells but must be brought about by other mechanisms. B cells are the best-characterized effectors of this function. Large, confluent inflammatory demyelinated lesions can be produced in rats by transferring encephalitogenic T cells along with a monoclonal antibody against myelin oligodendrocyte glycoprotein. The T cells cause inflammation, thereby opening the blood-brain barrier to the autoantibodies, which enter the CNS where they bind to myelin and destroy it via complement- or phagocyte-dependent mechanisms.46 “Simple” immunization of rodents with MOG can also produce the same result.2,53
In addition to producing humoral autoantibodies, brain-specific B cells target and present myelin antigens to specific T cells. During this presentation process, the B cells may stimulate the activation of specific T cells, with a possible tendency to shift them from Th1 (pro-inflammatory) to Th2 cytokine (anti-inflammatory) profiles.
The roles of autoreactive B cells in the pathogenesis of EAE are incompletely understood, mostly because of technical shortcomings. In contrast to T cells, there is no technology available to routinely clone autoantigen-specific B cells. As an alternative approach, B-cell “monoclonal” mice have recently been generated by gene replacement transgenesis.124 The germline repertoire of immunoglobulin genes in these mice has been replaced by the mature, rearranged gene encoding a MOG-specific autoantibody. Most, if not all, of the B cells in these mice express immunoglobulin receptors that are autoreactive to MOG. The mice spontaneously produce high titers of anti-MOG autoantibodies in their blood.
Chronic Relapsing EAE (CREAE). There is no natural, spontaneous animal model resembling MS. The immunological conditions leading to relapses
Page 94
and remissions of inflammatory demyelinating disease over time are most commonly examined in chronic relapsing versions of EAE. These episodic courses depend on the modes of immunization as well as on genetic factors innate to the host animals. Although the conditions that trigger relapses in CREAE models are not yet well understood, models are expected to provide clues to these essential aspects of MS, especially when refined by the use of suitable transgenic animals.
Primate EAE. It has recently been shown that a chronic relapsing EAE in a primate, the common marmoset, is more like MS than other EAE models.174 This form of EAE, induced in marmosets by immunization with MOG, produces lesions that are almost indistinguishable from fresh, acute human MS plaques.78 In both the human disease and this animal model, a zone of myelin destruction is seen at the margins of lesions; within the lesions, myelin sheaths are replaced by vesiculated membranous elements. MOG-specific antibodies, thought to be related to the deposition of antigen-specific antibody, are present over the vesiculated myelin. In both settings, oligodendrocytes were spared, and there was some evidence of myelin repair. Axonal pathology, however, was more conspicuous in MS cases than in this animal model. It has been suggested that processes mediated by T-cells initiate the demyelinating lesions and that other effector mechanisms are the principal offenders in damaging the myelin sheath. Mechanisms that initiate the lesion might be immunologically distinct from those that propagate disease. Antibodies might play an important role in these processes.174 The marmoset EAE model has also confirmed the encephalitogenic potential of autoreactive T-cell clones, whose precursors are preformed in the healthy immune repertoire. At the same time, however, these experiments also show that all T-cell clones are not equally autoaggressive.
Limitations of MS Animal Models
Experimental animal models of MS are based almost exclusively on the use of rodents, mostly rats and mice. Unfortunately, rodent and human immune systems differ to such a large degree that not all observations made in rodent EAE can be directly translated to human MS.
An important disadvantage of animal models is that they do not necessarily mirror the cellular or molecular pathology of MS. Some types of EAE, for example, produce brisk demyelination, whereas others produce little demyelination. Which is the best model? Since these features of MS are not yet fully understood, it is dificult to know how faithfully any given animal model of MS illustrates the human disease.
In addition, these models are not very tractable for studies on the electrophysiology and biophysics of neuronal function, a serious limitation in a disease such as MS in which symptoms and signs arise from impaired nerve function. Powerful research methods are now available for studying the physiology and
Page 95
biophysics of normal or injured nerve cells. These methods permit neuroscientists to study electrical signaling in both normal and injured neurons, but they require that these nerve cells, with reproducible abnormalities, be reproducibly located, within fractions of a millimeter, in specific parts of the nervous system so that they can be studied. In MS and in most currently available animal models, the pathology is patchy, and the location of demyelinated and injured neurons varies from case to case. For the electrophysiologist who studies neuronal signaling by precisely placing tiny microelectrodes within neurons, studying the physiology of demyelinated or otherwise injured neurons when their location varies from animal to animal is, indeed, a challenge. A model in which focal demyelination, or axonal injury, can be produced at specific locations that are consistent from animal to animal would be a great improvement.
Virus-Induced Models
Viruses can cause demyelination in several ways, the most straightforward of which is for viruses to lyse, or break open, oligodendrocytes, the myelinproducing cells. In some cases, however, the immune system is also involved. The mechanism by which virus-induced immune-mediated demyelination is carried out is not clear, but roles for molecular mimicry, bystander damage, and superantigen activation of T cells have all been proposed (see Box 2.3).
The best developed models of virus-induced demyelination are those caused by certain strains of TMEV and the mouse hepatitis virus (MHV) (reviewed in Kastrukoff et al.107). Probably the most fruitful of the remaining models are those of semliki forest virus (SFV) and herpes simplex virus (HSV) (Table 2.18). The advantages of SFV are its small, simple genome and ease of mutagenesis. Although the HSV genome is large and complex, the wealth of molecular information related to this virus and the ability to manipulate the viral genome make it an attractive model system, as well.
Theiler's Murine Encephalomyelitis Virus. The DA strain of TMEV produces an inflammatory demyelinating disease of the spinal cord with lesions that resemble MS. A variety of experimental studies of TMEV-induced demyelination suggest that as in MS, the immune system fosters demyelination. The inflammatory, demyelinating, and multifocal lesions of TMEV infection are mediated at least in part by T cells directed against viral antigens. The inflammatory response directly contributes to tissue damage in this MS-like model, since susceptibility is determined in part by immune response genes and immunosuppression abrogates demyelination.
One to two weeks after inoculation with the DA virus, there is a brisk inflammatory response in the brain with high levels of virus. This is generally a subclinical process since the mouse usually appears normal. After three weeks, the brain pathology virtually disappears, but mice develop a progressive spastic para-
Page 96
|
TABLE 2.18 Animal Viruses That Induce Demyelination |
|
|
Virus |
Consequences of Infection in Animals |
|
Theiler's murine ecephalomyelitis virus |
The TO subgroup strains of TMEV produces an inflammatory demyelinating disease of the spinal cord with lesions that resemble MS. A variety of experimental studies of TMEV-induced demyelination suggest that the immune system fosters the white matter disease. |
|
Mouse hepatitis virus |
The many different strains of MHV lead to a plethora of different diseases, including hepatitis, as well respiratory CNS disorders. The presence and extent of demyelination depend on the viral genotype, dosage, and route of inoculation, as well as the strain, age, and immune status of the infected mouse. Virus persists in glial cells of demyelinated mice. |
|
Semliki forest virus |
Experimental infection of mice and rats with specific strains of SFV leads to demyelination; other strains induce an encephalomyelitis. Demyelination depends on the specific strain of virus, the mouse strain, and the immune status of the host.68 Virus persists in the central nervous system. The role of the immune system in the demyelinating disease remains unclear. |
|
Herpes simplex virus |
Many strains of HSV produce a diffuse encephalitis in mice, but certain virus strains induce an inflammatory demyelinating disease in particular strains. Other strains of mice have multifocal demyelination that can relapse or persist in varied areas of the brain.106 The role of the immune system in this model is unclear but appears to contribute to the destruction of CNS tissue.211 |
|
Maedi visna virus |
Maedi visna is found only in sheep. There is no experimental rodent model of maedi visna infection, and therefore one needs to investigate sheep. The absence of markers for the sheep's immune system and of genetically modified sheep with knockouts of different arms of the immune system are clear limitations of the maedi visna model. The pathology of disease varies from an encephalomyelitis to a pure inflammatory demyelination that resembles MS.37 Virus persists during the disease with a restricted expression in microglial cells. It remains unclear whether demyelination is a result of direct viral lysis or is mediated by the immune response. |
|
Canine distemper virus |
CDV produces a variety of CNS diseases in dogs, including acute and chronic encephalitis and demyelinating disease. Virus persists in the chronic disease and appears to be present in oligodendrocytes in some cases. Work with this model has been limited since dogs are required as the host. The pathogenesis of the demyelinating disease remains unclear. |
paresis associated with an inflammatory demyelinating disease of the spinal cord. Although the titers of DA virus decrease over the first few weeks, virus persists in the central nervous system for the life of the mouse. The persistent virus is said to have a restricted, or incomplete, expression. In other words, viral genome is present, but the levels of infectious virus are low, with relatively little viral capsid
Page 97
protein produced. (The capsid is the protein shell surrounding the viral DNA or RNA and is generally required for viral infectivity.)
Advantages of the TMEV model include the simplicity of its genome, the detailed structural information about the virus, the ease with which it can be genetically manipulated, and finally, the extensive knowledge about the genes and immune system of mice, the natural host of TMEV (see Box 2.5.).
Despite all that is known about TMEV and despite the ease in manipulating this simple virus, the pathogenesis of the demyelinating disease is not yet fully understood. It is clear that viral persistence in the oligodendrocytes and microglia is critical to the development of TMEV-induced demyelination; that is, an ongoing virus infection is always associated with the white matter disease. It is also clear that the immune system contributes to the late demyelinating disease, but exactly how remains poorly understood. Part of the difficulty in dissecting the role of the immune system in the pathogenesis of TMEV (as well as some of the other animal models of virus-induced demyelination) is that it changes over time. Early after infection, the immune system controls the virus infection, but later in
The following features of TMEV make it an attractive model for studies of virus-induced immune-mediated demyelination: The virus is relatively small and simple, with only four structural proteins in the infectious particle. The genome is only approximately 8,100 nucleotides in length. A great deal is known about TMEV. Three strains are completely sequenced. The crystallographic structure of three strains of the virus has been solved, so that the location of every amino acid in the infectious particle is known. The B-cell epitopes that are the targets for neutralizing antibody have been identified and located on the infectious particle. Some of the epitopes on the virus that trigger proliferation of immune CD4+ T cells and some that act as targets for an antivirus cytolytic T-cell response are also known. Some components of the receptor for the virus have been identified. The virus is easily manipulated. Infectious clones of the virus are available so mutations can be quickly engineered into any region of the genome. There is extensive knowledge about mouse genetics and immunology. The mouse, which is the natural and experimental host of TMEV, provides a special benefit in studies of the pathogenesis of TMEV-induced demyelinating disease because so much is known about mouse genetics and the immune system. In addition, many genetically engineered mice are available, including those in which specific genes for different components of the immune system have been “knocked out.”
BOX 2.5
Advantages of the TMEV Model of Virus-Induced Demyelination
Page 98
the disease, it contributes to the demyelination. In addition, during the disease there appears to be a critical “balance” of the immune response that is necessary for the induction of demyelination: an inadequate immune response early in the disease can lead to the death of the mouse within the first couple of weeks and before the appearance of demyelination, while a very forceful immune response early can lead to clearance of the virus so that no virus persists and white matter disease fails to develop. In other words, viral persistence and demyelination occur only in association with a certain level of the antiviral immune response.
CD4+, as well as CD8+, T cells might be mediators of the late demyelinating disease. DA virus infection induces demyelination in both CD4+ and CD8+ T-cell knockout mice, suggesting that both CD4+ and CD8+ T cells mediate the late demyelinating disease.149 The targets for these immunopathogenic CD4+ and the CD8+ T cells are unknown. There is some evidence for epitope spreading, in which an increasing number of myelin antigenic epitopes become the target for a CD4+ T-cell response.141 However, epitope spreading appears to begin after demyelination has become established, so it is unclear how important this mechanism of immunopathology is to DA-induced demyelination, especially early in the white matter disease.
Mouse strains that are resistant to DA-induced demyelinating disease mount an antivirus cytolytic T-cell response and clear the virus. Mouse strains that are susceptible to the late disease do not mount this response, presumably allowing for virus persistence. L*, a small protein synthesized by demyelinating strains of TMEV via alternative translation, is critical for TMEV persistence and demyelination.38 L* inhibits the antivirus cytolytic T-cell response in susceptible mouse strains through an, as yet, unknown mechanism.122 Certain cells, for example, oligodendrocytes, may have cell-specific RNA-binding proteins that bind to the viral genome (as well as some nonviral messenger RNAs) and regulate whether L* or the viral capsid proteins are synthesized. The more L* that is synthesized, the more the expression of the virus is restricted.
TMEV induces apoptosis in certain cells, including neurons and macrophages.100 The relationship between apoptosis and DA-induced demyelination remains an open question.
Mouse Hepatitis Virus. There are many different strains of mouse hepatitis virus that lead to a plethora of different diseases, including hepatitis as well as respiratory and CNS disorders. JHM, S, and A59 strains of MHV induce demyelination. The extent of demyelination depends on the virus, including its geno-type, dosage, and route of inoculation, as well as on the condition of the infected mouse, including its strain, age, and immune status. Intracerebral inoculation of the JHM strain into weanling mice leads to demyelination in the mice that survive encephalitis. Virus persists in glial cells of demyelinated mice.
At present, there are some notable limitations to MHV pathogenesis studies. MHV has a remarkably large viral genome (32 kb), making this a complex
Page 99
pathogen. A system for efficient and rapid mutagenesis has not yet been perfected, and therefore manipulation of the viral genome is not straightforward.
Investigators initially thought that demyelination occurred as a result of viral lysis of oligodendrocytes independently of the immune system. More recent studies, however, suggested that it does contribute to the demyelination (reviewed in 1996 by Houtman et al.95). For example, investigators found that C57BL/6 mice that are immunosuppressed by gamma irradiation before being exposed to the JHM virus develop less severe demyelination. Adoptive transfer of JHM virus-infected splenic T cells to the infected irradiated mice leads to the development of significant demyelination. Other studies in rats showed that transfer of T cells from rats that have JHM virus-induced demyelinating encephalomyelitis leads to the development of experimental allergic encephalomyelitis-like lesions. Studies in CD4+ and CD8+ knockout mice115 demonstrated that both T-cell types are needed for clearance of the virus; however, CD4+ T cells contribute to central nervous system inflammation and demyelination. A suggestion of the latter study was that the CD4+ T cells influenced the expression of cytokines, specifically the RANTES cytokine, and led to macrophage entry into the CNS; treatment of the infected mice with anti-RANTES antibody resulted in a decrease in macrophage infiltration and demyelination. These studies and conclusions require confirmation.
The committee notes that the following animal models of virus-induced demyelinating disease are particularly likely to yield clues to the pathogenesis of MS:
-
HTLV-1 associated myelopathy. The HAM/TSP syndrome resembles MS. Because the principles relevant to this human disease might be similar to those in MS, investigating the pathogenesis of this disease could reveal insights into MS pathogenesis. The development of a widely available animal model for HAM/TSP is a high priority.
-
Postinfectious encephalomyelitis. PIE is of special interest since recurrences following this acute inflammatory white matter disease are so similar to MS attacks that the two diseases are indistinguishable, indicating a close relationship between PIE and MS. The availability of transgenic mice that carry a receptor for measles virus might provide an experimental model for the study of PIE.
-
Theiler's murine encephalomyelitis virus. The pathological lesions of TMEV are similar to MS plaques; therefore, continuing delineation of the mechanism by which the immune system contributes to the virus-induced demyelination might lead to a better understanding of the pathogenesis of demyelinating disease.
Page 100
-
Mouse hepatitis virus. Research on the MHV-induced demyelination model is presently limited by difficulties with site-directed mutagenesis methods. A high priority for research with this virus should be the generation of an infectious MHV cDNA clone and a refinement of mutagenesis techniques.
Genetically Engineered Models
Molecular genetic manipulation has become one of the most important tools for evaluating gene function in living organisms.52,79,147 These tools of molecular biology have extended the reach of researchers to a new level of understanding of neurodegeneration mechanisms. The development of animal models for neurodegenerative disorders by means of genetic engineering has revolutionized experimental neurology.27 The identification and cloning of genes involved in diseases such as Alzheimer's, Huntington's, and amyotropic lateral sclerosis provided the keys to develop mice that overexpress the human genes involved in these diseases (reviewed in 1999 by Brusa27). The nonobese diabetic (NOD) mouse is genetically susceptible to diabetes, and transgenic NOD mice have been developed to allow examination of the role of possible autoantigens in the development of diabetes, which like MS involves an inflammatory autoimmune pathology.227 The NOD mouse has also identified candidate molecules and processes that have influenced research in EAE and MS.65
Mutant mice have provided insights into all aspects of biology for generations, but only in the last two decades has it been possible to modify the expression of selected genes, an essential breakthrough for analyzing the role of specific genes in complex processes and diseases such as MS. In addition, identification of the genes that are activated or inactivated in both pathological and repair processes in the CNS will likely reveal new and unexpected targets for uniquely selective disease-modifying therapies in MS.
“Reverse genetics” and “forward genetics” offer contrasting approaches to the analysis of gene function. Forward genetics is an approach to identify genes that are not already implicated in a particular disease or process. Reverse genetics is an approach to identify the role of genes whose involvement in the disease or process being studied is already implicated.
In forward genetics, large numbers of mice are mutagenized (using techniques that mutagenize genes at random); their resulting phenotypes are analyzed to select mutants that exhibit spontaneous MS. The mutated genes in the selected mutants are then positionally cloned and identified. The homologous human clones can also be identified, a task that is becoming vastly simpler as the human genome project nears completion. The advantage of this approach is that the screen is not biased in any way and can reveal genes beyond those already known to be involved in MS. Another advantage of forward genetics is that once a gene is identified in a disease process, one can quickly do a screen for suppressors and
Page 101
enhancers of the phenotype. This will yield the entire biochemical pathway (as opposed to just one step) involved in disease pathogenesis.
One limitation to the forward genetics approach is that only a small percentage of the mutated genes will result in a phenotype relevant to MS. Consequently, large numbers of mutants have to be screened, which is generally expensive, time-consuming, and labor intensive. Yet the rewards—especially for complex diseases and processes that have resisted traditional approaches—are unparalleled.
In reverse genetics, mutant strains of mice that either overexpress or lack specific genes are generated through a variety of techniques. The classic approach to creating transgenic mice is to inject a foreign gene (“transgene”) into a fertilized egg, thereby inducing overexpression of the transgene. The egg bearing the transgene is implanted into a host mother. Progeny bearing DNA encoding the transgene are screened, as are the corresponding levels of RNA and protein. This transgene is randomly incorporated into a mouse chromosome and ultimately leads to production of the protein of interest.27 This approach has been used to transfer human genes (such as those for T-cell receptor, HLA DR2, and CD4) into mice to see if they develop spontaneous MS. One limitation of “knockin” mice is that genes can be inserted in uneven copy numbers in “replicate” animals or might be integrated into disparate sites in the genome.226
Gene expression can also be altered through knockout experiments. Knockout mice, or null mutants, are created using embryonic stem cells and homologous recombination to produce a cell line in which a certain gene has been removed, or ablated. When transferred into an early mouse embryo, these cells can participate in the generation of all cell lineages including germ cells, thereby transmitting their genotype to the next generation. Alternatively, embryonic stem cells can be used to create so-called knock-in mice by inserting a gene into a particular locus.
The transgenic gene-targeting approaches described above all rely on irreversible changes to the genome that are present from the onset of development throughout an animal's life. The function of the gene must be deduced from the phenotype of animals that have been deficient for the product of the disrupted gene throughout development. Yet, many genes play different roles at different stages of development and in different tissues. This presents serious drawbacks (reviewed in 1998 by Gingrich and Roder79 and in 1999 by Muller147). First, an animal with a gene alteration that lethally disrupts development obviously cannot be studied as an adult—even though the gene might play another critical role, for example, in neural repair. This is particularly relevant to MS because many of the genes that regulate embryonic neural development also regulate neural repair in adults. Another limitation of transgenic models is that changes in the regulation of other genes could yield misleading phenotypes, in part because of differences between effects of the gene at different developmental stages, gene redundancy (other genes might also play the role of the missing gene), or adaptive mecha-
Page 102
nisms that compensate for the missing gene. Even apparently unaltered phenotypes would thus not prove that the gene was not involved in the disease or process being studied. Another limitation of this approach is that in physiological responses, gene products tend to be produced in waves, whereas in transgenics, expression is usually “on” from the time of development.
Alternative approaches in which one gene, or parts of it, can be inactivated or activated in specific tissues or at specific times (“conditional” and “inducible” mutants) have recently been developed in this rapidly expanding array of genealtering techniques (reviewed in Brusa, 1999;27 Gingrich and Roder, 1998;79 and van der Neut, 1997215). This second generation of transgenic technology derives from the possibility of modulating the suppression of a transgene with external stimuli, by using a “biological switch” that can turn the foreign gene on and off.
Gene expression patterns in the nervous system are highly regionalized. For example, the enzymes involved in producing neurotransmitters and their receptors differ from one subpopulation of neurons to another. Other proteins are more widespread, such as the intermediate filament proteins NF (neurofilament) or GFAP (glial fibrillary acidic protein), or are ubiquitous, such as N-CAMs (neural cell adhesion molecules) and integrins.215 Thus, it would be particularly advantageous to develop tissue-specific mutants for MS research. The development of inducible systems will become an important tool in the many diverse aspects of research on the disease mechanisms and possibilities for repair in MS, including the potential administration of gene therapy.
Transgenic Mice and Demyelinating Disease. Transgenic overexpression of cytokines or gene targets of cytokines in the CNS offers a relatively non-intrusive mechanism to assess the role of individual cytokines in CNS development, function, and response to insult. A common technique for assessing the effects of a particular cytokine in EAE is to induce its expression directly in the CNS. This allows researchers to ask whether expression of the cytokine induces CNS pathology similar to that seen in MS. Directed expression of transgenes in the CNS relies on promoters that normally control the expression of CNS-specific genes. This includes the GFAP gene promoter, which drives expression in astrocytes, as well as the neurofilament promoter (neurons), and the myelin basic protein promoter (oligodendrocytes). To date, no one has isolated a microglial-specific promoter.
Only MBP promoters have been used to overexpress gamma-interferon in the CNS.47,94,176 Phenotypes of transgenic mice range from a lethal, “jimpy”-like hypomyelinating mouse,47* through progressive demyelinating disease,94 to mice with no outward phenotype that nevertheless showed progression from EAE to a
*A point mutation in the gene coding for myelin proteolipid protein causes male offspring of jimpy mice to have little or no myelination. Affected mice develop severe tremors and die prematurely at approximately 30 days.
Page 103
chronic demyelinating disease in contrast to control mice that recovered from EAE-induced demyelination.176 It is of interest that the same laboratory observed both extremes using the same constructs.13,176 This might be because the transgenes were unevenly integrated at different loci, since levels of expression did not obviously correlate with phenotype in one case.176 Asymptomatic transgenic mice did, however, show enhanced glial responses to CNS injury,13 and exacerbated ischemic infarction (Lambertsen et al., unpublished). These asymptomatic mice presumably reflect sub-threshold levels of cytokine. Nevertheless, crossing an asymptomatic MBP promoter-driven gamma-interferon transgenic mouse with MBP or MHC class I mice produced a more extreme jimpy phenotype.13 This might provide a clue to cytotoxic effects of beta-interferon-γ on oligodendrocytes,3 being perhaps dependent on local interferon-γ titers becoming sufficiently high to stimulate MHC I induction.151 It is not known whether similar mechanisms account for oligodendrocyte pathology in TNF-α and interferon-α transgenic mice.
The IL-3 and IL-12 transgenic mice provide a useful counterpart, there being no obvious suggestion of a direct effect on oligodendrocytes.39,160 These mice illustrate the potential for direct macrophage and microglial attack on oligodendrocytes, which might occur in TNF-α transgenic mice.4,33,204 It is not clear from any of these experiments whether activation of immune cells took place within the CNS or following cytokine exit to the periphery. A systemic effect must account for the fact that overexpression of the antiinflammatory cytokine IL-10 protected animals from EAE in two separate preparations.19,50 The fact that IL-4 transgenic mice did not show similar resistance19 might reflect insufficient expression within the CNS or strain background differences, given that IL-4 knockout exacerbated disease in another study.66
Most recently, a transgenic mouse has been “constructed” that expresses T-cell receptor genes from a human MBP-specific T-cell clone, along with relevant human MHC class II determinants and MBP. Under certain conditions, this “humanized” mouse developed spontaneous EAE that indeed showed inflammation with some demyelination.129
Summary of Genetic Models. In recent years, there have been many advances in the use of transgenes (including gene knockouts), as well as even more sophisticated models that make use of tissue-specific and time-dependent regulators. These models should facilitate the development of rational therapies and the transfer of knowledge from animal models to the prevention and treatment of human disease.226 However, although temporally regulated targeting controlled by the administration of an environmental inducer has become feasible with high efficiency for some organs, it remains to be further improved for other tissues, particularly the brain.147
New generations of inducible promoters will more faithfully mimic the in vivo kinetics and dynamics of cytokine production. Knock-in mice, in which
Page 104
transgenes are integrated into defined loci through homologous recombination, will likewise overcome the problems of uneven gene copy numbers in replicate animals and disparate sites of integration in the genome.
REFERENCES
1. . 1977. Epidemiology of multiple sclerosis. Br Med Bull.; 33: 9-14.
2. , , , . 1995. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol.; 63: 17-27.
3. , , . 1996. Reversible inhibitory effects of interferon-gamma and tumour necrosis factor-alpha on oligodendroglial lineage cell proliferation and differentiation in vitro. Eur J Neurosci.; 8: 1106-16.
4. , , , . 1998. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. Am J Pathol.; 153: 801-13.
5. , . 1999. Molecular mimicry and autoimmunity. N Engl J Med.; 341: 2068-74.
6. , . 1998. Observations on differences between interferons to treat multiple sclerosis. Journal of Clinical Research; 1: 381-392.
7. , , , . 1990. T cells responsive to myelin basic protein in patients with multiple sclerosis. Science.; 247: 718-21.
8. , , , . 1999. A prospective study on the natural history of multiple sclerosis: clues to the conduct and interpretation of clinical trials. J Neurol Sci.; 168: 96-106.
9. , , . 1996. Immunotherapy for multiple sclerosis: from theory to practice. Nat Med.; 2: 1074-5.
10. , . 1996. HLA class II genes: structure and diversity. Browning M, McMichael A, eds. HLA and MHC: Genes, Molecules and Function. Oxford: BIOS Scientific Publishers Limited; 97-109.
11. , , , . 1998. Clioquinol-zinc chelate: a candidate causative agent of subacute myelo-optic neuropathy. Mol Med.; 4: 665-670.
12. . 1997. Antimetabolites and an optic neuropathy epidemic in Cuba. Am J Clin Nutr.; 65: 1092-1093.
13. , , . 2000. Major histocompatibility complex heavy chain accumulation in the endoplasmic reticulum of oligodendrocytes results in myelin abnormalities. J Neurosci Res.; 59: 160-9.
14. , , , . 1997. Comparison of MRI criteria at first presentation to predict conversion to clinically definite multiple sclerosis. Brain.; 120 : 2059-69.
15. , . 1991. Immunology: a Short Course. NY: Wiley-Liss, Inc.
16. . 1997. Role of macrophages/microglia in multiple sclerosis and experimental allergic encephalomyelitis. J Mol Med.; 75: 165-73.
17. , , , . 1997. Myelin oligodendrocyte glycoprotein: a novel candidate autoantigen in multiple sclerosis. J Mol Med.; 75: 77-88.
18. , . 1999. Role of viral infection in the aetiology of multiple sclerosis. Status of current knowledge and therapeutic implications. CNS Drugs.; 12: 1-7.
19. , , , , , . 1998. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol.; 161: 3299-306.
Page 105
20. , , , , , . 1997. Direct ex vivo analysis of activated, Fas-sensitive autoreactive T cells in human autoimmune disease. J Exp Med.; 185: 1585-94..
21. , , . 1999. Methods for the analysis of quality-of-life and survival data in health technology assessment. Health Technol Assess.; 3: 1-152..
22. , , , , , . 2000. Sensory neuron specific sodium channel SNS is abnormally expressed in the brains of mice with experimental allergic encephalomyelitis and humans with multiple sclerosis. Proc Natl Acad Sci U.S.A.; 97: 11598-11602..
23. , , . 1987. Macromolecular structure of axonal membrane during acute experimental allergic encephalomyelitis in rat and guinea pig spinal cord. J Neuropathol Exp Neurol.; 46: 167-84..
24. , , , . 2000. Lesion heterogeneity in multiple sclerosis: a study of the relations between appearances on T1 weighted images, T1 relaxation times, and metabolite concentrations. J Neurol Neurosurg Psychiatry.; 68: 627-32..
25. , , , , . 1996. Heat shock proteins and multiple sclerosis: a review. J Neuropathol Exp Neurol.; 55: 389-402..
26. , . 1996. Mechanisms of immune injury in multiple sclerosis. Brain Pathol.; 6: 243-57..
27. . 1999. Genetically modified mice in neuropharmacology. Pharmacol Res.; 39: 405-19..
28. , , , . 1999. Embryonic stem cell-derived glial precursors: a source of myelinating transplants. Science.; 285: 754-6..
29. , , . 2000. Cost utility of drugs for multiple sclerosis. Systematic review places study in contrast. BMJ.; 320: 1474-5.; discussion 1475-6.
30. , , , . 1999. Cloning the antibody response in humans with inflammatory CNS disease: isolation of measles virus-specific antibodies from phage display libraries of a subacute sclerosing panencephalitis brain. J Neuroimmunol.; 94: 204-11..
31. , , , . 1997. Increases in soluble VCAM-1 correlate with a decrease in MRI lesions in multiple sclerosis treated with interferon beta-1b. Ann Neurol.; 41: 669-74..
32. , , . 1999. Cytokines, chaos, and complexity. Immunity.; 11: 507-13..
33. , , , , . 1998. Transgenic models to study the actions of cytokines in the central nervous system. Neuroimmunomodulation.; 5: 126-35..
34. , , , . 2000. Comparison of MRI pulse sequences for investigation of lesions of the cervical spinal cord. Neuroradiology.; 42: 669-75..
35. , , , . 1995. Plaque-associated expression of human herpesvirus 6 in multiple sclerosis. Proc Natl Acad Sci U S A.; 92: 7440-4..
36. . 1868. Histologie de la sclerose en plaques. Gaz Hosp.; 141: 554-555, 557-558..
37. , , , . 1998. Neuroinvasion by ovine lentivirus in infected sheep mediated by inflammatory cells associated with experimental allergic encephalomyelitis. J Neurovirol.; 4: 38-48..
38. , , , , . 1995. A picornaviral protein synthesized out of frame with the polyprotein plays a key role in a virus-induced immune-mediated demye linating disease. Nat Med.; 1: 927-31..
39. , , , , . 1996. Macrophage/microglial-mediated primary demyelination and motor disease induced by the central nervous system production of interleukin-3 in transgenic mice. J Clin Invest.; 97: 1512-24..
40. , , , , . 1992. Tumor necrosis factor alpha production as a possible predictor of relapse in patients with multiple sclerosis. Eur Cytokine Netw.; 3: 523-31..
Page 106
41. , , . 2000. The relationship of MHC-peptide binding and T cell activation probed using chemically defined MHC class II oligomers. Immunity.; 12: 241-50..
42. . 1991. Autoimmunity to chaperonins in the pathogenesis of arthritis and diabetes. Annu Rev Immunol.; 9: 567-89..
43. , , , . 1999. Therapy of relapsing multiple sclerosis. Treatment approaches for nonresponders. J Neuroimmunol.; 98: 29-36..
44. . 1997. Genetic epidemiology of multiple sclerosis. J Neurol Neurosurg Psychiatry.; 62: 553-61..
45. , , , . 2000. Relapses and progression of disability in multiple sclerosis. N Engl J Med.; 343: 1430-1438..
46. , , , . 1999. The immunobiology of multiple sclerosis: an autoimmune disease of the central nervous system. Neurobiol Dis.; 6: 149-66..
47. , , , , , . 1996. Targeted CNS expression of interferon-gamma in transgenic mice leads to hypomyelination, reactive gliosis, and abnormal cerebellar development. Mol Cell Neurosci.; 7: 354-70..
48. , , , , , . 1996. Identification of peptides specific for cerebrospinal fluid antibodies in multiple sclerosis by using phage libraries. Proc Natl Acad Sci U S A. ; 93: 11063-7..
49. , , , , , . 1999. Insights into the neurodegenerative process of Alzheimer's disease: a role for mononuclear phagocyte-associated inflammation and neurotoxicity. J Leukoc Biol.; 65: 416-27..
50. , , , , . 1999. Transgenic interleukin 10 prevents induction of experimental autoimmune encephalomyelitis. J Exp Med.; 189: 1005-10..
51. , , , . 1995. Persistent functional deficit in multiple sclerosis and autosomal dominant cerebellar ataxia is associated with axon loss. Brain .; 118 : 1583-92..
52. , , . 1994. Reverse genetics of the mouse central nervous system: targeted genetic analysis of neuropeptide function and reverse genetic screens for genes involved in human neurodegenerative disease. Prog Neurobiol.; 42: 319-31..
53. , , , . 1997. Induction of EAE in mice with recombinant human MOG, and treatment of EAE with a MOG peptide. J Neuroimmunol. ; 75: 169-73..
54. . 1997. An overview of the immune system: Immunologycial mechanisms in immune deficiency and autoimmunity. Leffell MS, Donnenberg AD, Rose NRE. Handbook of Human Immunology .em>. Boca Raton. CRC Press. 47-64..
55. , , , . 1995. Clustering of Na+ channels and node of Ranvier formation in remyelinating axons. J Neurosci.; 15: 492-503..
56. , . Paty DW, Ebers GC, eds. 1997a. Multiple Sclerosis. Philadelphia, PA. F.A. Davis Company. 5-28..
57. , . 1997b. Susceptibility: Genetics in multiple sclerosis. Paty DW, Ebers GC, eds. Multiple Sclerosis. Philadelphia, PA. F.A. Davis Company. 29-47..
58. , , . 1995. A genetic basis for familial aggregation in multiple sclerosis. Canadian Collaborative Study Group. Nature.; 377: 150-1..
59. . 2000. Cost utility of drugs for multiple sclerosis. Analysis goes too far. BMJ.; 320: 1475-6..
60. , , , . 2000. Adhesion molecules in multiple sclerosis: relation to subtypes of disease and methylprednisolone therapy. Arch Neurol.; 57: 546-51..
61. . 1998. Evoked potentials in clinical trials for multiple sclerosis. J Clin Neurophysiol. ; 15: 109-16..
62. , , . 2000. Glial fibrillary acidic protein: GFAP-thirty-one years (1969-2000). Neurochem Res.; 25: 1439-51..
63. , , , . 1990. Changed distribution of sodium channels along demyelinated axons. Proc Natl Acad Sci U S A.; 87: 6777-80..
Page 107
64. . 1999. Economic malpractice: when methods become an end instead of a means. Transplantation.; 68: 11-2..
65. , . 1999. Walking through the forest of transgenic models of human disease. Immunol Rev.; 169: 5-10..
66. , , , . 1998. A critical role for IL-4 in regulating disease severity in experimental allergic encephalomyelitis as demonstrated in IL-4-deficient C57BL/6 mice and BALB/c mice. J Immunol.; 160: 4822-30..
67. , . 1999. The glial scar and central nervous system repair. Brain Res Bull.; 49: 377-91..
68. , . 1987. Semliki Forest virus induced, immune mediated demyelination: the effect of irradiation. Br J Exp Pathol.; 68: 101-13..
69. , , , . 1999. The contribution of magnetic resonance imaging to the diagnosis of multiple sclerosis. Neurology.; 53: 448-56..
70. , , , . 1988. Criteria for an increased specificity of MRI interpretation in elderly subjects with suspected multiple sclerosis. Neurology.; 38: 1822-5..
71. , . 2000. Magnetisation transfer imaging in multiple sclerosis. J Neurovirol.; 6 Suppl: S115-20..
72. , , , , . 1998. HHV-6 and multiple sclerosis. Nat Med.; 4: 537..
73. , , , . 1999. Population based cost utility study of interferon beta-1b in secondary progressive multiple sclerosis. BMJ.; 319: 1529-33..
74. , , . 1980. Reorganization of the axon membrane in demyelinated peripheral nerve fibers: morphological evidence. Science.; 210: 661-3..
75. , , , . 1998. Expression and crystallization of the complex of HLA-DR2 (DRA, DRB1*1501) and an immunodominant peptide of human myelin basic protein. Proc Natl Acad Sci U S A.; 95: 11828-33..
76. , , , . 2000. Brain atrophy in relapsing-remitting multiple sclerosis and secondary progressive multiple sclerosis: longitudinal quantitative analysis. Radiology.; 214: 665-70..
77. , , , . 1999. Identification of autoantibodies associated with myelin damage in multiple sclerosis. Nat Med.; 5: 170-5..
78. , , , . 1995. Antibody facilitation of multiple sclerosis-like lesions in a nonhuman primate. J Clin Invest.; 96: 2966-74..
79. , . 1998. Inducible gene expression in the nervous system of transgenic mice. Annu Rev Neurosci.; 21: 377-405..
80. , , , . 1997. Longitudinal study of soluble adhesion molecules in multiple sclerosis: correlation with gadolinium enhanced magnetic resonance imaging. Neurology.; 48: 1557-65..
81. , . 1998. Neuronal regeneration: extending axons from bench to brain. Curr Biol.; 8: R310-2..
82. , . 1999. Microglia as mediators of inflammatory and degenerative diseases. Annu Rev Neurosci.; 22: 219-40..
83. , . 1999. Infectious etiology in multiple sclerosis: The debate continues. Trends in Microbiology.; 7: 475-7..
84. , . 2000. Practice parameter: the usefulness of evoked potentials in identifying clinically silent lesions in patients with suspected multiple sclerosis (an evidence-based review): Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology.; 54: 1720-5..
85. , , . 2000. Assessment of spinal cord damage in MS using MRI. J Neurol Sci.; 172: S36-9..
Page 108
86. , , , , . 1996. TCR usage in human and experimental demyelinating disease. Immunol Today.; 17: 152-9..
87. , , , . 1994. Human herpesvirus-6 infection in children. A prospective study of complications and reactivation. N Engl J Med.; 331: 432-8..
88. , . 1977. Pathophysiology of demyelinating disease. Brit. Med Bull.; 33: 21-27..
89. , , . 2000. The age-range of risk of developing multiple sclerosis: evidence from a migrant population in Australia. Brain.; 123: 968-74..
90. . 2000. [gamma][delta] cells: a right time and a right place for a conserved third way of protection. Annu Rev Immunol.; 18: 975-1026..
91. , , , . 1995. Illusory contours activate specific regions in human visual cortex: evidence from functional magnetic resonance imaging. Proc Natl Acad Sci U S A.; 92: 6469-73..
92. , , . 1998. Human autoimmune neuropathies. Annu Rev Neurosci.; 21: 187-226..
93. . 1999. Therapeutic strategies in multiple sclerosis. I. Immunotherapy. Philos Trans R Soc Lond B Biol Sci.; 354: 1697-710..
94. , , , , . 1997. Primary demyelination in transgenic mice expressing interferon-gamma. Nat Med.; 3: 1037-41..
95. , . 1996. Pathogenesis of mouse hepatitis virus-induced demyelination. J Neurovirol.; 2: 361-76..
96. , . 1988. Evoked potentials in suspected multiple sclerosis: diagnostic value and prediction clinical course. J. Neurol Sci.; 83: 191-210..
97. , . 1996. Multiple sclerosis: sunlight, diet, immunology and aetiology. Med Hypotheses.; 46: 67-74..
98. Institute of Medicine. Field MJ, Gold MR, editors. Summarizing Population Health. Directions for the Development and Application of Population Metrics. Washington, DC. National Academy Press. 1998.
99. , , . 1998. Isolation of Inoue-Melnick virus from cerebrospinal fluid of patients with epidemic neuropathy in Cuba. Arch Pathol Lab Med.; 122: 520-522..
100. , , . 1999. Differentiation of M1 myeloid precursor cells into macrophages results in binding and infection by Theiler's murine encephalomyelitis virus and apoptosis. J Virol.; 73: 3227-35..
101. , . 1974. Editorial: Koch's postulates and slow infections of the nervous system. Arch Neurol.; 30: 36-8..
102. , , , . 1984. Measles encephalomyelitis—clinical and immunologic studies. N Engl J Med.; 310: 137-41..
103. , , , . 1999. Elevated levels of antibody to myelin oligodendrocyte glycoprotein is not specific for patients with multiple sclerosis. Arch Neurol.; 56: 311-5..
104. , , , . 1996. Mechanism of suppression of cell-mediated immunity by measles virus. Science.; 273: 228-31..
105. , . 1998. Chemokine regulation of experimental autoimmune encephalomyelitis: temporal and spatial expression patterns govern disease pathogenesis. J Immunol.; 161: 2667-71..
106. , , , , , . 1992. Herpes si plex virus type I (HSV I)-induced multifocal central nervous system (CNS) demyelination in mice. J Neuropathol Exp Neurol.; 51: 432-9..
107. , . 1998. Virology. Paty DW, Ebers GC, eds. Multiple Sclerosis. Philadelphia. F.A. Davis Company. 370-402..
Page 109
108. , , , , , . 1993. Reactivity to myelin antigens in multiple sclerosis. Peripheral blood lymphocytes respond predominantly to myelin oligodendrocyte glycoprotein. J Clin Invest.; 92: 2602-8..
109. , , , . 1996. Western versus Asian types of multiple sclerosis: immunogenetically and clinically distinct disorders. Ann Neurol.; 40: 569-74..
110. , , . 1998. Underlying cause of death in Danish patients with multiple sclerosis: results from the Danish Multiple Sclerosis Registry. J Neurol Neurosurg Psychiatry.; 65: 56-9..
111. , , , . 1999. Mitoxantrone in progressive multiple sclerosis: MRI results of the European phase III trial. Neurology.; 52: A495..
112. . 1997. The Epidemiology of Multiple Sclerosis. Raine CS, McFarland HF, Tourtellotte WWE. Multiple Sclerosis: Clinical and Pathogenic Basis. London. Chapman & Hall. 91-139..
113. . 1997. On the role of veterans in the development of neurology in the United States: a personal reflection. Neurology.; 49: 323-33..
114. , . 1997. Epidemiology of multiple sclerosis in US veterans: VII. Risk factors for MS. Neurology.; 48: 204-13..
115. , , , . 2000. A central role for CD4(+) T cells and RANTES in virus-induced central nervous system inflammation and demyelination. J Virol.; 74: 1415-24..
116. . 1999. The pathology of multiple sclerosis and its evolution. Philos Trans R Soc Lond B Biol Sci.; 354: 1635-40..
117. , , , . 1988. Experimental allergic encephalomyelitis: the balance between encephalitogenic T lymphocytes and demyelinating antibodies determines size and structure of demyelinated lesions. Acta Neuropathol (Berl).; 75: 566-76..
118. , . Experimental models of multiple sclerosis. Compston A, Ebers GC, Lassmann H, Matthews B, Wekerle H, eds. 1998. McAlpine's Multiple Sclerosis. Third ed. London. Churchill Livingston. 409-434..
119. , , , . 2000. Axonal injury or loss in the internal capsule and motor impairment in multiple sclerosis. Arch Neurol.; 57: 65-70..
120. , , , . 2000. The motor cortex shows adaptive functional changes to brain injury from multiple sclerosis. Ann Neurol.; 47: 606-613..
121. , , . 1973. When is multiple sclerosis acquired? Harefuah.; 85: 206-08..
122. , , , , . 1999. A Theiler's virus alternatively initiated protein inhibits the generation of H-2K-restricted virus-specific cytotoxicity. J Immunol.; 162: 17-24..
123. , , , , . 1988. Augmentation of demyelination in rat acute allergic encephalomyelitis by circulating mouse monoclonal antibodies directed against a myelin/oligodendrocyte glycoprotein. Am J Pathol.; 130: 443-54..
124. , , , . 1998. B lymphocytes producing demyelinating autoantibodies: development and function in gene-targeted transgenic mice. J Exp Med.; 188: 169-80..
125. , . 1996. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology.; 46: 907-11..
126. , , , . 1998. Multiple sclerosis: lessons from neuropathology. Semin Neurol.; 18: 337-49..
127. , . 1997. The controversy surrounding the pathogenesis of the multiple sclerosis lesion. Mayo Clin Proc.; 72: 665-78..
Page 110
128. , , , , , . 2000. Spinal cord magnetic resonance imaging in suspected multiple sclerosis. Eur Radiol.; 10: 368-76..
129. , , , . 1999. A humanized model for multiple sclerosis using HLA-DR2 and a human T-cell receptor. Nat Genet.; 23: 343-7..
130. , , , . 1997. Absence of seven human herpesviruses, including HHV-6, by polymerase chain reaction in CSF and blood from patients with multiple sclerosis and optic neuritis. Acta Neurol Scand.; 95: 280-3..
131. , , . 1992. Immunological aspects of demyelinating diseases. Annu Rev Immunol.; 10: 153-87..
132. . 1998. Are there indicators of remyelination in blood or CSF of multiple sclerosis patients? Mult Scler.; 4: 228-31..
133. , , , . 1999. Transplanted embryonic stem cells survive, differentiate and promote recovery in injured rat spinal cord. Nat Med.; 5: 1410-2..
134. . 1977. Acute optic neuritis. Br J Hosp Med.; 18: 42-8..
135. , , . 1992. The pathological evolution of multiple sclerosis. Neuropathol Appl Neurobiol.; 18: 319-34..
136. . 1998. The lesion in multiple sclerosis: clinical, pathological, and magnetic resonance imaging considerations. J Neurol Neurosurg Psychiatry.; 64 Suppl 1: S26-30..
137. , , . 1997. Genetic Influences in Multiple Sclerosis. Raine CS, McFarland HF, Tourtellott WWE. Multiple Sclerosis: Clinical and Pathogenetic Basis.em>. London. Chapman & Hall. 205-219..
138. . Function of HLA class I restricted T cells. Browning M, McMichael A, eds. 1996. HLA and MHC: Genes, Molecules and Function. Oxford. BIOS Scientific Publishers Limited. 309-320..
139. , , , . 1998. A gliotoxic factor and multiple sclerosis. J Neurol Sci.; 154: 209-21..
140. , , , . 1997. Astrocyte-specific expression of human T-cell lymphotropic virus type 1 (HTLV-1) Tax: induction of tumor necrosis factor alpha and susceptibility to lysis by CD8+ HTLV-1-specific cytotoxic T cells. J Virol.; 71: 9143-9..
141. , , , . 1997. Persistent infection with Theiler's virus leads to CNS autoimmunity via epitope spreading. Nat Med.; 3: 1133-6..
142. , , , , . 1997. Acute exacerbation of multiple sclerosis increases plasma levels of S-100 protein. Acta Neurol Scand.; 96: 142-4..
143. , , , , . 1988. Acute measles in patients with and without neurological involvement: distribution of measles virus antigen and RNA. J Infect Dis.; 158: 433-42..
144. , , , . 1999. The psychosocial impact of multiple sclerosis: exploring the patient's perspective. Health Psychol.; 18: 376-82..
145. , , , . 1991. Increase of sodium channels in demyelinated lesions of multiple sclerosis. Brain Res.; 556: 311-6..
146. . Neuropathology and Pathophysiology of the Multiple Sclerosis Lesion. Paty DW, Ebers GC, eds. 1998. Multiple Sclerosis. Philadelphia, PA. F.A. Davis Company. 257-327..
147. . 1999. Ten years of gene targeting: targeted mouse mutants, from vector design to phenotype analysis. Mech Dev.; 82: 3-21..
148. , , , , , . 1994. The British Isles survey of multiple sclerosis in twins. Neurology.; 44: 11-5..
149. , , , , . 1998. CD4(+) and CD8(+) T cells make discrete contributions to demyelination and neurologic disease in a viral model of multiple sclerosis. J Virol.; 72: 7320-9..
Page 111
150. , , . 1991. A review of the aetiology of multiple sclerosis: an ecological approach. Ann Hum Biol.; 18: 95-112..
151. , , , , . 1997. Major histocompatibility complex (MHC) class I gene expression in single neurons of the central nervous system: differential regulation by interferon (IFN)-gamma and tumor necrosis factor (TNF)-alpha. J Exp Med.; 185: 305-16..
152. , . 1999. Beta interferons (1a and 1b) in relapsing-remitting and secondary progressive multiple sclerosis. Development and Evaluation Committee Report No. 98. Southampton. Wessex Institute for Health Research and Development.
153. . 1999. Progress in determining the causes and treatment of multiple sclerosis. Nature.; 399: A40-7..
154. , , , . 2000. Multiple sclerosis. N Engl J Med.; 343: 938-52..
155. , , , . 1998. Evoked potential abnormality scores are a useful measure of disease burden in relapsing-remitting multiple sclerosis. Ann Neurol.; 44: 404-7..
156. , , . 1996. Genetics of demyelinating diseases. Brain Pathol.; 6: 289-302..
157. . 1998. Molecular mimicry and immune-mediated diseases. FASEB J.; 12: 1255-65..
158. , , , . 1999. Measles virus infection in a transgenic model: virus-induced immunosuppression and central nervous system disease. Cell.; 98: 629-40..
159. . 1998. Comparison of drug treatments for multiple sclerosis. Ottawa. Canadian Coordinating Office for Health Technology Assessment.
160. , , , , , . 2000. Astrocyte-targeted expression of IL-12 induces active cellular immune responses in the central nervous system and modulates experimental allergic encephalomyelitis. J Immunol.; 164: 4481-92..
161. , , , , , . 1998. Innate and adaptive functions of the CD1 pathway of antigen presentation. Semin Immunol.; 10: 391-8..
162. , , , , , . 1998. A cost-utility analysis of interferon beta for multiple sclerosis. Health Technol Assess.; 2: iii-54..
163. , , , . 1999. Management of relapsing-remittting multiple sclerosis: diagnosis and treatment guidelines. European J Neurol.; 6 (suppl): S1-S35..
164. , . Diagnosis of multiple sclerosis 1998: do we need new diagnostic criteria? Siva et al. eds. 1998. Frontiers in Multiple Sclerosis .em>. London. Martin Dunitz. 2: 47-50..
165. , , , . 1987. S-100 protein and neuron-specific enolase in cerebrospinal fluid and serum: markers of cell damage in human central nervous system. Stroke.; 18: 911-8..
166. . 1995. Notes on the epidemiology of multiple sclerosis. J Formos Med Assoc.; 94: 300-8..
167. . 1997. Misdiagnosis of multiple sclerosis and beta-interferon. Lancet.; 349: 1916..
168. , , , . 1983. New diagnostic criteria for multiple sclerosis: guidelines for research protocols. Ann Neurol.; 13: 227-31..
169. , . 1978. The fine structure of chronically active multiple sclerosis plaques. Neurology .em>. 28: 68-75..
170. , , , , , . 1998. Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest.; 102: 1045-50..
171. , . 1994. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol 1994; 12: 487-520..
172. . 1997. Multiple sclerosis. Brain Pathology.; 7: 1237-1241..
Page 112
173. . 1997. The Norton Lecture: a review of the oligodendrocyte in the multiple sclerosis lesion. J Neuroimmunol.; 77: 135-52..
174. , , , . 1999. Demyelination in primate autoimmune encephalomyelitis and acute multiple sclerosis lesions: a case for antigen-specific antibody mediation. Ann Neurol.; 46: 144-60..
175. , , , , , . 1999. Neuroglial activation repertoire in the injured brain: graded response, molecular mechanisms and cues to physiological function. Brain Res Brain Res Rev.; 30: 77-105..
176. , , , . 1998. Interferon-gamma in progression to chronic demyelination and neurological deficit following acute EAE. Mol Cell Neurosci.; 12: 376-89..
177. , , . 2000. Cost utility of drugs for multiple sclerosis. Methods used don't calculate true benefit. BMJ.; 320: 1475.; discussion 1475-6.
178. . 1997. Soluble adhesion molecules (sVCAM-1 and slCAM-1) in cerebrospinal fluid and serum correlate with MRI activity in multiple sclerosis. Ann Neurol.; 41: 236-333..
179. , , , . 1995. Tumor necrosis factor-alpha messenger RNA expression in patients with relapsing-remitting multiple sclerosis is associated with disease activity. Ann Neurol.; 37: 82-8..
180. . 1992. Genetic linkage: interpreting lod scores. Science.; 255: 803-4..
181. . 1982. Sodium and potassium channels in regenerating and developing mammalian myelinated nerves. Proc R Soc Lond B Biol Sci.; 215: 273-87..
182. . Immunologic Diagnosis of Autoimmunity. Leffell MS, Donnenber AD, Rose NRE. 1997. Handbook of Human Immology. Boca Raton. CRC Press. 111-124..
183. , , . 1998. Event-related functional MRI: past, present, and future. Proc Natl Acad Sci U S A.; 95: 773-80..
184. , . 1998. The natural history of multiple sclerosis and its diagnosis. Phys Med Rehabil Clin N Am.; 9: 537-49..
185. , , , , . 1996. Patients with amyotrophic lateral sclerosis and other neurodegenerative diseases have increased levels of neur filament protein in CSF. J Neurochem.; 67: 2013-8..
186. , , , , . 2000. Cervical cord magnetic resonance imaging findings in systemic immune-mediated diseases. J Neurol Sci.; 176: 128-30..
187. . 1999. Disease-modifying drugs for relapsing-remitting multiple sclerosis and f ture directions for multiple sclerosis therapeutics. Arch Neurol.; 56: 1079-84..
188. . 1993. Familial recurrence risks and inheritance of multiple sclerosis. Curr Opin Neurol Neurosurg.; 6: 189-94..
189. , , , . 1996. Evidence for genetic basis of multiple sclerosis. The Canadian Collaborative Study Group. Lancet.; 347: 1728-30..
190. , , , . 1996. Detection of herpesviridae in postmortem multiple sclerosis brain tissue and controls by polymerase chain reaction. J Neurovirol.; 2: 249-58..
191. , , , , , . 1999. CD46 is a cellular receptor for human herpesvirus 6. Cell.; 99: 817-27..
192. , , , , . 1999. Serial evoked potential studies and MRI imaging in chronic progressive multiple sclerosis. J Neurol Sci.; 171: 79-83..
193. , , . 1997. The genetic analysis of multiple sclerosis. Trends Genet.; 13: 234-9..
194. , , . 1987. The prevalence of locally-synthesized virus antibodies in various forms of multiple sclerosis. J Neurol Sci.; 80: 343-9..
195. , , , , , . 1965. Problems of experimental trials of therapy in multiple sclerosis: Report by the panel on the evaluation of experimental trials of therapy in multiple sclerosis. Ann NY Acad Med.; 122: 552-568..
Page 113
196. , , , , . 2000. CCR5 delta32, matrix metalloproteinase-9 and disease activity in multiple sclerosis. J Neuroimmunol.; 102: 98-106..
197. , . 1991. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med.; 325: 467-72..
198. , , . 1999. Standardization of magnetization transfer imaging for multicenter studies. Neurology.; 53: S33-9..
199. . 2000. The contribution of spinal cord MRI to the diagnosis and differential diagnosis of multiple sclerosis. J Neurol Sci.; 172: S32-5..
200. . 1999. From enhancing lesions to brain atrophy in relapsing MS. J Neuroimmunol.; 98: 7-15..
201. . 1999. Biological markers in body fluids for activity and progression in multiple sclerosis. Mult Scler.; 5: 287-90..
202. , , , . 1999. . J Clin Invest.; 103: 807-15..
203. , , , . 1999. Chlamydia pneumoniae infection of the central nervous system in multiple sclerosis. Ann Neurol.; 46: 6-14..
204. , , , . 1998. Late-onset chronic inflammatory e cephalopathy in immune-competent and severe combined immune-deficient (SCID) mice with astrocyte-targeted expression of tumor necrosis factor. Am J Pathol.; 153: 767-83..
205. . 2000. Despite epitope spreading in the pathogenesis of autoimmune disease, highly restricted approaches to immune therapy may still succeed [with a hedge on this bet]. J Autoimmun.; 14: 278-82..
206. , . 1997. More mayhem from molecular mimics. Nat Med.; 3: 1321-2..
207. , , . 1997. Autoimmune pathogenesis of multiple sclerosis: role of autoreactive T lymphocytes and new immunotherapeutic strategies. Crit Rev Immunol.; 17: 33-75..
208. . 2000. Microglial response to brain injury: a brief synopsis. Toxicol Pathol.; 28: 28-30..
209. , , , . 2000. Diagnostic criteria for primary progressive multiple sclerosis: a position paper. Ann Neurol.; 47: 831-5..
210. . 1995. Cerebrospinal fluid. J Neurol Neurosurg Psychiatry.; 59: 349-57..
211. , . 1979. Morphology of central nervous system disease in immun suppressed mice after peripheral herpes simplex virus inoculation. Trigeminal root entry zone. Lab Invest.; 40: 178-82..
212. , , , , , . 1998. Axonal transection in the lesions of multiple sclerosis. New Engl J Med.; 338: 278-285..
213. , , , . 1999. Neurodegeneration in multiple sclerosis: relationship to neurological disability. The Neuroscientist.; 5: 48-57..
214. . . Browning M, McMichael A, eds. HLA and MHC: Genes, Molecules and Function .em>. Oxford. BIOS Scientific Publishers Limited. 1996.
215. . 1997. Targeted gene disruption: applications in neurobiology. J Neurosci Methods.; 71: 19-27..
216. , , , , , . 1995. Clonal expansion of myelin basic protein-reactive T cells in patients with multiple sclerosis: restricted T cell receptor V gene rearrangements and CDR3 sequence. Eur J Immunol.; 25: 958-68..
217. , , , , , . 1998. Increased reactivity to myelin oligodendrocyte glycoprotein peptides and epitope mapping in HLA DR2(15)+ multiple sclerosis. Eur J Immunol.; 28: 3329-35..
218. . 1982. Membranes, myelin, and the pathophysiology of multiple sclerosis. N Engl J Med.; 306: 1529-33..
Page 114
219. . 1998. Demyelinating diseases—new pathological insights, new therapeutic targets. N Engl J Med.; 338: 323-5..
220. . 2000. Multiple sclerosis as a neuronal disease. Arch Neurol.; 57: 22-4..
221. , , , . 1994. Glial cells and axo-glial interactions: implications for demyelinating disorders. Clin Neurosci.; 2: 202-10..
222. , , , . 1989. The natural history of multiple sclerosis: a geographically based study. I. Clinical course and disability. Brain.; 112: 133-46..
223. , , , , . 1999. Diffusion tensor imaging of lesions and normal-appearing white matter in multiple sclerosis. Neurology.; 52: 1626-32..
224. , , . 1999. A gender gap in autoimmunity. Science.; 283: 1277-8..
225. , , . 1994. Immunology of multiple sclerosis. Clin Neurosci.; 2: 229-45..
226. , , . 1999. Transgenes and knockout mutations in animal models of type 1 diabetes and multiple sclerosis. Immunol Rev.; 169: 93-104..
227. , . 1999. Insulin-dependent diabetes mellitus and its animal models. Curr Opin Immunol.; 11: 643-647..
228. , . 1995. A review of T-cell receptors in multiple sclerosis: clonal expansion and persistence of human T-cells specific for an immunodominant myelin basic protein peptide. Ann N Y Acad Sci 1995 Jul 7;756:241-58.; 756: 241-58..
229. , . 1995. Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell.; 80: 695-705..
230. , , , . 1993. Involvement of neurofilaments in motor neuron disease. J Cell Sci Suppl.; 17: 101-8..
231. , , , . 1999. HLA-DPB1*0501-associated opticospinal multiple sclerosis: clinical, neuroimaging and immunogenetic studies. Brain.; 122: 1689-96..
232. , , , , , . 1994. Increased frequency of interleukin 2-responsive T cells specific for myelin basic protein and proteolipid protein in peripheral blood and cerebrospinal fluid of patients with multiple sclerosis. J Exp Med.; 179: 973-84..
233. , , , , . 1998. T-cell vaccination for the treatment of multiple sclerosis. In Zhang J, editor. Immunotherapy in Neuroimmunologic Diseases. London, England. Martin Dunitz.





















































































