
Page 62
Bioengineering for the Science and Technology of Biological Systems
DOUGLAS A. LAUFFENBURGER
Division of Bioengineering & Environmental Health, Department of Chemical Engineering, and Biotechnology Process Engineering Center Massachusetts Institute of Technology Cambridge, Massachusetts
THE “-OMICS” ERA: SIMULTANEOUSLY “DATA RICH” AND “DATA POOR”
In the past two decades we have seen two dramatic revolutions in biology: first that of molecular biology followed by that of genomics. Certainly, these revolutions are closely related, for genomics would not even be possible without the tools created by molecular biology. Each, however, generates its own type of data and understanding. Molecular biology, on the one hand, ultimately offers reductionist knowledge concerning molecular mechanisms governing functions at all higher hierarchical levels: cell, tissue, organ, and organism. Genomics, on the other hand, at least promises global knowledge concerning relationships of genetically encoded information and operation of the physiological system for which it serves as the core program. These types of knowledge must converge, of course, because genetic information must be transcribed and translated into physicochemical molecular mechanisms in order for us to be able to actually carry out programmed operations.
Combining expectations for continually accelerating generation of both types of knowledge, scientists and engineers anticipate working in a “data-rich” era in the coming decades. Relatively speaking, there is no question that this should be the case, compared to the scattered mist of hard data previously available for biomolecular interactions. At the same time, the extent to which even an imminent avalanche of data of this sort can be presumed to cover the enormously complex and intricate network of interactions at every level of physiological hierarchy must be considered to be tiny for the foreseeable future. Moreover, even as the extent of information grows, it will remain merely information until it can be organized into meaningful conceptual understanding. These quite seri-
Page 63
ous limitations are relevant no matter how the term genomicsis broadened to comprise not solely the field aimed at discovering gene sequences but also those aimed at elucidating gene expression (the “transcriptome”) and protein structure and function (the “proteome”).
It is widely appreciated that gene sequence and expression data will be inadequate for understanding the operation of biological systems, for multiple reasons. First, even complete DNA sequence information obviously cannot indicate what genes are actually expressed under any given conditions. Messenger RNA levels for even the full transcriptome, likewise, do not adequately represent the levels of the proteins they spawn. Researchers have demonstrated mathematically, for instance, that the dynamic behavior of a gene regulation network may not be properly predicted solely from gene expression data (Hatzimanikatis et al., 1999a). Moreover, protein levels in themselves cannot satisfactorily characterize molecular mechanisms, since locations (i.e., in various intracellular compartments or multimolecular assemblies) and states (such as phosphorylation on various amino acid residues or cleavage from proforms) substantially alter what the proteins are doing. Thus, knowledge of the reagents (i.e., expression profiles) alone does not offer substantial insight into modes of interactions and mechanisms of operation, which determine ultimate functional behavior. Second, this litany of molecular players—DNA, RNA, proteins—neglects the capacity of other classes of biological molecules (e.g., oligosaccharides, lipids) to regulate biological functions in an epigenetic manner. Finally, living organisms are not autonomous, closed systems. Rather, they operate within the context of environment, which strongly influences every step along the hierarchy outlined above—as well as the genomic content itself.
These arguments should caution us all regarding both the pace and scope of what can be understood about biological systems by whatever combination of “-omics” might be proferred. Basically, my conclusion is that we will remain “data limited” for quite some time yet, despite at the same time surely becoming data rich.
Finally, it should be stressed that the issue raised here is relevant both to the future of biological science, in terms of understanding how biological systems work, and to the future of biotechnology, in terms of enabling creation of products and processes that benefit societal needs.
A BIOENGINEERING APPROACH TO PROGRESS IN THIS ERA
This situation is ideal for bringing to bear a classical engineering mindset on this new biological science and biotechnology—that of an “integrated systems,” “design principles and parameters” orientation to the analysis and synthesis of biological processes based on a fundamentally molecular foundation. Both basic scientific understanding and innovative technological development should be advanced by this kind of approach.
Page 64
~ enlarge ~
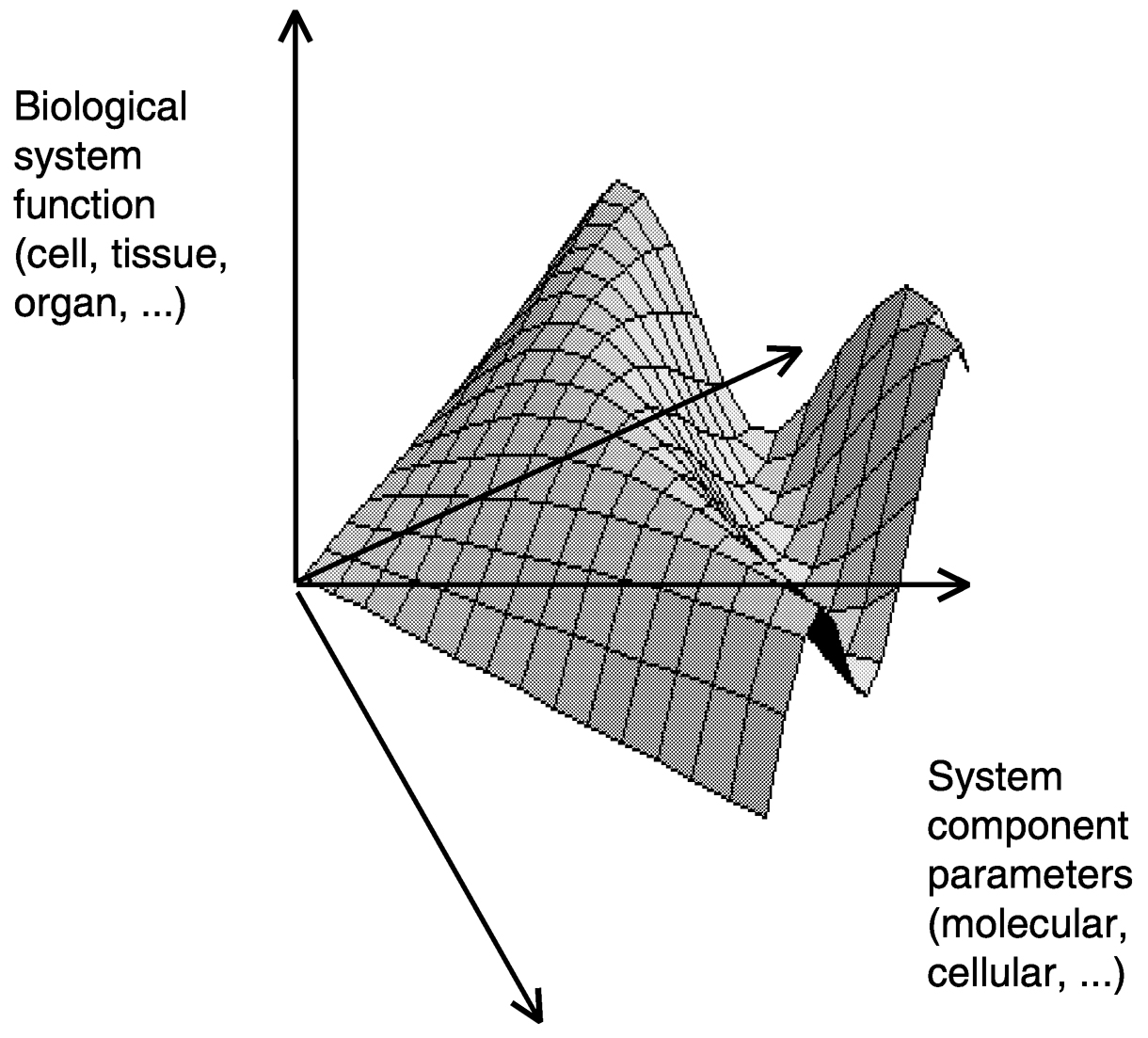
FIGURE 1 Illustration of an engineering perspective on biological systems as a dynamic function dependent on component properties.
What engineers generally bring to the research table is a predilection to analyze a complicated system in terms of principles useful for manipulating that system, with the goal of making it operate in some intended fashion (see Figure 1). This perspective emphasizes elucidating and working with an understanding about the system that is admittedly incomplete, but sufficient for “input/output” dependencies to be constructed with enough reliability at the desired level of description that it will do what is needed under most circumstances. Moreover, and just as important, engineers are taught to view a complicated system in terms of its essentially hierarchical nature: a relationship that is mechanistic at one level is phenomenological at another, with parameters empirically measured at one level being useful for prediction of system behavior at the next higher level and being ultimately derivable and predictable from properties of the constituents at the next lower level.
In essence, in this approach the data-limited nature of whatever system is being tackled is accepted, and an adequate balance is found between what is fundamentally known with certainty and what must be assumed or empirically related. The proof of adequacy, of course, lies within the objective of the exer-
Page 65
cise. For any objective short of universal predictive capability from first principles, this ought to suffice.
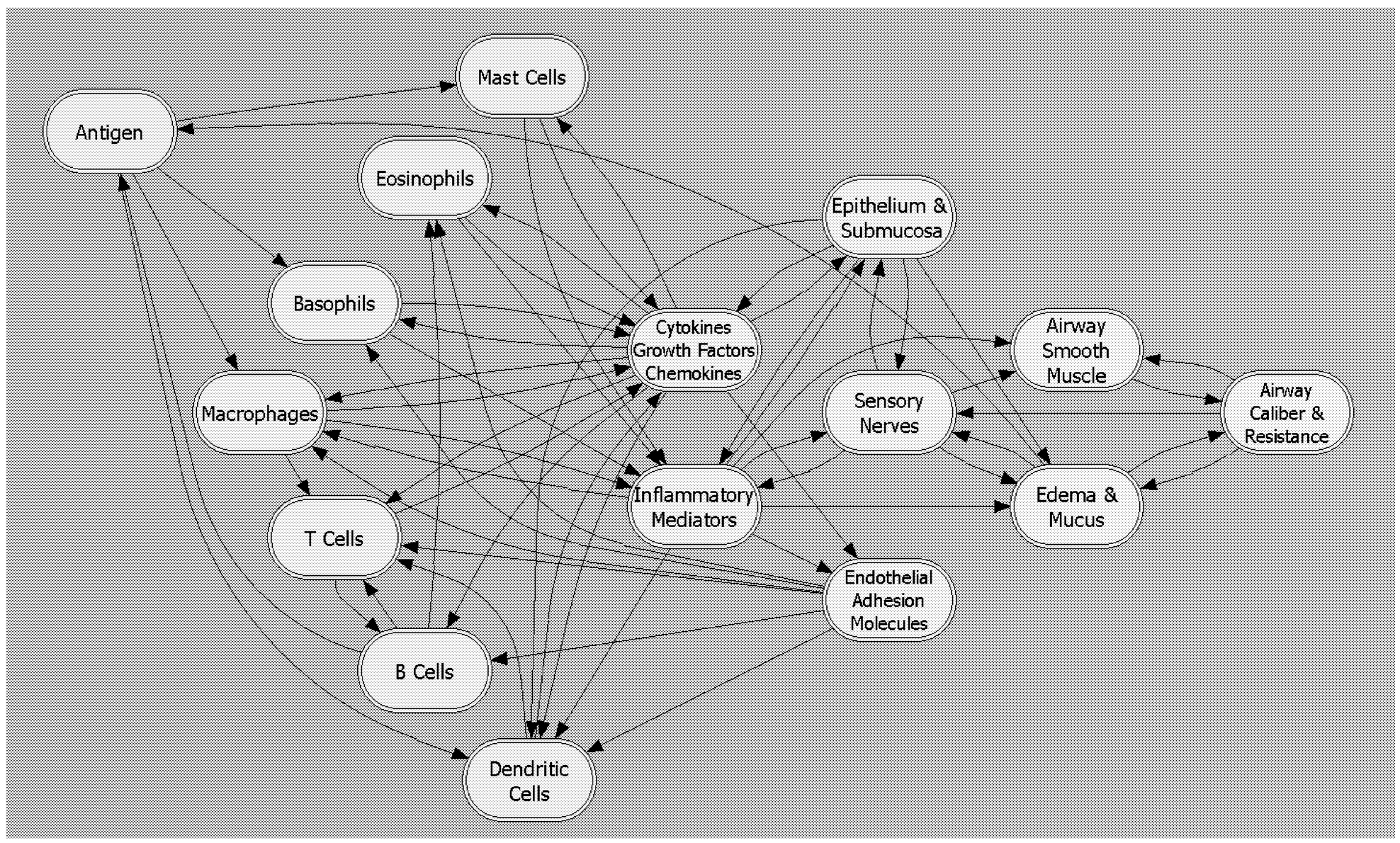
A number of examples of this approach can be offered. Some that provide insightful illustration are at perhaps the most ambitiously comprehensive end of the spectrum: the PhysioLab models being developed by Entelos, Inc. ( www.entelos.com) for a variety of pathophysiological conditions, with the objective of determining useful drug targets and interpreting clinical trial data. For instance, the Asthma PhysioLab model attempts to account for how the resistance to air flow in the lungs is governed by cell-level functions (e.g., airway smooth muscle cell contractility, airway epithelial cell permeability, white blood cell accumulation in tissues, and secretion of inflammatory mediators), which are, in turn, regulated by cytokine/receptor interactions and consequent intracellular signaling pathways (see Figure 2a).
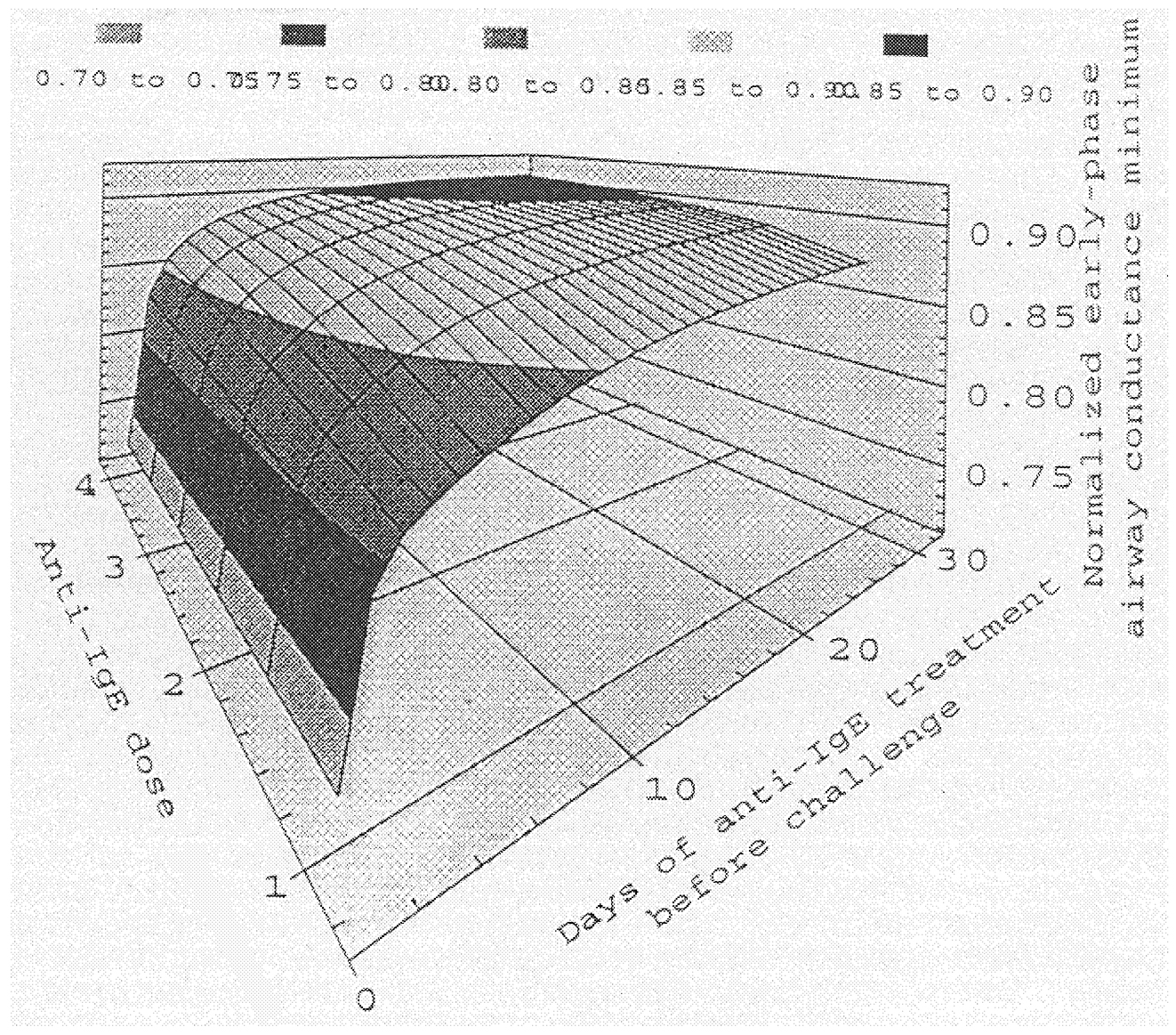
Computational simulations of this model yield particular realizations for how any variable in the system (levels of molecular species, degrees of cell behavioral activities, extents of tissue functions) depends on system component properties, measurable or assignable. In principle, the computational simulations could more generally produce a “state space” diagram, in which key system parameters (potentially characterizing asthmatic patients in comparison to healthy patients) govern whether airway resistance in response to an environmental challenge stays at a normal physiological state or jumps to a dangerous pathological state (see Figure 2b). In turn, effects of a putative drug regimen can be examined to determine whether it could bring the system back into the safe state. This approach is what will eventually bring the promise of pharmacogenomics to fruition. For any individual patient, information concerning foundational genetic variation (e.g., single-nucleotide polymorphisms) could be related to gene expression and protein property issues that are involved in this overall dynamic system. Now, it is presumptuous to expect that a fully complete model for all relevant physicochemical mechanisms at each level in the space-time hierarchy (e.g., gene expression regulation, signal transduction, cell functions [proliferation, death, differentiation, migration, secretion, contractility], tissue mechanics and transport) might even at the end of the 21st century be available with quantitatively determined parameters. However, it is cowardly to wait to pursue this kind of approach until complete information is on hand.
This tension between being data rich and data limited is, in fact, the history of engineering science and technology. Motor vehicles were manufactured and driven for decades without complete data on the combustion reactions taking place within the engine, yet they moved people and items very effectively, to the overall great benefit of society. Of course, realizing technologies derived from restricted basic scientific understanding has drawbacks: witness environmental issues surrounding the products of those engines. Advances in combustion chemistry and catalytic reactors have helped to reduce the drawbacks in that particular system, certainly, although in medical technologies cost/benefit analyses are sure-
Page 66
~ enlarge ~
FIGURE 2a Schematic framework for Entelos Asthma PhysioLab, showing cell, tissue, and organ components involved in the computation model. Underlying each component is a set of hierarchical models with increasing detail. SOURCE: Reprinted with permission from Entelos, Inc.
Page 67
~ enlarge ~
FIGURE 2b Example dynamic systems relationship from Entelos Asthma PhysioLab computer simulations, showing airway conductance minimum as a function of anti-IgE dose level and period. SOURCE: Reprinted with permission from Entelos, Inc.
ly even more difficult to resolve. The point is that the data-limited nature of any system must be considered cautionary, but not necessarily paralyzing.
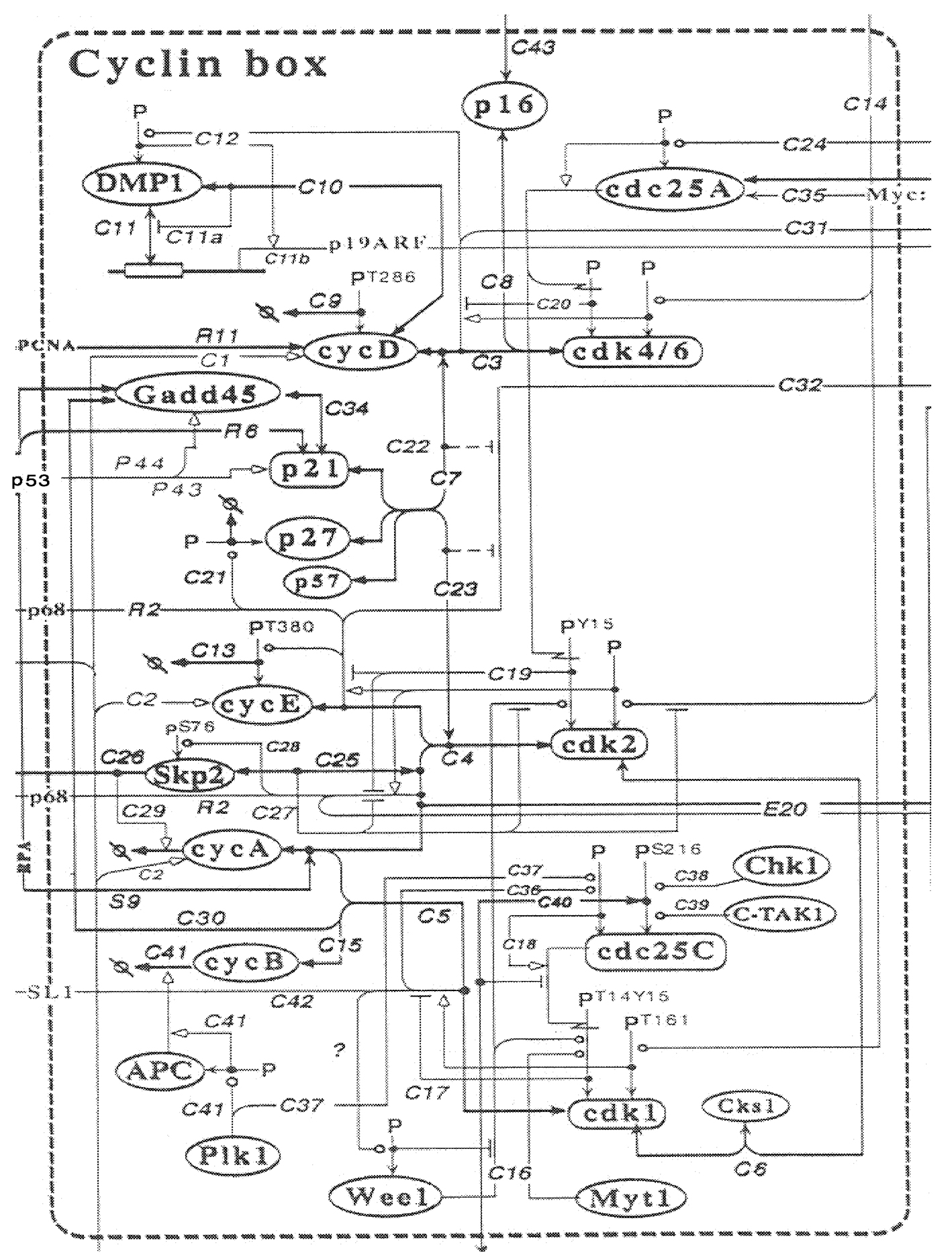
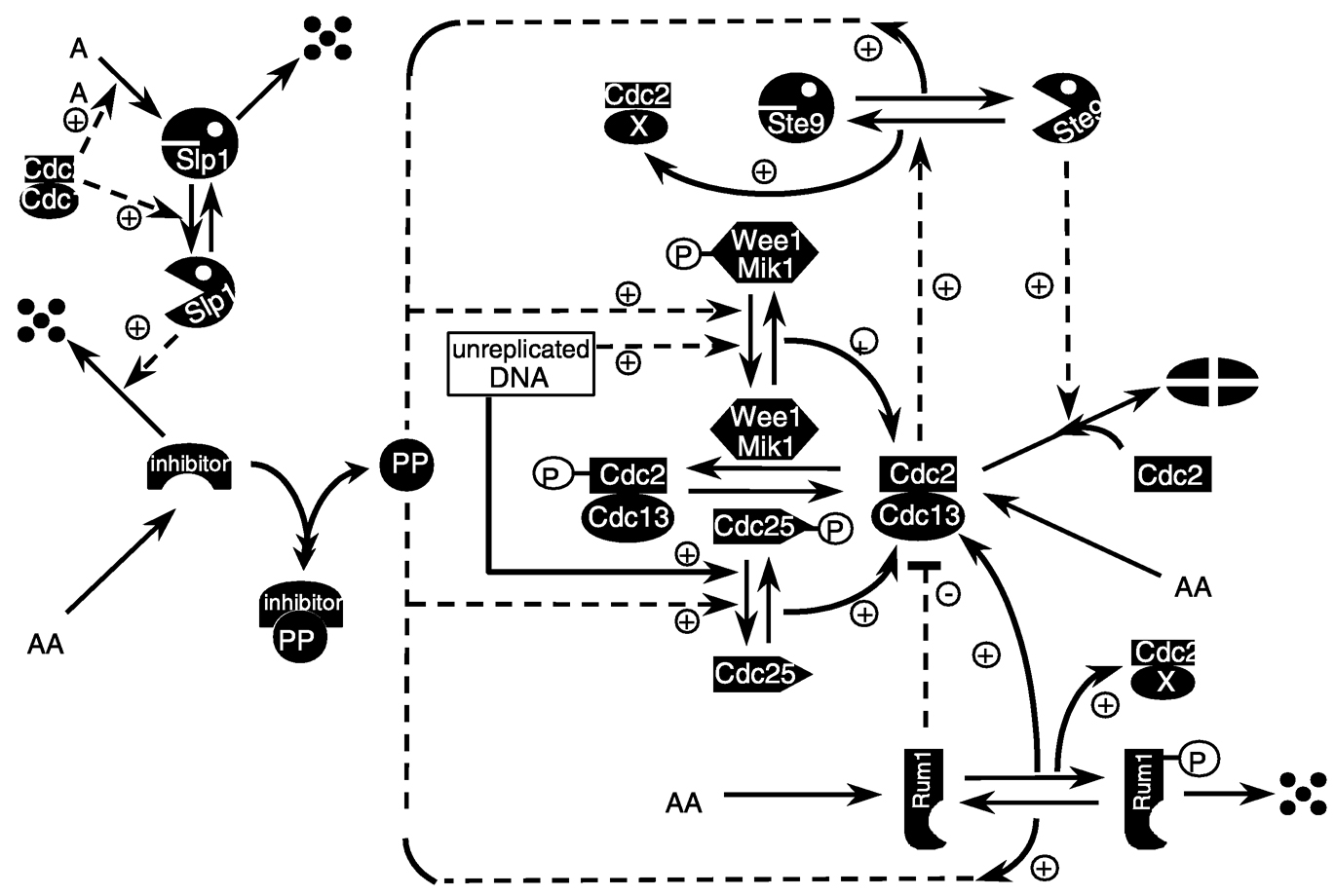
To be useful, these kinds of systems modeling and analysis approaches need not be aimed at a high-level physiological system. Complexities are just as daunting, yet the benefit of this approach for understanding and manipulation is as exciting, even at the level of an individual cell. An excellent example of this is the systems models for cell cycle control that have been developed over the past decade (e.g., Chen et al., 2000; Hatzimanikatis et al., 1999b). A “wiring diagram” that contextualizes the cell cycle molecular regulatory network has been provided (Kohn, 2000), and to look at it initially is to regret contemplating a systems modeling description (see Figure 3a). However, rather than rushing headlong into writing differential equations for all components and interactions, Tyson's and Bailey's groups at Virginia Polytechnic Institute and State Univer-
Page 68
~ enlarge ~
FIGURE 3aBiomolecular “circuit diagram” for the cell cycle. SOURCE: Reprinted with permission from the American Society for Cell Biology (Kohn, 2000).
Page 69
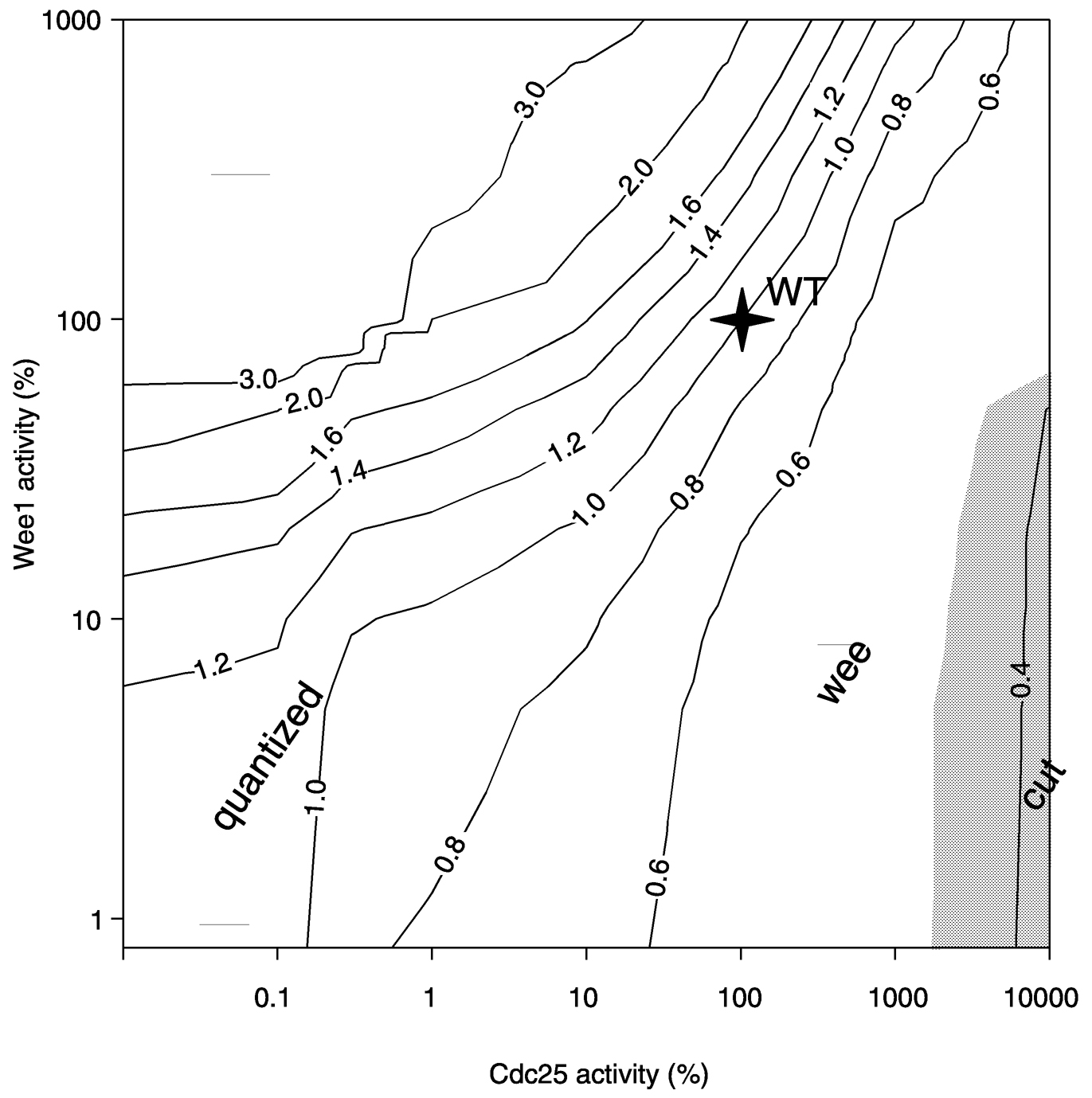
sity and ETH Zürich, respectively, have identified key underlying “modules” that are at the core of the dynamic behavior of this system and then incorporated additional network elements around this core to explain and predict more and more experimental data concerning wider aspects of its regulation (see Figure 3b). Again, one can aim for construction of dynamic systems behavior versus parameter relationships, such as a bifurcation diagram for understanding and predicting conditions under which cells will progress through a DNA synthesis checkpoint or not (see Figure 3c). It should be emphasized that an important element of biological systems analysis along these lines is likely to be a consideration of stochastic issues, since most biological data represent either individuals or individuals distributed across a population—whether at the level of cell, organ, or organism (e.g., Arkin et al., 1998).
This success provides a concrete example of the concept that a modular approach to modeling and understanding highly hierarchical biological systems should be productive (Hartwell et al., 2000; Lauffenburger, 2000). That is, it may be anticipated that crucial core functions of biological systems (e.g., metabolism, force generation) are performed by an identifiably restricted mechanism, but the vast proportion of the overall system's components are found to serve as control and safety networks in order to ensure robustness and reliability of core function
~ enlarge ~
FIGURE 3b Schematic illustration of mathematical model for the cell cycle. SOURCE: Reprinted with permission from the American Society for Cell Biology (Chen et al., 2000).
Page 70
~ enlarge ~
FIGURE 3c Example dynamic systems relationship. Model computations for yeast cell proliferation behavior as a function of activities of two key gene products. SOURCE: Reprinted with permission from the American Society for Cell Biology (Chen et al., 2000).
under highly variable environmental conditions (Carlson and Doyle, 1999). It is conceivable that much of the uncertainty regarding quantitative parameter values might, in fact, be rendered less problematic by this concept of functional and control modules, because their dynamic operation may turn out to be surprisingly insensitive to specific parameter values (von Dassow et al., 2000). This idea will be very interesting to examine with emerging powerful genetic (Rao and Verkman, 2000) and chemical (Schreiber, 2000) molecular intervention methodologies.
Neither of these impressive examples, however, embodies the full scope of the potential engineering systems approach that will need to be pursued, since
Page 71
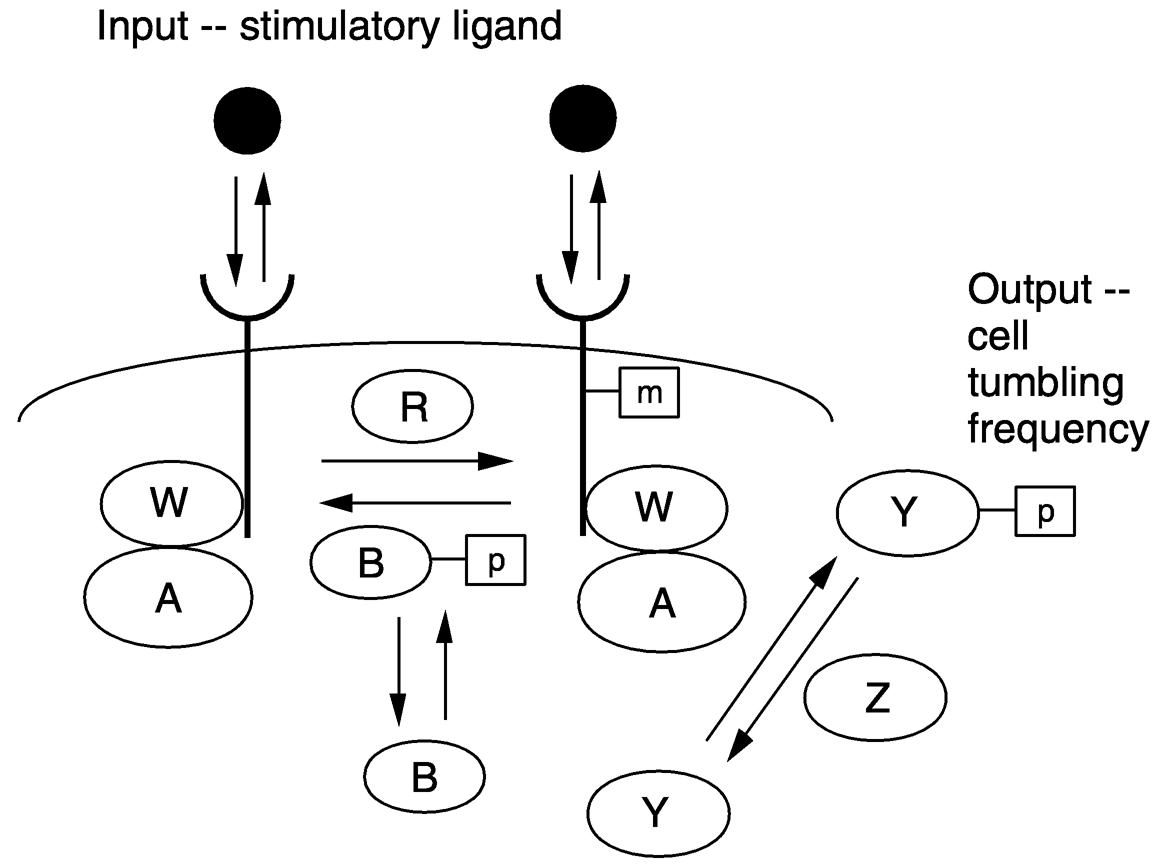
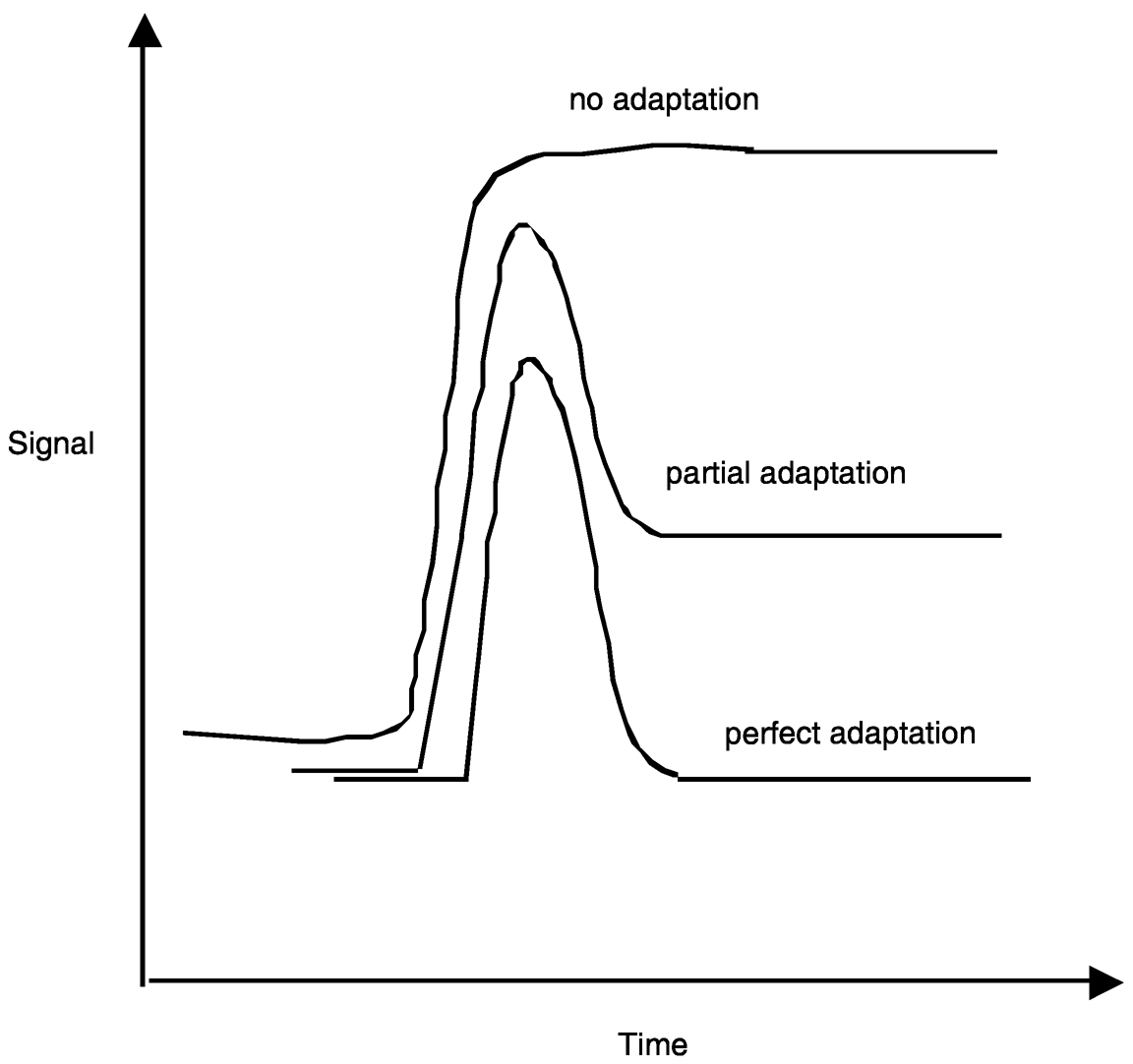
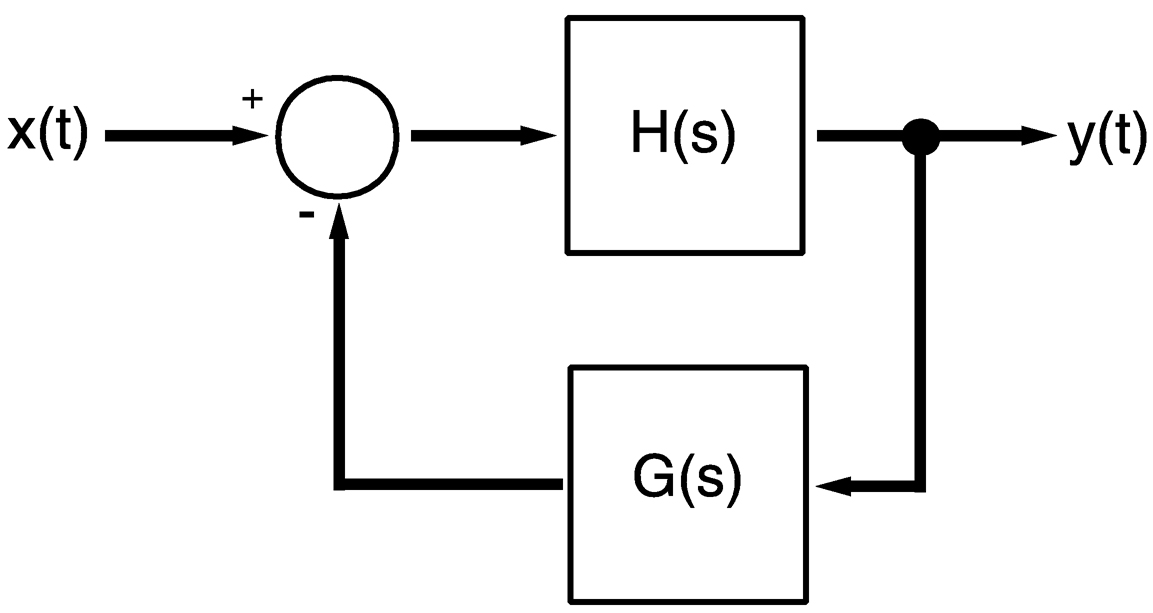
both focus on directly observable (at least in principle) physicochemical interactions. Since a large proportion of the physicochemical processes involved in molecular and cellular networks—and their corresponding parameters—will remain undetermined for years to come, systems modeling approaches less dependent on a detailed physicochemical framework should be valuable to pursue. The dynamic module approach noted above is one possibility. An early instance of this can be found in the signaling network regulating bacterial chemotaxis. For this phenomenon of biased cellular motor rotation, leading to directional cell movement, the dynamic network behavior of 14 protein components (see Figure 4a), which could require on the order of a few dozen parameters for quantitative physicochemical description, responds to a step change in input to yield a perfectly adapting output result (see Figure 4b). This input/output relationship then permits the full network to be replaceable by an integral feedback control loop that can be characterized by two phenomenological parameters (Yi et al., 2000; see Figure 4c). While at first glance this might seem to be a step backward, the physicochemical detail has not disappeared; rather, it is ultimately responsible for the dynamic behavior, including the quantitative parameter values, so the feedback control loop could be replaced by the detailed subsystem whenever the complete set of information becomes available.
~ enlarge ~
FIGURE 4a Signal transduction network for bacterial chemotaxis, showing how cell tumbling frequency (which governs chemotactic locomotion) is regulated by a stimulatory ligand. SOURCE: Reprinted with permission from Lauffenburger. Copyright (2000) National Academy of Sciences, U.S.A.
Page 72
~ enlarge ~
FIGURE 4b Dynamic signal behavior possibilities in response to step input; the network shown in Figure 4a yields “perfect adaptation” behavior. SOURCE: Reprinted with permission from Lauffenburger. Copyright (2000) National Academy of Sciences, U.S.A.
~ enlarge ~
FIGURE 4c Schematic of feedback control module, which can generate the dynamic signal behavior shown in Figure 4b.
Page 73
Another possibility is employment of more phenomenological relation-focused models, developed from analysis of large but not necessarily mechanism-oriented data sets. A classical reference point for this concept may be found in biomedical engineering treatments of organ-level physiological dynamics, such as an electrocardiogram, which use signal processing techniques to develop relationships useful for analysis and technology creation, despite possessing little direct connection to underlying fundamental mechanisms. An analogous methodology may be valuable for modeling at least some aspects of protein-protein and protein-gene regulatory networks as signal processing elements (Asthagiri and Lauffenburger, 2000; McAdams and Arkin, 1998). Finally, yet a different approach is that of a cybernetic perspective, in which physicochemical mechanistic details are largely replaced by “objective-based” algorithms that characterize programs by which cells might be presumed to manage molecular resources (Varner, 2000; Varner and Ramkrishna, 1999).
We have ourselves begun to embark on an effort to combine some of these concepts, attempting to analyze the cell behavioral response of apoptosis (programmed cell death) versus survival in response to death-promoting and survival-promoting factors by following a combination of gene expression and protein level/state/location dynamics. Out of hundreds of putative death-activating and survival-protective genes—and dozens of protein-based physicochemical kinetic and transport processes—we have selected a subset for microarray expression and proteomic experimental measurement following challenge by a matrix of input stimuli. The question posed is whether we can determine a signal processing algorithm utilized by the cells to make their decision based on information flows through key regulatory networks (e.g., Arkin and Ross, 1994).
Finally, it is important to emphasize that major advances are concomitantly needed in experimental methodologies for quantification of molecular and cellular processes. This need ranges from improvements in surface chemistry and fluorescence imaging for nucleic acid and peptide microarrays to isolation and characterization of large sets of proteins from small cellular samples, to creation of tissue-engineered in vitro organ surrogates for generating more nearly physiological cellular contexts (Griffith et al., 1997), to instrumentation enabling measurement of subtle but important functional properties of and within living organisms such as transgenic mice. In short, the same type of high-throughput acceleration of data gathering that has arisen at the gene expression level must be propagated to higher levels of the biological systems hierarchy.
ACKNOWLEDGMENTS
I would like to express my appreciation to a number of colleagues whose comments and perspectives in recent months have helped shape the thoughts outlined here, including Anand Asthagiri, Jay Bailey, Rick Horwitz, Kelvin Lee, and Steve Wiley.
Page 74
REFERENCES
, and . 1994. Computational functions in biochemical reaction networks. Biophysical Journal 67(2): 560–578.
, , and . 1998. Stochastic kinetic analysis of developmental pathway bifurcation in phage lambda-infected Escherichia colicells. Genetics 149: 1633–1648.
, and . 2000. Bioengineering models of cell signaling. Annual Review of Biomedical Engineering 2: 31–53.
, and . 1999. Highly optimized tolerance: A mechanism for power laws in designed systems. Physical Review E 60(2A): 1412–1427.
, , , , , and . 2000. Kinetic analysis of a molecular model of the budding yeast cell cycle. Molecular Biology of the Cell 11: 369–391.
, , , , , and . 1997. In vitroorganogenesis of liver tissue. Annals of the New York Academy of Sciences 831: 382–397.
, , , and . 2000. From molecular to modular cell biology. Nature 402(6761 Suppl): C47–C52.
, , and . 1999a. Proteomics: Theoretical and experimental considerations. Biotechnology Progress 15(3): 312–318.
, , and . 1999b. A mathematical description of regulation of the G1-S transition of the mammalian cell cycle. Biotechnology and Bioengineering 65(6): 631–637.
2000. Molecular interaction map of the mammalian cell cycle control and DNA repair systems. Molecular Biology of the Cell 10: 2703–2734.
2000. Cell signaling pathways as control modules: Complexity for simplicity? Proceedings of the National Academy of Sciences of the USA 97: 5031–5033.
, and . 1998. Simulation of prokaryotic genetic circuits. Annual Review of Biophysics and Biomolecular Structure 27: 199–224.
, and . 2000. Analysis of organ physiology in transgenic mice. American Journal of Physiology Cell Physiology 279(1): C1–C18.
2000. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 287: 1964–1969.
2000. Large-scale prediction of phenotype: Concept. Biotechnology and Bioengineering 69: 664–678.
, and . 1999. Mathematical models of metabolic pathways. Current Opinion in Biotechnology 10(2): 146–150.
, , , and . 2000. The segment polarity network is a robust development module. Nature 406: 188–192.
, , , and . 2000. Robust perfect adaptation in bacterial chemotaxis through integral feedback control. Proceedings of the National Academy of Sciences of the USA 97: 4649–4653.













