
Page 105
Appendix A
Study Methods
STRATEGIES FOR GATHERING INFORMATION
Traditional Literature Searches
Electronic Data Bases
The initial step in developing an evidence-based research base for the study described in this report was a targeted search of the National Library of Medicine's Medline. The basic subject search terms included Bayesian, sequential analysis, decision analysis, meta-analysis, and confidence profile method. These are the methods of analysis that the committee had been charged with assessing as they relate to the conduct of clinical trials with small sample sizes. The search also used a combination of each subject search term with descriptors such as models, statistical, clinical trials or randomized clinical trials, phase I, phase II, phase III, small sample, and n-of-1. For example, one set of subject search terms is “models, statistical[mh] AND sequential analysis AND (“clinical trials” OR “randomized clinical trials”) [mh] AND phase I.” (MeSH is the abbreviation for medical subject heading. [mh] is used as a tag to tell the search engine to look for the terms in the MeSH fields of the Medline database.) Abstracts from the initial search were reviewed for relevance to the committee's charge and the more prominent authors (defined by having published at least 10 articles) were deter-
Page 106
mined. Then a search by author name was done. A greater part of Appendix C, Selected Bibliography on Statistical and Small Clinical Trials Research, was the result of literature searches from the Medline database.
Databases such as Mathsci, Scisearch, and Pascal were used to verify the bibliographic information in the statistics literature that Medline does not cover. The Library of Congress Online Catalog and Social SciSearch were also searched for specific titles and authors provided by committee members.
Clinical Trials Listserv
A posting in a clinical trials listserv ( clinical-trial@listserv.acor.org) that requested literature on small-number clinical trials generated several suggestions from all over the United States and a few from other countries. Their abstracts were obtained from Medline and reviewed for relevance to the study. Several suggestions that originated from responses to this listserv posting are included in Appendix C.
Other Sources
Special attention was given to references included in benchmark publications on the subject of small-number clinical trials. Abstracts of these references were also obtained from Medline. Several of these references are included in Appendix C. Additionally, valuable suggestions for relevant literature came from statisticians in other units of The National Academies and the Washington, D.C. academic community. Their suggestions are also included in Appendix C.
Selections by Committee Members
The preliminary list of background literature was circulated to the committee members. During the first committee conference call, committee members agreed to identify and submit to Institute of Medicine staff references (included or not included in the preliminary list) that they believe should be reviewed by the entire committee. The committee members' selections were then compiled, and copies were included in the first committee meeting briefing book for study by the committee members. Additionally, through the duration of the project, other published literature and unpublished data that committee members believed should be reviewed by
Page 107
the entire committee were obtained and circulated to the committee. Literature selections from committee members are added in Appendix C. Literature reviewed by the entire committee and used to prepare this report are cited in the References section.
One-Day Invitational Conference
In fulfilling the second task of the committee, a 1-day conference was convened on September 28, 2000, in Washington, D.C. More than 200 professionals from federal research and regulatory agencies, academia, industry, and other areas of clinical research and health care practice were invited to participate. The conference focused on the state of the science, challenges, and strategies in the design and evaluation of clinical trials of drugs, biologics, devices, and other medical interventions with populations with small numbers of individuals. Methods including randomized clinical trials, sequential clinical trials, meta-analysis, and decision analysis were considered in terms of their potentials and their problems. Ethical considerations and statistical evaluations and comparisons were also covered. The reader is referred to the conference agenda, speakers, and participants at the end of this appendix for more details.
Speakers' Presentations
As part of the committee's information-gathering process, the conference speakers were asked to discuss recent developments, problems, and research needs in conducting clinical trials with small numbers of participants. In preparing their presentations, the speakers were asked to use two scenarios formulated by the committee during its first committee meeting, namely, (1) clinical trials to prevent bone loss in microgravity and (2) clinical trials on split liver transplantations in patients with human immunodeficiency virus infection and patients with fulminating liver failure. The conference format was organized to achieve balanced times for each speaker's presentation followed by a discussion of the committee with the speakers and invited participants. Overall, the speakers' presentations helped the committee frame the issues and questions in its task of reviewing the methodologies for clinical trials with small sample sizes, determining research gaps and needs, and making recommendations in the development of this area of medical research.
Page 108
Speakers' Recommended Literature
In a follow-up letter, the committee asked speakers to submit lists of their recommended readings in their specific fields of expertise. They were each asked for a minimum of 10 published articles: 5 of which they are neither an author nor a coauthor and 5 of which they are an author or a coauthor. The speakers' recommended readings are also included in Appendix C and, where relevant, in the References section.
Committee Website and Live Audiocast of Conference
The committee website ( http://www.iom.edu/smalln) included a page describing the September 28, 2000, conference. The page served the purpose of disseminating information about the conference to a broader public audience, beyond those professionals who were invited, who might have a stake in the study. The page also allowed comments, suggestions, and questions on the study in general and on the conference in particular to be electronically mailed to Institute of Medicine staff.
The conference was carried on a live audiocast for the benefit of those who were unable to attend. Two weeks before the conference date, a postcard announcement of the live audiocast together with the conference agenda and a one-page description of the conference were sent to invited individuals who had not registered to attend and a wider national audience unlikely to attend. This was done to ensure maximum dissemination of the conference proceedings. After the conference the speakers' visual and audio presentations were posted on the committee's website to allow the widest continuing public access in this phase of the committee's information-gathering activities before the subsequent release of its report to the public.
PREPARING THE REPORT
Discussion of Committee's Charge
At the first of three meetings, the committee reviewed the statement of task, which it accepted with minor changes to make it accurate with regard to statistical terminology and usage. The committee agreed that its composition was sufficient to accomplish the task given the work plan and time schedule of 6 months. In fulfilling the committee's charge, it was anticipated that the study report would serve as a state-of-the science guide to those interested in planning, designing, conducting, and analyzing clinical trials
Page 109
with small numbers participants. The audience for the report was defined to include federal agencies at the policy and program levels, researchers concerned with conducting clinical trials, clinicians, students, patients, and patient advocacy groups.
Discussion of Committee's Charge with Sponsor
In a telephone conference call during its first meeting, the committee discussed with the study sponsor the statement of task. The sponsor presented a summary of the issues that it wanted the committee to address: (1) a critique of the sponsor's proposed methodology on countermeasure evaluation; (2) suggestions for improvements to the proposed methodology or the use of an alternate methodology; and (3) methods for the evaluation of treatments, incorporation of data from surrogate studies, incorporation of treatment compliance information, and sequential decision making on treatment efficacy. The committee determined that, through the invitational conference and its final report, it would be able to meet the sponsor's charge to the committee.
Organization of the Report and Committee Working Groups
The committee agreed to organize the report around four chapters, which included an introduction ( Chapter 1), a chapter on design of small clinical trials ( Chapter 2), a chapter on statistical approaches to analysis of small clinical trials ( Chapter 3), and a concluding chapter on general guidelines on small clinical trials ( Chapter 4). Appendices include this one, on study methods, a glossary of statistical and clinical trials terms ( Appendix B), a selected bibliography on small clinical trials research ( Appendix C), and committee and staff biographies ( Appendix D). To facilitate writing of the report, the committee formed two working groups: the clinical trials group and the biostatistical group. The clinical trials group is primarily responsible for the content of Chapter 2, which focused on the design of clinical trials with various numbers of small sample sizes. The biostatistical group is primarily responsible for Chapter 3, which focused on the statistical analyses applied to clinical trials with small sample sizes. The introduction and Appendixes were written or assembled by study staff; the Statistical Analyses section in this appendix was written by the committee, however.
During the third meeting, the committee focused on refining the content of the report and the committee's principal findings and recommenda-
Page 110
tions. Due to the sponsor's imposed time limitation on the study, the committee had only the third meeting to discuss as a group the implications of the information collected. The committee felt that, given the time constraint, it was unable to engage in the amount of deliberation it wished it could to fully meet the task given to the committee for this important and developing area of research. The committee as a whole is responsible for the information included in the entire report.
CONFERENCE AGENDA
“Future Directions for Small n Clinical Research Trials”
National Academy of Sciences Lecture Room
2101 Constitution Avenue, N. W.
Washington, D. C.
September 28, 2000
|
8:25 a.m. |
Opening Remarks Suzanne T. Ildstad, Chair of the Committee |
|
8:30 |
Overview of the Science of Small n Clinical Research Trials Martin Delaney, Founding Director, Project Inform |
|
Design and Evaluation of Small n Clinical Research Trials |
|
|
9:00 |
Design of Small n Clinical Research Trials Curtis L. Meinert, Johns Hopkins University School of Public Health |
|
9:30 |
Quantitative Assessment of Countermeasure Efficacy for Long-Term Space Missions Alan H. Feiveson, Johnson Space Center, NASA |
|
10:00 |
Sequential Design Nancy Flournoy, The American University |
|
10:30 |
Welcome Kenneth I. Shine, M.D., President, Institute of Medicine |
|
10:40 |
Break |
|
11:00 |
Clinical Trials with n of > 1 Jean Emond, Center for Liver Disease and Transplantation, New York Presbyterian Hospital |
Page 111
|
11:30 |
Small Clinical Research Trials: Needs Beyond Space Denise L. Faustman, Massachusetts General Hospital |
|
12:00 |
Lunch Break |
|
Statistical Methodologies for Small n Clinical Trials |
|
|
1:00 p.m. |
Meta-Analysis and Alternative Methods Betsy J. Becker, Michigan State University |
|
1:30 |
Decision Analysis and Small Clinical Trials Stephen G. Pauker, New England Medical Center |
|
2:00 |
Bayesian Strategies for Small n Clinical Trials Peter F. Thall, M. D. Anderson Cancer Center, Houston |
|
2:30 |
n - of - 1 Clinical Trials Deborah R. Zucker, New England Medical Center |
|
3:00 |
Break |
|
Development and Monitoring of Small n Clinical Trials |
|
|
3:15 |
Research Needs in Developing Small n Clinical Trials Helena Mishoe, National Heart, Lung and Blood Institute, National Institute of Health |
|
3:45 |
Regulatory Issues with Small n Clinical Research Trials Jay Siegel, Office of Therapeutics Research and Review, Food and Drug Administration |
|
4:15 |
Ethical and Patient Information Concerns: Who Goes First? Lawrence K. Altman, New York University Medical School and New York Times medical correspondent and author of Who Goes First?: The Story of Self-Experimentation in Medicine |
|
4:45 |
Closing Remarks |
|
5:00 |
Reception in the Great Hall of the National Academy of Sciences |
Page 112
ADDITIONAL PARTICIPANTS
Gregory Campbell
Food and Drug Administration
Rockville, MD
Nitza Cintron
Wyle Laboratories
Houston, TX
Janis Davis-Street
NASA
Houston, TX
Lawrence Freedman
National Heart, Lung, and Blood Institute
Bethesda, MD
Carolin Frey
Children's National Medical Center
Washington, DC
Nancy Geller
National Heart, Lung, and Blood Institute
Bethesda, MD
Judith Hayes
Wyle Laboratories
Houston, TX
Eric Leifer
National Heart, Lung, and Blood Institute
Bethesda, MD
Linda Loerch
Wyle Laboratories
Houston, TX
Seigo Ohi
Howard University
Washington, DC
Kantilal Patel
Children's National Medical Center
Washington, DC
Clarence Sams
Wyle Laboratories
Houston, TX
David Samson
Blue Cross Blue Shield Association
Washington, DC
Victor S. Schneider
NASA Headquarters
Washington, DC
Steve Singer
Johns Hopkins University School of
Public Health
Baltimore, MD
Mario Stylianou
National Heart, Lung, and Blood Institute
Bethesda, MD
Marlies Van Hoef
Transplant Creations, L. C.
Falls Church, VA
Lakshmi Vishnuvajjala
Food and Drug Administration
Rockville, MD
Teng Weng
Food and Drug Administration
Rockville, MD
Gang Zheng
National Heart, Lung, and Blood Institute
Bethesda, MD
Page 113
STATISTICAL ANALYSES
Statistical Methods of Drawing Inferences
In the so-called frequentist mode of inference, there are three probabilities of interest, the p value, the Type I error probability, and the Type II error probability. In significance testing, as described by R. A. Fisher, the p value is the probability of finding results that are at least as far away from the null hypothesis as those actually observed in a given experiment, with respect to the null probability model for the experiment. Note that because the null hypothesis is assumed for the probability model, a p value can speak only to how probable or improbable the data are in their departure from expectation under the null hypothesis. The p value does not speak to how probable or improbable the null hypothesis is, because to do so immediately takes one outside the probability model generating the data under the null hypothesis (a Bayesian posterior probability addresses this –[see below]). Thus, it is entirely possible that a result can have a small p value, indicating that the data are unlikely to be so extreme in their departure from expectation under the null hypothesis, and yet the null hypothesis may itself be very likely. Anyone who has ever gotten a positive result on a screening examination and found that result to be a false positive understands this point: under the null hypothesis of no disease, the probability of a positive screening test result is very small (the p value is small and highly significant). With rare diseases, however, most of the positive results are false positives (i.e., the null hypothesis is actually true in most of these cases).
In the Neyman-Pearson paradigm of hypothesis testing, it is imagined that any experiment can be repeated indefinitely, and in each hypothetical replication the null hypothesis will be rejected according to a fixed decision rule. The Type I error describes the long-run proportion of times that the null hypothesis will be so rejected, assuming in each case that it is actually true. The Neyman-Pearson approach also considers various explicit alternative hypotheses and for each one quantifies the Type II error probability, which is the long-run proportion of times that the decision rule will fail to reject the null hypothesis when the given alternative is true.
The interpretation and use of Type I and Type II error probabilities are quite distinct because they are calculated under different hypotheses. The Type I error probability describes the largest p value that will be declared statistically significant by chance alone when the null hypothesis is true. Because the Type I error probability gives the long-run proportion of instances in which the decision procedure will err by rejecting a true null hypothesis,
Page 114
it is used to control the occurrence of false positives. The Type II error probability is related to the ability of the test to arrive at a proper rejection of the null hypothesis under any one of the non-null-hypothesis alternatives of interest. The statistical power of a hypothesis test is the probability complementary to the Type II error probability, evaluated at a given alternative, and generally depends on the decision criterion for rejection of the null hypothesis, the probability model for the alternative hypothesis, and the sample size.
The Type II error probability (or, equivalently, the statistical power) is important in the planning stage of an experiment, because it indicates the likelihood that a real phenomenon of interest will be declared statistically significant, thus helping the investigator avoid the time and expense of conducting an experiment that would be hopeless at uncovering the truth. The meaning of a failure to reject the null hypothesis by a low-power test procedure is difficult to interpret, because one cannot distinguish between no true effect or an experimental inability to find one even if it exists. (After an experiment is concluded, however, the Type II error probability has less relevance as a measure of the uncertainty than does a confidence interval or region for the parameter or parameters of interest.)
An important theoretical criticism of all of the above frequentist approaches is that the main questions of scientific inference are not addressed: given the results of an experiment, (i) what is the weight of the evidence in favor of one hypothesis and against another? (ii) what should the investigator believe is true? and (iii) what steps should now be taken? These three questions require different answers.
It is a common misconception that the p value is a valid measure of evidence against the null hypothesis—the (misguided) notion being that the smaller the p value, the stronger the evidence. Although in many common situations there is a useful correspondence between the p value and a proper notion of evidence, there are many other examples in which the p value utterly fails to serve as a cogent measure of weight of evidence. There is a strong argument that any measure of evidence must be a relative one (one hypothesis versus another). One sees immediately that the p value cannot be a proper measure of the weight of evidence against a null hypothesis relative to a given alternative, because the p value entirely ignores all alternatives, being solely concerned with null hypothesis probabilities. Thus, it is entirely possible for an observed p value of 0.0001 to be weak evidence against a given null hypothesis (e. g., if the probability of observing the data is even more unlikely to occur under a given alternative).
Although certain technical details are still under discussion, statisticians
Page 115
generally acknowledge that an appropriate measure of the weight of evidence of one hypothesis relative to that of another is provided by the likelihood ratio, which is the probability of the observed data under the first hypothesis divided by the probability of the observed data under the second one. (The probabilities here are point probabilities, not tail probabilities.) As a descriptive device, a likelihood ratio of 8 or more is sometimes taken as “fairly strong” evidence in favor of the first hypothesis relative to the second one, whereas a likelihood ratio of 32 or more would be taken as “strong” evidence. These adjectives and their boundaries are arbitrary, of course, but correspond to useful benchmarks in inferential settings. See Royall (2000) for further discussion and references to these ideas.
For example, in screening a population for a certain disease, suppose the screening test has a sensitivity of 0.90 and a specificity of 0.95. These are values that can be measured in a laboratory setting (e.g., by testing a large number of known patients with the disease and, in another series, a large number of healthy subjects without the disease). What is the weight of evidence in favor of disease as opposed to no disease given a positive test result? The answer is not 5 percent, which would be the p value (the probability of a rejection of the null hypothesis of no disease given no disease in truth is the complement of the specificity). The likelihood ratio is an arguably better measure of the weight of evidence provided by a positive test result: since the probability of a positive test result under the hypothesis of true disease is 0.90 while the probability of a positive test under the alternative of no disease is 1 − 0.95 = 0.05, the likelihood ratio is 0.90/0.05 = 18 in favor of disease. This is an objective measure of the relative likelihood of the data under the two hypotheses and may be taken as a quantitative measure of weight of evidence, which is “fairly strong” in the adjectival descriptive scheme given above.
Should a patient with a positive test result believe he or she has the disease? This is question (ii) above and takes one directly into the Bayesian framework (see Chapter 3). The Bayesian framework allows one to quantify degrees of subjective belief and to combine the “objective” data with “subjective” prior beliefs to arrive at an a posteriori degree of belief in a hypothesis or theory. In the simplest case, the fundamental result of Bayes's theorem states that the posterior odds in favor of one hypothesis relative to another hypothesis is given by the prior odds multiplied by the likelihood ratio. Thus, the Bayesian paradigm reveals the role of the weight of evidence as measured by the likelihood ratio (or the so-called Bayes factor in more complicated settings) as that which increases or decreases one's prior odds in light of the data to yield one's posterior odds (and thus the a posteriori
Page 116
degree of belief). In the screening example, whether or not the patient should believe he or she has the disease depends on the prior degree of belief. If there are no other signs or symptoms for this patient before the screening test, then the prior probability of disease may logically be taken as the general prevalence of the disease in the screened population, which may be low, say, 0.01 for the sake of discussion. In that case the prior odds (degree of belief) would be 0.01/0.99, or approximately 0.0101, which, when multiplied by the “fairly strong” evidentiary likelihood ratio of 18, yields a posterior odds of only 0.1818 in favor of disease, corresponding to a posterior degree of belief of only 15.4 percent in favor of disease. (Even without appealing to subjective degree of belief, this result indicates that about 85 percent of the positive results will be false-positive results in the general population.) On the other hand, if the patient has other symptoms that may be related to the disease (and might have prompted use of the screening test for diagnostic purposes), then the a priori odds on the disease would be correspondingly higher. If the patient or his physician believes that there is a 10 percent chance of disease before the test results are known, then the prior odds of 0.10/0.90 = 1/9 multiplied by the likelihood ratio of 18 yields posterior odds of 2, or an a posteriori degree of belief of 2/3 in favor of disease. This example also illustrates how two experts in a courtroom proceeding can look at the same body of evidence and arrive at opposite conclusions: a given weight of evidence may be convincing to someone with high prior odds, but unpersuasive to another with low prior odds.
Question (iii), what steps should be taken next?, is the subject of formal decision theory discussed in Chapter 3.
The committee concludes this section on statistical methods of drawing inferences with the statement that should be obvious by this point: there is no unique solution to the problem of scientific induction. Consequently, the various ways of drawing statistical inferences and the various probability measures should always be used with some caution, and with a view toward using the methods that are best suited to address the applied problem at hand.
Statistical Derivation of Prediction Limits
The following sections describe statistical derivation of nonparametric, normal, lognormal, and Poisson prediction limits and intervals in some detail, using the astronaut problem as an illustration.
Page 117
Nonparametric Prediction Limits
Recall that α is the false positive rate for a single endpoint and that α* is the experiment-wise false positive rate for all k endpoints. Under the conservative assumption of independence, the Bonferroni inequality gives α = α*/ k. For simplicity of notation, we do not index the p sets of measurements for each of the k endpoints, only for a single endpoint. Note that in practice, the endpoints may be correlated and the actual confidence level provided by the method will be somewhat higher than the estimated value.
To understand the implications of various design alternatives on the resulting experiment-wise confidence levels, let y(si; ni) denote the sith largest value (i.e., order statistic) from the ni astronauts on spaceflight i (i = 1, . . . , p) and let x (u,n) denote the uth order statistic from a group of control astronauts of size n. One can now express the previous discussion mathematically as
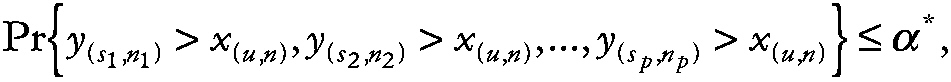
~ enlarge ~
where α* is the experiment-wise rate of false-positive results (e.g., α* = 0.05). To evaluate the joint probability note that the probability density function of the uth order statistic from a sample of size n [i.e., x (u,n)] is
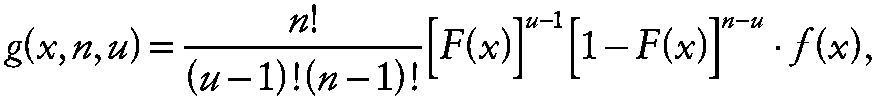
~ enlarge ~
where
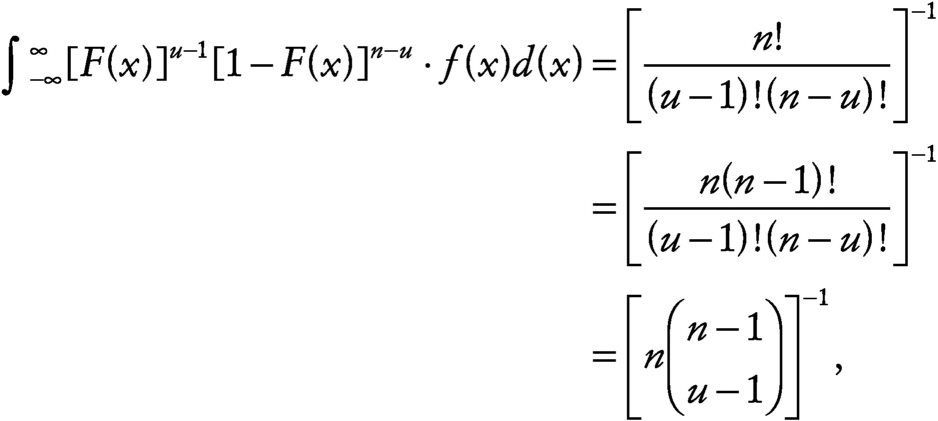
~ enlarge ~
(Sarhan and Greenberg, 1962). Since
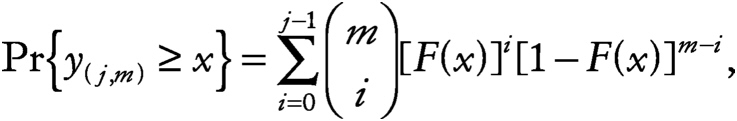
~ enlarge ~
Page 118
the joint probability is then
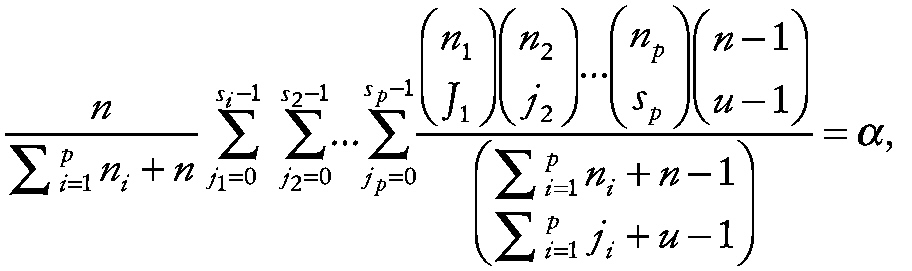
~ enlarge ~
(Chou and Owen, 1986; Gibbons, 1990, 1991, 1994). A lower bound on the probability of the sith largest experimental measurement (e.g., the median) in all p spaceflights exceeding the uth largest control measurement for any of the k outcome measures is given by α* = 1 − (1 − α)k . One minus this probability provides the corresponding confidence level. Ultimately, for practical applications one would typically like the overall confidence level to be approximately 95% (i.e., α* ≤ 0.05).
To determine if an outcome measure is significantly decreased in experimental astronauts relative to control astronauts, let x (l,n) denote the lth smallest measurement from the group of control astronauts of size n. Then the equation becomes
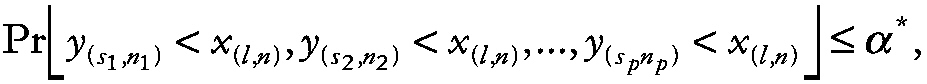
~ enlarge ~
which leads to
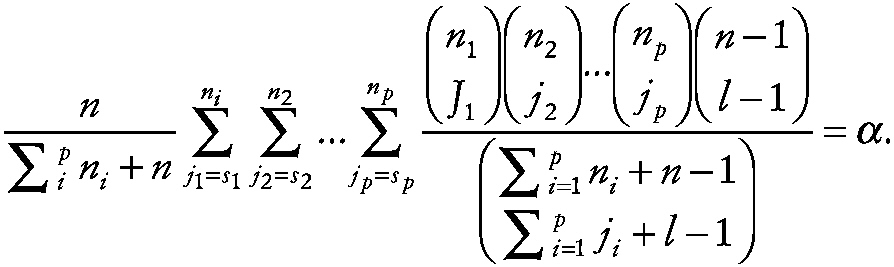
~ enlarge ~
The probability of the sith largest experimental measurement (e.g., the median) in all p spaceflights being simultaneously less than the lth smallest control measurement for any of the k outcome measures is given by α* = 1 − (1 − α)k , and 1 minus this probability provides the corresponding confidence level. For a two-sided interval, one can compute upper and lower limits each with probability α* /2. If si is the median of the ni experimental measurements in spaceflight i, then the upper prediction limit can be computed with probability approximately α*/2, and the lower limit is simply the value of the l = (n − u + 1)th ordered measurement.
Page 119
Parametric Prediction Limits
Although nonparametric prediction limits are most general, in certain cases the distributional form of a particular endpoint is well known and a parametric approach is well justified. Most relevant to this type of application are normal, lognormal, and Poisson prediction intervals. The reader is referred to the books by Guttman (1970), Hahn and Meeker (1991), and Gibbons (1994) for a general overview.
Normal Distribution
For the case of a normally distributed endpoint, one can derive an approximate prediction limit by noting that the experiment-wise rate of false positive results α* can be achieved by setting the individual endpoint rate of false-positive results α equal to
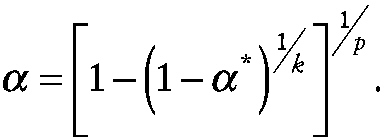
~ enlarge ~
As an example, let α* equal 0.05 and k equal 10 endpoints. For p equal to 1 (i.e., a single space mission),
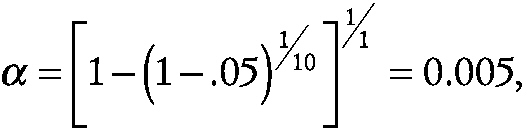
~ enlarge ~
which would lead to quite limited statistical power to detect a significant difference for any single endpoint. By contrast, with p equal 2 (i.e., two space missions),
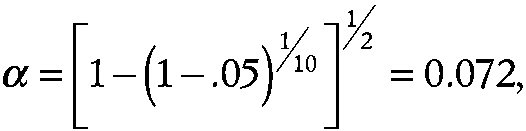
~ enlarge ~
which would require a much smaller effect size to declare a significant difference. With p equal to 3
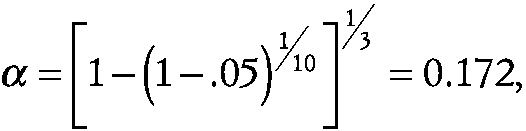
~ enlarge ~
which can be used to detect an even smaller difference. Assuming normality, the 100 (1 − α) percent upper prediction limit (UPL) for the mean of the ni experimental subjects in replicate i, for i = 1, . . . , p, is given by
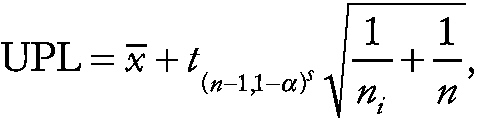
~ enlarge ~
Page 120
where x^„ and s are the means and standard deviation of the n control measurements, respectively, and t is the 1 − α upper percentage point of Student's t − distribution on n − 1 degrees of freedom. For example, if n is equal to 20, ni is equal to 5, p is equal to 3, and k is equal to 10, then the prediction limit is
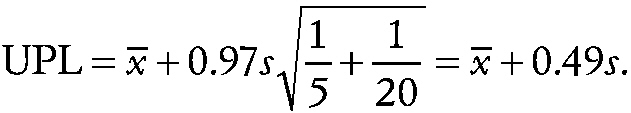
~ enlarge ~
By contrast, with p = 1, the corresponding prediction limit is almost three times larger, i.e.,
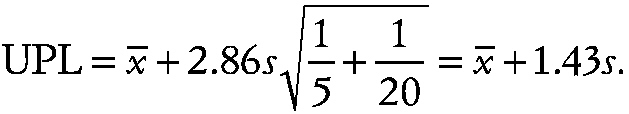
~ enlarge ~
The corresponding lower prediction limit (LPL) is given by
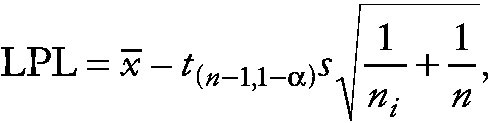
~ enlarge ~
and the two-sided prediction interval (PI) is
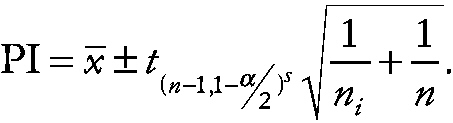
~ enlarge ~
Note that this solution is approximate because it ignores the dependency introduced by comparing each of the p source sets with the same control group. The magnitude of the correlation is
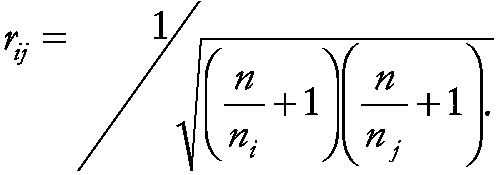
~ enlarge ~
For n equal to 20 and ni equal to nj which is equal to 5, the correlation (r) is 0.2. Incorporation of this correlation into the computation of the prediction limit is complex and requires computation of the relevant critical values from a multivariate t - distribution (Dunnett, 1955; Gibbons, 1994). The approximate prediction limits presented above, which assume independence, will slightly overestimate the true values that incorporate the dependence, but are much easier to compute.
Lognormal Prediction Limits
Note that it is often tempting to attempt to bring about normality by transforming the raw data and then applying the above method to the transformed data. A natural choice is to take natural logarithms of the raw data,
Page 121
compute the normal limit on the transformed data, and exponentiate the resulting limit estimate. Although this is a perfectly reasonable approach, it should be noted that the exponentiated limit is for the geometric mean of the ni experimental sources (i.e., the median) and not the arithmetic mean intensity, which will always be larger than the median if the data are, in fact, from a lognormal distribution. Nevertheless, this can be a useful strategy if the raw intensity data are highly skewed. Bhaumik and Gibbons (submitted for publication) have developed a lognormal prediction limit for the mean of ni future samples.
Poisson Distribution
For endpoints that are the result of a counting process, a Poisson distribution may be a reasonable choice for statistical purposes. To construct such a limit estimate, assume that y, the sum of n control measurements, has a Poisson distribution with mean μ. Having observed y one needs to predict y*, the sum of ni experimental measurements, which has a Poisson distribution with mean cμ In the present context, c is equal to ni/n. On the basis of a result of Cox and Hinkley (1974), Gibbons (1987) derived the corresponding UPL for y* as
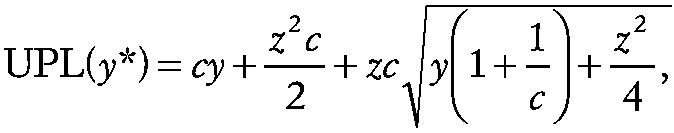
~ enlarge ~
where z is the 100(1 − α) percentage point of the normal distribution. In this context, the prediction limit represents an upper bound on the sum of the measurements of the ni experimental subjects in replicate i and α is defined in the first equation in the section Normal Distribution. The experimental condition is only considered significantly different from control only if the sum of the ni experimental measurements exceeds the UPL for y* in all p experimental replicates (e.g., space missions). The corresponding LPL
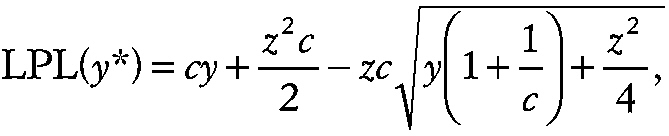
~ enlarge ~
and the two-sided PI is obtained by substituting z [1 − α/2] into the previous two equations.
The General Linear Hierarchical Regression Model
To describe the general linear hierarchical regression model in a general way for data that are either clustered or longitudinal, the terminology of
Page 122
multilevel analysis can be used (Goldstein, 1995). For this, let i denote the Level - 2 units (clusters in the clustered data context or subjects in the longitudinal data context), and let j denote the Level - 1 units (subjects in the clustered data context or repeated observations in the longitudinal data context). Assume that there are i = 1, . . . N Level - 2 units and j = 1, . . . ; ni Level - 1 units nested within each Level - 2 unit. The mixed-effects regression model for the ni x 1 response vector y for Level- 2 unit i (subject or cluster) can be written as
yi = Wiα + Xiβi + εi i = 1,...N,
where Wi is a known ni x p design matrix for the fixed effects, α is the p x 1 vector of unknown fixed regression parameters, Xi is a known ni x r design matrix for the random effects, and βi is the r x 1 vector of random individual effects, and εi is the ni x 1 error vector. The distribution of the random effects is typically assumed to be multivariate normal with mean vector 0 and covariance matrix ∑, and the errors are assumed to be independently distributed as multivariate normal with mean vector 0 and covariance matrix
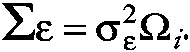
~ enlarge ~
Although Ωi carries the subscript i, it depends on i only through its dimension ni, that is, the number of parameters in Ωi will not depend on i. In the case of independent residuals, Ωi is equal to Ii, but for the case of longitudinal designs, one can define ω to be the s x 1 vector of autocorrelation matrix (Chi and Reinsel, 1989).
Different types of correlation structures have been considered including the first-order autoregressive process AR(1), the first-order moving average process MA(1), the first-order mixed autoregressive-moving average process ARMA(1,1), and the general autocorrelation structure. A typical assumption in models with correlation structures is that the variance of the errors is constant over time and that the covariance of errors from different time points depends only on the time interval between these time points and not on the starting time point. This assumption, referred to as the stationarity assumption, is assumed for the aforementioned forms. Another form of correlation structures is described by Mansour and colleagues (1985), who examine correlation structures that follow the first-order autoregressive process, however, where the assumption of stationarity is relaxed.
As a result of the above assumptions, the observations yi and random coefficients β have the joint multivariate normal distribution
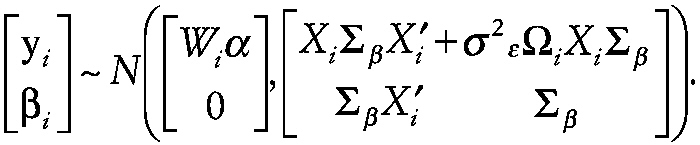
~ enlarge ~
Page 123
The mean of the posterior distribution of β, given yi, yields the empirical Bayes (EB) or EAP (expected a posteriori) estimator of the Level 2 random parameters,
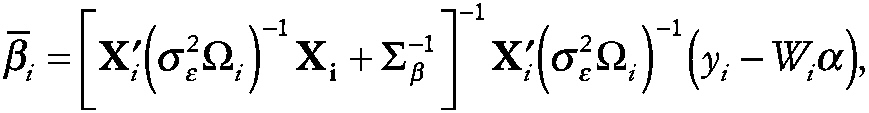
~ enlarge ~
with the corresponding posterior covariance matrix given by
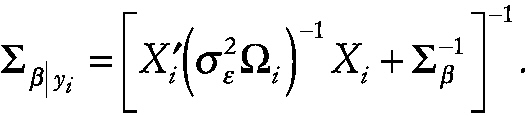
~ enlarge ~
Details regarding estimation of ∑β, α, σ2ε and ω were originally introduced by Laird and Ware (1982).
Empirical Bayes Methods and Risk-Based Allocation
Why are empirical Bayes methods needed for analysis of experiments with risk-based allocation? There are two reasons: first, the natural heterogeneity from subject to subject requires some accounting for random effects; and second, the differential selection of groups due to the risk-based allocation is handled perfectly by the “u - v” method introduced by Herbert Robbins. This section considers both of these ideas in a little detail.
Consider the following simple model. Let a be a prespecified nonnegative integer, and let X be a count variable observed pretreatment. Given an unobservable nonnegative random variable θ, which varies in an arbitrary and unknown manner in the population of subjects under study, assume that X has a Poisson distribution with mean θ. Now, let Y be another count variable, observed postreatment, and given both X and θ, we suppose that Y has a Poisson distribution with mean c0θ if X is ≤ a or mean c1θ if X is >a. In symbols, X|θ ∼ P(θ), θ∼G(θ) unknown, and Y|X, θ ∼ P(cI(X)θ), where the index I(X) = I[X > a] is the indicator that X exceeds a. This model reflects the risk-based allocation design: if X is ≤a the subject is at low risk and the standard treatment is given, where the treatment is assumed to operate multiplicatively on θ by the effect size c0, whereas if X is >a, the subject is at high risk and an experimental treatment is given, where the treatment is again assumed to operate on θ multiplicatively, but with a possibly different effect size, c1. (In the risk-based design, one does not need to assume the Poisson distribution for Y given X and θ, but only that the conditional mean of Y satisfies E[Y|X, θ] = cI(X)θ. The Poisson assumption is made for convenience in this discussion.) How can one estimate the effect sizes c0 and c1 in light of the fact that θ is an unobservable trait of the subjects with unknown distribution?
Page 124
Notice that θ usually cannot be ignored and a simple model for Y as a function of X is assumed. This is because the correct mean of Y given X alone is seen upon integration to be E[Y|X] = cI(X)E[θ|X], but the mean of θ for given values of X can be a highly complicated function and is generally unknown since the distribution G is unknown. The only distributions on θ for which E[θ|X] is a simple linear function of X is the family of gamma distributions, the so-called conjugate prior for the Poisson distribution. Unless there is an empirical basis for assuming the conjugate prior, however, the posterior mean of θ given X will not be linear. (This simple fact is what separates empirical Bayes methods from subjective Bayes methods, in which a gamma prior would often be assumed, gratuitously in many cases.)
A simple ratio of averages also cannot be taken because of the different treatments. If there were only a single treatment (the case in which a is equal to ∞), one could estimate c0 consistently by the ratio of the average Y to the average X, because E[Y] = E(E[Y|X, θ]} = E{c0θ} = c0E[θ] = c0E[X]. Where there are two treatment effects for X ≤ a and X > a, however, it can be shown that avg{Y]/avg{X} is an inconsistent (biased) estimator of both treatment effects. A better tool is needed.
Enter the Robbins u-v method. Robbins and Zhang (1991) proved the following theorem. If u(x) is any known function, then under the assumption X|θ ~ P(θ), with the distribution of θ arbitrary, the expected value of θ times u(X) is E[θu(X)] = E[Xu(X−1)]. This result is called the fundamental empirical Bayes identity for the Poisson case and is remarkable because it expresses an unknown expectation of an unobservable quantity on the left in terms of an expectation of an entirely observable, hence estimable, quantity on the right. 1 Here's how this result is applied to the problem. First, let u(x) = I[x ≤ a] be the indicator that standard treatment has been assigned. One finds an expression for treatment effect c0 as follows. First, note that E[Yθ(X)|X, q] = u(X)c0θ, because the expression is non-zero only for X ≤ a. Taking expectations of both sides with respect to the joint distribution of X and θ, and applying the fundamental identity, one finds
E[Yu(X)] = c0E[θu(X)] = c0E[Xu(X − 1)] = c0E(XI[X ≤ a+1]}.
Consequently, one has c0 = E{YI[X ≤ a]}/E[XI{X ≤ a +1]}, which can easily be estimated: simply sum the values of the endpoint Y among those subjects with X ≤ a and divide by the sum of the values of X among all those
1 The u-v method of estimation gets its name because Robbins considered problems in which for a given function u(x) there is another function v(x) such that E[θu(X)] is equal to E[v(X)]. In the Poisson case, the equation is v(x) = xu(x−1).
Page 125
with X ≤ a + 1 at the baseline. The Law of Large Numbers ensures that the ratio is a consistent estimator as the sample size of subjects increases.
As an illustration, consider the motorist example presented in Box 2-6 in Chapter 2. Suppose one wanted to find out if there was a multiplicative temporal effect on the accident-proneness parameter θ between year 1 and year 2 for the subset of drivers with zero accidents. This is the case in which a is equal to 0. The empirical Bayes estimate of c0 is given by the observed total number of accidents in year 2 (γ) for those with zero accidents in year 1 (X = 0), divided by the sum of the values of X among all those with X ≤ 1 in year 1, which is just the number of drivers with exactly one accident at the baseline. Thus, the prediction procedure discussed in Chapter 2 under the assumption that c0 is equal to 1 has been converted into an estimation procedure for treatment effect in an assured allocation design. If an active treatment rather than simply the passage of time was applied, its effect would be included in the parameter c0.
An analogous result applies for estimation of c1 among the high-risk subjects with a > 1. In this case c1 = E{YI[X > a]}/E{XI[X > a+1]}, which can be estimated by summing the values of the endpoint Y among those subjects with X > a, and dividing by the sum of the values of X among all those with X > a + 1 at the baseline. For example, the temporal effect on accident proneness for those drivers with one or more accidents in the baseline year can be estimated by the observed total number of accidents in the next year among those with one or more accidents in the baseline year divided by the total number of accidents suffered by all those with two or more accidents in the year baseline (and the denominator is an unbiased predictor of the numerator under the null hypothesis c1 is equal to 1).
The above theory is called semiparametric because of the parametric Poisson assumption and the nonparametric assumption concerning the distribution of θ. The committee closes this section with some remarks on a completely nonparametric estimation procedure with use of an auxiliary variable. The assumption that X has a Poisson distribution is now dropped, with only the assumption retained that whatever the distribution of X given θ E[X|θ] is equal to θ. This is really just a definition of θ. For the endpoint variable Y, assume a model for expectations that encompasses both an additive and a multiplicative effect model for standard treatment:
E0[Y | X,θ] = a + bX + cθ,
where the subscript on the expectation indicates standard treatment. The model allows the expectation of Y to depend on X because treatment mo-
Page 126
dality (e.g., the dosage of a standard therapeutic drug) may depend on the observed baseline measurement. In the risk-based design, one wants to use the model to estimate the mean response for those at higher risk (with, say, X > a) under standard treatment, but note that just as in the Poisson case, the model for Y given X alone is generally not a linear model in X, because generally E[θ|X] is not linear in X, unless θ has a conjugate prior relative to X.
Now assume that there is an auxiliary variable, X', also measured before treatment. X' may be a replicate measurement of X (e.g., a second blood assay), a concomitant measure of risk, or a baseline version of the endpoint Y. X' need not be independent of X, even conditionally given θ. It is assumed that X' is similar to Y in the sense that for some constants a', b', and c' ≠ 0,
E[X'|X,θ] = a'+b' X + c'θ,
It follows that the variable Y - (c/c')X' does have a simple linear expectation in X:
E0[Y − (c/c')X' | X] = a*+b* X,
where a* is equal to a − (c/c')a' and b* is equal to b − (c/c')b'. If the ratio c/c' is known, the model can be used with ordinary estimates of a* and b* from the subjects on standard treatment to estimate (predict) what the high-risk patients would have yielded for Y − (c/c')X'. The treatment effect is estimated using observed values of Y − (c/c1)X1 together with the relation
Treatment effect = E1[Y|X > a] − E0[Y|X > a] = E1[Y − (c/c')X'|X > a] − E0[Y − (c/c')X'|X > a]
which holds because the expectations of X' given X are the same under E0 or E1 as both X and X' are measured pretreatment. If the ratio c/c1 is not known, it can be estimated for the subjects under standard treatment by special methods (details omitted).
In the cholestyramine example (see Box 2-7), the allocation variable X was a baseline measurement of the total serum cholesterol level. The auxiliary variable X' was another measurement of the total serum cholesterol level taken 4 months after all subjects were given dietary recommendations but before randomization to cholestyramine or placebo. The analysis assumed the ratio c/c' was equal to 1, so that the simple change scores Y − X' were linearly related to X, even though Y itself would generally be a nonlin-
Page 127
ear function of X. Then the treatment effect of cholestyramine versus placebo among the high cholesterol subjects was estimated by the observed average of Y − X' among those taking cholestyramine minus the predicted value of Y − X' among the same patients if they had taken placebo, with the latter quantity estimated by a* + b*X evaluated at the average values of X among the subjects with high cholesterol levels. For further details, see Finkelstein et al. (1996) and Robbins (1993).
These results often surprise even the best statisticians. Robbins' general empirical Bayes theory is both elegant and nontrivial. In the Poisson case the point and interval predictions of the number of accidents to be had in year 2 by those drivers with zero accidents in year 1 are applications of general (i.e., semiparametric) empirical Bayes theorems for mixtures of Poisson random variables. The interested reader may want to consider the results in a little more detail. Let the random variable X denote the number of accidents in year 1 for a given motorist and let random variable Y denote the number of accidents in year 2 for the same motorist. Let random variable q denote the unobservable accident proneness parameter for that motorist. The model under consideration makes three assumptions:
(i) θ has an unknown distribution G with finite expectation;
(ii) given θ, the distribution of X is Poisson with mean θ; and
(iii) given X and θ, the distribution of Y is also Poisson with mean θ.
Assumption (iii) is the null assumption that road conditions and driving habits remain constant. The assumption implies that θ alone determines the distribution of Y, irrespective of X. X and Y are unconditionally correlated in ignorance of θ.
Now, let u(x) be any function of x. Two fundamental empirical Bayes identities for Poisson random variables due to Robbins can be stated:
First-order empirical Bayes identity for Poisson variables:
E[u(X)θ] = E[Xu(X − 1)],
where the expectation is taken with respect to the joint distribution of X and θ determined by (i) and (ii).
Second-order empirical Bayes identity for Poisson variables:
E[u(X)θ2] = E[X(X − 1)u(X − 2)].
Page 128
The proofs of these assertions are elementary: one demonstrates them first for conditional expectations given θ, pointwise for each θ. Then one takes expectations with respect to G (details omitted).
In the application, let u(x) equal I[x equal 0], the indicator function for x equal to 0. The key result for the point prediction is
E{YI[X = 0]} = P[X = 1].
To see this, write E[Yu(X)|X, θ] = u(X)E[Y|X, θ] = u(X)θ. Take expectations with respect to (X, θ) and use the first identity: E[Yu(X)] = E[u(X)θ] = E[Xu(X − 1)] = E{XI[X = 1]} = P[X = 1].
Thus in a population of n drivers, the predicted number of accidents in year 2 among zero-accident drivers in year 1 is
E{∑iYiI[Xi = 0]} = nP[X = 1],
and using the sample estimate of P[X = 1], namely (number of drivers with one accident)/n, one can conclude that the prediction is the number of drivers with exactly one accident in year 1. For the 95 percent prediction interval, the key result is that
E{[Yu(X) − u(X 1)]}2 = E{YI[X = 0] − I[X = 1]}2 = 2{P[X = 1] + P[X = 2]}.
The proof is as follows. Conditional on (X, θ),
E{[Yu(X) − u(X − 1)]2 |X,0} =
E[Y2 u(X)2 | X, 0] − 2u(X)u(X − 1)E[Y | X, θ] + u(X − 1)2 =
u(X)2 (θ + θ2 ) − 2u(X)u(X − 1)θ + u(X − 1)2.
The property var(Y|X, θ) = E[Y|X, θ] was used for Poisson variables in the first term. By using u(x) equal to I[x = 0], the right-hand side reduces to I[X = 0](θ + θ2) + I[X = 1]. Thus, unconditionally, E{[Yu(X) − u(X − 1)]2} = E{I[X = 0]θ} + E{I[X = 0]θ2} + P[X = 1], and using the first and second fundamental identities, this reduces to
E{XI[X = 1]} + E{X(X 1)I[X = 2]} + P[X = 1] = 2{P[X = 1] + P[X = 2]}.
Thus, in a population of n drivers, the mean squared error of prediction is
E[∑i {YiI[XI = 0] I[XI = 1]}2] = 2n{P[X = 1] + P[X = 2]},
which is consistently estimated by twice the number of drivers with exactly
Page 129
one or two accidents in year 1, as used in the 95 percent prediction interval in the report by Finkelstein and Levin (1990).
APPENDIX REFERENCES
, and . An upper prediction limit for the arithmetic mean of a lognormal random variable. Submitted for publication.
, and . 1989. Models of longitudinal data with random effects and AR(1) errors. Journal of the American Statistical Association 84: 452–459 .
, and . 1986. One-sided distribution-free simultaneous prediction limits for p future samples. Journal of Quality Technology 18: 96–98
, and . 1974. Theoretical Statistics . London: Chapman & Hall.
1955. A multiple comparisons procedure for comparing several treatments with a control. Journal of the American Statistical Association 50: 1096–1121.
, andd . 1990. Statistics for Lawyers . New York: Springer Verlag.
, , and . 1996. Clinical and prophylactic trials with assured new treatment for those at greater risk. Part II. Examples. American Journal of Public Health 86: 696–705.
1987. Statistical models for the analysis of volatile organic compounds in waste disposal sites. Ground Water 25: 572–580.
1990. A general statistical procedure for ground-water detection monitoring at waste disposal facilities. Ground Water 28: 235–243.
1991. Some additional nonparametric prediction limits for ground-water detection monitoring at waste disposal facilities. Ground Water 29: 729–736.
1994. Statistical Methods for Groundwater Monitoring . New York: Wiley.
1995. Multilevel Statistical Models , 2nd ed. New York: Halstead Press.
1970. Statistical Tolerance Regions: Classical and Bayesian . Darien, Conn: Hafner.
, and . 1991. Statistical Intervals: A Guide for Practitioners . New York: Wiley.
, and . 1982. Random effects models for longitudinal data. Biometrics 38: 963–974.
, , and . 1985. Maximum likelihood estimation of variance components in repeated measures designs assuming autoregressive errors. Biometrics 41: 287–294.
1993. Comparing two treatments under biased allocation. La Gazette des Sciences Mathematics du Quebec . 15: 35–41.
, and . 1991. Estimating a multiplicative treatment effect under biased allocation. Biometrika 78: 349–354.
and . 1962. Contributions to Order Statistics . New York: Wiley.



























