
6
Virulence-Factor Activity Relationships
INTRODUCTION
The term “virulence-factor activity relationship,” or VFAR (formerly referred to as virulence-activity relationship or VAR; NRC, 1999a), is rooted in a recognition of the utility of using structure-activity relationships (SARs) to compare the structure of newly identified or produced chemicals to known chemical structures to enable prediction of their toxicity and other physical properties. In essence, the committee believes the same principle can be applied to waterborne pathogens. It is important to state that many sections of this chapter necessarily include more extensive use of scientific terms and language than might typically be found in the body of a National Research Council (NRC) report. That is, rather than deleting, simplifying, or relegating such relevant technical language to an appendix, the committee decided to keep all information related to VFARs in one comprehensive chapter. This chapter should be read with that qualification in mind.
For microorganisms, there are many levels of structure, such as the cell or organism itself and the larger internal components that comprise the microorganism (e.g., nucleus, micronemes, flagellae). These morphological components can sometimes be used to identify pathogenic microorganisms. Beyond these relatively large structures, there are smaller, biochemical components of the organism, including proteins, carbohydrates, and lipids. Many of these biochemical building blocks are directly related to how a particular microorganism causes disease. Some examples of these include the outer coat of some bacteria (the lipid polysaccharide coat), attachment and invasion factors, and bacterial toxins. Thus, the central premise of VFARs is to relate the architectural and biochemical components of microorganisms to potential human disease.
Virulence can be defined as the quality of being poisonous or injurious to life (i.e., virulent). For an organism to be virulent, it must be able to infect its human host, reproduce, and/or cause a disease. This broad definition of virulence is more inclusive than the narrow definition commonly used by microbiologists (i.e., virulence is solely the severity of the disease produced after exposure and infection). Each of the microbiological attributes that contribute to virulence can in general be linked to specific architectural elements or biochemical compounds within the organism. Together, these elements and compounds can generally be termed “virulence factors,” and the blueprints for them are included in the genetic code of an organism. For this reason, a principal topic of this chapter is the genetic structure of various microorganisms because of its direct relationship to virulence factors.
Owing to recent advances in molecular biology, the genetic structures of many thousands of organisms (especially bacteria and viruses) have been identified, reported, and stored in what are called gene banks. Sophisticated computer programs allow for the sorting and matching of genetic structures and specific genes. The discipline that organizes and studies these genes is known as bioinformatics, while the study of genes and their function is known as genomics. In addition, a growing area of related interest is functional genomics, that is, understanding the specific role of genes in terms of the function of the organism. The ability to use these and related tools to address the microbial contamination of drinking water is illustrated by some of the following observations:
-
The genetic structures of most known waterborne pathogens have been characterized at least partially, with the information stored in gene databanks. The complete genome of several important waterborne pathogens, such as Vibrio cholerae (the agent of cholera), is now known, and many more will be characterized in the near future (Heidelberg et al., 2000).
-
Other related information is accumulating that allows the use of these databanks to determine or predict the ability of a microorganism to produce virulence factors, such as toxins, attachment factors, and other surface proteins, and genes that encode bacterial resistance to antibiotics.
-
On a more basic level, these data can be used to characterize similarities and differences between a microorganism of interest and known pathogens.
-
Data of this kind can also be used to identify sources of, and thus exposures to, microorganisms through molecular “fingerprinting.”
-
The functional genomics or bioinformatics expertise needed to establish a nationwide VFAR program already exists in the private and public sectors.
Thus, the committee concludes that a VFAR concept, with many parallels to the SAR concept used for chemicals, would be a powerful approach to examining emerging waterborne pathogens, opportunistic microorganisms, and other newly identified microorganisms.
STATEMENT OF THE PROBLEM
As noted in the committee’s first report (NRC, 1999a), the current approach to identifying and controlling waterborne disease is limited. It has followed a similar path since cholera was first linked to transmission via water (“from the Broad Street pump”) in London, England, nearly a century and an half ago, and since Koch first proposed his famous postulates regarding causation (see Okun, 1999). Typically, a disease outbreak is reported only when a significant portion of a community is recognized to have been affected, the responsible microorganism has been identified, and an epidemiological study is undertaken to determine possible sources of exposure to the agent in the community. If any of these three elements is lacking, the outbreak is generally missed and goes unreported. If the consumption of drinking water is identified as a potential source of exposure, a public health advisory to boil water may be issued. Alternatively, the culpable part of the system may be identified and isolated until the cause of contamination is eliminated. However, for most of the waterborne outbreaks in the United States, the etiology is never determined, the responsible microorganism is never identified, and public water systems are not easily fixed or shut off. The identification of pathogens is thus unnecessarily related to the recognition of an outbreak. Under the amended Safe Drinking Water Act, microbial contamination, regardless of whether it is associated with an outbreak or not, must be addressed.
Hundreds if not thousands of microorganisms have the potential to be spread through drinking water supplies and distribution systems. While data on health effects for many of these are described in the medical literature, there are no occurrence databases or even routine methodologies for developing these databases (NRC, 1999a). One of the princi-
pal dilemmas to be addressed is that current regulatory practice requires that methods to culture organisms of interest be developed before occurrence data can be gathered. Thus, a microorganism ordinarily must first be identified as a pathogen, and be capable of in vitro culture, before occurrence data are acquired. This long-standing paradigm makes it very difficult or impossible to develop a database of potential or emerging pathogens.
There is also no widely accepted approach for prioritization of waterborne pathogens, other than through expert judgment. For example, and as noted earlier, the U.S. Environmental Protection Agency (EPA) and the American Water Works Association Research Foundation have jointly sponsored a series of expert workshops since 1996 for the development of a decision process for prioritizing emerging waterborne pathogens that is nearing completion. These expert judgments must be made, of necessity, by a very small number of researchers in the discipline of health-related environmental microbiology. This approach to the process makes transparency very difficult to achieve, the importance of which is discussed in Chapters 2 and 5 of this report.
The committee believes strongly that if EPA continues to rely on exposure and health effects as two primary data categories for screening potential microbial drinking water contaminants, progress will continue to be unacceptably slow. Current efforts are able to address only one or two microorganisms every 5 to 10 years with the current CCL development and implementation approach. To illustrate the dilemma, consider that of the ten microorganisms and groups of related microorganisms on the 1998 CCL, nine are in the “research priorities” category (see Table 1–3) and will go unregulated in the first CCL cycle. Of these nine microbial contaminants, only one, Aeromonas hydrophila, is slated for delayed screening level monitoring during the first cycle of the Unregulated Contaminant Monitoring Regulation (UCMR) (EPA, 1999c) (see Table 1–4).
It is clear that a severe bottleneck exists in identifying and addressing important microbial contaminants in drinking water. Thus, a new approach to assessing pathogens could help overcome this ongoing problem.
VFAR ANALOGY TO SARS AND QUANTITATIVE SARS
A variety of terminology has developed in the literature to identify various classes of correlations useful for predicting the properties of
agents in environmental and health sciences.
For example, chemical properties are amenable to prediction through use of structure-activity relationships, which can be distinguished from property-activity relationships (PARs) and structure-property relationships (SPRs). Although careful classification along these lines certainly has heuristic value (e.g., Brezonik, 1990), few researchers adhere to these distinctions rigorously. Instead, only a few terms are commonly used and these are often applied to a wider range of correlation types than strict use of each expression would allow (Tratnyek, 1998). One example of this is the term linear free energy relationship (LFER), which originally referred to a specific type of correlation used by physical organic chemists but eventually came to represent the entire field of correlation analysis in organic chemistry (Shorter, 1973). Similarly, the term quantitative structure-activity relationship (QSAR) was originally coined for use in drug design but is now commonly used to refer to many types of correlations employed in the pharmaceutical, toxicological, and environmental sciences. By analogy to the above discussion, the committee has coined the term “virulence-factor activity relationship” and defined it as the known or presumed linkage between the biological characteristics of a microorganism and its real or potential ability to cause harm (pathogenicity).
FRAMEWORK
The central concept is to use microbial characteristics to predict virulence via what the committee terms a virulence-factor activity relationship. Microbial VFARs would function in much the same way as QSARs do, namely to assist in the early identification of at least several potential elements of virulence. Research increasingly has shown certain common characteristics among virulent pathogens, such as the production of specific toxins, specific surface proteins, and specific repair mechanisms that enhance their ability to infect and inflict damage in a host. Recently some of these “descriptors” (the terminology often used in QSARs) have been tied to specific genes, and it has become evident that the same can be done for others. Identification of these descriptors, either directly or through analysis of genetic databases, could become a powerful tool for estimating the potential virulence of a microorganism. This is particularly true for two important aspects of virulence: potency and persistence in the environment. The committee conceives of VFAR
as being the relationship that ties specific descriptors to outcomes of concern (see Figure 6–1).
FORMULATING VFARS
Conceptually, pathogens of interest must be related in that they exhibit pathogenicity through a common mechanism but are also likely to be distinguished through secondary characteristics that cause virulence to be variable. Since virulence is the target property to be predicted by the VFAR, it is by definition the dependent or “response” variable in a VFAR. Variability in the virulence of pathogens may be characterized by one or more independent variables (i.e., variability in the genetic makeup) —referred to as “descriptor variables” —that can be conven-
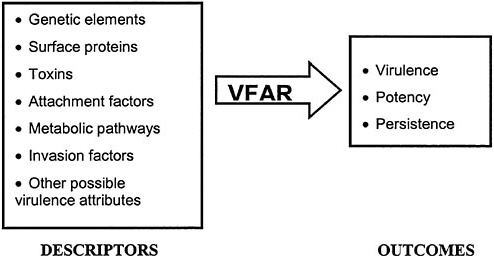
FIGURE 6–1 Schematic drawing of VFAR predicting outcomes of concern (virulence, potency, persistence) using the presence or quality of “descriptor” variables.
iently measured or otherwise determined. When correlation of the response and descriptor variables yields a consistent relationship, the result can be used as a (quantitative) model for comparing and predicting properties of related pathogens.
Note that there are many potential ways in which response and descriptor variables may be defined, and this gives flexibility to the VFAR approach, such that it should in principle be able to accommodate many complicating factors. For example, when variability in a pathogen’s virulence is related substantially to host factors (e.g., when the host is in an immuno-compromised state) then an “interaction effect” could generate cases that do not obey a VFAR. However, if the response variable (virulence) is defined in such a way that it is unaffected by the behavior of opportunistic pathogens, or if descriptor variables are used that incorporate opportunistic behavior, then a VFAR can incorporate this effect and outliers can be avoided. Such subtleties suggest that developing and validating robust and reliable VFARs will require considerable research, but the committee believes that the promise of VFARs should make them a high priority for such research.
Response (Outcome) Variables
As noted previously, the response variable of concern in VFARs is virulence. Narrowly defined, pathogenicity can be characterized as the ability to cause disease and clinical virulence as a measure of the severity of disease. A broader definition is used in this report, where virulence (with respect to VFARs) incorporates both the concept of pathogenicity and the narrower concept of clinical virulence. Viewed in this manner, it may be useful to include attributes of persistence in the environment as contributing to virulence. It is also conceivable that pathogenicity, clinical virulence, and environmental persistence could be considered separate response variables that work together to contribute to the broadly defined “virulence” of a pathogenic organism.
There are a number of potential metrics of virulence (broadly defined) that may be used as a quantitative outcome measure. These include the duration of symptomatic illness and the intensity of symptoms (perhaps using a disability-weighted scale).
Descriptor Variables
Descriptor variables, in this context, are those attributes of a microorganism that may prove useful in predicting their virulence. For example, the presence of toxins, adherence factors, adhesins, invasins, capsular components, fimbria, hemolysins, metabolic pathways, and antibiotic resistance could prove to be effective descriptors of microbial virulence. Alternatively, association with certain families of pathogenic microorganisms may be sufficient as a descriptor (e.g., for viruses), and species and genotype may be all that is necessary for protozoa. As our knowledge of pathogens improves, the definition and calibration of specific descriptors will evolve as well.
For many pathogens, the specific mechanisms or virulence descriptors that underlie the range of virulence from one genotype to the next are not well understood. Because of this circumstance, it has already been demonstrated in waterborne pathogens that a genetically based VFAR approach could be particularly powerful. For example, recent studies suggest that various isolates or species of Cryptosporidium are virulent to varying degrees in humans (e.g., Okhuysen et al., 1999; Widmer et al., 1998). The ability to recognize and differentiate the genomic content of these different isolates or species, and thus recognize differences in virulence, is based upon the same intellectual concepts that underlie the recognition of toxin-encoding bacterial genes. The power of a VFAR approach is that it has the ability to genuinely reflect the true biological diversity found in human pathogens, even when the exact mechanisms that shape this diversity are not yet understood (Morgan et al., 1999a; Sulaiman et al., 2000).
The committee anticipates that the VFAR paradigm is robust enough to accommodate the reality that sometimes the mere presence of a protozoan in drinking water is not of public health concern. For example, there is now abundant evidence that the species Cryptosporidium parvum is, in fact, made up of a number of genotypes, each with different virulence where the human population is concerned (Xiao et al., 2000). Furthermore, one study (Morgan et al., 1999a) used genetic methods to identify eight different species of Crytposporidium: parvum (many mammals), muris (rodents, cattle), felis (cats), wrairi (guinea pigs), meleagridis and baileyi (birds), serpentis (reptiles), and nasorum (fish). The same study demonstrated seven genotypes of parvum: genotype 1 infects humans only; genotype 2 infects cattle, sheep, goats, and humans; genotype 3 infects mice and bats; genotype 4 infects pigs; genotype 5 infects koalas and kangaroos; genotype 6 infects dogs; and genotype 7
infects ferrets. Subsequent studies indicate the existence of an eighth genotype (Sulaiman et al., 2000). Of these, only genotypes 1 and 2 are believed to infect immuno-competent humans, but several genotypes have been found to infect immuno-compromised individuals (Morgan et al., 1999b). In addition, several non-parvum species (C. felis, C. meleagridis) have been found to infect people with AIDS (Morgan et al., 2000).
Genomics and Proteomics
Proteomics, a discipline within functional genomics, is the study of protein sets made (expressed) when the genomic blueprint of an organism is actually translated into functional molecules. When faced with changing environmental conditions, organisms will respond by making different sets of proteins to help them survive. For example, it has been estimated that Vibrio cholerae is capable of making approximately 3,900 different proteins depending on environmental conditions (Heidelberg et al., 2000). These proteins are the actual molecules that build other important structural molecules, such as lipids, deoxyribonucleic acid (DNA), and ribonucleic acid (RNA), and are capable of having both structural and catalytic or enzymatic functions. It is known that some important bacterial toxins (such as Shiga toxin, discussed later in this chapter) are maximally produced under very specific conditions (Acheson et al., 1991). Faced with a hostile environment, many bacteria will shift production of a protein set that is associated with growth to another set associated with a viable but nonculturable state or to the formation of spores as discussed later in this chapter. Thus, knowledge of the set of proteins being made by an organism can impart information far more revealing that that gained from studying the expression of a single protein.
The committee anticipates that because the state of the art of genomics is currently more advanced than that of proteomics, the initial emphasis in VFAR formulation will be genetic. While much is already known about the growing field proteomics, the committee believes it would be premature to discuss or make recommendations about how much research and data will be needed to examine this aspect of developing VFARs, particularly under changing environmental conditions. Nonetheless, the logical extension of identifying and understanding the entire genome of an organism is ascertaining how this is translated into the expression of proteins and other structural building blocks. In this
regard, the committee anticipates that the same rationale that exists for using genomics also exists for proteomics. For example, in a subsequent section of this chapter the committee discusses the use of DNA chips that act as sensors for finding the characteristic DNA elements that encode a particular virulence factor. These chips function via a binding interaction between a section of DNA spotted onto the chip and the complementary strand of a target DNA molecule, such as one from a pathogenic organism. Protein chips that bind the actual virulence protein factors could work in an analogous fashion. Under such a scenario, binding molecules known to attach to specific bacterial toxins (e.g., monoclonal antibodies) could be spotted onto a chip and used to sift through the proteins expressed by a novel bacterium to see if a protein of concern is made by the targeted organism.
CURRENT LEVEL OF GENETIC CHARACTERIZATION
In this section, three existing, major bodies of endeavor that have relevance to the development and implementation of VFARs are discussed.
Microbial Genome Projects and Comparative Databases
The first major endeavor to be discussed in this section is the set of single-organism genome projects and the large genomic databases that are used for comparing the genes of one organism with those of others. The genome projects are comprehensive attempts to sequence the entire genomes of organisms, such as yeast, pathogenic microorganisms, and humans. Computerized analysis and the growing use of automated polymerase chain reaction (PCR) techniques have allowed for tremendous gains in the study of microbial genomics as well as of whole organisms. The databases that exist to store such information are large and expanding daily. For example, the Institute for Genomic Research (TIGR) maintains a collection of databases containing DNA, protein and gene expression, and taxonomic data for microbes, plants, and even humans (see http://www.tigr.org for further information). TIGR also provides links to worldwide genome sequencing projects.
A number of microorganisms are listed in Table 6–1 whose genomes have already been studied; the results of much of this work are available in the published literature. A number of these organisms are associated
TABLE 6–1 Examples of Microbial Genome Databases for Waterborne Pathogens
|
Microorganism |
Size (Million Base Pairs) |
|
Campylobacter jejuni |
1.7 |
|
Encephalitozoon cuniculi |
2.9 |
|
Enterococcus faecalis |
3.0 |
|
Escherichia coli |
4.6 |
|
Giardia lamblia |
12 |
|
Helicobacter pylori |
1.66 |
|
Klebsiella pneumoniae |
|
|
Legionella pneumophila |
4.0 |
|
Leptospira interrogans serovar icterohaemorrhagiae |
4.8 |
|
Mycobacterium avium |
4.7 |
|
Pseudomonas aeruginosa |
5.9 |
|
Salmonella paratyphi A |
4.5–4.8 |
|
Salmonella typhi |
|
|
Salmonella typhimurium |
|
|
Shigella flexneri |
4.7 |
|
Vibrio cholerae |
4.0 |
|
SOURCE: TIGR (see http://www.tigr.org). |
|
with waterborne disease. For example, studies on the Giardia genome have recently been published (Adam, 2000), and the complete sequence of Vibrio cholerae was recently announced with great acclaim (Heidelberg et al., 2000). The Cryptosporidium genome is being sequenced by investigators at the University of Minnesota (http://www.cbc.umn.edu/ResearchProjects/AGAC/Cp/index.htm), with other important work being conducted in the United Kingdom (http://www.mrclmb.cam.ac.uk/happy/CRYPTO/Ref.html), California (http://medsfgh.ucsf.edu/id/CpTags/), and elsewhere. Notably, funding for the Vibrio cholerae and Cryptosporidium genome projects was provided by the National Institute of Allergy and Infectious Diseases (NIAID) at the National Institutes of Health (see http://www.niaid.nih.gov/dmid/genomes/genome.htm for a listing of genome projects currently supported by NIAID).
On May 30,2000, an important report entitled Interagency Report on the Federal Investment in Microbial Genomics was published by the Biotechnology Research Working Group—a subcommittee of the National Science and Technology Council (BRWG, 2000). The charge for
the Subcommittee on Biotechnology was to summarize the activities in microbial genetics of a number of federal agencies; identify each federal agency’s areas of interests; and identify opportunities for, and limitations to, research in microbial genetics.1 One of the main findings of this report is that the current effort in microbial genomics in each federal agency is based on the mission of the specific agency, and, thus, there are clear research gaps—or opportunities for cooperation—in the area of microbial genomics.
Notably, despite EPA’s clear mandate for the surveillance of water supplies and developing a rational and transparent scheme for regulating pathogenic water contaminants to date, it has not yet participated in these interagency genome project efforts. Indeed, the research gaps and opportunities identified in this important report (BRWG, 2000) did not even mention the field of waterborne pathogens, possibly because the participating federal agencies do not address this particular area of public health and EPA has not yet participated in this forum. However, the report did identify the problem of pathogens that are difficult to culture—the importance of which is discussed elsewhere in this chapter—high-lighting the potential for synergism between federal agencies. In this regard, the committee strongly recommends the future participation of EPA in such cooperative interagency programs.
The U.S. Department of Energy (DOE) has been involved extensively in building genetic databases on microorganisms. Although much of DOE’s focus has been on bioremediation and not on waterborne pathogens per se, the methodologies for building, using, and interpreting genetic databases are applicable (DOE, 2000). More specifically, much of DOE’s research in this field has recently turned to address functional genomics. As noted previously, breakthroughs in genome sequencing, along with characterization of proteins through use of supercomputers, make this possible.
The uses of large genetic databases are many, but most important to this report is their role in identifying similar genes—and thus virulence
factors—in different organisms. As discussed later in this chapter, many well-recognized waterborne pathogens share the same or very similar genes that encode virulence factors. However, it is not possible to predict all of the insights that may result from the use of these databases. To illustrate this point, when the genome of Vibrio cholerae was compared to other organisms in the database (Heidelberg et al., 2000), the largest number of similarities was found to Escherichia coli (the common enteric pathogen), but the second largest number was unexpectedly to Haemophilus influenzae (a respiratory pathogen). Although the latter is not known to be transmitted via water, this finding aptly demonstrates the surprising ability of pathogens to share common components that may or may not relate to overall virulence.
Molecular Methods for Characterization of Waterborne Microorganisms
The second major arena of endeavor to be discussed is the use of molecular methods, particularly PCR, for the detection and characterization of waterborne pathogens (NRC, 1999a; Wiedenmann et al., 1998). Perhaps because the recognized and known bacterial pathogens are culturable, the major recent focus has been on microorganisms that are not yet culturable (e.g., protozoan agents, certain viruses). In this regard, PCR is an attractive diagnostic procedure because it is rapid, sensitive, and pathogen specific (DiGiovanni et al., 1999; Johnson et al., 1995; Kostrzynska et al., 1999; Rochelle et al., 1997b). Independent of this report, Rochelle et al. (1997b) have recommended that PCR should become a more widely accepted method of pathogen detection and monitoring within the water industry and should be used in parallel with conventional techniques (e.g., cultivation) to improve detection capabilities for existing and newly emerging pathogens.
Although it is beyond the scope of this report to discuss genetics in a substantive way, a very concise overview of molecular genetics as related to virulence factors is appropriate. All living organisms carry with them a complete genetic code, the genome, which serves as a master blueprint for all cellular structures and activities for the lifetime of the cell or organism. An organism’s genome consists of DNA and associated protein molecules, except for RNA viruses, which use RNA rather than DNA. Each DNA molecule contains many genes—the basic functional units of heredity and what makes each organism individually distinct. Genes in turn are comprised of specific sequences of four nucleo-
tide bases (adenine, thymidine, cytosine, and guanine represented as A, T, C, and G, respectively). Each strand of DNA is bound to a complementary strand of DNA. Every individual base in one DNA strand is matched in the complementary strand by the base that binds to it to form a base pair (bp). These pairs of DNA bases are formed from As and Ts or from Gs and Cs (see Figure 6–2). Copies of individual genes are made by an organism by separating the two strands, and then making a new complementary strand of DNA using one strand as a template. By convention, an organism’s genome size is expressed as the total number of base pairs (see Table 6–1). Some organisms use slightly different nucleotides, but the principle is the same. Genes specify the exact genetic instructions required to create individual organisms with unique traits (e.g., eye color in humans, the toxins of Escherichia coli O157:H7). They also provide the information needed to produce an incredible variety of proteins through an indirect process that utilizes a transient intermediary molecule called messenger RNA (mRNA). When the DNA code is to be translated, the two intertwined strands of DNA are separated, and an RNA copy is made of one of the strands. This RNA copy is then sent to the cell “machinery” that translates the code into proteins. Proteins provide the structural components of cells and tissues, and enzymes for essential biochemical reactions, but they can also act as toxins as illustrated below.
PCR techniques are based on the principle that genetic elements such as DNA or RNA, which are present at very low concentrations in water or other materials, can be copied many times by specific enzymes (polymerases) and subsequently detected via fairly standard biochemical methods. This amplification process is fundamentally the same process used by living creatures to duplicate their genes. A primer, or small stretch of DNA known to match a portion of the target organism’s genome, is added to the concentrated water (or other sample) to be tested. If it finds a match (i.e., if the organism’s DNA is present), the polymerase chain reaction can take place, and the products can be detected after amplification. It is typical for PCR to amplify the targeted DNA by a factor of 10,000 to a millionfold. The products of the copy (amplification) process are called amplicons.
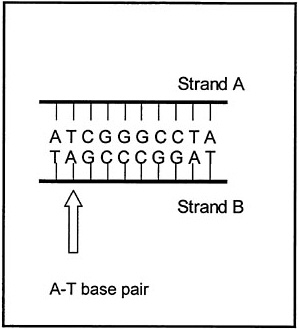
FIGURE 6–2 Illustration of two complementary strands of DNA forming A-T and G-C base pairs.
Protozoa
Several recent papers have described a variety of PCR-based protocols for the detection of waterborne pathogens, including Cryptosporidium (Champliaud et al., 1998; DiGiovanni et al., 1999; Kimbell et al., 1999; Kostrzynnska et al., 1999; Sulaiman et al., 1999; Xiao et al., 1999a,b), microsporidia (Dowd et al., 1998a,b), Cyclospora (Jinneman et al., 1996), and Giardia (Rochelle et al., 1997a,b) in environmental samples. A comprehensive review of the use of PCR techniques for the detection of protozoa has recently been published (Morgan and Thompson, 1998).
The primers of PCR protocols for Cryptosporidium parasites are based on undefined genomic sequences (Laxer et al., 1991; Morgan and Thompson, 1998) or specific genes (Johnson et al., 1995; Leng et al., 1996; Rochelle et al., 1997a,b). Recent molecular studies have shown
that there are extensive genetic differences among different Cryptosporidium species, as well as within C. parvum (Sulaiman et al., 2000). These inter- and intraspecies differences have been used in the development of molecular diagnostics tools for Cryptosporidium spp. and for genotype differentiation. For example, Xiao et al. (1999a,b) evaluated several PCR techniques to determine whether they could accurately detect C. parvum in environmental samples. The authors concluded that two nested-PCR-restriction fragment length polymorphisms (RFLPs) based on the small-subunit ribosomal RNA (rRNA) (Xiao et al., 1999a,b) and dihydrofolate reductase genes (Gibbons et al., 1998) were more sensitive than single-round PCR or PCR-RFLP protocols for detection and Cryptosporidium species differentiation.
Using a slightly different approach, Kozwich et al. (2000) have capitalized on the observation that a double-stranded RNA molecule, which is apparently found only in C. parvum but not in other Cryptosporidium species (Khramtsov et al., 2000); can be used as a “signature” sequence that is specific for this organism. Using a novel solid-phase capture material and PCR, they were able to detect levels of oocysts as low as one per liter in environmental samples. This technology provides a simple reading that looks very much like the commercially available pregnancy test.
For the water industry to make accurate human health risk assessments for C. parvum, it is essential to have methods to detect viable, infectious oocysts in water samples. DiGiovanni et al. (1999) described a new strategy for the detection of infectious C. parvum oocysts in water samples, which combines immunomagnetic separation (IMS) techniques for recovery of oocysts with in vitro cell culturing and PCR. This method, by its very nature, requires that the oocysts be viable if cell culture and subsequent PCR are to detect anything. Assays that use dyes appear to overestimate the viability of oocysts after disinfection with ozone, as judged by mouse infectivity studies (Bukhari et al., 2000).
Not all of the molecular methods of interest are based on PCR techniques. A fluorescent in situ hybridization (FISH) technique has been developed recently that shows considerable promise as an indicator of C. parvum oocyst viability (Vesey et al., 1998). The basic premise is that a viable oocyst, but not a dead one, has genetic elements that can be found with a colored tag that is visible when examined under a microscope. Technically, a fluorescent DNA probe is targeted to the 18S rRNA of C. parvum on the premise that 18S rRNA is usually present in viable organisms and is degraded by cellular ribonucleases (RNases) in dead or dying cells. Another proposed target for this kind of simple visual con-
firmation of viability is β-tubulin mRNA (Widmer et al., 1999). While existing FISH techniques are limited to measuring the viability of C. parvum oocysts and not their infectivity (Neuman et al., 2000), as our understanding of the Cryptosporidium genome becomes more sophisticated, the latter may become possible.
Organisms other than Cryptosporidium have also been investigated with PCR methods. Giardia-specific primers used in published reports amplify a 183-bp product from the small subunit rRNA gene (Weiss et al., 1992); 218- and 171-bp amplicons from a giardin gene (Mahbubani et al., 1992); and a 163-bp product from a heat shock protein gene (Abbaszadegan et al., 1993). Rochelle et al. (1997a) reported a PCR assay based on primers that are specific for G. lamblia in water samples and that amplify a 218-bp product. A PCR-based test to detect Cyclospora has been developed by Jinneman et al. (1996). This method amplifies a region of the Cyclospora 18S ribosomal RNA gene. Specific nucleotide differences in the amplified segment can be used to differentiate amplicons of two closely related genus of coccidian parasites (Cyclospora sp. and Eimeria sp.) by digestion with the restriction endonuclease Mnl I. Dowd et al. (1999b) assessed two methods for isolating microsporidia DNA from water for use in PCR amplification of microsporidia target sequences. Both of the DNA isolation methods when combined with PCR showed the ability to detect less than 10 spores of human pathogenic microsporidia in water.
Capitalizing on these advances, Orlandi and Lampel (2000) of the U.S. Food and Drug Administration have published an extraction-free, filter-based PCR template preparation technique for the detection of Cryptosporidium, Cyclospora, and pathogenic microsporidia. This methodology does not require DNA extraction; is rapid, efficient, and reproducible; and can be used in multiplex PCR applications designed to detect multiple parasitic protozoa.
Viruses
The most frequently identified viruses in drinking water and/or associated with waterborne outbreaks are hepatitis A, Norwalk virus and other caliciviruses, coxsackievirus, rotavirus, and echovirus (Jaykus, 1997). The cultivatable enteroviruses, which include poliovirus, echovirus, and coxsackievirus, make up only a small percentage of the viruses present in wastewater. The standard culture method is to inoculate mammalian cells with these viruses or potentially contaminated water
and watch for damage to the cells (a “cytopathic effect”). Unfortunately, this method has been shown to be insensitive for many viruses, which replicate but do not destroy cells. Thus, PCR is now being utilized in cell culture to detect noncytopathic viruses, and it has been used to screen for viruses in groundwater (Abbaszadegan et al., 1999). In addition, new viruses (e.g., caliciviruses) are constantly being identified that are nonculturable causes of gastroenteritis. These human enteric viruses will have to be investigated in water using PCR methods or their equivalent.
Hepatitis E virus (HEV) is the most common cause of acute hepatitis in humans in many parts of the world, especially Asia, and is also found in wild and domestic animals. Although mortality rates for this infection in humans are generally low (<1 percent) as many as 25 percent of pregnant women who acquire this infection will die (Aggarwal and Krawczynski, 2000). Waterborne epidemics as well as person-to-person spread of this virus have been reported. Recent genetic sequencing studies have found that human and pig HEV are very similar genetically in the United States as well as in Nepal (Meng et al., 1997, 1998; Tsarev et al., 1999). Thus, zoonotic transmission seems very possible (Smith, 2001). Although the cultivation of HEV has recently been reported (Smith, 2001), reverse transcriptase PCR is the method of choice in water and has been used successfully to detect HEV RNA in sewage (Jothikumar et al., 1993; Pina et al., 1998).
The Norwalk-like caliciviruses (NLVs) are noncultivatable enteric viruses (also known as small round structured viruses) that have been reported to cause numerous waterborne outbreaks (Jaykus, 1997; Kaplan et al., 1982; Schaub and Oshiro, 2000). A diverse group of RNA viruses, they are a common cause of gastroenteritis, with diarrhea and or vomiting lasting approximately two days. These viruses may be quite prevalent in the environment and have been reported in sewage at 107 RNA-containing particles per liter using PCR techniques (Lodder et al., 1999).
Bacteria
Because of their relative ease in manipulation in the laboratory, rapid growth, and sophistication of our understanding of bacterial genomes, most of the published literature dealing with the molecular characterization of pathogens is focused on cultivable bacteria. Indeed, the insights developed in this particular field have led to the whole-organism genomic sequencing projects discussed earlier in this chapter.
However, it is now apparent that even bacteria of very major public health concern are not always culturable. This important issue is also discussed at length later in this chapter under virulence factor “persistence.” One of the best paradigms in this regard is the agent of cholera, Vibrio cholerae. This organism’s favored natural environment is estuarine areas. It can enter a viable but not culturable dormant state wherein it has greatly reduced needs for nutrients and oxygen (see reviews by Colwell and Huq, 1994; Sanchez and Taylor, 1997). The ingestion of these nonculturable forms by humans is believed to allow for reversion to the highly pathogenic form, leading to cholera disease and potential outbreaks. These forms can be detected via PCR methods, as well as epifluorescent microscopy and acridine orange staining. Of note, it has been shown directly that PCR methods can detect the presence of cholera toxin genes even when the organisms are in a viable but nonculturable state (Colwell and Huq, 1994). It is not generally appreciated that a number of other highly pathogenic microorganisms such as Campylobacter jejuni, Salmonella enteritidis, Escherichia coli, Helicobacter, and Legionella have the same ability to enter into nonculturable form. These examples help support the conclusion of the Biotechnology Research Working Group—a subcommittee of the National Science and Technology Council (BRWG, 2000) —and this committee, that methods other than culture must be used to fully evaluate microbial contamination of drinking water.
A recent example of the successful application of PCR methods and the use of genetics for waterborne pathogens was reported by Kingombe et al. (1999) for the bacteria Aeromonas. Aeromonas hydrophila is included on the 1998 CCL (EPA, 1998a) and as previously noted is the only CCL microorganism on the UCMR (EPA, 1999f,g) that will be monitored in the first cycle of the UCMR. Yet the methodology proposed to evaluate and identify this pathogen in water is based on a cultivation technique (Havelaar et al., 1987) and will yield little information on the virulence or the pathogenic nature of the isolates (EPA, 1999f). Aeromonas spp. are ubiquitous in nature and only recently have the bacteria been associated with human illness. A large number of virulence factors for this microorganism have been described in the literature. The similarity between the cytolytic enterotoxin gene of A. hydrophila and the aerolysin genes of Aeromonas spp. was recently used to characterize the distribution of virulence in water (Kingombe et al., 1999), with 58 percent of the isolates associated with potential health threats. The par-
ticularly clever aspect of this work by Kingombe et al. (1999) is their use of molecular techniques to detect virulence factors in a variety of Aeromonas species that are of concern to humans.
There are more than 100 serotypes of Escherichia coli that are capable of producing Shiga toxins (STXs) (Nataro and Kaper, 1998). The genes encoding for STXs are located on bacteriophages and thus, may be spread from one serotype to another. Law (2000) reports that the pathogenicity of any given E. coli is likely related to a number of virulence factors in addition to the STX gene. This finding supports the premise that it may be possible to identify the potential for emerging types of pathogenic bacteria by understanding the array of virulence genes that are present in environmental waters.
Gene Microarrays and Genomics
Gene chip technology (microarrays), has been used in research laboratories since the late 1990s to study gene expression and is the subject of recent and intense scientific interest. DNA microarrays are immobilized pieces of single-stranded manufactured DNA, typically spotted onto glass or nylon substrates, that are used to capture key genetic targets.
The premise is that the DNA is spotted using an arrayer device that places it on the slide in a known position. The arrayer device can be an expensive robotic device or a simple ink-jet spotter (e.g., see “How to Build Your Own Microarrayer” at http://cmgm.stanford.edu/pbrown/mguide). The DNA spotted onto the chip is designed to bind a sequence of DNA that may or may not be in the sample to be tested. Currently, most studies using this technology are examining the entire spectrum of genes of an organism (its genome) to see how the entire set of genes varies under different metabolic conditions or when stressed by a malignancy or other circumstance (e.g., Aach et al., 2000; DeRisi and Iyer, 1999). However, that need not be the only way in which this technology is used.
For example, a chip could catch several thousand bits of DNA from pathogenic microorganisms that are, or may be, waterborne. The chip would be incubated in a water sample that had been treated to liberate the DNA from any microorganisms. If the pathogenic microorganism of concern was present in the sample, its DNA would bind to the bit of DNA that matched its own on the chip (i.e., a complementary strand). This bound DNA could then be amplified using PCR and subsequently
labeled (e.g., with fluorescent molecules) and detected using a variety of means such as spectroscopy. Alternatively, DNA spotted onto the slide could act as a “molecular beacon,” or a piece of DNA that becomes intensely fluorescent once the target DNA binds to it (Tan et al., 2000). This once conceptual method has already been used clinically to identify specific bacteria found in the bloodstream of hospital patients (Anthony et al., 2000).
By 1998, microarrays were manufactured to assay 500 to 5,000 genes (Marshall and Hodgson, 1998; Ramsay, 1998), and chips that can assay up to 100,000 genes are predicted to be available in the near future (Lander, 1999). Although the experimental power of such arrays is considerable, current costs for complete systems range from $25,000 to $135,000. Commercially available systems are currently being produced for what is known as “gene discovery” (i.e., rapid identification of targets in infectious disease).
The committee anticipates that in a very short period of time, microarrays could be developed that are labeled with all of the genes for a variety of virulence factors identified within enteric bacteria, pathogenic viruses, opportunistic protozoa, and other (waterborne) microorganisms. Examples of some of these are provided in the next section of this chapter. These gene chips could be used to assay environmental and drinking water samples for the presence of genetic virulence factors of concern. If such virulence factors were present, the sample could be assayed further if needed to better identify the microorganism. Thus, the commercial and public health implications are enormous.
VIRULENCE AND POTENCY RESPONSE—HEALTH ASPECTS
Introduction
Historically, it has been well recognized that the virulence of a microbial pathogen can be correlated with some observable feature. In this section, several examples of the current state of knowledge concerning the molecular basis for variation in virulence, and what is known about some of the underlying mechanisms of virulence itself, are discussed. This section is intended to give the reader an overview of some of the microbial virulence factors that have already been identified and could act as virulence factors in the future development of a VFAR.
Viral Examples
Influenza—Mutation is Associated with Virulence
Influenza can be considered as a prototypic emerging disease. Although it is not known to be waterborne, it is discussed here to support the contention that specific virulence attributes can be mapped to an important and well-understood pathogen that is widely recognized by the public. Influenza virus is zoonotic (i.e., it infects and is maintained in other animals, chiefly birds and swine) and is capable of mutating so frequently that immunity to one strain will not be protective against another strain. It can also result in an innocuous infection or a lethal one. In 1918, influenza was responsible for a worldwide pandemic that killed more than 20 million people (Webster et al., 1992). More recently, a well-publicized outbreak in Hong Kong in 1997 resulted in the deaths of 6 of 18 people who were confirmed to have been infected with a new serotype of influenza (H5N1) (Snacken et al., 1999). For influenza, it became clear that the outer surface proteins of the virus are closely linked to the degree of disease that humans suffer.
Serotypes of influenza are classified by the neuraminidase (N) and hemagglutinin (H) protein molecules on the surface of the virus. These are encoded by the RNA-based genome of the virus. Several isolates of influenza can infect the same animal at the same time, leading to gene reassortment events in which the progeny virus are a mixture of the two parental strains. Each of the major families of the neuraminidase and the hemagglutinin molecules on the surface of the virus has a specific subtype name, such as N1, N2, H1, H2, and so forth. Every year, a decision is made by U.S. public health authorities regarding which subtypes to concentrate on for vaccine production based upon infection information from elsewhere in the world. Once a decision is made to manufacture a specific vaccine using specific serotypes, the vaccine must be grown, inactivated, and packaged for delivery. In the case of the Hong Kong outbreak, the new isolate was so virulent that it killed the chickens and chicken eggs that are typically used to grow influenza virus for vaccine production (Snacken et al., 1999). Thus, the Hong Kong isolate was so virulent that to protect the human population adequately would have been extremely difficult. Fortunately, its high virulence was apparently not matched by high transmissibility, and it did not spread beyond Hong Kong.
Influenza thus represents a virus whose surface molecules correlate with virulence, but researchers do not yet know why this is the case. Nonetheless, genetic recognition of the molecules associated with virulence can serve as an important public health tool.
Hepatitis E—An Emerging Waterborne Infection in the United States
Hepatitis E virus is the third leading cause of hepatitis, after hepatitis A and B. It occurs worldwide, but rarely in developed countries such as the United States (Labrique et al., 1999). As discussed earlier in this chapter, HEV is predominantly a waterborne disease since direct transmission from person-to-person is uncommon (~2 percent of cases; Labrique et al., 1999). Disturbingly, it now appears that transmission is occurring in the United States. Tsang and colleagues (2000) reported a case of HEV in California. A man, who had not traveled outside the United States for more than 10 years, drank water from a well and a lake one month before becoming ill. In El Paso, Texas, 0.4 percent of pregnant women (the group most at risk of death during acute infection) were reported to have had serological evidence of prior exposure to this agent (Redlinger et al., 1998). In Iowa, 4.9 percent of patients with non-A, non-B hepatitis were seropositive for HEV (Karetnyi et al., 1999). In other developed countries such as the United Kingdom, it has also become apparent that HEV is a cause of acute hepatitis and is underdiagnosed (McCrudden et al., 2000). Since HEV is principally a waterborne disease elsewhere in the world, it is appropriate to be concerned about waterborne transmission in the United States.
Two people in the United States have been diagnosed as having HEV with strains that are genetically different from the other HEVs that occur in less developed countries (Kwo et al., 1997; Meng et al., 1997, 1998b). However, these strains were nearly identical genetically to the recently discovered swine HEV, which is found throughout the United States in swine populations. Other mammals can also be infected with HEV in the United States. In a recent nationwide survey by the Centers for Diseases Control and Prevention, 806 rodents (26 species in 15 genera) were tested and 60 percent of rats were seropositive for HEV (Favorov et al., 2000). Tying these data to information about transmission of HEV in the United States is the fact that actual clinical differences have been reported between HEV strains within a genotype (Labrique et al., 1999; Ticehurst, 1999; Tsarev et al., 1999).
Thus, it is probable that modern molecular surveillance of U.S. drinking water could detect HEV of animal and potentially human origin. Furthermore, analysis of strains and genotypes in the detected HEV would probably lead to further information regarding the potential for this pathogen to infect humans who ingest such water. Moreover, tracking different genotypes could assist in establishing dispersion patterns of HEV into the aquatic environment.
Poliovirus and Other Enteroviruses
Enteroviruses account for 10 million to 15 million infections per year in the United States (Sawyer, 1999) and are easily detected in surface waters (Melnick and Gerba, 1980). They are spread by waterborne transmission, direct person-to-person contact, and common-source exposures such as swimming pools. Paralytic poliomyelitis is often the dreaded consequence of infection by a common enterovirus, poliovirus. This enteric virus has historically been transmitted both in waterborne epidemics and through person-to-person contact. The majority of children infected with polio have a simple undifferentiated febrile illness or no symptoms, while others will develop aseptic meningitis or paralysis. Despite virtual eradication of this endemic disease in the western hemisphere through universal vaccination, it unfortunately remains common in Africa and parts of Asia. Since 1980, all cases of poliomyelitis in the United States have actually been caused by vaccine strains of polio, which is exceedingly rare (Strebel et al., 1992). Any region with wild-type poliovirus can act as a reservoir for reintroducing virulent poliovirus to areas that no longer have endemic transmission problems, and this has happened frequently over the past three decades (Kubli et al., 1987). Thus, continued surveillance as well as vaccination for this virus is clearly warranted throughout the United States.
Three serotypes of poliovirus exist (Bodian et al., 1949). They share some antigens but are characterized by marked intertypic differences (Melnick, 1996). Protection against this virus in conferred when a person has antibodies to the three main structural proteins VP1, VP2, and VP3 that make up the viral surface of all three serotypes. A fourth protein, VP4, is internal. Vaccination with one serotype does not, however, confer adequate protection against the other serotypes of polio (Melnick, 1955). Most of the epitopes, or sites that are recognized by antibodies, lie on VP1. These VP proteins vary markedly between serotypes and even within a given serotype. The tendency for poliovirus to infect the
nervous system (and not just the intestine) is called neurotropism. Although all three serotypes of poliovirus can infect the nervous system, some isolates are more or less neurotropic. Indeed, the original Sabin (live, oral) vaccine was developed and used because it lacked neurotropism (Sabin, 1985). Specific mutations that are associated with the lack of neurotropism have been identified (Mento et al., 1993; Ren et al., 1991).
The discovery of enteroviruses was closely linked to the poliovirus control effort. Animal and tissue culture virus isolation studies, looking for poliovirus isolates, often revealed other viruses of the poliovirus family, or enteroviruses (Pallansch and Anderson, 1998). These include other groupings, such as the coxsackievirus A, coxsackievirus B, and echovirus groups. Since it became clear that all of these viral agents actually belonged to one family, all new isolates have simply been classified as enteroviruses and numbered sequentially. These agents cause diseases marked by rash (hand-foot-and-mouth disease, herpangina); cardiac disease (myocarditis and pericarditis, which can be fatal); respiratory tract infections; central nervous system disease including paralysis identical to that of poliovirus; and a variety of other diseases. Epidemiologic and observational studies have suggested that the development of insulin-requiring diabetes mellitus is related to infection with enteroviruses (e.g., Roivainen et al., 1998). The entire genome of any given enterovirus, including polioviruses, is encoded by a single strand of RNA that is about 7,500 nucleotides long, and the entire genome of many enteroviruses is already known. Moreover, the structure and physical properties of enteroviruses are nearly identical to those of polioviruses (Pallansch and Anderson, 1998). Thus, the very major variances in virulence and tissue tropism of all of the enteroviruses are encoded in this one relatively short molecule.
At this time, the mere detection in the United States of a wild-type poliovirus in a drinking water supply would be of very major public health importance because endemic transmission is believed to have ended in this hemisphere. Similarly, the monitoring of public drinking waters for the presence of enteroviruses is likely to lead to important information regarding the causal role of these viruses in a number of chronic diseases such as diabetes mellitus. While traditional cell culture techniques required the (prohibitively expensive) use of at least four cell types to screen for all enteroviruses (Dagan and Menegus, 1986), the committee notes that powerful PCR methods have recently been developed to detect enteroviruses in surface waters collected under the Information Collection Rule (Chapron et al., 2000). This methodology has
been developed at the same time that similar PCR techniques have been evaluated and found to be cost-effective for the rapid diagnosis of enterovirus infections in humans (Nigrovic and Chiang, 2000). Thus, the use of VFAR molecular methods for monitoring enterovirus infections would (1) take advantage of the immense knowledge base about virulence characteristics that exists at the viral level and (2) tie into new clinical technologies that could be used to confirm actual human disease characteristics with specific virulence attributes.
Bacterial Examples
Although the previous discussion may have suggested that the knowledge base for viral pathogens is extensive, the most robust knowledge about pathogens actually lies in the realm of bacterial pathogens. This set of examples, like the viral ones, begins with a pathogen that is generally recognized by all members of the public—streptococcal infection.
Streptococcal Infections
Group A streptococci (Streptococcus pyogenes) are the bacteria that cause “strep throat,” scarlet fever, and rheumatic fever, as well as the emerging “flesh-eating” syndromes with rapidly spreading infection and loss of limb and life (necrotizing fasciitis). They have very well characterized exterior proteins that are recognized as distinct serotypes (Lancefield, 1928).2 These serotypes are determined by exterior cell wall M, T, and R antigens, which are glycoproteins (proteins with sugars attached) encoded by the bacterial genome (Stollerman, 1998). The M protein, a major surface antigen, is the predominant virulence attribute that predicts pathogenicity. It acts to prevent ingestion of the bacteria by human phagocytic cells.
Acute glomerulonephritis, or renal failure, frequently follows a group A streptococcal infection, and a very limited number of M and T serotypes are associated with this form of kidney disease. Similarly, only specific serotypes commonly cause rheumatic fever. Thus, there is an excellent correlation between the presence of specific markers of
virulence—the M and T antigens—and actual adverse human outcomes. Rheumatic heart disease was once one of the leading causes of death in the United States, and it remains a major cause of morbidity and death in many developing countries (Stollerman, 1998).
Streptococci also produce a very large number of toxins. Some of these cause the bright red rash of scarlet fever, whereas others destroy and literally dissolve human tissues. One set of these toxins causes toxic shock syndrome and belongs to a family of toxins that is shared with an otherwise unrelated bacterium Staphylococcus aureus. These pyrogenic toxins are relatively small (molecular weight 20,000 to 30,000) and are similar on a genetic basis (Bohach et al., 1989). They also provide an excellent example of how a virulence attribute in one organism predicts virulence in an organism of a different genus and species. Thus, there are well-known virulence attributes that predict pathogenicity within species (M proteins) and across species (toxic shock toxins).
Enteric Bacteria
Most of the bacteria that cause diarrhea or dysentery (bloody diarrhea) do so after a set of interactions with the human host. These interactions include (1) ingestion by the host; (2) survival after passage through the acidic environment of the stomach; (3) attachment to the lining of the host’s intestine (epithelium); and (4) production of compounds that induce the host to secrete fluid that leads to diarrhea and/or to envelop the invading bacteria so it can enter the host cell. Bacteria are far more complex than typical viruses and have the capacity to devote energy and part of their genome to accomplish each of the above tasks successfully. Moreover, bacteria have the ability to share the genes for these tasks with each other.
This sharing of toxin genes and other virulence factors means that bacteria detected within the United States may have their native representatives that are just as harmful. For example, Shigella bacteria (a cause of dysentery) are now rarely of major concern in the United States because hygienic standards are generally high. Shigella dysenteriae produces a highly lethal toxin called Shiga toxin (Acheson, 1998; Acheson et al., 1991; Conradi, 1903; Keusch, 1998; Keusch et al., 1972). Other Shigella species (e.g., S. sonnei, flexneri, boydii) are unlikely to make this toxin in similar quantities (Keusch, 1998). STX is composed of one A unit and five B subunits. The five B subunits first attach to a host cell, the toxin penetrates; and then the A subunit kills the cell. Although
originally described from the bacterium Shigella dysenteriae type 1, an essentially identical set of toxins is made by Escherichia coli O157:H7, which is why this and other E. coli strains that make the toxin are of such public health concern.
Escherichia coli O157:H7 is frequently carried in cattle, has been implicated in many raw beef or hamburger-related epidemics, and has been implicated as the causative lethal bacterium involved in a number of waterborne disease outbreaks (Anonymous, 1999; Swerdlow et al., 1992). Haas et al. (2000) estimated that the median infectious dose is 5.96×105 organisms in drinking water and that ingestion of only 4,000 organisms would result in a 1 percent attack rate. Nevertheless, average ingested doses below this level can still pose a significant public health threat, particularly if exposure of large population occurs. For example, Tuttle et al. (1999) found that the median “most probable” number of organisms in contaminated hamburger patties involved in a recent widespread outbreak in Canada was only 67.5.
A very closely related toxin called Shiga toxin 2 (STX2) is also produced by E. coli and has three major known biological variants (WHO, 1998). The majority of STX-type genes are actually present on a bacteriophage that infects the E. coli bacteria, fostering transfer between bacteria. It is these toxins that are thought to cause the bloody diarrhea and frequent death of people who have shigellosis or who are infected with E. coli strains that produce these toxins. The mere presence of these genes and the ability to make this toxin may not, however, always be sufficient to cause disease. The bacteria often attach to the human host intestine after having entered the digestive tract, and this requires a set of genes that facilitate this type of interactions.
The attachment of these bacteria to the intestinal wall is through a characteristic attaching and effacing (A/E) cytopathic lesion. The ability to attach and efface is encoded by 41 genes in a “pathogenicity island” or “locus for enterocyte effacement” (LEE). This island or locus encodes the adhesin called intimin on the eaeA gene, as well as other factors that are needed for secretion of these molecules into the space between the bacterium and the host cell. This group of genes can be shared among all of the bacteria that adhere to the intestine via this mechanism (Acheson, 1998; Goosney et al., 1998; WHO, 1998).
Some enteric bacteria invade the host cell and evade the immune response, essentially “hiding” within the host. This is a very dangerous property of such bacteria, which is very strongly correlated with virulence. These bacteria include Shigella, Salmonella, Yersinia, Listeria, and others. Some of these enteric bacteria literally blow a hole in the
host cell as they move into an adjacent cell and accomplish this by causing host proteins to jell behind them and force them forward like a jet-propelled object. These genes are shared between bacteria that are as unrelated as Shigella (Goosney et al., 1998) and Listeria (Chakraborty et al., 1995). The genes that encode for such destructive characteristics are known and are highly associated with virulence. They also represent an example of how a VFAR relationship, which ties specific genes that encode for virulence factors, can correspond to pathogenic effects in humans.
Summary
It is beyond the scope of this chapter to comprehensively review the very extensive knowledge base that already exists regarding microbial genes that are associated with virulence. However, several examples have been discussed and serve to help make the following points:
-
Because of their public health risks, for more than a century scientific interest has existed in pathogens and their associated virulence factors, and the scientific foundation for these measures of virulence is appreciable.
-
Virulence is often clearly and easily linked to known genetic elements.
-
The genetic elements associated with virulence vary somewhat among isolates of specific pathogens, and this variation has been linked directly to the genetic variation within many species of bacteria or viruses (e.g., polioviruses, influenza, Shigella toxins).
-
These genetic elements are frequently shared among waterborne pathogens having a similar ecological niche that warrants public health concern (e.g., Salmonella, Shigella, Vibrio, Escherichia bacteria).
-
The ease of identification of these genetic elements, their sequencing, and comparison has increased dramatically in the past decade due to advances in molecular biology and bioinformatics.
PERSISTENCE RESPONSES
When pathogens are released into the aquatic environment they may die, multiply, or enter a dormant state. Each of these has implications for
subsequent human exposure and health. The factors responsible for pathogen decay are reviewed below. It is important to note at the outset that the assessment of pathogen survival is dependent on the assay used for analysis. This relationship has been particularly well documented for bacteria (Roszak and Colwell, 1987). Further discussion of this issue is provided later in the chapter. It is one of the committee’s central assertions that the assessment of persistence (survival) in the environment using molecular techniques may be superior to some of the older methods.
Mechanisms Responsible for Decay
The factors that can cause removal of pathogens from the aquatic environment can be grouped broadly into abiotic and biotic factors, based on the mediation of other organisms in the process. In any environment, overall pathogen loss rates reflect the combination of these factors (Auer and Niehaus, 1993; Canale et al., 1993; Chamberlin and Mitchell, 1978).
Abiotic Factors
A number of abiotic factors may be responsible for microbial loss in the aquatic environment. Perhaps the most straightforward is sedimentation. Although environmental pathogens have quite low settling velocities by virtue of their size and density, many organisms can become attached to particles in the water column and increase their effective settling velocity to that of the aggregate. This process has been demonstrated clearly for Cryptosporidium (Medema et al., 1998), and similar phenomena are likely to occur with other microorganisms.3
Although, the physical removal of microorganisms via sedimentation represents a loss process from the water column, it does not result in total elimination of the pathogens from the aquatic environment. In fact, there is strong circumstantial evidence that the transport of pathogens into sediments may create a reservoir for recontamination of the water column. Furthermore, microorganisms in sediments may have lower net inactivation rates than microorganisms in the water column (Burton et
al., 1987; Davies et al., 1995; Laliberte and Grimes, 1982; Matson et al., 1978; Palmer, 1988; Smith et al., 1978).
The effect of visible light on increasing decay rates of pathogens (and indicator organisms) in water is well documented (Chamberlin and Mitchell, 1978; Davies-Colley et al., 1994; Mancini, 1978; Muela et al., 2000). It is known that the penetration of sunlight into natural waters can produce a variety of photochemical oxidants, including hydroxyl radicals and hydrogen peroxide. Thus, the promotion of pathogen decay by light may involve direct, as well as indirect (oxidant-mediated), mechanisms (Arana et al., 1992).
Biotic Factors
Interactions between pathogens and other organisms in water may result in loss of viability of the pathogens. Among the more commonly documented processes is the predation of bacteria by indigenous protozoan organisms (Davies et al., 1995; Enzinger and Cooper, 1976; Sibille et al., 1998). The obligate parasitic bacteria Bdellovibrio may also serve to diminish pathogenic (and indicator) bacterial levels (Fry and Staples, 1974). Predation of bacteria by bacterioviruses (phage) may also occur in natural waters (Bergh et al., 1989). Although the evidence for this is not well documented, it is known that phage exist for many pathogenic bacteria.
However, a recent review by Wommack and Colwell (2000) focusing primarily on viral interactions with indigenous bacterioplankton (most of which are nonpathogenic), stated that “less than 20% of bacterioplankton and phytoplankton [in natural waters] is attributable to viral infection.” They concluded that mortality loss from infection tends to increase as the density of the host bacteria increases. Thus, it would be anticipated that the persistence of pathogenic bacteria (or other potential hosts, such as algae or protozoa) would be impacted minimally by the potential for viral infection.
Mechanisms Affording Protection to Microorganisms
There are several potential mechanisms that protect microorganisms against adverse environmental conditions. Foremost among these is association with solids that confer protection from die-off (Gerba et al., 1978). The mechanism whereby this protection occurs has not been
well-characterized but may result from protection against biotic antagonists or from transport of inhibitory substances to the solid-associated microbe. Some microorganisms are able to enter dormant stages (e.g., bacterial spores), which affords a measure of protection against adverse environmental conditions. For example, the relatively strong persistence of nonpathogenic Clostridium perfringens in water has been well documented (Cabelli, 1977; Fujioka and Shizumura, 1985).
The formation of cysts in protozoa such as Giardia (Adam, 1991) or oocysts by Cryptosporidium (O’Donoghue, 1995) results in a life stage that is able to survive for extended periods in the environment. Helminthic ova also represent a similar dormant or protective stage, and Ascaris lumbricoides ova have been documented to survive for years and even decades in the soil (Bergstrom and Langeland, 1981; Buts, 1969; Kizeval’ter and Derevitskaia, 1968; Kransnonos, 1978).
In some cases, microorganisms may occupy habitats that offer unique protection against environmental agents. For example, it is has been established that intracellular association with aquatic protozoa may be important in protecting Legionella pneumophila from adverse environmental conditions (Barker et al., 1992; Fields et al., 1984). This association may also assist in the resuscitation of “viable nonculturable” Legionella (see more below) (Steinert et al., 1997). Other bacteria (and perhaps other nonbacterial pathogens) may be harbored protectively in this manner. For example, King et al. (1988) reported that the protozoan Tetrahymena could harbor and protect cells of Escherichia coli, Citrobacter freundii, Enterobacter agglomerans, E. cloacae, Klebsiella pneumoniae, K. oxytoca, Salmonella typhimurium, Yersinia entercolitica, Shigella sonnei, Legionella gormanii, and Campylobacter jejuni for at least several hours. The relative importance of pathogen harboring by indigenous protozoa versus diminution of pathogen levels through predation in aquatic environments deserves further study. The committee contends that the molecular methods discussed in this chapter are likely to prove useful in assessing the viability of “harbored” pathogens.
Although the concept is controversial (Bogosian et al., 1996; Bogosian et al., 1998; Kell et al., 1998), a number of researchers have indicated that bacteria can enter a dormant stage, generally termed “viable nonculturable.” This was first shown in studies on Vibrio cholerae, in which the viable nonculturable state apparently plays an important role in the maintenance of the pathogen in the water column (Brayton et al., 1987; Colwell, 1996; Ravel et al., 1995). (This topic is discussed earlier in this chapter; however, the focus there is on the value of PCR tech-
niques in detecting nonculturable bacteria rather than on persistence responses.)
In some cases, however, the pathogenicity of the putative viable nonculturable organisms may be low, as reported by Caro et al. (1999) for Salmonella typhimurium. In this dormant state, assessment of microbial levels by plate counts may indicate little occurrence (unless resuscitation has been triggered), while other assays such as total microscopic count or nucleic acid assay may indicate higher levels. Table 6–2 summarizes some published reports of the occurrence of viable nonculturable states in bacteria.
Range of Decay Rates
Regardless of the mechanism(s) promoting loss of viability of microorganisms in aquatic systems, several researchers have reported the observed rate of disappearance under different conditions as summarized below.
TABLE 6–2 Viable Nonculturable States in Bacteria
|
Microorganism |
References |
|
Campylobacter jejuni |
Buswell et al., 1998 Rollins and Colwell, 1986 |
|
Coliforms |
McFeters et al., 1986 |
|
Escherichia coli O157:H7 |
Rigsbee et al., 1997 |
|
Klebsiella pneumoniae |
Byrd et al., 1991 |
|
Enterobacter aerogenes Agrobacterium tumefaciens Streptococcus faecalis Micrococcus flavus |
|
|
Salmonella enteriditis |
Chmielewski and Frank, 1995 |
|
Salmonella dysenteriae |
Islam et al., 1993 |
|
Enterococcus faecalis |
Lleo et al., 1998 |
|
Legionella pneumophila |
Steinert et al., 1997 |
|
Vibrio vulnificus |
Weichartand Kjelleberg, 1996 |
Determination of Viability
For studies that measure decay rates in microorganisms, the particular assays used for assessment of viability become important. In most cases, but particularly for data obtained before the mid-1990s, the assessment of viability was based on culturing methods such as agar plate growth of bacteria and most-probable-number assays for bacteria or viruses (in the form of the TCID50 determination [i.e., tissue culture infectious dose, or dose required to infect 50 percent of the tissue culture in which a sample is inoculated]).
As noted previously, however, bacteria may form viable nonculturable stages that by definition are not readily enumerated using culture techniques. Therefore, reliance on culture techniques may incorrectly estimate the true decay rates. In more recent years, some investigators have used molecular genetic techniques such as PCR (Abbaszadegan et al., 1999; DiGiovanni et al., 1999; Shieh et al., 1997; Sturbaum et al., 1998) to assess occurrence or decay of pathogens in environmental systems. Although PCR and other molecular methods may allow more efficient data collection, they may also overestimate the occurrence or persistence of viable microorganisms (Deere et al., 1996; Dupray et al., 1997). Thus, any reports of survival times (or occurrences) of pathogens in water should be accompanied by a description of the methods used to assess viability.
Data for Established Pathogens
As part of a mid-1980s reevaluation of the coliform standards for drinking water, a comprehensive review of the decay of indicators and pathogens in water was performed under the sponsorship of EPA. A summary of these decay values is provided in Table 6–3. The original tabulated values of times required for 50, 90, 99, or 99.9 percent reduction are indicated—this is preferable to conversion to a single metric (e.g., half-life), since in many cases the underlying data differed from ideal first-order decay. The information in this table reflects microorganism survival under diverse conditions ranging from raw water (of various sources) to finished drinking water (although in no circumstances was there any disinfectant residual present).
Data for More Recent Pathogens
There have been studies of additional microorganisms since the efforts summarized below in Table 6–3. Although a comprehensive review of such studies is beyond the objective of this report, a brief synopsis of findings for some emerging pathogens is appropriate. DeRegnier et al. (1989) suspended Giardia muris in river water and lakewater at ambient temperature to monitor viability using propidium iodide and animal infectivity. As measured by infectivity, cysts remained viable at least 40 days. It should be noted, however, that the small number of test animals did not likely permit measurement of inactivation beyond one log. The authors concluded
…G. muris cysts suspended in environmental water remained viable for 2 to 3 months, and their survival was enhanced by exposure to low water temperature, despite the fact that the cysts were suspended in the fecal biomass within the sample vial.
TABLE 6–3 Survival Times for Pathogens in Raw or Finished Watersa
Robertson et al. (1992) used a differential dye inclusion assay to monitor viability decay of two strains of Cryptosporidium parvum oocysts. One was originally isolated from deer and cultured in sheep, and the other was a bovine isolate. The organisms were held in membrane diffusion chambers in river water under ambient conditions. Their results indicate that 1-log inactivation is estimated to occur at 100 and 180 days for the two strains examined. More recently, Jenkins et al. (1997) determined that the survival of oocysts in fecal material (as measured by vital dyes) correlates well with the ability of the oocysts to excyst. However there is continuing controversy about the suitability of dye incorporation assays versus other techniques with respect to assessing oocyst viability (Belosevic et al., 1997; Bukhari et al., 2000). As noted earlier in this chapter, FISH techniques that use mRNA or other targets may be superior to estimate microbial viability. Enriquez et al. (1995) reported that adenovirus held for 60 days in dechlorinated tap water produced only a 2-log reduction at 23°C as measured through tissue culture assays.
Intrinsic Factors Influencing Decay
A key question is to what degree the persistence or decay of pathogens in water can be predicted quantitatively and how this information could be used in the construction of VFARs. Based on the preceding information, it is clear that a wide range of variation exists in the removal rates of pathogens in aquatic systems. However, beyond some broad generalizations, a fully quantitative model that incorporates effects of adverse conditions on a range of pathogens has eluded investigators. It is encouraging, however, that in some cases for specific microorganisms, an overall predicted model can be developed (Auer and Niehaus, 1993; Auer et al., 1998; Canale et al., 1993; Chamberlin and Mitchell, 1978; Mancini, 1978).
Nonetheless, there are identifiable differences between microorganisms that should allow for a semiquantitative assessment of environmental persistence; these are
-
ability to sorb suspended solids;4
-
ability to form dormant stages, including viable nonculturable;
-
ability to survive and/or multiply within aquatic protozoa or other microbial hosts;
-
ability to survive freezing;
-
ability to survive desiccation;
-
ability to survive wastewater treatment and to reenter drinking water; and
-
ability to survive in anaerobic sediment.
INTERPRETATION OF ISSUES
Data Information and Management Issues
Although the technology, methodology, and even the genetic databanks exist, the application of a VFAR approach to assess waterborne pathogens would require considerable effort and expenditure of resources by EPA in conjunction with the Centers for Disease Control and Prevention, National Institutes of Health, and other federal and state health organizations (NRC, 1999b). Such an interagency “Waterborne Microbial Genomics” (WMG) project would also require extensive expertise in bioinformatics, molecular microbiology, environmental microbiology, and infectious disease. Initially, existing gene banks would have to be screened and evaluated for key targets. For example, the National Center for Biotechnology Information jointly established by the National Library of Medicine and the National Institutes of Health maintains GenBank (www.ncbi.nlm.nih.gov). However, the available sequence data are not error free, and greater quality control and quality assurance would have to go into screening the genetic information. Of course, new data on genetic sequences would have to be added to the WMG as they are reported in the clinical literature. As a start, this would require the evaluation of literature for references to microorganisms having the potential to be waterborne, found in feces or urine, and naturally occurring in the water environment.
Furthermore, protocols would have to be established and tested regarding data entry, validation, and use in the development of microbial VFARs. Background levels and determination of prevalence, persistence, and quantity of key target genes would have to be gathered for analysis and interpretation of health risk and/or exposure potential. Outbreaks and contamination events would provide useful information to enhance the database. Once key parameters had been established, the
possibility of screening hundreds of water samples for thousands of key microbial hazards could be achieved through development of custom chip arrays.
The use of appropriate data and sophisticated data management would be crucial to the development and validation of VFARs. Because VFARs have the potential to be very powerful, they will require thorough validation and careful use, with attention to the limits of their validity. Validation can proceed in a variety of ways, such as the statistical measure of how well a VFAR fits into the descriptor or response data, the use of sensitivity analysis to validate VFAR data that are already available, using a VFAR with new data after it has been established with older data, and comparing the predictions of one VFAR to those of another. All of these will require the development of appropriate data sets and data management tools. The committee also notes that the use of prototype classification methods to help select PCCL contaminants for inclusion on a CCL can obviously be applied to VFARs. That is, training sets of descriptor and response variables could be developed and used in conjunction with prototype classification methods to help derive VFARs. This again implies that such training sets would have to be appropriate and robust.
The committee fully recognizes that just the initial establishment of such a program (excluding maintenance and expansion) is likely to require at least a five-year commitment and significant cooperation and expenditure of resources by EPA and other participating organizations. However, the opportunities for rapid identification of microbial hazards in water afforded by such a program would greatly improve the ability of EPA to quickly and successfully protect public health and improve water quality.
FEASIBILITY
For the VFAR concept to be ultimately adopted and used by EPA in its drinking water program, it must be feasible. Several aspects of feasibility are discussed below, including scientific validity and applicability; actual technological feasibility; application of these technologies to studying disease in humans (validation); the degree to which these methodologies are being universally adopted within the scientific community; and the need for their development and use to adhere to the principles of transparency, public participation, and other sociopolitical considerations reviewed in Chapter 2. To one extent or another, each of these elements
affects the ability of the VFAR concept to be developed, used, or validated. Since these elements either are present or can reasonably be expected to be available in the near future, the committee strongly concludes that the development and use of VFARs is indeed feasible. Having carefully noted some caveats and limitations in the preceding text, the committee remains enthusiastic about the utility of developing and using VFARs in the protection of the nation’s drinking water.
First, the underlying concept must be scientifically valid and robustly applicable. As previously noted, the relationship between specific microbial attributes and human disease has been known for more than a century. This linkage has become increasingly documented and precise as our knowledge of microorganisms and human disease has dramatically improved in the past few decades. While the illustrative examples provided in this chapter are by no means exhaustive, they certainly speak to the power of specific microbial attributes to predict virulence in humans. It is not only possible, but in fact now routine, to associate very specific human disease outcomes with the presence, absence, or variability of specific microbial characteristics (or “descriptors” in the language of SARs). To state that the concept is robustly applicable, the committee means that it is neither limited nor narrow, but is in fact valid across a very large number of microorganisms and remains valid when small variations in a single organism are explored in great depth. The fact that this is indeed the case, and is considered a paradigm of biomedical science, provides a convincing demonstration of the validity and robustness of the concept. Thus, the committee deems that the first necessary condition for feasibility exists.
For the VFAR concept to be useful, it must be able to extend a known relationship between a virulence attribute and human disease to a situation in which the attribute is found in a new or unexpected circumstance or in microorganisms that have not heretofore been recognized as potentially pathogenic. The profound scientific revolution associated with the unraveling of DNA’s double helix speaks to this second aspect of feasibility—the disciplines of bioinformatics, proteomics, and genomics. The ability to rapidly, and completely, sequence the genome of entire organisms, and to use bioinformatic techniques to compare gene sequences of different organisms, provides this mechanism for comparison. The committee adds that the time and cost required for sequencing a microorganism have both declined markedly in just the past few years. Independent of any judgment made by this committee, a number of biotechnology and pharmaceutical companies have chosen to aggressively pursue opportunities that rely on comparative genomics, which the com-
mittee regards as confirming its judgment of the adequacy, power, and affordability of the methodology. The committee thus warrants that the necessary condition of technological feasibility also exists.
Although the committee is unclear as to whether compelling logic currently exists for the development of VFARs based on genomic techniques that can be extended to proteomics, it fully acknowledges the potential for this in the near future. The possibility for proteomics to also play a role in the development of VFARs adds both depth and an additional dimension to ways in which VFARs might operate in the future within EPA’s drinking water program.
A third element by which to judge feasibility is the likelihood that adverse human outcomes (e.g., diseases) will continue to be discovered in association with the action or presence of a microbial contaminant, microbial gene, or gene product in the clinical setting. This clinical linkage between diseases and specific microorganisms or their products has been a hallmark of medical sciences for the past two centuries. There is no indication that the accelerating pace of these medical discoveries is abating; one need only consider prions and mad cow disease, Ebola virus, Escherichia coli O157:H7, Helicobacter pylori, and nanobacteria to be reminded of important pathogens that were unknown to science until a few decades ago. As discussed previously, the intent of the VFAR is to characterize, categorize, and make scientific the potential linkage, as outlined in Figure 6–3 below.
This external element represents both a validation of the linkage for those organisms already known to cause disease and an element that will be helpful in validating the discovery of emerging waterborne pathogens through the use of VFARs. For “established” waterborne microoorganisms such as Vibrio cholerae, Salmonella, and the pathogenic protozoa, this linkage was, and is, made easily. That is, these microorganisms are already easily cultured or visible in human specimens—no new technological or scientific advances are required for them to be linked to human disease. In the case of emerging microorganisms that are often unsuspected agents of disease, or are difficult to detect using traditional methods of culture or microscopy, it is likely that the novel molecular detection techniques discussed in this chapter (e.g., PCR, gene chips) will continue to lead to new medical associations. Identification of some viruses (e.g., herpes simplex virus) or specific microbial antigens through PCR techniques has now become commercially viable and widespread in the medical setting, replacing earlier methods. Many diagnostic kits are now available to detect antigens shed by pathogenic bacteria and viruses in urine, blood, and spinal fluid. Viruses that were once “too expensive”
FIGURE 6–3 Linkage between microbial virulence factors and human disease.
to look for on a routine basis (e.g., rotavirus) are now tested routinely for in the clinical microbiology laboratory using simple ELISA (enzyme-linked immunosorbent assay) methods. Indeed, ELISA kits that recognize Giardia and Cryptosporidium antigens are already available (e.g., Alexon-Trend’s ProSpecT® assays). Thus, the committee can foresee the likelihood that the same technologies needed for constructing VFARs will already be used in the clinical setting to detect microorganisms or their products, further strengthening the utility of the VFAR relationship.
A fourth element regarding feasibility is the likelihood that EPA’s movement to adopt VFARs for use in its drinking water program will be congruent with the direction that other government, private, and public agencies are taking. In simplest terms, is such an effort likely to be adopted solely by EPA, or is it likely to be a direction that other agencies will follow? Resoundingly, all evidence points to a massive public and private investment in genomics, bioinformatics, and proteomics, which are the key disciplines behind developing and using VFARs. Further-
more, the specific needs of EPA are likely to be recognized by others as being of crucial importance to shared problems. For example, the “identification” problem that exists for EPA—identifying potentially pathogenic microbial water contaminants that are difficult or impossible to culture—has already been recognized by other government agencies as a high-priority area for research (BRWG, 2000). There is every reason to believe that the path EPA must follow to develop VFARs will be similar to one already blazed by many other agencies. Indeed, it can be argued persuasively that EPA will benefit very substantially from the synergistic efforts of other agencies and independent outside researchers as recommended in the committee’s second report (NRC, 1999a). In all likelihood, the resources of EPA will have to be directed primarily to (1) focusing its own internal efforts and the attention of other government agencies on waterborne pathogens and (2) integrating a tremendous knowledge base (in large part developed outside the agency) with the purpose of VFAR construction, validation, and use in EPA’s drinking water program. Thus, adoption of the technologies behind VFARs by the wider scientific community considerably improves the feasibility of any related EPA efforts to develop and use VFARs. The high-priority issues identified in this report are well recognized by other agencies that have similar needs but different applications in mind.
Lastly, the committee’s (Chapter 2) recommendations that the process for selecting drinking water contaminants for future CCLs be systematic, scientifically sound, transparent, and involve broad public participation should also be met in the development and use of VFARs.
CONCLUSIONS AND RECOMMENDATIONS
Despite the identification and discussion of some necessary caveats and limitations, the committee concludes that the construction and eventual use of VFARs in EPA’s drinking water program are feasible and merit careful consideration. More specifically, the committee makes the following recommendations:
-
Establish a scientific VFAR Working Group on bioinformatics, genomics, and proteomics, with a charge to study these disciplines on an ongoing basis and periodically inform the agency as to how these disciplines can affect the identification and selection of drinking water contaminants for future regulatory, monitoring, and research activities. The committee acknowledges the importance of several practical considera-
-
tions related to the formation of such a working group within EPA, including how it should be administered and supported (e.g., logistically and financially) or where it could be located. However, the committee did not have sufficient time in its meetings to address these issues or make any related recommendations.
-
The findings of this report, and especially those of the Biotechnology Research Working Group (BRWG, 2000) should be made available to the VFAR Working Group at its inception. The committee views the activities of such a working group as a continuing process in which developments in the fields of bioinformatics, genomics, and proteomics can be assessed rapidly and adopted for use in EPA’s drinking water program.
-
The working group should be charged with the task of delineating specific steps and related issues and time lines needed to take VFARs beyond the conceptual framework of this report to actual development and implementation by EPA. All such efforts should be made in open cooperation with the public, stakeholders, and the scientific community
-
With the assistance of the VFAR Working Group, EPA should identify and fund pilot bioinformatic projects that use genomics and proteomics to gain practical experience that can be applied to the development of VFARs while it simultaneously dispatches the charges outlined in the two previous recommendations.
-
EPA should employ and work with scientific personnel trained in the fields of bioinformatics, genomics, and proteomics to assist the agency in focusing efforts on identifying and addressing emerging waterborne microorganisms.
-
EPA should participate fully in all ongoing and planned U.S. government efforts in bioinformatics, genomics, and proteomics as potentially related to the identification and selection of waterborne pathogens for regulatory consideration.











































