
Colloquium
The role of antimicrobial peptides in animal defenses
Robert E. W. Hancock* and Monisha G. Scott
Department of Microbiology and Immunology, University of British Columbia, Vancouver, BC, Canada V6T 1Z3
It is becoming clear that the cationic antimicrobial peptides are an important component of the innate defenses of all species of life. Such peptides can be constitutively expressed or induced by bacteria or their products. The best peptides have good activities vs. a broad range of bacterial strains, including antibiotic-resistant isolates. They kill very rapidly, do not easily select resistant mutants, are synergistic with conventional antibiotics, other peptides, and lysozyme, and are able to kill bacteria in animal models. It is known that bacterial infections, especially when treated with antibiotics, can lead to the release of bacterial products such as lipopolysaccharide (LPS) and lipoteichoic acid, resulting in potentially lethal sepsis. In contrast to antibiotics, the peptides actually prevent cytokine induction by bacterial products in tissue culture and human blood, and they block the onset of sepsis in mouse models of endotoxemia. Consistent with this, transcriptional gene array experiments using a macrophage cell line demonstrated that a model peptide, CEMA, blocks the expression of many genes whose transcription was induced by LPS. The peptides do this in part by blocking LPS interaction with the serum protein LBP. In addition, CEMA itself has a direct effect on macrophage gene expression. Because cationic antimicrobial peptides are induced by LPS and are able to dampen the septic response of animal cells to LPS, we propose that, in addition to their role in direct and lysozyme-assisted killing of microbes, they have a role in feedback regulation of cytokine responses. We are currently developing variant peptides as therapeutics against antibiotic-resistant infections.
A nimals are exposed to millions of potential pathogens daily, through contact, ingestion, and inhalation. Their ability to avoid infection depends on their mechanisms of innate immunity. There has been a tendency to emphasize the role of the humoral and/or cellular immunological system in defense against infection; however, it is equally clear that this system is not triggered rapidly enough to protect against exposure to pathogens. In the past decade, the role of cationic antimicrobial peptides has become increasingly apparent (1), and there is a growing body of evidence that their role in defense against microbes is as important to the host as antibodies, immune cells, and phagocytes. In the fruit fly Drosophila, for example, cationic peptides are the major form of defense against infection and are induced, in response to challenge by bacteria or lipopolysaccharide (LPS), by a regulatory pathway similar to that used by the mammalian immune system, involving Toll receptors and the transcription factor NFκB (2).
The Nature of Cationic Antimicrobial Peptides
We use the term (cationic) antimicrobial peptides to describe gene-encoded peptides comprising between 12 and 50 amino acids, with at least two excess positive changes due to lysine and arginine residues and around 50% hydrophobic amino acids. They are found in all species of life, ranging from plants and insects to animals, including molluscs, crustaceans, amphibians, birds, fish, mammals, and humans (1, 3). More than 500 such peptides have been discovered. They fit into at least four structural classes, namely β-sheet, comprising two to three β-strands stabilized by disulphide bridges, amphipathic α-helices, extended structures, and loop structures (Table 1). Despite these different folding patterns, there appear to be two types of three-dimensional configurations, an amphipathic structure with opposing hydrophobic and polar/cationic faces and a cationic double-wing structure with two regions of positive charge bracketing a hydrophobic core [ref.1 and A. Rozek, C. Friedrich & R.E.W.H. (unpublished results)].
Examples of mammalian cationic antimicrobial peptides are presented in Table 1. In addition to these classes, there is a variety of animal cationic proteins including bactericidal permeability increasing protein, lactoferrin, transferrin, cathepsin G, cystatin, CAP18, pepsinogen C, ribosomal protein S30, etc., whose antibacterial activities can be traced to a cationic peptide sequence within these basic proteins.
These peptides are termed antimicrobial because they have unusually broad spectra of activity. These can include an ability to kill or neutralize Gram-negative and Gram-positive bacteria, fungi (including yeasts), parasites (including planaria and nematodes), cancer cells, and even enveloped viruses like HIV and herpes simplex virus. Nevertheless, many peptides are quite selective for microbes over eukaryotic cells. Not all peptides have all of the above activities. However, a single 13-aa peptide, indolicidin, for example, is able to kill bacteria, fungi, and HIV.
Antimicrobial peptides tend to be found in those parts of animals that are most likely to come into contact with pathogens from the environment. Thus, they are found on the skin, ear, and eye, on epithelial surfaces, including the tongue, trachea, lungs, and gut, and in the bone marrow and testes. They are also the most prevalent protein species of neutrophils, being associated with azurophilic granules and comprising a major nonoxidative killing mechanism of these dedicated antimicrobial phagocytes.
Mechanism of Production in Animals
The mucous layer of animals covers and protects all epithelial tissues against microbial, mechanical, and chemical insults and forms an initial line of defense. The main constituents are the mucins and other glycoproteins, proteinase inhibitors, and cationic peptides, like defensins. Healthy-tissue epithelial cells have been shown to express β-defensin genes at a low level. However, some defensin genes can be induced on treatment with proinflammatory cytokines, LPS, or bacteria. For example, production of human β-defensin (HBD)-2 in keratinocytes is strongly induced on contact of keratinocytes with Gram-negative bacteria or proinflammatory cytokinessuch as tumor necrosis factor (TNF)-α or IL-1β (4). HBD-2 mRNA expression has been observed in the skin, foreskin, lungs, and trachea, but not in the
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defence in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviations: TNF, tumor necrosis factor; LPS, lipopolysaccharide; LTA, lipoteichoic acid; PG, peptidoglycan; LPB, LPS-binding protein.
|
* |
To whom reprint requests should be addressed. E-mail: bob@cmdr.ubc.ca. |
Table 1. Some representative mammalian peptides
|
Peptide |
Class |
|
|
HNP-1 (α-defensin) |
β-sheet |
AC1YC2RIPAC3lAGERRYGTC3lYQGRLWAFC2C1 |
|
HBD-2 (β-defensin) |
β-sheet |
MRVLYLLFSFLFIFLMPLPGVFGGIGDPVTC1LKSGAIC2HPVFC3PRRYKQIGTC2GLPGTKC1C3KKP |
|
Protegrin |
β-sheet |
RGGRLC1YC2RRRFC1VC2VGR |
|
Indolicidin |
Extended |
ILPWKWPWWPWRR-NH2 |
|
Bac5 |
Extended |
RFRPPIRRPPIRPPFYPPFRPPIRPPIFPPIRPPFRPPLGPFP |
|
Bactenicin |
Loop |
RLC1RIVVIRVC1R |
|
LL-37 |
α-helical |
LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES |
|
Cecropin P1 |
α-helical |
SWLSKTAKKLENSAKKRISEGIAIAIQGGPR |
|
* Single-letter amino acid code. † Subscript numbers represent amino acids that are joined by cysteine disulfides. |
||
kidney, salivary glands, small intestine, and liver. This is in contrast to HBD-1, which is constitutively expressed mainly in the urogenital tract and kidney. Several other inducible β-defensins are produced in the epithelia, including tracheal antimicrobial pep tide (TAP), isolated from bovine respiratory mucosa, lingual antimicrobial peptide, and the enteric β-defensin (5). TAP is expressed in columnar epithelial cells of the conducting airways, and the mRNA of this β-defensin peptide is upregulated in response to LPS. The production of HBD-2 by human epithelium resembles the ancient defense mechanisms of plants and insects (2), because there is evidence in these situations for the involvement of the transcription factor NFκB and Toll receptors. Such a mechanism elicits an immediate antimicrobial response to the same microorganisms that have had contact with the epithelial cells, and these responses are related to, but completely independent of, the leukocyte-dependent immune defense mechanisms. Other obvious differences between the cationic antimicrobial peptide response and the immune response in animals are the highly specific nature of immune responses, the relative slowness of immune responses because of the requirement for clonal cell expansion, and the self vs. nonself discrimination built into the immune response. In contrast, inducible peptide responses result in an ability to act against a wide spectrum of pathogens, occur within minutes rather than days, and lack real-self vs. nonself discrimination (although the peptides appear to have relatively low activity against host cells).
Evidence for Their Role in Host Defenses
The evidence for a role of cationic antimicrobial peptides in innate host defenses has become quite convincing. It includes data demonstrating that some peptides are inducible by bacterial products and convincing animal model and transgenic animal experiments indicating that the peptides protect against infection in experimental animals. The inducibility of peptides has been summarized above. The kinetics of induction are highly suggestive of a role in early defenses against infection.
The animal model data demonstrating protection by a variety of different peptides applied both topically and systemically were summarized recently (6). Such studies have confirmed the antibacterial (vs. both Gram-negative and Gram-positive bacteria), antifungal, synergistic and antiendotoxic (see below) nature of antimicrobial peptides. These results have been confirmed by studies showing systemic protection by nisin against Streptococcus pneumoniae infections of mice (7), local protection by the protegrin-related peptide IB367 against polymicrobic oral mucosaitis in hamsters (8), protection against lethal Pseudomonas aeruginosa infections of burn-wound sites in mice by peptide D4B (9), and protection by LL-37 (CAP 18)-derived peptides against lethal endotoxemia and P. aeruginosa infection in mice (10). Our own recent studies have demonstrated that peptides (including the fish peptide pleurocidin) protect Coho salmon against lethal vibriosis (Aeromonas salmonicida infection), when administered continuously at low levels by using a device called an osmotic pump.
An alternative method of doing these types of studies involves a “gene-therapy” treatment of mice with an adenovirus vector containing the DNA for the human peptide LL-37 (11). Such mice showed a dramatic increase in serum and lung LL-37 and demonstrated significantly fewer bacteria and a lower inflammatory response after sublethal challenge and a dramatic increase in resistance to endotoxin and Escherichia coli challenges.
Although the role of such peptides in defense against infections has been emphasized, many other intriguing properties have been ascribed to selected cationic peptides, including induction of the wound-repair proteoglycans termed Syndecans (12), stimulation of nonopsonic phagocytosis (13), chemoattraction of IL-8-stimulated neutrophils (14), and penetration of the blood–brain barrier (15).
It is well known (1) that cationic antimicrobial peptides are major components of certain phagocytic cells, especially neutrophils and alveolar macrophages. They appear to be involved in nonoxidative killing by such cells (16). Although oxidative killing of bacteria by phagocytes is often emphasized, nonoxidative killing can be very effective, because neutrophils from chronic granulomatis disease patients, which lack an oxidative response, are still able to kill most bacteria (16). Indeed, such patients are only substantially more susceptible to infections by Burkholderia cepacia, one of the bacteria that are naturally resistant to cationic antimicrobial peptide action (17).
Mechanism of Action
Cationic antimicrobial peptides have been described as membrane-active agents (1). This is certainly true for many peptides, but we and others have recently described data that indicate that the membrane is not necessarily the target for many, or perhaps even most, cationic peptides (3, 18, 19 and 20). To summarize the proposed mechanism of action for Gram-negative bacteria, the peptides interact with and cross both cell envelope membranes and then kill cells by a multihit mechanism that involves action on more than one anionic target. The initial uptake across the outer membrane is via self-promoted uptake (3, 13) proposed by us 20 years ago to explain uptake of polycationic antibiotics like aminoglycosides and polymyxins across the outer membrane. In this mechanism, the peptides initially interact with the polyanionic surface LPS and competitively displace the divalent cations that bridge and partly neutralize the LPS. This causes disruption of the outer membrane (visualized as surface blebbing), and it is through these disrupted outer membranes that peptide molecules are proposed to pass (i.e., the peptides self promote their own uptake). Next, the peptides associate with the negatively charged phospholipid membrane and insert into the membrane so they are oriented parallel to the membrane. It is then proposed that the peptide molecules, when they reach a certain critical concentration, form informal transmembrane channels that we have termed aggregate channels but that others ( 21) call
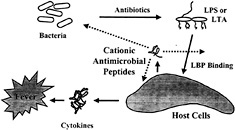
Fig. 1. Model outlining the major events in induction of sepsis by bacteria and the points at which cationic peptides are proposed to intervene.
supramolecular peptide/lipid complexes (or toroidal channels). In some cases, these channels may cause sufficiently severe permeability problems to inhibit or perhaps even kill cells. The lifetime of such channels is short (18) and, when they collapse, some of the peptide molecules are left in the inner monolayer and then are proposed to disassociate and interact with cytoplasmic polyanions such as DNA (19). Some of the potential targets in cells include DNA, membrane permeability, and autolysins. Moreover, the relative importance of each of these targets may vary from peptide to peptide.
Nevertheless, one consequence of this physical mechanism of action (based on ionic and hydrophobic interactions) is that it is rather difficult for bacteria to become resistant to such peptides, and making mutants is not at all easy (22). Mutation of the PhoP/PhoQ system is reputed to lead to resistance to cationic antimicrobial peptides but actually only increases minimum inhibitory concentrations about 2- to 4-fold. Very few naturally resistant bacteria occur; they include B. cepacia (which has an outer membrane composition that does not permit self-promoted uptake) and Proteus and Serratia sp., which make proteases that cleave some but not all cationic peptides.
Role in Counteracting Sepsis
More than a half million patients suffer from sepsis every year in North America. Sepsis is associated with the presence of pathogenic microorganisms or their toxins in the blood (Fig. 1). It can result from infections with either Gram-negative or Gram-positive bacteria. Gram-negative sepsis is usually caused by the release of a bacterial outer membrane component, endotoxin (LPS). The toxicity of LPS is contained within its lipid A portion. Gram-positive sepsis is also presumed to be caused by the release of bacterial cell wall components. A number of Gram-positive cell wall constituents, including lipoteichoic acid (LTA) (23), peptidoglycan (PG) (24), Streptococcus rhamnose-glucose polymers (25), and Staphylococcus capsular polysaccharide (26), have been shown to stimulate the production of inflammatory mediators in vitro. When injected into animals, these Gram-positive cell wall components elicit many of the characteristic features of septic shock, including cytokine production, leukocytopenia, circulatory failure, multiple organ dysfunction syndrome, and mortality (27, 28). Antibiotics used to treat the bacterial infection can actually be harmful in that they can stimulate the release of endotoxin ( 29), or, in the case of Gram-positive bacteria, peptidoglycan and LTA (30, 31). Both LTA and PG are released spontaneously into the culture medium during growth of Gram-positive bacteria. Moreover, β-lactam antibiotics such as penicillin enhance the release of LTA and PG. Thus, the release of LTA and PG from Gram-positive bacteria may also promote septic shock during bacterial infections and subsequent antibiotic treatment. The increasing incidence of Gram-positive-induced septic shock (33) indicates that there is a need to develop therapeutic strategies to prevent the activation of inflammatory cells by components of Grampositive cell walls.
The mechanism by which LPS activates macrophages is now understood in some detail (ref.33; Fig. 1). LPS-binding protein (LBP), an acute-phase reactant that is present in the blood, binds LPS and transfers it to CD 14, a protein that exists as a soluble form in blood and as a glycosyl phosphatidylinositollinked molecule on the surface of monocytes and macro-phages. LPS·CD14 complexes are thought to initiate intracellular signaling reactions by binding to Toll-like receptors (TLRs) on macrophages and other cells (33). TLR4 appears to be required for LPS to initiate signaling and to induce inflammatory responses. LPS·CD14 complexes cause activation of the NF-κB transcription factor as well as activation of the ERK, JNK, and p38 mitogen-activated protein kinases, all of which mediate the production of inflammatory cytokines (34). Despite their structural differences, both LTA and PG also activate macrophages and polymorphonuclear leukocytes in association with CD14 and TLR4/TLR2 (35, 36, 37 and 38). Thus, substances that bind to bacterial components and ablate their ability to bind surface receptors would be good candidates as antiendotoxic agents. A fragment of the cationic protein bactericidal permeability increasing protein (rBPI-21) is currently in clinical trials for this purpose (6).
As described above, cationic peptides act on Gram-negative bacteria by initially binding to their surface polyanionic LPS, followed by self-promoted uptake across the outer membrane. We have shown that some cationic peptides have a high affinity for LPS (17, 39). Coincident ally, such peptides inhibit the production of cytokines such as TNF-α and IL-6 by macrophages stimulated with LPS. We have been able to demonstrate such effects with peptides representing all structural classes, including such animal peptides as defensins, indolicidin, and protegrin (40). Furthermore, studies with the cationic proteins bactericidal permeability increasing protein, CAP37, and lactoferrin (41, 42), have indicated that these molecules have an analogous ability to antagonize the ability of LPS to stimulate cytokine production in macrophages.
We went on to study the potential in vivo relevance of these observations. The peptides CEMA and CEME were able to prevent lethal endotoxemia in the galactosamine-sensitized mouse model (39). With the former more active peptide, it was shown that the LPS-stimulated induction of the important sepsis-mediating cytokine, TNF-α, could also be dramatically suppressed in the blood of galactosamine-sensitized mice.
It was also possible to assess binding of cationic antimicrobial peptides to the Gram-positive surface molecule LTA by using a variation of the dansyl polymyxin displacement assay that is used to assess LPS binding (43). LTA, like LPS, has both a polyanionic and lipidic nature, and thus was able to interact with dansyl polymyxin and cationic peptides, although the kinetics of binding indicated a lower affinity for binding to LTA compared with LPS. From these studies, it appeared that the action of cationic peptides against Gram-positive bacteria did not appear to be related to their ability to bind LTA, as the relative ability of these peptides to bind LTA did not correspond to their minimum inhibitory concentration values. For example, the α-helical cecropin/melittin hybrid peptide CEME was the most effective peptide at killing S. aureus and other Gram-positive bacteria, even though many of the peptides studied had a higher affinity for S. aureus LTA than CEME. In contrast, the ability of CEME-related peptides to bind LTA appears to be important for reducing the ability of LTA to stimulate inflammatory reactions. Thus LTA from different species of Gram-positive bacteria stimulated TNF-α and IL-6 production by the murine macro-phage cell line RAW 264.7, and cationic antimicrobial peptides
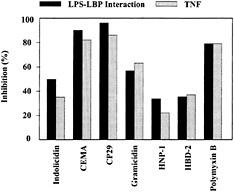
Fig. 2. Inhibition of LPS–LBP interaction and LPS-induced TNF-α production by structurally different cationic peptides. (A) Biotinylated LPS (45 ng/ml) was added to wells with immobilized LBP in the presence or absence of the indicated cationic peptides (10 µg/ml). The peptides were added to the wells at the same time as the LPS, and residual LPS binding was assessed by ELISA. (B) RAW 264.7 cells were incubated with E. coli O55:B5 LPS (100 ng/ml) in the presence or absence of the indicated peptides (20 µg/ml) for 6 h. TNF-α released into the culture supernatant was measured by ELISA. Data are from ref.39.
were able to significantly inhibit this production of cytokines. The peptides were also effective in human blood at reducing production of TNF-α in response to LTA, although the levels of inhibition were lower than those observed in in vitro studies with RAW macrophage cells. The effects of the peptides on soluble LTA are significant, because bacteria release LTA during normal growth, and LTA release is enhanced by β-lactam antibiotics (31). Using a system in which macrophages are separated from bacteria by a membrane filter, we demonstrated that growing E. coli O111:B4, P. aeruginosa PAO, and S. aureus could, in a dose-dependent manner, stimulate the production of TNF-α, and in the case of the Gram-negative bacteria only, IL-6, because of their shedding into the medium of molecules that were able to cross the filter and interact with macrophages (39, 43). These stimulatory molecules are probably shed LPS or LTA, respectively, as indicated by the ability of cationic peptides to block cytokine production in this situation.
The mechanism of peptide blocking of the stimulation of macrophages by LPS was studied (40). Because it is known that the action of LPS is enhanced by binding to a serum protein, LBP, one possible mechanism is clearly the ability of the peptides to bind with high affinity to LPS. Indeed, in the case of LPS, the peptides can bind LPS (17, 39) and inhibit LPS·LBP interaction (Fig. 2). However, although coincubation of peptides, LPS, and LBP resulted in successful inhibition of LPS binding to LBP, the antimicrobial peptides were less successful in dissociating LPS·LBP complexes. In other experiments, it had been observed that cationic antimicrobial peptides could block the ability of LPS to stimulate TNF-α production by serum-bathed macro-phages, even when added up to 1 hour after the LPS (39). This indicated that there might be another mechanism by which the peptides were acting, possibly a direct interaction with the cytokine-producing cells.
There have been a few strong indicators that cationic peptides can interact directly with eukaryotic cells. Generally speaking, the interaction of most peptides with eukaryotic membranes is inhibited by the lack of negatively charged lipids on the surface of such cells, by the rather low membrane potential (−15mV) across the plasma membrane of eukaryotic cells, and by the presence of cholesterol in the plasma membranes of such cells (compared to bacteria that have an abundance of anionic surface phospholipids such as phosphatidyl glycerol, a transmembrane potential of −140mV, and no cholesterol). Other studies have demonstrated directly that peptide PR-39 rapidly enters human microvascular endothelial cells (12). Moreover, it is known that some tumor cell lines (but not the macrophage cell line described here) can be killed by cationic peptides at high concentrations (44).
To investigate whether cationic antimicrobial peptides might influence sepsis through a novel mechanism involving direct interaction with macrophage cells, we chose the peptide CEMA, a peptide that becomes α-helical on contact with membranes (45). Although it is derived from insect peptides, it is a paradigm for this class of peptides and has both strong antimicrobial activity against Gram-negative bacteria and good antiendotoxic activity both with cultured macrophages and in mouse models (38). Gene array technology was used to examine the differential gene expression in RAW macrophages stimulated with LPS and the cationic peptide, CEMA [M.G.S., C. M. Rosenberger, M. R. Gold, B. B. Finlay & R.E.W.H. (unpublished results)]. A considerable number of genes were found to be up-regulated by LPS, including several already published, confirming the potential value of the gene array technology. For example, LPS was observed to affect a large number of cell-cycle mediators such as cyclins, cyclin-dependent kinases, retinoblastoma proteins, and related transcription factors, confirming the antimitogenic effects of LPS on macrophages (46). There were also several genes not found previously to be regulated by LPS, including brain factor 1 (up-regulated) DP-1, Ski, and dystroglycan (down-regulated).
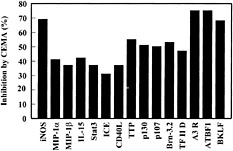
Fig. 3. Effect of CEMA on LPS-induced gene expression in RAW 264.7 cells. RAW 264.7 cells were stimulated for 4 h with media alone, S. typhimurium LPS (100 ng/ml), or S. typhimurium LPS (100 ng/ml) and CEMA (50 µg/ml). The RNA was isolated from the cells and used to make 32P-labeled cDNA probes, which were hybridized to the CLONTECH Atlas arrays, and after a 3-day exposure, they were analyzed with a Phosphorimager and CLONTECH ATLAS software. The average percent inhibition of gene transcription by CEMA as measured by a change in fold intensity is shown in the graph. The following selected genes are shown: iNOS, inducible nitric oxide synthase; MIP-2 α, macrophage inflammatory protein (chemokine); MIP-1 β, macrophage inflammatory protein (chemokine); IL-15, interleukin-15; (cytokine), Stat3, acute-phase response factor; ICE, interleukin-converting enzyme; CD40L, CD40 ligand; TTP, tristetraprolin: (destabilizes TNF mRNA); p130 and p107, retinoblastoma proteins; Brn-3.2 POU, transcription factor 1; TF II D, transcription factor; A3R, adenosine A3 receptor; ATBF1, AT motif-binding factor (transcription factor); BKLF, CACCC Box-binding transcription factor. Data are from M.G.S., C. M. Rosenberger, M. R. Gold, B. B. Finlay & R.E.W.H. (unpublished results).
Many of the genes that showed increased expression after LPS treatment could be blocked by at least 30% by CEMA (e.g., Fig. 3). These included many known inflammatory mediators such as IL-1β, IL-15, macrophage inflammatory proteins, and inducible nitric oxide synthase. Interestingly, we saw a similar induction pattern with LTA and the bacterial DNA motif CpG, although some genes were differentially induced. The gene array data were confirmed in selective instances by assaying the stimulated macrophages for protein production in the supernatants by ELISA and by direct Northern blot analysis of transcripts. For example, the protein levels of TNF-α, IL-6, and MIP-1α were greatly increased (0.3 ng/ml to 6–12 ng/ml) by LPS stimulation, and CEMA inhibited these responses by 78, 86, and 45%, respectively (46).
In contrast, CEMA did not have a pronounced effect on LPS-induced down-regulation of gene expression. Also, CEMA did not block the ability of LPS to induce the expression of some genes, including the genes for CD14, ICAM-1, LFA-1α, HMG-box transcription factor, MAPKK1, c-rel proto-oncogene, and Mdm2, representing many different families of genes. This indicates that the peptides have a selective effect on gene induction by LPS. Therefore, we looked for a previously unsuspected effect of cationic peptides on macrophage gene expression. We observed that CEMA had a wide range of effects on macrophage gene expression. It up-regulated a number of genes encoding transcription factors, kinases, and cell-cycle regulators (e.g., Fig. 4), suggesting that cationic peptides not only dampen the antimitotic effect of LPS but also may directly affect cell-cycle regulation. Several cell-surface antigens and adhesion proteins were also up-regulated by CEMA. CEMA also affected several apoptosis-related genes, increasing the expression of PD-1 and decreasing the expression of BAG-1 and neuronal death protein. Thus, CEMA has pleiotropic effects on macrophages.
It appears that cationic antimicrobial peptides can suppress LPS stimulation of cytokine production in macrophages by interfering with LPS binding to serum LBP and probably other molecules/receptors and possibly also by directly influencing the expression repertoire of macrophages. Because it is known that LPS may induce host-defense cationic antimicrobial peptides and do so at the same time that cationic antimicrobial peptides suppress the effects of LPS on macrophages leading to sepsis, we propose that these peptides are part of a feedback mechanism for regulating cytokine responses to bacterial products (Fig. 1).
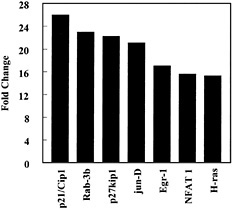
Fig. 4. Influence of CEMA on gene expression in RAW 264.7 cells. Gene arrays were used to compare transcription in unstimulated cells and cells stimulated with CEMA (50 µg/ml) for 4 h. The average fold change is shown for the following genes: p21/Cip1, cyclin-dependent kinase (cdk)-inhibitor protein 1; Rab-3b, ras-related protein; p27kip1, G1 cyclin-cdk protein kinase inhibitor; jun-D, c-jun-related transcription factor; Egr-1, Zn-finger transcription factor; NFAT 1, transcription factor; H-ras, transforming G-protein. Data are from M.G.S., C. M. Rosenberger, M. R. Gold, B. B. Finlay & R.E.W.H. (unpublished results).
There is substantial interest in identifying novel strategies to overcome not only sepsis but also the underlying infection. Presently, there are no effective compounds that have been proven to overcome the lethal nature of sepsis. Many new strategies, including neutralizing antibodies, soluble cytokine receptors, and various endotoxin-binding factors, have been tested with mixed results. The above-described abilities of the cationic peptides warrant further studies of their potential as part of an antisepsis treatment.
Peptide/Lysozyme and Peptide/Peptide Synergy
As discussed above, the mechanism of action of peptides leads to disruption of the outer membrane barrier and perturbation of the cytoplasmic membrane. Possibly as a consequence of these actions, antimicrobial peptides can show synergy with conventional antibiotics against both Gram-negative (17) and Gram-positive (20) bacteria. Our early studies with rabbit defensins led us to question whether peptides always acted on their own or rather in synergy with other host defense components (13). We observed that the ability of rabbit defensins to permeabilize the outer membrane of P. aeruginosa increased as the pH was lowered (as would occur in the phagolysosome after ingestion of P. aeruginosa by neutrophils), even though the antibacterial activity of defensins is antagonized at this pH. Therefore, we considered the possibility that peptides actually worked in synergy with the protein lysozyme, a slightly basic enzyme that is excluded from its target, the peptidoglycan, by the outer membrane. Indeed, we could demonstrate that peptides promoted the ability of lysozyme to lyse Gram-negative bacteria, and that there was excellent synergy between lysozyme and a range of peptides against several bacteria. Because lysozyme is present in most parts of the body, it is possible that such synergy is critical to the action of cationic antimicrobial peptides in their natural hosts. Indeed, in fish challenged with bacteria, both lysozyme and cationic peptides appear to be rapidly induced.
A second type of synergy that has been observed is between peptides. This was first demonstrated with the frog peptides magainin and PGLa (47), and we have recently confirmed this with the mammalian peptides protegrin 1 and indolicidin (unpublished results). Study of the kinetics of interaction of peptides shows that they act cooperatively. Thus, we assume that two peptides reinforce this cooperative interaction resulting in positive cooperativity.
Potential as Therapeutics
There is no question that, with the increasing antibiotic resistance problem, there is a need to develop new classes of antibiotics. Cationic antimicrobial peptides have many of the desirable features of a novel antibiotic class (6). In particular, they have a broad spectrum of activity, kill bacterial rapidly, are unaffected by classical antibiotic resistance mutations, do not easily select antibiotic resistant variants, show synergy with classical antibiotics, neutralize endotoxin, and are active in animal models. Despite this, many issues remain to be solved. For example, these peptides have relatively high molecular weights compared with most antibiotics and will have to be produced recombinantly to keep prices down (1). Although several processes for doing this have been described, to our knowledge they have not yet been successfully performed on an industrial scale. Another issue is toxicity. Some cationic antimi-
crobial peptides are very toxic for mammalian cells (e.g., bee venom melittin), whereas others show little or no acute cytotoxicity. However, more subtle toxicities have not been studied, although we assume, based on the presence of natural peptides in vivo at concentrations of, e.g., 44 µg/ml in the saliva of an individual with peritonitis, that they can be tolerated at high levels. Another issue would be their lability to proteases in the body. In this regard, there are strategies for protecting the peptides from proteases, including liposomal incorporation or chemical modification.
With this in mind, two very promising clinical trials are underway. The protegrin derivative IB-367 (Intrabiotics, Mountain View, CA) is being examined for its potential against oral mucosaitis, a polymicrobial ulcerative disease of cancer patients. The peptide MBI-226 (Micrologix Biotech, Vancouver) is being investigated for sterilizing catheter insertion sites, thus preventing serious infections caused by colonization of such catheters by skin bacteria. Results from clinical trials to date have indicated efficacy, and MBI-226 has been given fast-track status by the Federal Drug Administration.
Thus cationic antimicrobial peptides are not only important components of the innate defenses of all animals against infections, but synthetic variants thereof hold great potential as a weapon against antibiotic-resistant bacteria. The great sequence and structural diversity offered by peptides (i.e., 20 possible amino acids in each position) will provide many possibilities for drug design.
We thank the many collaborators and laboratory personal who contributed to this work, but particularly Carrie Rosenberger, Brett Finlay, and Mike Gold, who are collaborating on transcriptional array experiments, and Hong Yan and Don Woods, who are performing the animal model infections experiments. Our peptide work is supported by the Canadian Bacterial Diseases Network and the Canadian Cystic Fibrosis Foundations SPARX program. Robert Hancock is the recipient of a Medical Research Council (MRC) of Canada Distinguished Scientist Award, and Monisha Scott has an MRC Scholarship.
1. Hancock, R. E. W. & Lehrer, R. ( 1998) Trends Biotechnol. 16, 82–88.
2. Hetru, C., Hoffmann, D. & Bulet, P. ( 1998) in Molecular Mechanisms of Immune Responses in Insects, eds. Brey, P. T. & Hultmark, D. (Chapman & Hall, London), pp. 40–66.
3. Hancock, R. E. W. & Chapple, D. S. ( 1999) Antimicrob. Agents Chemother. 43, 1317–1323.
4. Schroder, J.-M. & Jurgen, H. ( 1999) Int. J. Biochem. Cell Biol. 31, 645–651.
5. Diamond, G., Russell, J. P. & Bevins, C. L. ( 1996) Proc. Natl. Acad. Sci. USA 93, 5156–5161.
6. Hancock, R. E. W. ( 1998) Exp. Opin. Invest. Drugs 7, 1354–3784.
7. Goldstein, B. P., Wei, J., Greenberg, K. & Novick, R. ( 1998) J. Antimicrob. Chemother. 42, 277–278.
8. Loury, D. J., Embree, J. R., Steinberg, D. A., Sonix, S. T. & Fiddes, J. C. ( 1999) Oral Surg. Oral Med. Oral Pathol. 87, 544–551.
9. Gamelli, R. L., He, L., Liu, H. & Ricken, J. D. ( 1998) Arch. Surg. 133, 715–720.
10. Kirikae, T., Hirata, M., Yamasu, H., Kirikae, F., Tamura, H., Kayama, F., Nakatsuka, K., Yokochi, T. & Nakano, M. ( 1998) Infect. Immun. 66, 1861–1868.
11. Bals, R., Weiner, D. J., Moscioni, A. D., Meegalla, R. L. & Wilson, J. M. ( 1999) Infect. Immun. 67, 6084–6089.
12. Chan, Y. R. & Gallo, R. L. ( 1998) J. Biol. Chem. 273, 28978–28985.
13. Sawyer, J. G., Martin, N. L. & Hancock, R. E. W. ( 1988) Infect. Immun. 56, 693–698.
14. Chertov, O., Michiel, D. F., Xu, L, Ming Wang, J., Tani, K., Murphy, W. J., Longo, D. L., Taub, D. D. & Oppenheim, J. J. ( 1996) J. Biol. Chem. 271, 2935–2940.
15. Schluesener, H. & Meyermann, R. ( 1995) J. Neurosci. Res. 42, 718–723.
16. Elsbach, P. & Weiss, J. ( 1988) in Inflammation: Basic Principles and Clinical Correlates, eds. Gallin, J. I., Goldstein, I. M. & Snyderman, R. (Raven, New York), pp. 445–470.
17. Scott, M., Yan, H. & Hancock, R. E. W. ( 1999) Infect. Immun. 67, 2006–2009.
18. Wu, M., Maier, E., Benz, R. & Hancock, R. E. W. ( 1999) Biochemistry 38, 7235–7242.
19. Zhang, L., Benz, R. & Hancock, R. E. W. ( 1999) Biochemistry 38, 8102–8111.
20. Xiong, Y., Yeaman, M. R. & Bayer, A. S. ( 1999) Antimicrob. Agents Chemother. 43, 1111–1117.
21. Matsuzaki, K., Mitani Y., Akada, K. Y., Murase, O., Yoneyama, S., Zasloff, M. & Miyajima, K. ( 1998) Biochemistry 37, 15144–15153.
22. Steinberg, D. A., Hurst, M. A., Fujii, C. A., Kung, A. H. C., Ho, J. F., Cheng, F.-C., Loury, D. J. & Fiddes, J. C. ( 1997) Antimicrob. Agents Chemother. 41, 1738–1742.
23. Heumann, D., Barras, C., Severin, A., Glauser, M. P. & Tomasz, A. ( 1994) Infect. Immun. 62, 2715–2721.
24. Mattsson, E., Rollof, J., Verhoef, J., Van Dijk, H. & Fleer, A. ( 1994) Infect. Immun. 62, 3837–3843.
25. Soell, M., Lett, E., Holveck, F., Scholler, M., Wachsmann, D. & Klein, J.-P. ( 1995) J. Immunol. 154, 851–860.
26. Soell, M., Diab, M., Haan-Archipoff, G., Beretz, A., Herbelin, C., Poutrel, B. & Klein, J.-P. ( 1995) Infect. Immun. 63, 1380–1386.
27. De Kimpe, S. J., Kengatharan, M., Thiemermann, C. & Vane, J. R. ( 1995) Proc. Natl. Acad. Sci. USA 92, 10359–10363.
28. Kengatharan, K. M., De Kimpe, A., Robson, C., Foster, S. J. & Thiemermann, C. ( 1998) J. Exp. Med. 188, 305–315.
29. Goto, H. & Nakamura, S. ( 1980) Jap. J. Exp. Med. 50, 35–43.
30. Soto, A., Evans, T. J. & Cohen, J. ( 1996) Cytokine 8, 300–304.
31. van Langevelde, P., van Dissel, J. T., Ravensbergen, E., Appelmelk, B. J., Schrijver, I. A. & Groeneveld, P. H. ( 1998) Antimicrob. Agents Chemother. 42, 3073–3078.
32. Bone, R. C. ( 1994) Arch. Intern. Med. 154, 26–34.
33. Ulevitch, R. J. & Tobias, P. S. ( 1999) Curr. Opin. Immunol. 11, 19–22.
34. DeFranco, A. L., Crowley, M. T., Finn, A., Hambleton, J. & Weinstein, S. L. ( 1998) Prog. Clin. Biol. Res. 397, 119–136.
35. Dziarski, R., Tapping, R. I. & Tobias, P. S. ( 1998) J. Biol. Chem. 273, 8680–8690.
36. Weidemann, B., Schletter, J. Dziarski, R. Kusumoto, S., Stelter, F., Rietschel, E. T., Flad, H.-D. & Ulmer, J. ( 1997) Infect. Immun. 65, 858–864.
37. Heumann, D., Glauser, M. P. & Calandra, T. ( 1998) Curr. Opin. Microbiol 1, 49–55.
38. Takeuchi, O., Hoshino, K., Kawai, T., Sanjo, H., Takada, H., Ogawa, T., Takeda, K. & Akira, S. ( 1999) Immunity 11, 443–451.
39. Gough, M., Hancock, R. E. W. & Kelly, N. M. ( 1996) Infect. Immun. 64, 4922–4927.
40. Scott, M. G., Vreugdenhil, A. C. E., Buurman, W. A., Hancock, R. E. W. & Gold, M. R. ( 2000) J. Immunol. 164, 000–000.
41. Elsbach, P. & Weiss, J. ( 1998) Curr. Opin. Immunol. 10, 45–49.
42. Elass-Rochard, E., Legrand, D., Salmon, V., Roseanu, A., Trif, M., Tobias, P. S., Mazurier, J. & Spik, G. ( 1998) Infect. Immun. 66, 486–491.
43. Scott, M. G., Gold, M. R. & Hancock, R. E. W. ( 1999) Infect. Immun. 67, 6445–6453.
44. Moore, A. J., Devine, D. A. & Bibby, M. C. ( 1994) Peptide Res. 7, 265–269.
45. Friedrich, C., Scott, M. G., Karunaratne, N., Yan, H. & Hancock, R. E. W. ( 1999) Antimicrob. Agents Chemother. 43, 1542–1548.
46. Vadiveloo, P. K. ( 1999) J. Leukocyte Biol. 66, 579–582.
47. Matsuzaki, K., Mitani, Y., Akada, K. Y., Murase, O., Yoneyama, S., Zasloff, M. & Miyajima, K. ( 1998) Biochemistry 37, 15144–15153.






