
Colloquium
Structure and function of pectic enzymes: Virulence factors of plant pathogens
Steven R. Herron*, Jacques A. E. Benen†, Robert D. Scavetta‡, Jaap Visser†, and Frances Jurnak*§
* Department of Physiology and Biophysics, University of California, Irvine, CA 92697;
† Department of Molecular Genetics of Industrial Microorganisms, Wageningen Agricultural University, Dreijenlaan 2, 6703 HA Wageningen, The Netherlands; and
‡ Department of Pharmacology, C236, University of Colorado Health Sciences Center, 4200 East Ninth Avenue, Denver, CO 80262
The structure and function of Erwinia chrysanthemi pectate lysase C, a plant virulence factor, is reviewed to illustrate one mechanism of pathogenesis at the molecular level. Current investigative topics are discussed in this paper.
P lant cell walls are primarily polysaccharide in composition. A simple but major pathogenic mechanism in plants involves degradation of the cell wall by a battery of polysaccharidases secreted by pathogens. Most of the degradative enzymes are glycoside hydrolases, which degrade the cellulose and pectate matrices by the addition of water to break the glycosidic bonds. The pectate network is also degraded by polysaccharide lyases, which cleave the glycosidic bonds via a β-elimination mechanism. To better understand the latter virulence mechanism, research has been carried out on pectate lyase C, a pectolytic enzyme secreted by the pathogenic bacterium Erwinia chrysanthemi. The story of pectate lyase C illustrates how structural techniques have contributed to a detailed understanding of polysaccharide recognition and the lyase cleavage mechanism. In the process, a novel protein structural fold and a unique catalytic role for an arginine have been discovered. The structural results have also provided the first atomic description of a pectate fragment, which differs considerably from the popular view in conformation as well as the mode of interactions with Ca2+ ions. Finally, the growing structural database of pectolytic enzymes is enabling researchers to elucidate subtle structural differences that are responsible for the specific recognition of a unique oligosaccharide sequence from a heterogeneous mix in the plant cell wall. Such knowledge will ultimately lead to a better understanding of the characteristics that render the host susceptible to attack by a particular pathogen.
Difference in Outer Barriers of Plant and Mammalian Cells
To be successful in attacking a host cell, a pathogen must pass the outer barrier of a cell. In plants, the outer barrier is the cell wall, composed primarily of polysaccharides. In mammals, the outer barrier is a membrane, composed primarily of lipids. Polysaccharides do serve a function at the mammalian membrane, either as cell surface components involved in molecular recognition or as the primary components of the gelatinous intercellular milieu. Given the prevalence of polysaccharides in the plant cell wall, it is not surprising to find that many plant pathogens secrete a battery of saccharidases as a major mode of attack. The analogous mechanism in mammalian systems is the secretion of enzymes that degrade the intercellular matrix to allow the pathogen access to the cell membrane and cellular cytoplasm.
Degradative Enzymes of Plant Cell Wall Components
The plant cell wall is composed of two types of polysaccharide matrices: the pectate network and the cellulose network (1, 2). The pectate network consists of the smooth region composed of homogalacturonans and the hairy region composed of highly branched rhamnogalacturonans. The cellulose network consists of microfibrils composed of 1-4-linked β-D-glucan units as well as xyloglucan and arabinoxylan, the two hemicelluloses that coat the cellulose microfibrils to prevent excessive aggregation. Not only does the basic saccharide unit differ in the two types of network, but the saccharide units in the pectate network also have a uronic acid or an esterified uronate moiety at the C-6 position of the saccharide units. Enzymes, which degrade the pectate network, belong to two classifications: glycoside hydrolases (3) and polysaccharide lyases (4). Glycoside hydrolases incorporate a water molecule via a general acid catalysis during the cleavage of the glycosidic bond between the two saccharide units. In contrast, polysaccharide lyases cleave the glycosidic bond via a β-elimination reaction that removes a proton. The final product contains an unsaturated bond between C-4 and C-5 of the saccharide unit at the nonreducing end. Enzymes, which degrade the cellulose network, all function as glycoside hydrolases. Generally, hydrolases have an acidic pH optima, using aspartic and glutamic acid groups during catalysis, whereas lyases have a basic pH optima, using catalytic amino acids that are still under active investigation.
The degradation of plant cell walls by secreted enzymes from pathogens has been a fertile area of research for many years (5). Although it is well known that certain pathogens are effective against specific hosts, the mechanisms of host–pathogen interactions remain elusive. In certain organisms, such as Erwinia chrysanthemi, the genetic organization and regulation of many secretory saccharidases have been elucidated (6, 7). One surprising finding is that many pathogenic organisms secrete multiple isozymes of the same enzyme but the transcription of the genes is often independently regulated. Since 1990, research has been directed at the protein level, to understand the structure and enzymatic mechanism of the degradative cell wall enzymes. Additional efforts are being made to elucidate the molecular basis for protein recognition of the composition and conformation of the individual saccharide units in the substrate. Another major question, yet to be resolved, is whether there is isozymespecific recognition of a unique polysaccharide sequence and if this specificity plays an important role in pathogen–host cell interactions. The work will be illustrated by the research on Erwinia chrysanthemi pectate lyase C (PelC).
Three-Dimensional Structures of Plant Cell Wall Degradative Enzymes
PelC is one of several isozymes secreted by E. chrysanthemi that cleave the pectate component of the plant cell wall, causing
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviation: PelC, pectate lyase C.
|
§ |
To whom reprint requests should be addressed at: Department of Physiology and Biophysics, Room 350-D Med Sci I, University of California, Irvine, CA 92697-4560. E-mail: jurnak@uci.edu. |
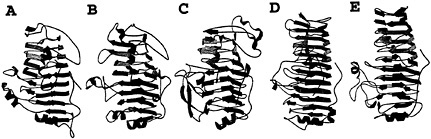
Fig.1. Five examples of plant cell wall degradative enzymes that fold into a parallel β helix motif. The predominant secondary structural features of the proteins are illustrated as β strands, and the coils represent α helices. (A) E. chrysanthemi pectate lyase C. (B) E. chrysanthemi pectate lyase E. (C) A. niger pectin lyase B. (D) E. carotovora polygalacturonase. (E) A. aculeatus rhamnogalacturonase A.
soft-rot disease in a variety of crops (8). The pelC gene has been cloned into Escherichia coli, with the latter constructs exhibiting maceration activity comparable to E. chrysanthemi (9). By using the recombinant form from E. chrysanthemi, the threedimensional structure of PelC has been determined to fold into a novel topology, not observed before 1993 or predicted during the first 30 years of crystallography (10, 11). PelC folds into a large right-handed coil termed the parallel β helix. There are eight coils in the helix, and each is comprised of three β strands, connected by three turns with unique features. When the coils are stacked, the structure has the appearance of three parallel β sheets, stabilized by an extensive network of interstrand hydrogen bonds. Another notable feature of the PelC fold is the internal organization of the amino acids that form the core of the parallel β helix. All of the amino acids, which are oriented toward the interior, are regularly aligned with amino acids from neighboring coils, giving rise to long ladders of hydrophobic, aromatic, or polar amino acids. In contrast, the exterior amino acids are randomly oriented and comprise loops, of varying length and composition, which protrude from the central core. The initial postulate that the protruding loops form the substrate and active site has proven to be correct, as will be discussed in a later section. From the perspective of an effective plant virulence factor, the most important feature of the parallel β helix fold is the stability that it confers upon an enzyme that must function in the hostile extracellular environment.
Most enzymes that degrade the plant cell wall are members of the glycoside hydrolase superfamily. Of the 62 families, organized according to sequence similarities, three-dimensional structures for members of 28 families have been reviewed (12). The enzymes that degrade the cellulose network of the plant cell wall are represented by a diversity of protein folds. The predominant structural fold, represented by endoglucanases and xylanases, is the (β/α)8 barrel. In this topology, eight parallel β strands, arranged in a circular, barrel-like motif, are connected by α helices that cover the exterior of the β barrel. The polysaccharide substrate site is located at the carboxy terminal base of the barrel, with the catalytic residues often found on β strands 4 and 7 (13). The structures of other cellulases fold into antiparallel β motifs as well as into domains comprised entirely of α helices. For example, cellobiohydrolase folds into a distorted antiparallel β barrel, sometimes called a β sandwich (14). Loops of different lengths and conformations fold over one face of the β sandwich and form the substrate-binding tunnel. Another example is endoglucanase CelD from Clostridium thermocellum, a cellulase that is structured into a motif best described as an (α/α)6 barrel (15). In contrast to the cellulase and hemicellulase families, the enzymes that degrade the pectate network all share the same parallel β helix topology initially found in PelC (Fig. 1). These include two additional pectate lyases, E. chrysanthemi PelE (16), Bacillus subtilis Pel (17); two pectin lyases from Aspergillus niger, PLA (18) and PLB (19); one rhamnogalacturonase, Aspergillus aculeatus RGase A (20); and two polygalacturonases, Erwinia carotovora polygalacturonase (21) and A. niger endopolygalacturonase II (22). Although the mechanism of pectic cleavage differs for the hydrolases and the lyases, the substrate binding sites are all found in a similar location within a cleft formed on the exterior of the parallel β helix. Moreover, parallel β helix folds, with analogous substrate-binding clefts, have been found in enzymes that degrade oligosaccharides found on cell surface receptors, such as P22 tailspike endorhamnosidase (23), and in the intercellular matrix, such as chondroitinase B (24).
Structural Approaches to the Elucidation of the Enzymatic Mechanism of PelC
Before 1990, publication of a plethora of biochemical data usually preceded the publication of a three-dimensional structure. The two types of information are mutually beneficial. A structure explains the biochemical data and the biochemical data helps to elucidate the active site of an enzyme or the functionally relevant parts of a protein. However, in the last decade, threedimensional structures have been determined at such a rapid pace, as a consequence of dramatically improved x-ray diffraction and NMR techniques, that it is now more common to solve a structure, in the absence of any supporting biochemical data. Thus, new strategies are being developed to extract functional information from static three-dimensional images. PelC has been among the first group of protein structures solved, without the benefit of relevant biochemical data to interpret the functional aspects of the three-dimensional structure. When the PelC structure was reported, details of its enzymatic mechanism could be summarized in the following few sentences. PelC is composed of 353 amino acids, with a molecular mass of 37,676 daltons and two disulfide bonds (9). As shown in Fig. 2, PelC catalyzes the endo- and exolytic cleavage of the α-1-4 glycosidic bond of polygalacturonic acid, generating an unsaturated trimer end product (25). Ca2+ is essential for the in vitro reaction and is likely required in the in vivo reaction in the plant cell wall in which the Ca2+ concentration is estimated to be as high as 1.0 mM. PelC lyase activity can be detected from a pH of 6.2 to a pH of 11.2, with an optimal pH at 9.5 (26). No amino acids, which participate in catalysis or substrate binding, had been identified by classical techniques. Consequently, neither the active site nor the substrate-binding pocket could be deduced with certainty from the initial PelC structure. The native PelC structure does provide two clues about the location of the region involved in catalysis (10). First, a Ca2+-like, heavy atom derivative binds in
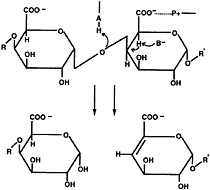
Fig. 2. Schematic diagram of α-1,4-polygalacturonic acid cleavage by a β-elimination mechanism. The pectate lyases are expected to contribute a minimum of three groups to the catalytic mechanism: P+, which neutralizes the charge on the carboxylic acid group; B, a general base that abstracts the proton from C-5; and A, which is involved in the transfer of the final proton to the glycosidic oxygen, leaving a double bond between C-4 and C-5.
a site that has coordination compatible with a Ca2+ binding site. This has been the first indication that the essential Ca2+ might bind directly to the protein, rather than to the polygalacturonic acid substrate, as previously believed. The hypothesized Ca2+ binding site was ultimately shown to be analogous to the Ca2+ site captured directly in the B. subtilis Pel structure (17). The second revelation is that all surface charges are localized to an elongated groove surrounding the Ca2+ site. The shape, length, and charge of the groove are complementary to a negatively charged, oligogalacturonic moiety, suggesting the region was the substrate-binding pocket.
The next insight into the enzymatic mechanism has occurred with the report of the PelE structure (16). PelE is one of the E. chrysanthemi isozymes but belongs to a different subfamily of pectate lyases. The PelE subfamily is characterized by a single disulfide bond and cleaves the polygalacturonic acid substrate to a dimer. PelE shares 22% sequence identity with PelC. Not surprisingly, PelE folds into the same parallel β helix topology and has a similar Ca2+ binding pocket. However, in contrast to PelC, the charged amino acids on PelE are randomly distributed on the surface and are not localized into an elongated groove. This finding suggests that either the active site region has been deduced incorrectly from the PelC structure or that the optimal in vivo substrate for PelE is not the same as for PelC. To extract additional clues about the location of the catalytic site, the structures of PelC and PelE have been superimposed, and the similarities as well as the differences have been analyzed. In general, key amino acids that are essential to a structural or functional property of an enzyme are invariant and the region around an active site is structurally conserved. The superposition of the PelC and PelE structures has permitted the correction of the evolutionary-based amino acid sequence alignment for the two proteins as well as for the entire superfamily (27). The superfamily includes 14 extracellular pectate lyases, 7 pectin lyases, and 12 plant pollen and style proteins. The corrected sequence alignment has identified 10 invariant amino acids, 5 of which are amino acids that could potentially be involved in catalytic activity. In PelC nomenclature, these five potentially catalytic amino acids include Asp-131, Asp-144, His-145, Thr206, and Arg-218. The five chemically inert amino acids include Gly-6, Gly-12, Gly-13, Trp-142, and Pro-220. With most enzymes, the invariant amino acids cluster around the same general region, that of the active site. However, the situation is different with PelC. The 10 invariant amino acids cluster in two distinctly different regions (Fig. 3). Asp-131, Arg-218, and Pro-220 are grouped around the Ca2+ binding site that has been postulated to be part of the active site. The other seven amino acids cluster on the opposite side of the parallel β helix and are too distant to be part of a catalytic site near the Ca2+ ion. Instead of identifying the active site, the cluster analysis has posed new questions. If PelC has two active sites, which one is responsible for pectolytic activity and what is the function of the second active site?
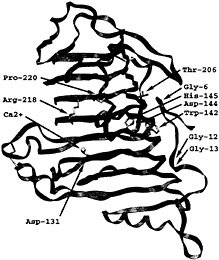
Fig. 3. Stereoview of the PelC locations of 10 invariant amino acids within the extracellular pectate lyase superfamily. The two clusters of invariant amino acids are located on opposite sides of the parallel β helix, separated by approximately 25 Å across the diameter and by approximately 90 Å around the circumference of the parallel β helix. The α-carbon backbone of PelC is illustrated as a green ribbon and the Ca2+ ions as yellow spheres. The invariant amino acids are labeled at the α-carbon and are represented by rods using the International Union of Pure and Applied Chemistry coloring code: carbon atoms are gray; oxygen atoms, red; and nitrogen atoms, blue.
Site-directed mutagenesis has been used to conclusively identify the pectolytic active sites (28). Both invariant and highly conserved amino acids have been targeted for mutations. Moreover, several mutations have been made at each position. In all, 32 PelC mutants have been prepared, purified, and characterized. Those amino acids clustering around the Ca+2 site have been shown to simultaneously affect maceration of plant tissue and pectolytic activity. Moreover, all mutations in two amino acids, Asp-131 and Arg-218, reduce the pectolytic activity to less than 0.5% of the wild type, suggesting that these amino acids have some type of catalytic role. Mutations in the invariant amino acids in the second cluster, including Trp-142, Asp-144, His-145, and Thr-206, have also been prepared, but most of the mutants cannot be purified in high yields. From preliminary experiments, it appears that most of these mutant proteins remain bound to the membrane fraction and are in an unfolded state, as demonstrated by their sensitivity to trypsin. Only very
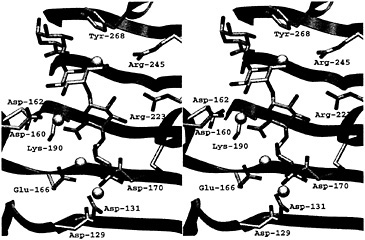
Fig. 4. Stereoview of the active site of the PelC R218K-(Ca2+)4-pentaGalpA complex. The color code is the same as that used in Fig. 3.
small quantities of W142H, D144N, and T206A could be purified and characterized. The latter mutants retain pectolytic activity, demonstrating that the second cluster of invariant amino acids is not involved in the enzymatic cleavage of a polysaccharide. The function of the second cluster remains a mystery.
Structure of the PelC R218K-(Ca2+)4-pentaGalpA Complex
With the active site of PelC now identified, attention has turned toward the elucidation of the atomic details of the protein– saccharide interactions in the substrate binding site and the details of the enzymatic mechanism. In an ideal world, a crystallographer prefers to study the structure of a complex between a native protein and a transition state analogue or an inhibitor, to work out atomic details of the active site of the enzyme. Unfortunately, no efficient inhibitors of the pectate lyases are known. Therefore, an alternate approach, that of forming complexes between catalytically impaired mutants of PelC and an oligosaccharide fragment, has been used. X-ray diffraction data for several mutant PelC-oligosaccharide complexes have been collected. However, only one data set, using the R218K PelC mutant and pentagalacturonic acid (pentaGalpA), has yielded a view of an ordered oligosaccharide composed of four of the five GalpA units (29). The PelC mutantoligosaccharide structure held many new surprises (Fig. 4). First, instead of finding only one Ca2+ ion, as in the native PelC structure, a total of four Ca2+ sites have been found. All Ca2+ ions are bridging acidic groups on the protein to uronic acid moieties on the oligosaccharide. One Ca2+ ion also forms a link between two adjacent uronic acid moieties. The Ca2+-oligosaccharide interactions are substantially different from the popular “eggbox” model in which Ca2+ ions cross-link the uronic moieties of adjacent chains of pectate together in the plant cell wall (1). The second surprising feature is that the conformation of the tetraGalpA fragment is different from the pectate conformation previously predicted (30, 31). Instead of having a 21 or 31 helical conformation, the pectate fragment is a mixture of both 21 and 31 helices. Third, the complex represents an enzymesubstrate interaction and thus provides a detailed view of a Michaelis intermediate in the reaction pathway. The scissile bond has been assigned to the glycosidic bond between the third and fourth GalpA units from the reducing end of the ordered tetrasaccharide by coupling the structural results with enzymatic cleavage patterns of oligogalacturonates of defined lengths. In the substrate cleft, all invariant and conserved amino acids in the pectate lyase family could be assigned either a role in catalysis or a role in specific recognition of the Gal pA units. The GalpA units on either side of the scissile bond form the most interactions with the protein. A conserved Arg-223 recognizes the galactose epimer and a Ca2+ network surrounds each uronic acid group. The invariant aspartic acid, Asp-131 in PelC, is instrumental in coordinating strongly to the primary Ca2+, which in turn forms a linked network with other Ca2+ ions and uronic acid moieties. The most surprising finding is that Arg-218, invariant in the pectate lyase superfamily, is the amino acid that initiates proton abstraction from C-5 in the GalpA unit adjacent to the scissile bond. Such a role for an arginine has not been observed previously but is consistent with the high pH optimum of the in vitro pectolytic reaction. That arginine can serve as a proton abstractor is made possible by the local electronic environment surrounding C-5 and the adjacent uronic acid moiety. As predicted by Gerlt and Gassman (32), positive charges, in the form of a Ca2+ ion and a conserved lysine, Lys-190, neutralize the uronic acid group and stabilize an enolic intermediate. Together, these positive charges lower the pKa of the C-5 proton to levels that approximate the pKa of an arginine. Overall, the structure of the R218K-(Ca2+)4-pentaGalpA complex provides many details about the enzymatic reaction as well as the nature of the protein–saccharide interaction. Yet there remain unanswered questions that can best be addressed with structural techniques. One major puzzle is the identification of the group responsible for transferring a proton to the glycosidic bond, simultaneous with bond cleavage. The answers must await structures of new complexes that mimic alternate stages of the reaction.
Significance of Multiple Isozymes for Pathogenesis
One enigma, yet to be resolved in the pectate lyase field, is the function of multiple, independently regulated pectate lyase isozymes and the role, if any, in pathogenesis. The composite results of the structural studies suggest that the answer may lie in the heterogeneous nature of the substrate. The pectate
component is composed of repeating units of negatively charged galacturonic acid. The uronic acid group on C-6 is frequently esterified with a methyl group. Methylation neutralizes the negative charge of an individual GalpA unit and, consequently, alters the surface charge of the pectate polymer. The percentage and the positional sequence of methylated GalpA units (mGalpA) vary during the life cycle of the plant and from one type of plant to another. Thus, a pathogen that secretes a battery of pectic enzymes, each of which uses a similar catalytic mechanism, but recognizes a different sequence of methylated and nonmethylated oligogalacturonate units, would be expected to have a broader host range. The structural studies provide tentative evidence that the pectate lyase isozymes cleave the same in vitro substrate but differ in the preferred in vivo substrate. In the majority of enzyme families, the most conserved region, in terms of primary and tertiary structure, is the active site and the surrounding region. The pectate lyase family appears to be somewhat different. Comparisons of the pectate lyase structures have revealed that, although the immediate region around the catalytic site is highly conserved, the substrate groove extending on either side is structurally the most diverse (16). The diversity arises from the variation in the sequence and length of the loops protruding from the parallel β barrel. One set of loops, termed the T3 loops, form the binding pockets required for recognition of multiple saccharide units in the substrate. The surface charges in PelC are localized to an elongated groove that complements the shape and length of an oligosaccharide composed of repeating GalpA units (10). In contrast, the analogous region in PelE is highly charged only around the primary Ca2+ binding site, with a predominance of neutral groups lining the remaining portion of the groove (16). Analogous differences are found in three-dimensional models constructed from the sequences of other pectate lyase isozymes. Thus, polygalacturonic acid may serve as a suitable in vitro substrate for all isozymes because the saccharide binding pockets near the active site recognize only galacturonic acid moieties. The structural differences between PelC, PelE, and other isozymes at more distant subsites suggest that the optimal in vivo substrate may differ in length, charge, and possibly type of saccharide unit from one isozyme to another. Differences among isozyme substrates need not be as great as a different type of saccharide unit. Smaller differences, such as the degree of methylation of the pectate component, may be sufficient to explain the physiological significance of multiple pectate lyase isozymes.
Model of the PLB-mGalpA4Complex
Given the difficulty of obtaining defined lengths and specific methylated sequences, it will be difficult to ascertain the optimal substrate of each pectic isozyme through structural studies of additional complexes. Instead, some information may be gleaned from modeling saccharide units in substrate subsites. The first step is to elucidate the structural determinants that are necessary to distinguish between a methylated and a nonmethylated uronic acid moiety on GalpA units. This goal can be achieved by comparing the substrate subsites in PelC with those in a pectin lyase. Pectin lyases share a similar enzymatic mechanism with the pectate lyases but recognize a different substrate, that of pectin, the fully methylated, neutral form of pectate. Given the sequence and functional similarities, it is not surprising that the two known pectin lyase structures, PLA (18) and PLB (19), are homologous to the pectate lyases. However, no structural complexes containing pectin are currently available. To answer the question about the atomic determinants of methylation, the PLB structure has been superimposed upon the R218K-(Ca2+)4-pentaGalpA structure, minimizing the atomic distances between all conserved and invariant amino acids shared by the two structures. The superposition has allowed for a feasible placement of an oligosaccharide substrate in the PLB substrate. The Ca2+ ions from the R218L model have been removed, and methyl groups have been added to the uronate moiety. The contacts between PLB and the modeled pectin fragment have been visually and energetically optimized to avoid unfavorable contacts. The resulting model is shown in Fig. 5 and Fig. 6; a schematic representation of the modeled PLB-pectin contacts is shown in Fig. 7. Three striking features emerge from the model. First, there is no ambiguity in positioning the methylated uronate group. For three of the four mGalpA units, a hydrophobic pocket, large enough to accommodate a methyl group, exists around one oxygen of the uronate moiety and a polar environment surrounds the other oxygen. The hydrophobic pockets surrounding the methyl groups are composed primarily of tryptophan and tyrosine residues, the unusual cluster of which was noted in the original report of the PLB structure. The second notable feature of the model is the conservation of a positive charge between the uronate moieties of the third and four saccharide units. In pectin lyase, the positive charge is conferred by Arg-176, a conserved arginine in the pectin lyase subfamily. In pectate lyases, the positive charge is conferred by a Ca2+ ion, which is essential for catalytic activity within the pectate lyase subfamily. The high degree of conservation of the positive charge at analogous positions in the structures suggests that the positive charge is likely to play a significant role during catalysis, possibly stabilizing a negative intermediate or participating in the transfer of a proton to the glycosidic bond. The third notable feature is confirmation of the roles of the three amino acids that are invariant in the pectate lyase superfamily: Asp-154, Arg-236, and Pro-238 in PLB nomenclature. As in the pectate lyases, Pro-238 is found in the unusual cis-conformation, which helps to orient Arg-236 toward the α proton on C-5 of the saccharide unit adjacent to the scissile bond. Although it is unusual, the involvement of Arg-236 in a catalytic step has been confirmed by site-specific mutational data presented in Table 1. Both R236Q and R236K PLB mutants are catalytically inactive. The invariant aspartic acid, Asp-154, does not have a catalytic role but, rather, as suggested from the structures, appears to stabilize the position of a positive charge near the scissile bond. The lack of a catalytic role for the invariant aspartic acid has also been confirmed by
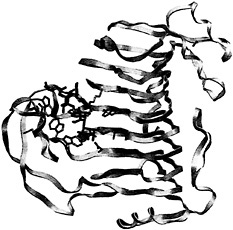
Fig. 5. Overview of the PLB-(mGalpA)4 model. Pectin lyase B is shown as yellow ribbons. The substrate and the interacting amino acids are represented by rods using the color code described in Fig. 3.
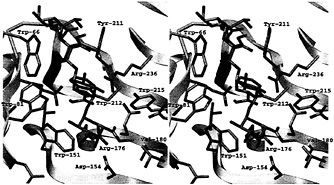
Fig. 6. Stereoview of the active site of the PLB-(mGalpA)4 model. The view of the active site is the same as that used in Fig. 4 to allow for comparisons. The hydrophobic pockets around three of the four methyl groups esterified to the uronate moieties are visible. The color code is the same as that used in Fig. 6.
mutational data (Table 1). Both PLB mutants, D154E and D154N, retain significant catalytic activity. The role of Arg-176 remains to be confirmed through mutational experiments.
Conclusions
The story of PelC, although incomplete, illustrates how structural information can contribute to an understanding of the multifaceted steps of pathogenesis. In the process, a novel protein structural fold and a unique catalytic role for an arginine have been discovered. The structural results have also provided the first atomic description of a pectate fragment, finding the conformation as well as the mode of interactions with Ca2+ ions to be considerably different from the popular view. The growing structural databases of pectolytic enzymes should allow researchers to elucidate the structural motifs that recognize specific types of saccharides. The initial attempt to elucidate the structural principles that permit an enzyme to distinguish between pectate and pectin is the first step in understanding how saccharidases recognize a unique oligosaccharide sequence from a heterogeneous mix in the plant cell wall. Such knowledge should ultimately lead to a better understanding of the unique properties of the host that render it more vulnerable to attack by a given pathogen.
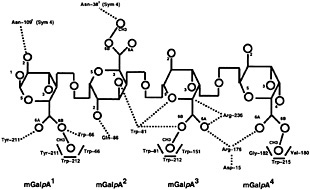
Fig. 7. Schematic representation of the pectin lyase B with (GalpA)4 at a distance of ≤3.0 Å. mGalpA (1) is the reducing saccharide and mGalpA (4) is the nonreducing terminus. Hydrogen bonds are represented with dotted lines and hydrophobic interactions, with boldface lines. Oxygen atoms and methyl groups are represented by circles, with the corresponding number, and the carbon atoms are assumed at the intersection of bonds designated in boldface lines.
Methods
PLB-(mGalpA)4Modeling. Amino acids in PLB, corresponding to the invariant amino acids in the pectate lyase superfamily, were superimposed upon their counterparts in the structure of the PelC R218K-(Ca2+)4-pentaGalpA complex. The superposition was improved by LSQMAN (33). The Ca2+ ions were removed from the R218K structure, and methyl groups were added to one of the uronate oxygens. AUTODOCK was used to maximize the favorable interactions and to allow carbohydrate flexibility (34). The pectin substrate was first introduced into the PLB active cleft as two dimers to improve the fit. Then, the two fragments were rejoined and subjected to several rounds of energy minimization. All model figures were prepared with SETOR (35).
Table 1. Specific activities and kinetic parameters of wild-type and mutated PLB
|
Enzyme |
Specific activity, units/mg |
Km, mg/ml |
Vmax, units/mg |
pH optimum |
|
PLB wild type |
698 |
6.3 |
1,429 |
8.5 |
|
PLB D154E |
306 |
7.8 |
1,143 |
8.5 |
|
PLB D154N |
91 |
14.8 |
512 |
>9.5 |
|
PLB R236Q |
0 |
– |
– |
|
|
PLB R236K |
1.4 |
– |
– |
8.5 |
DNA Manipulations. Cloning experiments for the preparation of PLB mutants were performed in E. coli DH5α (36) by using standard protocols (37). Restriction enzymes were used as described by the supplier (GIBCO/BRL). Nucleotide sequences were determined by using a Cy5 AutoCycle Sequencing Kit (Amersham Pharmacia) with universal and reverse primers or gene specific primers. The reactions were analyzed with an ALFred DNA sequencer (Amersham Pharmacia). Computer analysis was done by using the program GENERUNNER (Hastings Software). The cloning of A. niger N400 pelB PLB was described previously (38). A translational promoter gene fusion was constructed by using the pyruvate kinase promoter (39). The pki-pelB fusion (pPK-PLB) was described previously (38).
Site-Directed Mutagenesis. Site-directed mutagenesis of PLB was carried out by using the Altered Sites II kit (Promega) and synthetic oligonucleotides (Isogen, Maarsen, The Netherlands). The procedure was performed as described (40). The pki-pelB promoter gene fusion was excised with pPK-PLB by using restriction endonucleases BamHI and HindIII and was ligated into BamHI- and HindIII-digested pALTER I, resulting in plasmid pIM3550. Plasmid DNA was isolated and sequenced to confirm the desired mutations and to check the gene for undesired mutations. Those plasmids showing the correct sequence and the expected mutation were used to transform A. niger strain NW1888 (cspAI, pyrA6, leu-13, prtF28), a derivative of A. niger N400 (CBS. 120.49). Transformations were carried out as described (41).
Culture Conditions and Enzyme Purifications. Mutant PLB producing transformants were selected by growing individual transformants in minimal medium as described (42). Large scale cultivation of transformants producing mutant PLB was performed as outlined (43) in multiple 300 milliliter batches in one liter Erlenmeyer flasks incubated in an orbital shaker (250 revolutions per minute) at 30 °C. Wild-type and mutant PLB were purified essentially as described by Kester and Visser (43) with the following modification. After dialysis of the solubilized ammonium sulfate precipitate, the dialysate was loaded onto a Source Q anion exchanger (15.5-milliliter bed volume) (Pharmacia) preequilibrated with 10 mM Tris·HCl (pH 7.5). Proteins were eluted with a 0–200 mM sodium chloride linear gradient in 10 mM Tris ·HCl (pH 7.5). PLB-containing fractions were pooled and exhaustively dialyzed against 20 mM sodium phosphate (pH 6.0). Enzyme solutions were stored at − 20°C. The purity of the mutant enzyme was confirmed by SDS/PAGE and Coomassie brilliant blue staining. The concentration of purified mutant enzymes was determined by measuring the absorbance at a wavelength of 280 nm, using a molar absorption coefficient ( ε) of 50,220 M−1·cm−1 for PLB, as calculated from the tryptophan, tyrosine, and cysteine content (44).
Enzyme Assay and Determination of Kinetic Parameters. Standard PLB assays were carried out in 50 mM Tris·HCl and 0.06 M sodium chloride (pH 8.5), containing 3 mg/ml (wt/vol) lime pectin with 75% methyl esterification (Copenhagen Pectin Factory, Lille Skensved, Denmark) in a total volume of 1.0 ml. The assay buffer was equilibrated at 30°C, and the reaction was started by the addition of 20 µl of enzyme solution. The activity was determined by measuring the increase in absorbance at 235 nm (ε = 5200 M−1·cm−1). The kinetic traces were corrected for spontaneous chemical β-elimination. Km and Vmax values were determined from triplicate initial rate measurements in the same way as described for the standard assays, with the exception that the pectin concentrations varied from 0.5 to 7.0 mg/ml.
The research was supported by the U.S. Department of Agriculture (Grant 98-35304). The research was conducted in part at the Stanford Synchrotron Radiation laboratory, which is operated by the Office of Basic Energy Science of the U.S. Department of Energy.
1. Carpita, N. C. & Gibeaut, D. M. ( 1993) Plant J. 3, 1–30.
2. Albersheim, P., Darvill, A., O'Neill, M., Schols, H. A. & Voragen, A. G. J. ( 1996) in Progress in Biotechnology: Pectins and Pectinases, eds. Visser, J. & Voragen, A. G. J. (Elsevier, Amsterdam), Vol. 14, pp. 47–53.
3. Davies, G. & Henrissat, B. ( 1995) Structure ( London) 3, 853–859.
4. Kiss, J. ( 1974) Adv. Carbohydr. Chem. Biochem. 29, 229–230.
5. He, S. Y., Lindeberg, M. & Collmer, A. ( 1993) in Biotechnology in Plant Disease Control ed. Chet, I. (Wiley–Liss, New York), pp. 39–64.
6. Reverchon, S., Nasser, W. & Robert-Baudouy, J. ( 1991) Mol. Microbiol. 5, 2203–2216.
7. Tamaki, S. J., Gold, S. Robeson, M., Manulis, S. & Keen, N. T. ( 1988) J. Bacteriol. 170, 3468–3478.
8. Barras, F., Van Gijsegem, F. & Chatterjee, A. K. ( 1994) Annu. Rev. Phytopathol. 32, 201–234.
9. Keen, N. T., Dahlbeck, D., Staskawicz, B. & Belser, W. ( 1984) J. Bacteriol. 159, 825–831.
10. Yoder, M. D., Keen, N. T. & Jurnak, F. ( 1993) Science 260, 1503–1507.
11. Yoder, M. D., Lietzke, S. E. & Jurnak, F. ( 1993) Structure ( London) 1, 241–251.
12. Davies, G. & Henrissat, B. ( 1995) Structure ( London) 3, 853–859.
13. Jenkins, J., Leggio, L. L., Harris, G. & Pickersgill, R. ( 1995) FEBS Lett. 362, 281–285.
14. Divne, C., Ståhlberg, J., Teeri, T. T. & Jones, T. A. ( 1998) J. Mol. Biol. 275, 309–325.
15. Juy, M., Amit, A., Alzari, P., Poljak, R. J., Claeyssens, M., Bguin, P. & Aubert, J.-P. ( 1992) Nature ( London) 357, 89–91.
16. Lietzke, S. E., Keen, N. T., Yoder, M. D. & Jurnak, F. ( 1994) Plant Physiol. 106, 849–862.
17. Pickersgill, R., Jenkins, J., Harris, G., Nasser, W. & Robert-Baudouy, J. ( 1994) Nat. Struct. Biol. 1, 717–723.
18. Mayans, O., Scott, M., Connerton, I., Gravesen, T., Benen, J., Visser, J., Pickersgill, R. & Jenkins, J. ( 1997) Structure ( London) 5, 677–689.
19. Vitali, J., Schick, B., Kester, H. C. M., Visser, J. & Jurnak, F. ( 1998) Plant Physiol. 116, 69–80.
20. Petersen, T. N., Kauppinen, S. & Larsen, S. ( 1997) Structure ( London) 5, 533–544.
21. Pickersgill, R., Smith, D., Worboys, K. & Jenkins, J. ( 1998) J. Biol. Chem. 273, 24660–24664.
22. van Santen, Y., Benen, J. A. E., Schröter, K. H., Kalk, K. H., Armand, S., Visser, J. & Dijkstra, B. W. ( 1999) J. Biol. Chem. 274, 30474–30480.
23. Steinbacher, S., Seckler, R., Miller, S., Steipe, B., Huber, R. & Reinemer, P. ( 1994) Science 265, 383–386.
24. Huang, W., Matte, A., Li, Y., Kim, Y. S., Linhardt, R. J., Su, H. & Cygler, M. ( 1999) J. Mol. Biol. 294, 1257–1269.
25. Preston, J. F., Rice, J. D., Ingram, L. O. & Keen, N. T. ( 1992) J. Bacteriol. 174, 2039–2042.
26. Tardy, F., Nasser, W., Robert-Baudouy, J. & Hugouviewx-Cotte-Pattat, N. ( 1997) J. Bacteriol. 179, 2503–2511.
27. Henrissat, B., Heffron, S. E., Yoder, M. D., Lietzke, S. E. & Jurnak, F. ( 1995) Plant Physiol. 107, 963–976.
28. Kita, N., Boyd, C. M., Garrett, M. R., Jurnak, F. & Keen, N. T. ( 1996) J. Biol Chem. 271, 26529–26535.
29. Scavetta, R. D., Herron, S. R., Hotchkiss, A. T., Kita, N., Keen, N. T., Benen, J. A., E., Kester, H. C. M., Visser, J. & Jurnak, F. ( 1999) Plant Cell 11, 1081–1092.
30. Morris, E. R., Powell, D. A., Gidley, M. J. & Rees, D. A. ( 1982) J. Mol. Biol. 155, 507–533.
31. Walkinshaw, M. D. & Arnott, S. ( 1981) J. Mol. Biol. 153, 1055–1085.
32. Gerlt, J. A. & Gassman, P. G. ( 1992) J. Am. Chem. Soc. 114, 5928–5934.
33. Kleyegt, G. J. & Jones, T. A. ( 1995) Structure ( London) 3, 535–540.
34. Morris, G. M., Goodsell, D. S., Huey, R. & Olson, A. J. ( 1996) J. Comput. Aided Mol. Des. 10, 293–304.
35. Evans, S. V. ( 1993) J. Mol. Graphics 11, 134–138.
36. Woodcock, D. M., Crowther, P. J., Doherty, J., Jefferson, S., DeCruz, E., Noyer-Weidner, M., Smith, S. S., Michael, M. Z. & Graham, M. W. ( 1989) Nucleic Acids Res. 17, 3469–3478.
37. Sambrook, J., Fritsch, E. F. & Maniatis, T. ( 1989) in Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 1–125.
38. Kusters-van Someren, M., Flipphi, M., de Graaff, L. H., van den Broeck, H., Kester, H. C. M., Hinnen, A. & Visser, J. ( 1992) Mol. Gen. Genet. 234, 113–120.
39. de Graaff, L. H., van den Broeck, H. & Visser, J. ( 1992) Curr. Genet. 22, 21–27.
40. Armand, S., Wagemaker, M. J., Sánchez-Torres, P., Kester, H. C. M., van Santen, Y., Dijkstra, B. W., Visser, J. & Benen, J. A. E. ( 2000) J. Biol. Chem. 275, 691–696.
41. Kusters-van Someren, M. A., Harmsen, J. A. M., Kester, H. C. M. & Visser, J. ( 1991) Curr. Genet. 20, 293–299.
42. Parenicová, L., Benen, J. A. E., Kester, H. C. M. & Visser, J. ( 1998) Eur. J. Biochem. 251, 72–80.
43. Kester, H. C. M. & Visser, J. ( 1994) FEMS Microbiol. 120, 63–68.
44. Edelhoch, H. ( 1967) Biochemistry 6, 1948–1954.








