
Colloquium
Molecular and cell biology aspects of plague
Guy R. Cornelis*
Microbial Pathogenesis Unit, Christian de Duve Institute of Cellular Pathology, and Faculté de Médecine, Université Catholique de Louvain, Avenue Hippocrate, 74, UCL 74.49, B-1200 Brussels, Belgium
A 70-kb virulence plasmid (sometimes called pYV) enables Yersinia spp. to survive and multiply in the lymphoid tissues of their host. It encodes the Yop virulon, a system consisting of secreted proteins called Yops and their dedicated type III secretion apparatus called Ysc. The Ysc apparatus forms a channel composed of 29 proteins. Of these, 10 have counterparts in almost every type III system. Secretion of some Yops requires the assistance, in the bacterial cytosol, of small individual chaperones called the Syc proteins. These chaperones act as bodyguards or secretion pilots for their partner Yop. Yop proteins fall into two categories. Some are intracellular effectors, whereas the others are “translocators” needed to deliver the effectors across the eukaryotic plasma membrane, into eukaryotic cells. The translocators (YopB, YopD, LcrV) form a pore of 16–23 Å in the eukaryotic cell plasma membrane. The effector Yops are YopE, YopH, YpkA/YopO, YopP/YopJ, YopM, and YopT. YopH is a powerful phosphotyrosine phosphatase playing an antiphagocytic role by dephosphorylating several focal adhesion proteins. YopE and YopT contribute to antiphagocytic effects by inactivating GTPases controlling cytoskeleton dynamics. YopP/YopJ plays an anti-inflammatory role by preventing the activation of the transcription factor NF-KB. It also induces rapid apoptosis of macrophages. Less is known about the role of the phosphoserine kinase YopO/YpkA and YopM.
bacterial pathogenesis | Yersinia | Yops | translocation | type III secretion
Y ersinia pestis has in the past caused social devastation on a scale unmatched by any other infectious agent. Although it is presently not a major public health problem, there are still at least 2,000 cases of plague reported annually, and plague has recently been recognized as a re-emerging disease by the World Health Organization. The pathogenicity of Yersinia results from its impressive ability to overcome the defenses of the mammalian host and to overwhelm it with massive growth. Multiplication of Y. pestis is largely extracellular (1). In infected mice, significant levels of interferon γ (IFN-γ) and tumor necrosis factor α (TNF-α) arise only just before death. In contrast, prompt and marked synthesis of these cytokines is observed upon infection with avirulent strains (2). All these observations suggest that the pathogenicity arsenal of Y. pestis protects the bacterium from phagocytosis and slows down the onset of the inflammatory response. The closely related food-borne pathogens Yersinia pseudotuberculosis and Yersinia enterocolitica cross the intestinal barrier and multiply in the abdominal lymphoid tissues. Although they cause infections that are generally self-limited, they share with Y. pestis the Yop virulon, the core of the Yersinia pathogenicity arsenal. This Yop virulon allows extracellular Yersinia docked at the surface of a host cell to inject specialized proteins, called Yops, across the plasma membrane. The injected Yops disturb the dynamics of the cytoskeleton and block the production of pro-inflammatory cytokines, thereby favoring the survival of the invading Yersinia. The Yop virulon is thus a complex weapon for close combat with cells of the immune system (for an exhaustive review see ref.3). It is the archetype of the so-called “type III secretion” virulence mechanisms now identified in more than a dozen major animal or plant pathogens (for review see ref.4).
A Device to Inject Bacterial Proteins Across Eukaryotic Cell Membranes
The Yersinia Ysc Secretion Apparatus. “Yop secretion” was discovered around 1990 by trying to understand the mysterious phenomenon of Ca2+ dependency: when incubated at 37°C in the absence of Ca2+ ions, Yersinia bacteria do not grow but, instead release large amounts of proteins called Yops into the culture supernatant (5). Although it is generally referred to as Yop “secretion,” it is not a physiological secretion but rather a massive leakage resulting from the artificial opening of an otherwise tightly controlled delivery apparatus. Despite the fact that it is presumably artifactual, this observation turned out to be of paramount importance because it allowed the genetic analysis that led to the identification of 29 ysc (Yop secretion) genes involved in the process of Yop release.
Among the 29 Ysc proteins, 10 (YscC, -J, -N, -O, -Q, -R, -S, -T, -U, and -V) appear to have counterparts in almost every type III secretion apparatus. YscC belongs to the family of secretins, a group of outer membrane proteins involved in the transport of various macromolecules and filamentous phages across the outer membrane. Similar to other secretins, it forms a ring-shaped structure with an external diameter of about 200 Å and an apparent central pore of about 50 Å (6). At least one disulfide bond is essential for its assembly (7), and its proper insertion in the outer membrane requires the presence of an ancillary lipoprotein called YscW (6). Four proteins (YscD, -R, -U, and -V, formerly called LcrD) have been shown, and two other proteins (YscS and -T) have been predicted to span the inner membrane. The secretion process absolutely requires YscN, a 47.8-kDa protein with ATP-binding motifs (Walker boxes A and B) resembling the β catalytic subunit of F0F1 proton translocase and related ATPases (8). YscJ is a lipoprotein that has not been localized yet, but its counterpart in Pseudomonas syringae has been shown to span the inner and outer membranes (9). Little is known about the YscL, YscQ, and Ysc proteins, which are less conserved. Finally, the two proteins YscO and YscP are themselves released upon Ca2+ chelation, suggesting that they belong to the external part of the apparatus (10, 11, 69). Fig. 1 summarizes current knowledge of the Yop virulon.
Assembly of the bacterial flagellum also involves a type III secretion system. This system has no secretin but it has counterparts to the nine other conserved Ysc proteins (YscJ, -N, -O, -Q, -R, -S, -T, -U, and -V). All these proteins belong to the most internal part of the basal body—i.e., the MS ring, the C ring, and the ATPase (reviewed in ref.12), which is in
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviation: TNF-α, tumor necrosis factor α.
|
* |
E-mail: cornelis@mipa.ucl.ac.be. |
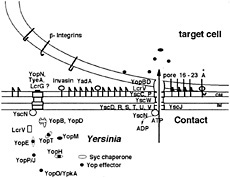
Fig. 1. The Yop virulon. When Yersinia are placed at 37°C in a rich environment, the Ysc secretion channel is installed. Proteins YscD, -R, -S, -T, -U, and -V are localized in the inner membrane (IM), whereas YscC and YscP are exposed at the bacterial surface. Lipoprotein YscW stabilizes YscC. YscN belongs to the family of ATPases. A stock of Yop proteins is synthesized, and some of them are capped with their specific Syc chaperone. As long as there is no contact with a eukaryotic cell, a stop-valve, possibly made of YopN, TyeA, and LcrG, blocks the Ysc secretion channel. On contact with a eukaryotic target cell, the bacterium attaches tightly by interaction between its YadA and Inv adhesins and β-integrins, and the secretion channel opens. The Yops are then transported through the Ysc channel, and the Yop effectors are translocated across the plasma membrane, guided by the translocators YopB, YopD, and LcrV.
good agreement with the localization proposed for the homologous Ysc proteins. Thus, the similarity between the Ysc apparatus and the flagellum export apparatus resides in their most inner part. While the Salmonella and Shigella “injectisomes” can be visualized by electron microscopy (13, 14), such visualization is not yet the case for the Yersinia Ysc apparatus. Little is known about the actual mechanism of export, but it is generally assumed that the Ysc apparatus serves as a hollow conduit through which the exported proteins travel to cross the two membranes and the peptidoglycan barrier, in one step. Whether proteins travel folded or unfolded has not yet been demonstrated but, given the size of channel, it is likely that they travel at least partially unfolded.
Translocation of Effectors Across Animal Cell Membranes. Purified secreted Yops have no cytotoxic effect on cultured cells, although live extracellular Yersinia have such an activity. Cytotoxicity nevertheless depends on the capacity of the bacterium to secrete YopE and YopD, and YopE alone is cytotoxic when microinjected into the cells (15). This observation led to the hypothesis that YopE is a cytotoxin that needs to be injected into the eukaryotic cell's cytosol by a mechanism involving YopD to exert its effect (15). This hypothesis was demonstrated by confocal laser scanning microscopy (16) and by the adenylate cyclase reporter enzyme strategy, an approach that is now widely used in “type III secretion” (17): infection of eukaryotic cells with a recombinant Y. enterocolitica producing hybrid proteins consisting of the N terminus of various Yops (other than YopB and YopD) fused to the catalytic domain of a calmodulin-dependant adenylate cyclase (Yop-Cya proteins) leads to an accumulation of cyclic AMP (cAMP) in the cells. Since there is no calmodulin in the bacterial cell and culture medium, this accumulation of cAMP signifies the internalization of Yop-Cya into the cytosol of eukaryotic cells (17). The phenomenon is strictly dependant on the presence of YopD and YopB. Thus, extracellular Yersinia inject Yops into the cytosol of eukaryotic cells by a mechanism that involves at least YopD and YopB (18, 19). Yops are thus a collection of intracellular “effectors” (YopE, YopH, YopM, YpkA/YopO, YopP/YopJ, and YopT) and “translocators” (including YopB and YopD) which are required for the translocation of the effectors across the plasma membrane of eukaryotic cells ( 20).
This model of intracellular delivery of Yop effectors by extracellular adhering bacteria is now largely supported by a number of other results, including immunological observations. During a mouse infection by wild-type Y. enterocolitica, the epitope formed by amino acid residues 249–257 of the YopH effector protein is presented by MHC class I molecules, as cytosolic proteins are, and not by MHC class II molecules, as antigens are that are processed in phagocytic vacuoles (21).
A Pore Formed by Translocators. The translocators YopB and YopD have hydrophobic domains, suggesting that they could act as transmembrane proteins (16, 17 and 18, 22). In agreement with this possibility, Yersinia has a contact-dependent lytic activity on sheep erythrocytes, depending on YopB and YopD (19, 23), which suggests that the translocation apparatus involves some kind of a pore in the target cell membrane by which the Yop effectors pass through to reach the cytosol. This YopB- and YopD-dependent lytic activity is higher when the effector yop genes are deleted, suggesting that the pore is normally filled with effectors (19, 23). The idea of a translocation pore is further supported by the observation that the membrane of macrophage-like cells infected with an effector polymutant Y. enterocolitica becomes permeable to small dyes (23). If the macrophages are preloaded before the infection with a low-molecular weight fluorescent marker, they release the fluorescent marker but not cytosolic proteins, indicating that there is no membrane lysis but rather insertion of a small pore (diameter 16–23 Å) into the macrophage plasma membrane (23). The hypothesis of a channel is reinforced by the observation that artificial liposomes that have been incubated with Yersinia contain channels detectable by electrophysiology (24). All these events are dependent on the presence of the translocators YopB and YopD. These two hydrophobic Yops seem thus to be central for the translocation of the effectors and for the formation of a channel in lipid membranes. They presumably play different roles in pore formation. Indeed, YopB alone can disturb artificial membranes, whereas YopD cannot. Moreover, YopD has been shown to end up in the cytosol of eukaryotic cells (25).
YopB and YopD are encoded by a large operon that also encodes LcrV, LcrG, and the chaperone SycD. LcrV is a secreted Yop that has a different name for historical reasons. The fact that LcrG and LcrV are encoded together with translocators suggests that they could also be involved in translocation. Not surprisingly, LcrV interacts with YopB and YopD (26), is surfaceexposed before target cell contact (27), and is also required for translocation (26). In contrast with YopB, YopD, and LcrV, LcrG is not a released protein, but its exact localization in the bacterium remains elusive. It is required for efficient translocation of Yersinia Yop effector proteins into the eukaryotic cells but it is not required for pore formation. It binds to heparan sulfate proteoglycans (28), but the significance of this binding is not clear yet.
The Cytosolic Chaperones. Type III secretion often involves a new type of small cytosolic chaperone (29, 30 and 31) (Fig. 1). In Yersinia, these chaperones are called “Syc” (for specific Yop chaperone) (31). Generally, they are encoded by a gene that is located close to the gene encoding the Yop protein they serve, and this is a useful indication to recognize them. These chaperones may not form a single homogeneous group but rather could belong to two
different subfamilies, one devoted to effectors and one devoted to translocators.
SycE, the chaperone of YopE, is the archetype of the first family (31). The other representatives of this family in Yersinia are SycH (30), SycT (32), and SycN (33, 34). They are small (14–15 kDa), acidic (pI 4.4–5.2) proteins with a putative C-terminal amphiphilic α-helix. They specifically bind to their cognate Yop and, in their absence, secretion of this Yop is severely reduced, if not abolished. Until now, research has focused mainly on SycE and SycH, but their exact roles remain elusive. Three hypotheses have been proposed, based on different types of observations. SycE and SycH have been shown to bind to their partner Yop (YopE and YopH) at a unique site spanning roughly residues 20–70 (35). Surprisingly, when this site is removed, the cognate Yop is still secreted and the chaperone becomes dispensable for secretion (36). This observation indicates that the binding site itself creates the need for the chaperone and suggests that the chaperone acts as a “bodyguard” protecting this site from premature associations that would lead to degradation. In agreement with this first hypothesis, SycE has been shown to protect YopE from intrabacterial degradation: the half-life of YopE is longer in wild-type bacteria than in sycE mutant bacteria (37, 38). The partners in these hypothetical premature associations could be the translocators (36), but such interactions could not be demonstrated. Moreover, the hypothesis of premature associations with translocators is not sufficient to explain the need for SycE. Indeed, YopE can be secreted by the plant pathogen Xanthomonas campestris (see below) and, although X. campestris does not synthesize proteins resembling the Yersinia translocators, SycE is still necessary to ensure intrabacterial stability of YopE in X. campestris (39).
According to a second hypothesis, discussed below, SycE could act as a secretion pilot leading the YopE protein to the secretion locus.
Finally, a recent observation suggests a third hypothesis. Both SycE and SycH are required for efficient translocation of their partner Yops into eukaryotic cells (35). However, when YopE is delivered by a Yersinia polymutant strain that synthesizes an intact secretion and translocation apparatus but no other effector than YopE, it appears that YopE is delivered even in the absence of its chaperone and chaperone-binding site (70). Thus, the SycE chaperone appears to be needed only when YopE competes with other Yops for delivery. This observation suggests that the Syc chaperones could be involved in some kind of hierarchy for delivery. This third hypothesis about the role of the Syc chaperones fits quite well with the observation that only a subset of the effectors seems to have a chaperone, but it still needs to be strengthened. Little is known about the role of SycT and SycN. However, there is an unexpected complexity for SycN in the sense that it requires YscB working as a cochaperone (34, 40).
SycD is the archetype of the second group of “type III chaperones”. In its absence, translocators YopB and YopD are not secreted and they are less detectable inside the bacterial cell (30, 41). SycD appears to be different from SycE and SycH in the sense that it binds to several domains on YopB, reminiscent of SecB, a molecular chaperone in Escherichia coli that is dedicated to the export of proteins and has multiple binding sites on its targets (41). IpgC, the related chaperone from Shigella flexneri, can prevent the intrabacterial association between translocators IpaB and IpaC (29). The similarity between IpgC and SycD suggested that SycD could play a similar role and would thus prevent the intrabacterial association of YopB and YopD. However, this turned out not to be the case (41). Because YopB and YopD have also the capacity to bind to LcrV, one could speculate that SycD prevents the premature association, not between YopB and YopD but rather between YopB, YopD, and LcrV, but this possibility has not been shown yet.
Recognition of the Transported Proteins. Effectors delivered by type III secretion systems have no classical cleaved N-terminal signal sequence (5). Instead, it was demonstrated in 1990 that Yops are recognized by their N terminus and that no sequence is cleaved off during Yop secretion (5). The minimal region shown to be sufficient for secretion was gradually reduced to 17 residues for YopH (35), to 15 residues for YopE (35), and to 15 residues for YopN (42).
A systematic mutagenesis of the secretion signal by Anderson and Schneewind (42, 43) led to doubts about whether this signal was encoded in the protein. No point mutation could be identified that specifically abolished secretion of YopE, YopN, and YopQ. Moreover, some frameshift mutations that completely altered the peptide sequences of the YopE and YopN signals also failed to prevent secretion. Anderson and Schneewind (42, 43) concluded from these observations that the signal leading to the secretion of these Yops could be in the 5′ end of the messenger RNA rather than in the peptide sequence. Secretion would thus be cotranslational, and translation of yop mRNA might be inhibited either by its own RNA structure or as a result of its binding to other regulatory elements. If this is correct, one would expect that no Yop could be detected inside bacteria. However, while this is reported to be true for YopQ (43), it is certainly not true for other Yops, such as YopE. To determine whether this N-terminal (or 5′-terminal) signal is absolutely required for YopE secretion, Cheng et al. (37) deleted codons 2–15 and they observed that 10% of the hybrid proteins deprived of the N-terminal secretion signal were still secreted. They inferred that there is a second secretion signal and they showed that this second, and weaker, secretion signal corresponds to the SycE-binding site. Not surprisingly, this secretion signal is functional only in the presence of the SycE chaperone (37). Whether this signal plays a role in vivo remains to be elucidated.
Control of the Injection. Yersinia secrete their Yops in vitro under conditions of Ca2+ deprivation. What is the triggering signal in vivo? Most probably contact with a eukaryotic cell. Several reports have shown that Yop delivery by Yersinia is a “directional” phenomenon in the sense that most of the load is delivered inside the eukaryotic cell and that there is little leakage (22). According to the assays used, there is some discrepancy on the degree of “directionality” (18), but there is no doubt that the bulk of the released Yops load ends up inside the eukaryotic cell, indicating that contact must be the signal. Pettersson et al. (44) provided a nice visual demonstration of the phenomenon. By expressing luciferase under the control of a yop promoter, they showed that active transcription of yop genes is limited to bacteria that are in close contact with eukaryotic cells. However, although contact is clearly the triggering event, it is not clear yet whether a specific receptor is involved. Pore formation in artificial membranes (24) tends to suggest that there is none.
Effector Yops and Host Response
The Array of Yop Effectors. Six effector Yops have been characterized: YopE, YopH, YopM, YopJ/YopP, YopO/YpkA, and YopT (Fig. 2). Only two of them have a known enzymatic activity: YopH is a powerful phosphotyrosine phosphatase resembling eukaryotic phosphatases. The catalytic activity is exerted by the C-terminal domain (≈200 residues), which contains a phosphate-binding loop including a critical cysteine residue (Cys-403) (45). YpkA/YopO is a serine-threonine kinase (46) which shows some similarity with the COT (Cancer Osaka Thyroid) oncogene product, a cytosolic serine/threonine protein kinase expressed in hematopoietic cells and implicated in signal transduction by growth factors. YpkA catalyzes auto-
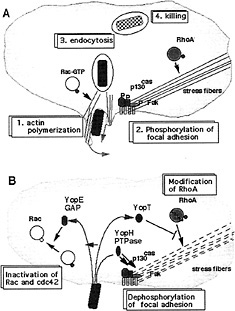
Fig. 2. Inhibition of phagocytosis by YopE, YopH, and YopT. (A) Phagocytosis of an invading bacterium by a macrophage. The process involves phosphorylation of focal adhesion proteins (p130cas, Fak, Fyn, paxillin) and actin polymerization controlled by GTPases such as RhoA and Rac. Phagocytosis is followed by killing of the bacterium. (B) Resistance to phagocytosis by Yersinia. On contact, Yersinia injects Yop effectors. YopH dephosphorylates proteins from the focal adhesion (PTPase, phosphotyrosine phosphatase); YopE inactivates Rac and cdc42 by stimulating their GTPase activity (GAP, GTPase-activating protein); YopT deactivates RhoA.
phosphorylation of a serine residue in vitro. Infection of HeLa cells with a multiple yop mutant overproducing YpkA leads to a morphological alteration of the cells, different from those mediated by YopE and YopH. The cells round up but do not detach from the extracellular matrix. Inside the HeLa cells the YpkA protein is targeted to the inner surface of the plasma membrane (47). No target protein corresponding to YpkA/YopO has been identified yet.
YopM is a strongly acidic protein containing leucine-rich repeats (LRRs) whose action and target remain unknown. It belongs to a growing family of type III effectors that has several representatives in Shigella (ipaH multigene family) and Salmonella (48). YopM has been shown to traffic to the cell's nucleus by means of a vesicle-associated pathway (49), but its action in the nucleus remains unknown.
The Cytoskeleton Is a Target of YopE, YopH, and YopT. Three effectors, of six identified so far, exert a negative role on cytoskeleton dynamics and, by doing so, contribute to the strong resistance of Yersinia to phagocytosis by macrophages (ref.15; N. Grosdent and G.R.C., unpublished observations). Studies using HeLa cells have shown that YopH dephosphorylates p130cas, paxillin, and the focal adhesion kinase (FAK) (50, 51 and 52), leading to disruption of the focal adhesion and a reduced invasinmediated engulfment by HeLa cells (a phenomenon called “invasion”). YopH is specifically targeted to the focal complexes; residues 223–226, which are known to be surface-exposed, are involved in this process. Deletion of these targeting residues affects the anti-invasion effect. These observations also apply to phagocytosis by the J774 macrophage–monocyte cell line, at least in the absence of opsonization (53). In the latter cells, a catalytically inactive YopH coprecipitates not only with p130cas but also with FYB (54).
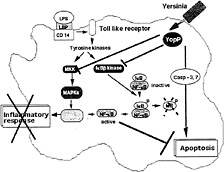
Fig. 3. Effects of YopP/YopJ. Bacterial lipopolysaccharide (LPS), bound to the LPS-binding protein (LBP), interacts with its receptor CD14 and coreceptor from the Toll-like family, which leads to phosphorylation cascades resulting in the activation of mitogen-activated protein kinases (MAPKs) and of the kinase of the inhibitor of NF-κB (IκB). Phosphorylation of IκB is followed by its degradation, and NF-κB migrates to the nucleus and activates transcription of proinflammatory cytokines, including TNF-α. Translocated YopP/YopJ prevents the activation of the two phosphorylation cascades, and thus blocks the release of TNF-α. YopP/YopJ also induces macrophage apoptosis. See text for details and references.
YopE has been known for a long time to disrupt actin filaments (15, 55), but its exact target has not yet been identified. However, YopE is homologous to the N-terminal domain of SptP from Salmonella, another type III effector, and it has been shown recently that this N-terminal domain of SptP acts as a GTPase-activating protein (GAP) for Rac-1 and Cdc42 (56). It is thus likely that YopE exerts its negative effect on the dynamics of the cytoskeleton by exerting the same GAP activity.
Finally, YopT exerts a dramatic depolymerizing effect on actin (32) by modifying RhoA, a GTPase that regulates the formation of stress fibers (57). The exact nature of the modification is not yet known.
YopP/YopJ Down-Regulates the Inflammatory Response. As shown schematically in Fig. 3, YopP/YopJ is a key player in the down-regulation of the inflammatory response that is observed during Yersinia infection. In vitro, YopP/YopJ has been shown to counteract the normal proinflammatory response of various cell types. Its injection reduces the release of TNF-α by macrophages (58) and of IL-8 by epithelial (59, 60) and endothelial (G.R.C. and S. Tötemeyer, unpublished results) cells. It also reduces the presentation of adhesion molecules ICAM-1 and E-selectin at the surface of endothelial cells (G.R.C. and S. Tötemeyer, unpublished results) and hence presumably reduces the recruitment of neutrophils to the sites of infection. All these events result from the inhibition of the activation of NF-κB, a transcription factor known to be central in the onset of inflammation (59, 61). The inhibition of NF-κB activation was recently shown to result from YopP/YopJ-mediated inhibition of IKKβ, a kinase that phosphorylates IκB, the inhibitor of NF-κB (62). By preventing
phosphorylation of IκB, YopP/YopJ prevents its degradation and the translocation of NF-κB to the nucleus. The inhibition of NF-κB activation is accompanied by a lack of activation of the mitogen-activated protein (MAP) kinases (MAPKs) c-Jun-N-terminal kinase (JNK), p38, and extracellular signalregulated kinase (ERK) 1 and 2 (58, 63, 64) that is observed upon infection of macrophages by a Yersinia producing YopP/YopJ. Lack of activation of these MAPKs results from the inhibition of the upstream MAPK kinases (MAPKKs) by binding of YopP/YopJ (62).
Last but not least, YopP/YopJ also induces apoptosis in macrophages (65, 66). This apoptosis is accompanied by cleavage of the cytosolic protein BID, the release of cytochrome c, and the cleavage of caspase-3 and -7 (C. Geuijen, W. Declerq, A. Boland, P. Vandenabeele, and G.R.C., unpublished results). The release of cytochrome c and the cleavage of BID can both be inhibited by caspase inhibitors, suggesting that YopP/YopJ interferes with a signaling pathway upstream of the mitochondria (C. Geuijen, W. Declerq, A. Boland, P. Vandenabeele, and G.R.C., unpublished results). The reduction in the release of TNF-α is not simply the consequence of apoptosis, because it occurs even when apoptosis is prevented by inhibiting the activity of caspases (61). On the contrary, apoptosis may result from the loss of the anti-apoptotic factor NF-κB (61); however, this hypothesis still awaits demonstration. It is thus not yet clear whether YopP/YopJ causes apoptosis by activating a death mechanism or by inhibiting an NF-κB-dependent survival mechanism. Interestingly, YopP/YopJ share a high level of similarity with AvrXv and AvrBsT from X. campestris and a protein from the nitrogen-fixing Rhizobium.
Inhibition of Antigen-Specific T and B Lymphocytes Responses. While they colonize and multiply in Peyer's patches or lymph nodes, Yersinia must also encounter lymphocytes. Artificial in vitro systems demonstrated that B and T lymphocytes are indeed targets for Yersinia injections (ref.67; A. P. Boyd and G.R.C., unpublished results). Yao et al. (68) observed that T and B cells transiently exposed to Yersinia were impaired in their ability to be activated by means of their antigen receptors. T cells are inhibited in their ability to produce cytokines, and B cells are unable to up-regulate surface expression of the costimulatory molecule B7.2, in response to antigenic stimulation. This block of activation results from the inhibition of early phosphorylation events (68). Through the analysis of various mutants, YopH appeared to be the main effector involved in these events. Thus YopH not only contributes to the evasion of the innate immune response but it could also incapacitate the host adaptive immune response.
I thank S. Bleves, G. Denecker, and C. Josenhans for a critical reading. Our work on Yersinia is supported by the Belgian Fonds National de la Recherche Scientifique Médicale (Convention 3.4595.97), the Direction Générale de la Recherche Scientifique-Communauté Française de Belgique (Action de Recherche Concertée 99/04–236), and the Interuniversity Poles of Attraction Program–Belgian State, Prime Minister's Office, Federal Office for Scientific, Technical and Cultural Affairs (PAI 4/03).
1. Simonet, M., Richard, S. & Berche P. ( 1990) Infect. Immun. 58, 841–845.
2. Nakajima, R. & Brubakker, R. R. ( 1993) Infect. Immun. 61, 23–31.
3. Cornelis, G. R., Boland, A., Boyd, A. P., Geuijen, C, Iriarte, M., Neyt, C. Sory, M.-P. & Stainier, I. ( 1998) Microbiol. Mol. Biol. Rev. 62, 1315–1352.
4. Hueck, C. J. ( 1998) Microbiol. Mol. Biol. Rev. 62, 379–433.
5. Michiels, T., Wattiau, P., Brasseur, R., Ruysschaert, J. M. & Cornelis, G. R. ( 1990) Infect. Immun. 58, 2840–2849.
6. Koster, M., Bitter, W., de Cock, H., Allaoui, A., Cornelis, G. R. & Tommassen, J. ( 1997) Mol. Microbiol. 26, 789–798.
7. Jackson, M. W. & Plano, G. V. ( 1999) J. Bacteriol. 181, 5126–5130.
8. Woestyn, S., Allaoui, A., Wattiau, P. & Cornelis G. R. ( 1994) J. Bacteriol. 176, 1561–1569.
9. Deng, W. L. & Huang, H. C. ( 1999) J. Bacteriol. 181, 2298–2301.
10. Payne, P. L. & Straley, S. C. ( 1998) J. Bacteriol. 180, 3882–3890.
11. Payne, P. L. & Straley, S. C. ( 1998) J. Bacteriol. 181, 2852–2862.
12. Minamino, T. & Macnab, R. M. ( 1999) J. Bacteriol. 181, 1388–1394.
13. Kubori, T., Matsushima, Y., Nakamura, D., Uralil, J., Lara-Tejero, M., Sukhan, A. & Galan, J. E. ( 1998) Science 280, 602–605.
14. Blocker, A., Gounon, P., Larquet, E., Niebuhr, K., Cabiaux, V., Parsot, C. & Sansonetti, P. ( 1999) J. Cell. Biol. 147, 683–693.
15. Rosqvist, R., Forsberg, A. & Wolf-Watz, H. ( 1991) Infect. Immun. 59, 4562–4569.
16. Rosqvist, R., Magnusson, K. E. & Wolf-Watz, H. ( 1994) EMBO J. 13, 964–972.
17. Sory, M.-P. & Cornelis, G. R. ( 1994) Mol. Microbiol. 14, 583–594.
18. Boland, A., Sory, M.-P., Iriarte, M., Kerbourch, C., Wattiau, P. & Cornelis, G. R. ( 1996) EMBO J. 15, 5191–5201.
19. Hakansson, S., Schesser, K., Persson, C., Galyov, E. E., Rosqvist, R., Homble, F. & Wolf-Watz, H. ( 1996) EMBO J. 15, 5812–5823.
20. Cornelis, G. R. & Wolf-Watz, H. ( 1997) Mol. Microbiol. 23, 861–867.
21. Starnbach, M. N. & Bevan, M. J. ( 1994) J. Immunol. 153, 1603–1612.
22. Persson, C., Nordfelth, R., Holmström, A., Hakansson, S., Rosqvist, R. & Wolf-Watz, H. ( 1995) Mol. Microbiol. 18, 135–150.
23. Neyt, C. & Cornelis, G. R. ( 1999) Mol. Microbiol. 33, 971–981.
24. Tardy, F., Homblé, F., Neyt, C., Wattiez, R., Cornelis, G. R., Ruysschaert, J.-M. & Cabiaux, V. ( 1999) EMBO J. 18, 6793–6799.
25. Francis, M. S. & Wolf-Watz, H. ( 1998) Mol. Microbiol. 29, 799–813.
26. Sarker, M. R., Neyt, C., Stainier, I. & Cornelis, G. R. ( 1998) J. Bacteriol. 180, 1207–1214.
27. Pettersson, J., Holmström, A., Hill, J., Leary, S., Frithz-Lindsten, E., von Euler-Matell, A., Carlsson E., Titball, R., Forsberg, A. & Wolf-Watz, H. ( 1999) Mol. Microbiol. 32, 961–976.
28. Boyd, A. P., Sory, M.-P., Iriarte, M. & Cornelis, G. R. ( 1998) Mol. Microbiol. 27, 425–436.
29. Ménard, R., Sansonetti, P. J., Parsot, C. & Vasselon, T. ( 1994) Cell 79, 515–525.
30. Wattiau, P., Bernier, B., Deslee, P., Michiels, T. & Cornelis, G. R. ( 1994) Proc. Natl. Acad. Sci. USA 91, 10493–10497.
31. Wattiau, P. & Cornelis, G. R. ( 1993) Mol. Microbiol. 8, 123–131.
32. Iriarte, M. & Cornelis, G. R. ( 1998) Mol. Microbiol. 29, 915–929.
33. Iriarte, M. & Cornelis, G. R. ( 1999) J. Bacteriol. 181, 675–680.
34. Day, J. B. & Plano, G. V. ( 1998) Mol. Microbiol. 30, 777–788.
35. Sory, M.-P., Boland, A., Lambermont, I. & Cornelis, G. R. ( 1995) Proc. Natl. Acad. Sci. USA 92, 11998–12002.
36. Woestyn, S., Sory, M.-P., Boland, A., Lequenne, O. & Cornelis, G. R. ( 1996) Mol. Microbiol. 20, 1261–1271.
37. Cheng, L. W., Anderson, D. M. & Schneewind, O. ( 1997) Mol. Microbiol. 24, 757–765.
38. Frithz-Lindsten, E., Rosqvist, R., Johansson, L. & Forsberg, A. ( 1995) Mol. Microbiol. 16, 635–647.
39. Rossier, O., Wengelnik, K., Hahn, K. & Bonas, U. ( 1999) Proc. Natl. Acad. Sci. USA 96, 9368–9373.
40. Jackson, M. W., Day, J. B. & Plano, G. V. ( 1998) J. Bacteriol. 180, 4912–4921.
41. Neyt, C. & Cornelis, G. R. ( 1999) Mol. Microbiol. 31, 143–156.
42. Anderson, D. M. & Schneewind, O. ( 1997) Science 278, 1140–1143.
43. Anderson, D. M. & Schneewind, O. ( 1999) Mol. Microbiol. 31, 1139–1148.
44. Pettersson, J., Nordfelth, R., Dubinina, E., Bergman, T., Gustafsson, M., Magnusson, M. & Wolf-Watz, H. ( 1996) Science 273, 1231–1233.
45. Guan, K. & Dixon, J. E. ( 1990) Science 249, 553–556.
46. Galyov, E. E., Hakansson, S., Forsberg, A. & Wolf-Watz, H. ( 1993) Nature ( London) 361, 730–732.
47. Hakansson, S., Galyov, E. E., Rosqvist, R. & Wolf-Watz, H. ( 1996) Mol. Microbiol. 20, 593–603.
48. Miao, E. A., Scherer, C. A., Tsolis, R. M., Kingsley, R. A., Adams, G., Baumler, A. J. & Miller, S. I. ( 1999) Mol. Microbiol. 34, 850–864.
49. Skrzypek, E., Cowan, C. & Straley, S. C. ( 1998) Mol. Microbiol. 30, 1051–1065.
50. Black, D. S. & Bliska, J. B. ( 1997) EMBO J. 16, 2730–2744.
51. Persson, C, Carballeira, N., Wolf-Watz, H. & Fällman, M. ( 1997) EMBO J. 16, 2307–2318.
52. Black, D. S., Montagna, L. G., Zitsmann, S. & Bliska, J. B. ( 1998) Mol. Microbiol. 29, 1263–1274.
53. Persson, C., Nordfelth, R., Andersson, K., Forsberg, A., Wolf-Watz, H. & Fällman, M. ( 1999) Mol. Microbiol. 33, 828–838.
54. Hamid, N., Gustavsson, A., Andersson, K., McGee, K., Persson, C., Rudd, C. E. & Fallman, M. ( 1999) Microb. Pathog. 27, 231–242.
55. Rosqvist, R., Forsberg, A., Rimpilainen, M., Bergman, T. & Wolf-Watz, H. ( 1990) Mol. Microbiol. 4, 657–667.
56. Fu, Y. X. & Galan, J. E., ( 1999) Nature ( London) 401, 293–297.
57. Zumbihl, R., Aepfelbacher, M., Andor, A., Jacobi, C. A., Ruckdeschel, H., Rouot, B. & Heeseman, J. ( 1999) J. Biol Chem. 274, 29289–29293.
58. Boland, A. & Cornelis, G. R. ( 1998) Infect. Immun. 66, 1878–1884.
59. Schesser, K., Spiik, A.-K., Dukuzumuremyi, J.-M., Neurath, M. F., Pettersson, S. & Wolf-Watz, H. ( 1998) Mol. Microbiol. 28, 1067–1079.
60. Schulte, R., Wattiau, P., Hartland, E. L., Robins-Browne, R. M. & Cornelis, G. R. ( 1996) Infect Immun. 64, 2106–2113.
61. Ruckdeschel, K., Harb, S., Roggenkamp, A., Hornef, M., Zumbihl, R., Kohler, S., Heesemann, J. & Rouot, B. ( 1998) J. Exp. Med. 187, 1069–1079.
62. Orth, K., Palmer, L. E., Qin Bao, Z., Stewart, S., Rudolph, A. E, Bliska, J. B. & Dixon, J. E. ( 1999) Science 285, 1920–1923.
63. Ruckdeschel, K., Machold, J., Roggenkamp, A., Schubert, S., Pierre, J., Zumbihl, R., Liautard, J.-P., Heesemann, J. & Rouot, B. ( 1997) J. Biol. Chem. 272, 15920–15927.
64. Palmer, L. E., Pancetti, A. R., Greenberg, S. & Bliska, J. B. ( 1999) Infect. Immun. 67, 708–716.
65. Mills, S. D., Boland, A., Sory, M.-P., Van der Smissen, P., Kerbourch, C., Finlay, B. B. & Cornelis, G. R. ( 1997) Proc. Natl. Acad. Sci. USA 94, 12638–12643.
66. Monack, D. M., Mecsas, J., Ghori, N. & Falkow, S. ( 1997) Proc. Natl. Acad. Sci. USA 94, 10385–10390.
67. Chaux, P., Luiten, R., Demotte, N., Vantomme, V., Stroobant, V., Traversari, C., Russo, V., Schultz, E., Cornelis, G. R., Boon, T. & van der Bruggen, P. ( 1999) J. Immunol. 163, 2928–2936.
68. Yao, T., Mecsas, J., Healy, J. I., Falkow, S. & Chien, Y.-H. ( 1999) J. Exp. Med. 190, 1343–1350.
69. Stainier, I., Bleves, S., Josenhans, C., Karmani, L., Kerbourch, C., Lambermont, I., Tötemeyer, S., Boyd, A. & Cornelis, G. R. ( 2000) Mol. Microbiol., in press.
70. Boyd, A. P., Lambermont, I. & Cornelis, G. R. ( 2000) J. Bacteriol., in press.






