
Colloquium
Striking a balance: Modulation of the actin cytoskeleton by Salmonella
Jorge E. Galán* and Daoguo Zhou
Section of Microbial Pathogenesis, Boyer Center for Molecular Medicine, Yale School of Medicine, New Haven, CT 06536-0812
Salmonella spp. have evolved the ability to enter into cells that are normally nonphagocytic. The internalization process is the result of a remarkable interaction between the bacteria and the host cells. Immediately on contact. Salmonella delivers a number of bacterial effector proteins into the host cell cytosol through the function of a specialized organelle termed the type III secretion system. Initially, two of the delivered proteins, SopE and SopB, stimulate the small GTP-binding proteins Cdc42 and Rac. SopE is an exchange factor for these GTPases, and SopB is an inositol polyphosphate phosphatase. Stimulation of Cdc42 and Rac leads to marked actin cytoskeleton rearrangements, which are further enhanced by SipA, a Salmonella protein also delivered into the host cell by the type III secretion system. SipA lowers the critical concentration of G-actin, stabilizes F-actin at the site of bacterial entry, and increases the bundling activity of the host-cell protein T-plastin (fimbrin). The cellular responses stimulated by Salmonella are short-lived; therefore, immediately after bacterial entry, the cell regains its normal architecture. Remarkably, this process is mediated by SptP, another target of the type III secretion system. SptP exert its function by serving as a GTPase-activating protein for Cdc42 and Rac, turning these G proteins off after their stimulation by the bacterial effectors SopE and SopB. The balanced interaction of Salmonella with host cells constitutes a remarkable example of the sophisticated nature of a pathogen/host relationship shaped by evolution through a longstanding coexistence.
W hen examining the strategies used by microbial pathogens to colonize and multiply within a host, it is useful to make the distinction between pathogens that may only accidentally encounter a particular host and those that have sustained a longstanding association. The distinction is relevant because in the first case, the terms of the host/pathogen interactions have not been shaped by evolutionary forces. Therefore, infections by this type of microorganisms oftentimes lead to serious or lethal disease. In contrast to “accidentally encountered ” pathogens, the interaction between “host-adapted” or “quasi-adapted” pathogens and their hosts has been shaped by evolution, resulting in balanced encounters that most often lead to infections that are either subclinical or self-limiting. In some of these cases, the process of coevolution and adaptation has effectively precluded this type of microorganisms from exploring other niches. To be sure, the evolutionary forces may have dictated that infection leads to a certain degree of host pathology that is not acceptable for society, and therefore the outcome of such interaction is referred to as “disease. ” However, this outcome does not mean that the interactions between these microorganisms and their hosts are “unbalanced” in evolutionary terms. It may simply mean that a certain degree of host damage is required for these pathogens to replicate and to move on to a new host and/or for the host to expel them.
Salmonella enterica is an excellent example of a bacterial pathogen whose interactions with vertebrate animals have been shaped by millions of years of close coexistence. There are different serotypes of Salmonella enterica that cause different diseases. The type of disease caused by these bacteria range from self-limiting gastroenteritis (commonly referred to as “food poisoning”) to more systemic illness such as typhoid fever. The outcome of Salmonella infections depends on both the serotype of the infecting Salmonella as well as the species and/or the immunological status of the infected hosts. Central to the pathogenesis of Salmonella is the function of a specialized protein secretion system, termed type III, that is encoded within a pathogenicity island located at centisome 63 of its chromosome (1). The nucleotide composition of this pathogenicity island suggests that these genes have been acquired by horizontal transfer from another microorganism. This event most likely took place early in the evolution of Salmonella, resulting in a significant niche expansion for these bacteria, perhaps marking the beginning of its close association with vertebrate hosts. This type III protein secretion system directs the secretion and translocation into the host cell of a number of bacterial proteins that have the capacity to modulate a variety of host cellular functions (2, 3). Among these functions is the ability of Salmonella to modulate the host cell actin cytoskeleton to induce its own internalization into nonphagocytic cells, which is essential for its pathogenicity. This process is the result of the coordinated activities of several bacterial effector proteins that alternatively stimulate and down-regulate host cell responses in a remarkable “yin and yang.” In this article, we will discuss the molecular mechanisms that lead to Salmonella internalization into non-phagocytic cells. However, this paper is not intended to be a comprehensive review of the literature on Salmonella–host cell interaction. Instead, we have chosen to focus on key aspects of the entry process that better illustrate the remarkable balance between a pathogen and its host.
The Centisome 63 Type III Secretion System: Salmonella's Key to Enter into Host Cells
At the center of the mechanisms used by Salmonella to gain access into nonphagocytic cells is the type III secretion system encoded at centisome 63 of its chromosome (1). Salmonella encodes another type III secretion system at centisome 31 that is only functional at later stages of the pathogenic cycle after the bacteria has entered host cells (4, 5). Type III secretion systems are widely distributed among plant and animal pathogenic bacteria that share the property of engaging host cells in an intimate manner (for detailed reviews see refs. 2 and 3). Composed of more than 20 proteins, these systems stand among the most complex protein secretion systems known. Such complexity is caused by their specialized function, which is not only to secrete proteins from the bacterial cytoplasm but
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviation: GAP, GTPase activating protein.
|
* |
To whom reprint requests should be addressed. E-mail: jorge.galan@yale.edu. |
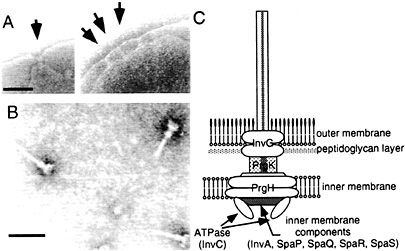
Fig. 1. Needle complex of S. typhimurium type III secretion system. (A) Electron micrographs of osmotically shocked S. typhimurium showing needle complexes in the bacterial envelope (arrows). (B) Electron micrographs of purified needle complexes. (C) Schematic representation of the needle complex and its components. The location of the different components is hypothetical and more proteins may be present in the complex. [Figure reprintzed with permission from ref. 2 (Copyright 1999, American Association for the Advancement of Science).]
also to deliver them to the inside of the eukaryotic host cell. Perhaps a more important factor contributing to their complexity is the temporal and spatial restrictions that govern their activity. Thus, the function of type III secretion systems requires poorly characterized signals that cue the bacteria to secrete and deliver proteins at the appropriate time and in the appropriate environment.
A number of components of the type III secretion system assemble into an organelle, appropriately termed the “needle complex,” that spans both the inner and outer membranes of the bacterial envelope (ref.6; Fig. 1). The architecture of the needle complex resembles that of the flagellar hook-basal body (7, 8). It is composed of two pairs of inner and outer rings that presumably anchor the structure to the inner and outer membranes of the bacterial envelope. The rings are connected by a rod-like structure, which together form the base of the needle complex. A needle-like structure of ≈80 nm in length protrudes outwards from the base of the needle complex. The entire structure is ≈100 nm in length and ≈40 nm in diameter at its widest section. The identification of the needle complex has provided important clues in understanding the transit of type III secreted proteins through the bacterial envelope. However, this information has been less useful in elucidating the mechanisms by which this system mediates the translocation of the secreted proteins into the host cell. The latter process depends on the function of a subset of type III secreted proteins that, although dispensable for protein secretion, are essential for the translocation of effector proteins into the host cell. In Salmonella, those proteins are SipB, SipC, and SipD (9, 10, 11 and 12). The mechanisms by which these “protein translocases” mediate the delivery of effector proteins into the eukaryotic host cell are not understood. However, functionally equivalent proteins in other type III secretion systems (e.g., Yersinia spp.) have been proposed to form a pore or channel through which the effector proteins cross the eukaryotic cell membrane (13, 14).
Another important component of type III secretion systems is a family of small molecular weight acidic polypeptides that bind a specific subset of cognate cytosolic proteins (15). Although absolutely required for the function of the type III secretion systems, the actual mechanisms by which these proteins exert their chaperone-like function is poorly understood and is the subject of some controversy. At least two functions have been proposed for these proteins: (i) partitioning factors that prevent the premature association of type III secreted proteins within the bacterial cytoplasm, and ( ii) secretion pilots that “guide” the cognate secreted protein to the secretion machinery. It is possible that different chaperones may exert different functions. The Salmonella invasion-associated type III secretion system encodes at least three such chaperone-like proteins: SicP (16), SicA (12), and SigD (17, 18). SicP serves as chaperone for the effector protein SptP (16). Consistent with this role, SicP binds SptP, which in its absence completely is degraded within the bacterial cytoplasm. Thus, SicP seems to function as a partitioning factor for SptP, perhaps preventing it from interacting with an as-yet-unidentified protein. SicA, however, appears to play a more complex role (19). One of its functions is to prevent the association of SipB and SipC in the bacterial cytosol that would target these proteins for degradation. Absence of SicA results in the degradation of both SipB and SipC. Interestingly, in the absence of both SicA and SipC, SipB is not only stable but also is secreted at wild-type levels, indicating that SicA is not essential for SipB secretion per se. In addition, absence of SicA results in the lack of expression of several genes that encode type III effector proteins, suggesting another function for this protein (19). It is possible that some of the chaperones associated with type III secretion systems exert a role in the temporal regulation of substrate secretion by the secretion machinery. Indeed, evidence has been presented that indicates that there is a hierarchy in the secretion of type III proteins in Salmonella (20, 21).
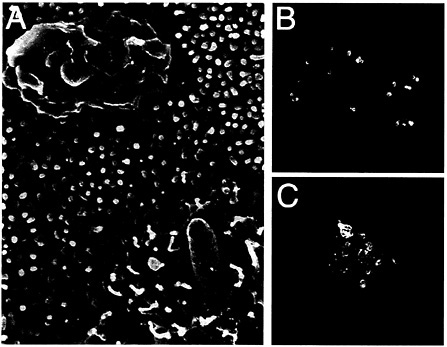
Fig. 2. Interaction of S. typhimurium with intestinal epithelial cells. (A) Scanning electron micrograph of S. typhimurium-infected intestinal epithelial Caco-2 cells (S. Olmsted, C. Ginocchio, C. Wells, and J.E.G., unpublished data). (B and C) Actin cytoskeleton rearrangements in S. typhimurium-infected intestinal epithelial Caco-2 cells. Filamentous actin was stained with rhodamine phalloidin (red) and S. typhimurium with an FITC-conjugated antibody (green).
Revving Up Rho GTPases: Signaling for Entry and Beyond
Salmonella entry into host cells strictly depends on the function of the actin cytoskeleton as addition of drugs that interfere with actin dynamics effectively block bacterial internalization (22, 23). Furthermore, immediately after contact with host cells, Salmonella induces rapid and marked actin cytoskeleton rearrangements (Fig. 2). It was recognized early on that these changes resemble the membrane ruffles induced by the stimulation of cells with growth factors or the expression of activated oncogenes (24, 25). An important insight into the understanding of the cellular responses that lead to bacterial internalization came from the finding that Cdc42 and Rac, two members of the Rho subfamily of actin-organizing small GTP-binding proteins, were essential for Salmonella entry into host cells (26). These small molecular weight GTP-binding proteins can cycle between two states, a GDP-bound (inactive) and a GTP-bound (active) conformation that can bind a variety of downstream effector proteins. Therefore, they can act as effective molecular switches

Fig. 3. Actin cytoskeleton reorganization induced by the expression of SopE and SopB in cultured mammalian cells. Cos-1 cells were transfected with plasmids expressing SopE or SopB or with the empty vector and were stained with rhodamine phalloidin to visualize the actin cytoskeleton.
to control signaling events in a temporal and spatial manner. Expression of dominant-negative mutants of these GTPases either completely (Cdc42N17) or partially (Rac-1N17) blocked bacterial entry into cultured cells. Stimulation of these GTPases by Salmonella also leads to the activation of the downstream mitogen-activated protein kinases Jnk and p38 ( 26, 27). This finding is significant for Salmonella pathogenesis because the type III secretion-dependent activation of the mitogen-activated protein kinase pathways leads to the stimulation of the transcription factors NF-KB and AP-1 ( 27) and to the production of proinflammatory cytokines ( 27, 28 and 29). These events are presumed to be essential for the establishment of the inflammatory diarrhea that follows Salmonella infection.
How does Salmonella engage the Rho-GTPase signaling pathways? The answer to this question became clear when it was found that one of the proteins delivered by the invasionassociated type III secretion system, termed SopE, was capable of directly activating both Cdc42 and Rac-1 (30). SopE was found to catalyze the exchange of GDP for GTP on several (but not all) members of the Rho GTPase subfamily of small G proteins (Rac-1, Rac-2, Cdc42, RhoG, and RhoA, but not RhoB or RhoC). Consistent with this activity, microinjection or transient expression of SopE in cultured cells leads to marked actin cytoskeleton rearrangements and membrane ruffling that resemble the changes induced by Salmonella infections (Fig. 3). These cytoskeleton rearrangements can be blocked by coexpression of dominant-negative forms of either Cdc42 or Rac-1 (30). These results position SopE as a key stimulator of signaling events leading to bacterial entry.
Introduction of a loss-of-function mutation in sopE reduced but did not abolish the ability of Salmonella to enter host cells (31, 32). Because expression of dominant-negative Cdc42 abolished the ability of Salmonella to enter into host cells, these results indicated that, in addition to SopE, there must be another bacterial protein capable of stimulating small GTPase signaling (26). Recently, such a factor was identified as SopB, another protein delivered by Salmonella into host cells by its invasionassociated type III secretion system (D.Z., L.-M. Chen, S. Shears, and J.E.G., unpublished data). SopB is an inositol phosphate polyphosphatase with the potential to generate several products with second messenger activity (17, 34). Consistent with its role in the actin-mediated bacterial internalization process, transient expression of SopB in mammalian cells leads to actin cytoskeleton rearrangements resembling those induced by the bacteria (D.Z., L.-M. Chen, S. Shears, and J.E.G., unpublished data, and Fig. 3). Furthermore, a Salmonella strain defective in both sopE and sopB is completely defective in its ability to stimulate actin cytoskeleton rearrangements and membrane ruffling. Interestingly, expression of a dominant-negative mutant of Cdc42 but not Rac-1 effectively blocked SopB-mediated actin cytoskeleton rearrangements. Consistent with this finding, internalization of a sopE mutant strain of Salmonella (i.e., a strain internalized only by the function of SopB) was blocked by expression of a dominant-negative mutant of Cdc42 but not Rac-1 (D.Z., L.-M. Chen, S. Shears, and J.E.G., unpublished data). These results indicate that unlike SopE, which exerts its effect on both Cdc42 and Rac-1 (see above), SopB appears to restrict its function to Cdc42. At this point it is not known how SopB stimulates Cdc42 activation. It is possible that some of the phosphoinositide products of the SopB activity
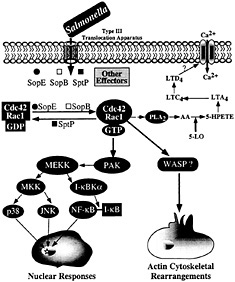
Fig. 4. Host-cell signal transduction pathways stimulated by Salmonella through the invasion-associated type III secretion system.
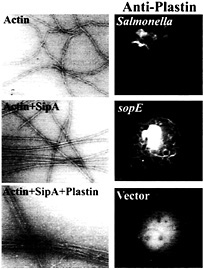
Fig. 5. Role of the Salmonella type III secreted protein SipA and the host-cell protein T-plastin (fimbrin) in bacterial-induced actin remodeling. (Left) Panels show electron micrographs of negatively stained actin, actin plus SipA, and actin plus SipA and plastin. Note the increased bundling activity of T-plastin in the presence of SipA. (Right) Panels show the recruitment of T-plastin to the membrane ruffles induced by either Salmonella or the transient expression of the bacterial effector SopE. Cells were transfected with hemagglutinin-epitope tagged T-plastin (fimbrin) and either infected with S. typhimurium or cotransfected with a plasmid expressing SopE. Cells were stained with a monoclonal antibody directed to the epitope tag. For details on these experiments, see ref. 42.

Fig. 6. SptP mediates the recovery of the normal organization of the actin cytoskeleton after S. typhimurium internalization. Ref52 cells were infected with either wild-type or a ∆sptP mutant strain for 3 h, and the actin cytoskeleton was visualized by rhodamine phalloidin staining. Bacteria were visualized by 4′,6-diamidino-2-phenylindole (DAPI) staining. Phalloidin and DAPI images were captured individually and merged using Adobe PHOTOSHOP. [Figure reprinted with permission from ref. 21 (Copyright 1999, Macmillan Magazines, Ltd.), http://www.nature.com.]
either may activate an endogenous cellular exchange factor (e.g., Dbl) or may directly stimulate nucleotide exchange.
Downstream Signaling: Effectors of Cdc42 and Rac-1 Function
Stimulation of Rho GTPases leads to a variety of cellular responses, including actin remodeling, stimulation of nuclear responses, and modulation of cell cycle progression (35, 36). Although the mechanisms by which small GTP-binding proteins modulate all these activities are not well understood, it is assumed that they exert their functions by engaging a variety of downstream effectors that bind the GTP-bound (active) form of these regulators. In many instances, mutation analysis of these GTPases has allowed the identification of specific residues, termed “effector loops,” that are involved in engaging specific downstream signaling pathways. Stimulation of Rho GTPases by Salmonella leads to at least two clearly defined responses: (i) actin cytoskeleton rearrangements resulting in bacterial uptake, and (ii) the stimulation of the mitogen-activated protein kinase pathways leading to nuclear responses. It has been shown that these responses are mediated by different downstream effectors (ref.37; Fig. 4). For example, the stimulation of nuclear responses depends on the activity of p21-activated kinase (PAK) (37). Consistent with this involvement, Salmonella infection or transient SopE expression lead to a robust and rapid activation of PAK (37). Furthermore, expression of a dominant-negative kinase-defective mutant form of PAK effectively blocks Salmonella or SopE-induced JNK activation. However, expression of dominant-negative PAK does not block Salmonella-induced actin cytoskeleton rearrangements or bacterial internalization, indicating that the nuclear and morphological responses induced by the bacteria are mediated by different downstream effectors.
The identity of the downstream targets of Cdc42 and Rac-1 that mediate bacterial-induced actin rearrangements are not known. However, the effector loop of Cdc42 that is necessary to stimulate bacterial-induced actin rearrangements has been identified (37). This loop specifically binds effector proteins that contain a conserved 16-aa motif termed CRIB (CDC42/Rac interacting binding) or PBD (p21-binding domain). Therefore, it is expected that the effector protein(s) involved in the stimulation of actin rearrangements must contain such a domain. A very good candidate is the Wiskott Aldrich Syndrome protein (WASP), which has been shown to be involved in Cdc42-mediated modulation of the actin cytoskeleton (38). Another downstream effector potentially involved in actin remodeling is phospholipase A2 (PLA2). Previous studies have shown that Salmonella infection of cultured intestinal cells leads to the activation of PLA2 and to the subsequent production of arachidonic acid metabolites and Ca2+ fluxes, events required for bacterial entry (39). Subsequent studies have demonstrated that Rac stimulation leads to the activation of PLA2 and the production of arachidonic acid metabolites (40). It is therefore likely that the calcium fluxes stimulated by Salmonella are also the consequence of the activation of Cdc42 and Rac by SopE and SopB.
Fine-Tuning the Actin Cytoskeleton Rearrangements: The Role of the Actin-Binding Protein SipA
Microinjection or transient expression in host cells of the bacterial-encoded exchange factor SopE leads to actin cytoskeletal rearrangements and membrane ruffling. These morphological changes resemble the actin reorganization induced by Salmonella infections but they differ in one important aspect. Whereas the morphological changes induced by the transient expression of SopE are distributed throughout the cells, those stimulated by Salmonella are localized to the point of bacterial host cell contact. Although localized delivery of effector proteins by the bacteria may account in part for signal localization, evidence indicates that at least one Salmonella type III secreted effector protein is actively engaged in this process. A strain of Salmonella typhimurium that carries a loss-of-function mutation in sipA, a gene that encodes a type III secreted protein, is significantly impaired in its ability to induce localized actin cytoskeleton reorganization, despite the fact that this strain is not impaired in its ability to deliver other effector proteins (41). The sipA mutant induces diffuse actin cytoskeleton rearrangements and consequently is impaired in its ability to enter cells. A search for a cellular target of this bacterial effector protein revealed that SipA binds to filamentous (F) actin (41). The binding of SipA to actin results in a significant reduction in the concentration of monomeric actin required for polymerization (critical concentration) and a marked increase in the stability of F-actin (Fig. 5). In addition, SipA binding to F-actin increases the bundling activity of T-plastin (fimbrin), an actin-binding protein that is actively recruited to membrane ruffles on Salmonella infection or transient expression of SopE (ref.42; Fig. 5). These activities may contribute to the cytoskeletal changes induced by Salmonella in various ways. Lowering the critical concentration of actin may facilitate actin nucleation on bacterial stimulation of Cdc42 and Rac. Furthermore, increasing the bundling activity of plastin as well as stabilizing F-actin may enhance the formation of membrane ruffles, resulting in more pronounced outward extension of these structures and more efficient bacterial uptake.
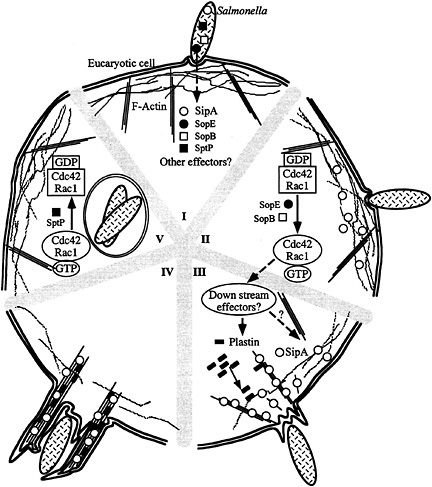
Fig. 7. Model for S. typhimurium interaction with host cells. (I–V) Contact with host cells activates the invasion-associated type III secretion system resulting in the delivery of effector proteins (e.g., SopE, SopB, SipA, and SptP) (I). Delivery of different effectors may not happen at the same time. Introduction of the exchange factor SopE and the inositol polyphosphatase SopB results in the activation of Cdc42 and Rac-1 (II), the stimulation of downstream signaling pathways, and the recruitment of plastin and other ruffling-associated molecules (III) initiating the actin cytoskeleton reorganization. The bacterial effector protein SipA helps this process by lowering the critical concentration of actin, stimulating the bundling activity of plastin, and stabilizing F-actin (IV). This activity results in a localized and more pronounced outward extension of the ruffling process (IV). Subsequent to the stimulation of Cdc42 and Rac-1 by SopE and SopB, Salmonella delivers another effector protein, SptP, which reverses the activation of these small G proteins by stimulating their intrinsic GTPase activity and therefore facilitating cell recovery (V).
An additional type III secreted protein, SipC, recently has been reported to have actin-nucleating activity (43). It is possible that this activity may aid bacterial entry by contributing to the actin nucleation events that lead to membrane ruffling and bacterial uptake. However, SipC by itself is not sufficient to induce actin cytoskeleton rearrangements because an S. typhimurium sopE sopB double-mutant strain is unable to stimulate these cellular responses despite having an intact type III secretion system and wild-type SipC expression (D.Z., L.-M. Chen, S. Shears, and J.E.G., unpublished data). The requirement of SipC for the translocation of all type III secreted proteins (9, 10) has precluded the use of standard genetic analysis to evaluate the contribution of SipC's actin-nucleating activity to bacterial internalization. The isolation of a mutant defective in actin nu-
cleation but proficient in protein translocation will be required to assess the contribution and significance of the actin-binding activity of SipC in bacterial entry.
Putting on the Brakes: A Lesson on Self-Restraint
In his pioneering electron microscopic description of the interaction of S. typhimurium with the intestinal epithelium of infected animals, Akio Takeuchi made the observation that the morphological changes of the brush border induced by the bacteria were reversible (44). He observed that shortly after infection, the brush border of intestinal epithelial cells infected by Salmonella regained their normal appearance despite the presence of numerous bacteria within an intracellular compartment. The molecular bases for the reversion of these marked morphological changes remained unknown until recently. The finding that Salmonella induces its own internalization by activating the small GTP binding proteins Cdc42 and Rac suggested the possibility that the endogenous cellular mechanisms controlling the down-regulation of these GTPases may mediate the reversion of the bacterial-induced responses. However, micro-injection into cultured cells of purified SopE in amounts equivalent to those injected by the bacterial type III secretion system led to actin cytoskeleton rearrangements and membrane ruffling that continued on for periods of time that far exceeded those resulting from bacterial infection (30). This result suggested a more active participation of Salmonella in mediating cellular recovery after bacterial infection. The finding that cells infected with a S. typhimurium strain carrying a null mutation in sptP failed to recover the integrity of their actin cytoskeleton after bacterial infection further supported this hypothesis (ref.21; Fig. 6). SptP is secreted by the invasion-associated type III secretion system and contains two modular domains: (i) an amino terminal domain with sequence similarity to the YopE and ExoS toxins of Yersinia spp. and Pseudomonas aeruginosa, respectively, and (ii) a carboxyl terminal domain homologous to Yersinia YopH and eukaryotic tyrosine phosphatases (10, 45).
How does SptP exert its function? A mechanism for its biochemical function was suggested by the observation that the actin cytoskeleton rearrangements stimulated by the exchange factor SopE could be effectively blocked by the comicroinjection of SptP, suggesting a Cdc42/Rac-1 antagonistic function for this protein (21). G proteins have an intrinsic GTPase activity that allows them to turn themselves off after activation by switching to the GDP-bound (inactive) conformation (46). However, such intrinsic activity is very low unless in the presence of GTPase activating proteins (GAPs), which can stimulate the intrinsic GTPase activity by several orders of magnitude (47). Consistent with its antagonistic function, SptP was found to be a potent GAP for Cdc42 and Rac-1 but not for other GTPases from the Rho or more distantly related families (21). The GAP activity is encoded in the amino terminal domain of SptP. Consistent with this finding, it now has been reported that the related bacterial toxins ExoS and YopE are also GAPs for Rho GTPases (33). Although different GAPs often do not share strong sequence similarity, they do share a short sequence motif that contains a conserved arginine that is essential for catalysis (47). SptP, ExoS, and YopE exhibit such a motif indicating a similarity between the catalytic mechanism of these bacterial proteins and those of eukaryotic GAP proteins (21). Consistent with this hypothesis, an SptP mutant in which the invariant arginine has been changed to an alanine is totally devoid of GAP activity. This mutant also is unable to reverse the actin cytoskeleton rearrangements that follow Salmonella infection, therefore confirming the role of the SptP GAP activity in this process. SptP not only reverses the actin cytoskeletal changes but also prevents the potential harm to the host cell derived from excessive signaling through Cdc42 and Rac that may lead to apoptosis (21). This function may allow the bacteria to preserve the integrity of its intracellular niche long enough to permit its replication and to allow reprogramming of gene expression, a necessary step to continue with the next phase of its pathogenic life cycle.
Lessons Learned from Salmonella
Salmonella entry into host cells requires the coordinated action of several bacterial effector proteins that, on delivery into the host cell, exert their activity in a temporally coordinated manner (Fig. 7). Thus, activation of Cdc42 and Rac by SopE and SopB is followed by actin cytoskeleton rearrangements that are further modulated by the activity of the actin-binding protein SipA. The bacterial-induced cellular responses subsequently are reversed by another bacterial effector protein, SptP, which opposes the activities of SopB and SopE. In addition, it is likely that the effector molecules themselves may have regulatory domains that control their activity. The existence of other putative effector proteins of unknown function that also are delivered into host cells by the centisome 63 type III secretion system indicates that we are just beginning to understand the complexities of this system. The interaction of Salmonella with host cells is therefore an eloquent example of the sophisticated nature of the mechanisms used by bacterial pathogens that have sustained long-standing associations with their hosts. Such sophistication is the result of evolutionary forces operating over extended periods of time, leading to a rather balanced interaction that allows bacterial replication while preventing excessive harm to the host. Because the study of microbial pathogens commonly focuses in the examination of the events that lead to overt harm to the host, it is frequently overlooked that the interaction of these “adapted pathogens” with their hosts most often does not lead to overt disease. In fact, the Salmonella example indicates that the bacteria can be actively engaged in preventing overt harm. This fact is particularly important when considering the design of novel therapeutic and prevention strategies because inadvertent interference with “down-modulators of virulence” could result in more harm to the host. As we increase our understanding of the molecular mechanisms that govern the interaction of adapted pathogens with their hosts, we will undoubtedly find more examples of “pathogen self-restraint. ”
We thank members of J.E.G.'s laboratory for careful review of this manuscript. Work in J.E.G. 's laboratory that was discussed in this article was supported by Grants AI30492 and GM52543 from the National Institutes of Health.
1. Galán, J. E. ( 1999) Curr. Opin. Microbiol. 2, 46–50.
2. Galán, J. E. & Collmer, A. ( 1999) Science 284, 1322–1328.
3. Hueck, C. J. ( 1998) Microbiol. Mol. Biol. Rev. 62, 379–433.
4. Ochman, H., Soncini, F. C., Solomon, F. & Groisman, E. A. ( 1996) Proc. Natl. Acad. Sci. USA 93, 7800–7804.
5. Shea, J. E., Hensel, M., Gleeson, C. & Holden, D. W. ( 1996) Proc. Natl. Acad. Sci. USA 93, 2593–2597.
6. Kubori, T., Matsushima, Y., Nakamura, D., Uralil, J., Lara-Tejero, M., Sukhan, A., Galán, J. E. & Aizawa, S.-I. ( 1998) Science 280, 602–605.
7. Aizawa, S. I. ( 1996) Mol. Microbiol 19, 1–5.
8. Macnab, R. M. ( 1996) in Escherichia coli and Salmonella typhimurium: Cellular and Molecular Biology, ed. Neidhardt, F. C. (Am. Soc. Microbiol., Washington DC), 2nd Ed., pp. 123–145.
9. Collazo, C. & Galán, J. E. ( 1997) Mol. Microbiol. 24, 747–756.
10. Fu, Y. & Galán, J. E. ( 1998) Mol. Microbiol. 27, 359–368.
11. Kaniga, K., Trollinger, D. & Galán, J. E. ( 1995) J. Bacterial. 177, 7078–7085.
12. Kaniga, K, Tucker, S. C., Trollinger, D. & Galán, J. E. ( 1995) J. Bacteriol. 177, 3965–3971.
13. Hakansson, S., Schesser, K., Persson, C, Galyov, E. E., Rosqvist, R., Homble, F. & Wolf-Watz, H. ( 1996) EMBO J. 15, 5812–5823.
14. Neyt, C. & Cornelis, G. R. ( 1999) Mol. Microbiol. 33, 971–981.
15. Wattiau, P., Woestyn, S. & Cornelis, G. R. ( 1996) Mol. Microbiol. 20, 255–262.
16. Fu, Y. & Galán, J. E. ( 1998) J. Bacteriol. 180, 3393–3399.
17. Norris, F. A., Wilson, M. P., Wallis, T. S., Galyov, E. E. & Majerus, P. W. ( 1998) Proc. Natl. Acad. Sci. USA 95, 14057–14059.
18. Hong, K. H. & Miller, V. L. ( 1998) J. Bacteriol. 180, 1793–1802.
19. Tucker, S. & Galán, J. E. ( 2000) J. Bacteriol., in press.
20. Collazo, C. & Galán, J. E. ( 1996) Infect. Immun. 64, 3524–3531.
21. Fu, Y. & Galán, J. E. ( 1999) Nature ( London) 401, 293–297.
22. Kihlstrom, E. & Nilsson, L. ( 1977) Acta Pathol. Microbiol. Scand. 85, 322–328.
23. Buckholm, G. ( 1984) Acta Pathol. Microbiol. Immun. Scand. 92, 145–149.
24. Galán, J. E., Pace, J. & Hayman, M. J. ( 1992) Nature ( London) 357, 588–589.
25. Francis, C. L., Ryan, T. A., Jones, B. D., Smith, S. J. & Falkow, S. ( 1993) Nature ( London) 364, 639–642.
26. Chen, L. M., Hobbie, S. & Galán, J. E. ( 1996) Science 274, 2115–2118.
27. Hobbie, S., Chen, L. M., Davis, R. & Galán, J. E. ( 1997) J. Immunol. 159, 5550–5559.
28. Eckmann, L., Stenson, W. F., Savidge, T. C., Lowe, D. C., Barrett, K. E., Fierer, J., Smith, J. R. & Kagnoff, M. F. ( 1997) J. Clin. Invest. 100, 296–309.
29. Jung, H. C., Eckmann, L., Yang, S.-K., Panja, A., Fierer, J., Morzycka-Wroblewska, E. & Kagnoff, M. F. ( 1995) J. Clin. Invest. 95, 55–65.
30. Hardt, W.-D., Chen, L.-M., Schuebel, K. E., Bustelo, X. R. & Galán, J. E. ( 1998) Cell 93, 815–826.
31. Hardt, W.-D., Urlaub, H. & Galán, J. E. ( 1998) Proc. Natl. Acad. Sci. USA 95, 2574–2579.
32. Wood, M. W., Rosqvist, R., Mullan, P. B., Edwards, M. H. & Galyov, E. E. ( 1996) Mol. Microbiol. 22, 327–338.
33. Goehring, U. M., Schmidt, G., Pederson, K. J., Aktories, K. & Barbieri, J. T. ( 1999) J. Biol. Chem. 274, 36369–36372.
34. Galyov, E. E., Wood, M. W., Rosqvist, R., Mullan, P. B., Watson, P. R., Hedges, S. & Wallis, T. S. ( 1997) Mol. Microbiol. 25, 1903–1912.
35. Hall, A. ( 1998) Science 279, 509–514.
36. Van Aelst, L. & D'Souza-Schorey, C. ( 1997) Genes Dev. 11, 2295–2322.
37. Chen, L. M., Bagrodia, S., Cerione, R. A. & Galán, J. E. ( 1999) J. Exp. Med. 189, 1479–1488.
38. Symons, M., Derry, J. M. J., Karlak, B., Jiang, S., Lemahieu, V., McCormick, F., Francke, U. & Abo, A. ( 1996) Cell 84, 723–734.
39. Pace, J, Hayman, M. J. & Galán, J. E. ( 1993) Cell 72, 505–514.
40. Peppelenbosch, M. P., Qiu, R. G., de Vries-Smits, A. M., Tertoolen, L. G., de Laat, S. W., McCormick, F., Hall, A., Symons, M. H. & Bos, J. L. ( 1995) Cell 81, 849–856.
41. Zhou, D., Mooseker, M. & Galán, J. E. ( 1999) Science 283, 2092–2095.
42. Zhou, D., Mooseker, M. S. & Galán, J. E. ( 1999) Proc. Natl. Acad. Sci. USA 96, 10176–10181.
43. Hayward, R. D. & Koronakis, V. ( 1999) EMBO J. 18, 4926–4934.
44. Takeuchi, A. ( 1967) Am. J. Pathol. 50, 109–136.
45. Kaniga, K., Uralil, J., Bliska, J. B. & Galán, J. E. ( 1996) Mol. Microbiol. 21, 633–641.
46. Bourne, H. R., Sanders, D. A. & McCormick, F. ( 1990) Nature ( London) 348, 125–132.
47. Scheffzek, K., Ahmadian, M. R. & Wittinghofer, A. ( 1998) Trends Biochem. 23, 7257–7262.








