
Colloquium
A framework for interpreting the leucine-rich repeats of the Listeria internalins
Michael Marino*, Laurence Braun†, Pascale Cossart†, and Partho Ghosh*‡
* Department of Chemistry and Biochemistry, University of California, San Diego, 9500 Gilman Drive, La Jolla, CA 92093-0314; and
† Institut Pasteur, Unité des Interactions Bactéries-Cellules, 28 rue du Dr. Roux, 75015 Paris, France
The surface protein InlB of the bacterial pathogen Listeria monocytogenes is required for inducing phagocytosis in various non-phagocytic mammalian cell types in vitro. InlB causes tyrosine phosphorylation of host cell adaptor proteins, activation of phosphoinositide 3-kinase, and rearrangements of the actin cytoskeleton. These events lead to phagocytic uptake of the bacterium by the host cell. InlB belongs to the internalin family of Listeria proteins, which also includes InlA, another surface protein involved in host cell invasion. The internalins are the largest class of bacterial proteins containing leucine-rich repeats (LRR), a motif associated with protein–protein interactions. The LRR motif is found in a functionally diverse array of proteins, including those involved in the plant immune system and in the mammalian innate immune response. Structural and functional interpretations of the sequences of internalin family members are presented in light of the recently determined x-ray crystal structure of the InlB LRR domain.
A number of intracellular pathogens have the ability to invade mammalian cells that are normally nonphagocytic by inducing phagocytic behavior (1). The cellular machinery required for phagocytosis appears to be present in many types of non-phagocytic cells but in a constitutively unassembled or inactive state. Certain pathogens possess means by which to induce assembly of the phagocytic machinery necessary to bring about their own uptake. Among these pathogens is the facultative intracellular bacterium Listeria monocytogenes, a cause of meningitis and abortion in humans (2). L. monocytogenes induces its own phagocytosis in a large number of nonphagocytic cell types in vitro through the actions of the bacterial surface proteins InlA and InlB (also called internalin and internalin B, respectively) (3).
The related proteins InlA and InlB act on different sets of cell types. Although InlA is required for inducing phagocytosis in the enterocyte-like epithelial cell line Caco-2 (4), InlB is required for inducing phagocytosis in some hepatocyte, endothelial, epithelial, or fibroblast-like cell lines (e.g., Vero, HeLa, HEp-2, Chinese hamster ovary) (5, 6, 7, 8, 9 and 10). The different cell type specificities likely reflect the fact that InlA and InlB have different receptor specificities. The receptor for InlA is E-cadherin (11), and, whereas the receptor for InlB has not been identified, it is clear that it is not E-cadherin. Nevertheless, InlB has been shown to have potent effects on host cell signaling events. InlB promotes activation of host cell phosphoinositide (PI) 3-kinase and tyrosine phosphorylation of certain adaptor proteins (Gab1, Shc, and Cbl) implicated in the membrane localization of phosphoinositide 3-kinase (PI 3-kinase) (12, 13). PI 3-kinase is known to have effects on actin polymerization ( 14), a process that is required for L. monocytogenes invasion of host cells (12). Reorganization of the actin cytoskeleton presumably leads to changes in the membrane that cause phagocytosis of the bacterium.
Internalin Family
InlA and InlB belong to the internalin family of Listeria proteins, which includes at least seven additional members in L. monocytogenes (InlC, InlC2, InlD, InlE, InlF, InlG, and InlH) (4, 15, 16, 17 and 18). Internalin proteins are also found in Listeria ivanovii, a pathogen of ruminants (19, 20), and also in nonpathogenic species of the genus Listeria. Except for InlC, which is a soluble and secreted protein, the L. monocytogenes internalins have sequence motifs that are consistent with bacterial cell surface localization. In contrast to InlB, which has been shown to be important to proliferation in hepatocytes in vivo (21, 22), the role of InlA in virulence has not been clearly demonstrated (23, 24). The biochemical functions and exact virulence properties of the other L. monocytogenes internalins remain to be determined.
Proteins of the internalin family share several sequence motifs, the most distinctive of these being leucine-rich repeats (LRR). The LRR motif is associated with protein–protein interactions and is found in both bacterial and eukaryotic proteins (25, 26 and 27). The majority of bacterial LRR proteins are found in Listeria and belong to the internalin family, but LRR proteins have also been identified in Yersinia spp. (28), Shigella flexneri (29), Salmonella typhimurium (30), and Burkholderia (Pseudomonas) solanacearum (31). Among eukaryotes, the LRR motif is found in proteins encoded by the disease resistance (R) genes of the plant immune system (32, 33 and 34) and by the toll and toll-like genes of Drosophila and mammals, which are involved in innate immune responses (35, 36). The LRR motif is not limited to virulence- or immunity-related proteins but is instead found in a large group of proteins with highly diverse functions and cellular localizations (25).
The LRR regions of InlA and InlB play critical roles in interactions with mammalian cells. The LRR region of InlA is necessary to promote invasion of permissive cells in vitro (37). Likewise, the LRR region of InlB is necessary for the activation of phosphoinositide 3-kinase, rearrangement of the actin cytoskeleton, and invasion of permissive cells in vitro (38). The recently determined x-ray crystal structure of the InlB LRR domain (39) provides a framework for the interpretation of sequences of internalin family members.
Internalin LRR
The internalin LRR regions are highly regular, with almost all repeats being 22 residues in length (Fig. 1). The LRRs are tandemly repeated from 6 times in InlG to 16 times in InlA. The 7.5 LRRs present in InlB give it its elongated and curved shape (Fig. 2). The repeats are similar to those observed in the LRRs of porcine and human ribonuclease inhibitors (RI) (40, 41 and 42), the
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviations: LRR, leucine-rich repeat; RI, ribonuclease inhibitor.
|
‡ |
To whom reprint requests should be addressed. E-mail: pghosh@ucsd.edu. |

Fig. 1. Sequence alignments of L. monocytogenes internalin LRR regions. Stars above the sequences denote conserved internalin LRR residues, and bars denote the position of β-strands and extent of 310-helices observed in the InlB structure (39). Residues predicted to form the concave or convex face of these structures are enclosed in boxes. Hydrophobic, negatively charged, and positively charged residues predicted to be surface exposed are highlighted in yellow, red, and cyan, respectively. Sequences for InlA, InlB, InlC, InlC2, InlD, InlE, InlF, InlG, and InlH are shown. The repeats are 22 residues in length except for repeat 4 of InlC and repeat 6 of InlA, which are 21 residues, and repeat 5 of InlF, which is 23 residues. These small deletions and insertions are predicted to occur at loop regions.
U2LRR fragment of U2 small nuclear ribonucleoprotein ( 43), and the Ran GTP-ase activating protein rnalp (44). As in these structures, each repeat alternates between a short β-strand and an opposing antiparallel helical segment; the β-strands and helices are connected to each other by coils. The main chain wraps around in a right-handed sense, with the β-strands forming a parallel β-sheet and the helices stacking on each other, giving rise to the elongated shape of the LRR.
Although the β-strands are a highly conserved structural feature of LRR proteins, the helical segments are more divergent. The β-strands are almost always composed of three residues that are in precise register. On the other hand, the helical segments are variable in length, register, and type. In fact, one repeat of U2LRR contains a second β-strand instead of a helix. In InlB, the helical segments are short (three to five residues) and are exclusively composed of 310-helices. U2LRR possesses 23-and 25-residue repeats that also form 310-helices. This contrasts with RI and rnalp, which have long (10–14 residues) α-helices and also have longer repeat lengths (28 residues or greater). Apparently the more tightly wound 310-helix accommodates formation of short repeats more favorably than does the α-helix.
The curvature of the LRR region arises from the β-strands of adjacent repeats being packed more closely together than opposing helices. The β-strands are within interrepeat hydrogen bonding distance, whereas the helices form intrarepeat hydrogen bonds. The β-strands form the concave face and the helices the
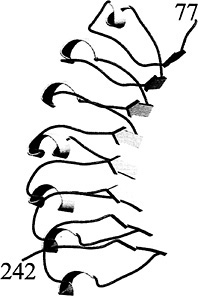
Fig. 2. Structure of the InlB LRR region (residues 77–242). The right-handed coil of the LRR alternates between β-strands and 310-helices. The β-strands form the concave face of the molecule and have a superhelical twist not observed in other LRR proteins. This figure and Fig. 4 were generated with MOLSCRIPT (48) and rendered with RASTER3D (49).
convex face of the molecule (Fig. 2). This curvature would appear to limit the possible number of tandem LRRs, because a closed circle would be formed. However, the InlB β-strands are twisted and give the entire structure a right-handed superhelical twist, thereby placing no obvious limits on the possible number of tandem repeats (39). The β-strands of RI, U2LRR, and rnalp are not as extensively twisted and indeed place a limit on the number of possible tandem repeats. It is not clear at present what gives rise to this difference.
The coil regions connecting β-strands and 310-helices constitute the great majority (14–17 residues) of the 22-residue InlB LRR. Even though these regions lack conventional secondary structure, they form highly regular structures (rms deviation of 0.32 Å in Cα positions). This regularity may in part be caused by the presence of water molecules that act as extensions of secondary structure. These waters are intercalated between repeats and form bridging hydrogen bonds between main-chain atoms of adjacent repeats. Structural determination of InlB to high resolution (1.86 Å) allowed visualization of three distinct spines of water molecules that run the length of the repeat region (39). Some of these hydrogen bond-mediating waters are also observed in the 2.0-Å resolution structure of human RI (42). A regular pattern of hydrogen bond-mediating waters is expected to be present in other LRR proteins and to be observed in high-resolution structures of other LRR proteins.
LRR Flanking Sequences
The LRR regions are flanked by sequences that are conserved in the internalin family and that most likely play a role in providing stability. At the N terminus of the LRR region is a segment of ≈40 residues termed the “N-terminal cap” in InlB, because it provides a hydrophilic cap on the hydrophobic core of the first LRR (39). The hydrophobic core would be exposed to solvent at either end of the LRR were it not for other portions of the protein. The N-terminal cap forms numerous polar and hydrophobic interactions with the first LRR. The most prominent of these is a hydrophobic interaction with Tyr at position 20 of the first LRR, a highly conserved residue in the internalin family (Fig. 1).
In addition to its structural role, the N-terminal cap has been suggested to be involved in function based on the observation of two highly solvent-exposed calcium ions that bind to this region (39). The calciums are not required for formation of protein structure and may instead act as metal ion bridges between InlB and mammalian cell surface receptors or binding proteins. The N-terminal cap and LRR regions of InlB are sufficient to elicit mammalian cell effects and to induce phagocytosis (38).
At the C terminus of the LRR region is a conserved sequence of ≈100 residues termed the interrepeat (IR) region (17). The IR region is dispensable for the mammalian cell signaling activities of InlB but is required in InlA for inducing phagoctyosis (37). The IR region has not yet been visualized, but crystals of intact InlB have been obtained that should elucidate the role of this as well as other regions of internalins (M.M. and P.G., unpublished data). At least a portion of the IR region is likely to provide a hydrophilic cap on the last LRR.
LRR Pattern: Structural Residues
A total of 10 positions of the 22-residue internalin LRR are highly conserved and serve structural rather than functional roles. Seven positions (2, 5, 7, 12, 15, 18, and 21) of the repeat contain mainly Leus or Iles that point inward and compose the hydrophobic core of the protein (Fig. 3). Other hydrophobic residues are accommodated at these positions, including Val, Phe, Met, and Ala (Fig. 1). Position 10 also faces inward and usually contains Asns or Glns, which form the so-called “Asn ladder” (45). The Asn hydrogen bonds to main-chain atoms in its own repeat and to those in the preceding repeat. This explains why position 10 in the first repeat is not constrained to be an Asn or Gln.
The seven hydrophobic positions along with the Asn-ladder position define the internalin LRR sequence motif (Fig. 4). This motif corresponds almost identically to the 22-residue repeat
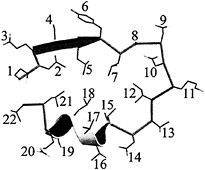
Fig. 3. Structure of a single InlB LRR. Positions 2, 5, 7, 10, 12, 14, 15, 17, 18, and 21 are conserved for structural reasons. The remaining positions are solvent exposed and variable. The register of the InlB LRR is that described for porcine RI (40).
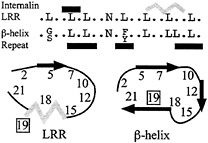
Fig. 4. Consensus motifs of the internalin LRR and the β-helix repeat. The two differ primarily at position 19. Black bars above the sequences represent β-strands, and the gray bar represents the 310-helix. Black arrows in the cartoons represent β-strands and the gray zigzag the 310-helix. The register for the β-helix repeat has been adjusted for purposes of comparison.
motif identified for proteins of the right-handed β-helix family (Fig. 4) (46, 47). This correspondence led to a suggestion that the internalins would form a β-helix structure rather than the observed LRR structure (47). The β-helix structure is found in pectate lyases and forms a distinctive L-shaped repeat containing three β-strands. The greatest difference between the two repeat motifs occurs at position 19, which faces outward and is variable in the internalin LRR but is occupied by an inward-facing conserved hydrophobic residue in the β-helix motif. The close correspondence of these two motifs suggests a means by which to probe the basis for their formation.
Two structural positions of the InlB LRR are unique to the internalin family. Position 14 is outward facing and is usually occupied by an Asp that hydrogen bonds to main-chain atoms at the beginning of 310-helices (Fig. 3). Other residues capable of hydrogen bonding, including Ser, Thr, Asn, and Gln, are also found at this position (Fig. 1). Position 17 is generally occupied by a small amino acid, usually Gly, Pro, Ala, or Ser. It is located on the 310-helix, and a larger residue at this position would sterically clash with the preceding 310-helix. These two positions add to the eight inward-facing ones to yield the 10 positions conserved for structural reasons.
LRR Pattern: Functional Residues
More than half the residues of the internalin LRR face outward, are variable, and can serve to define protein–protein interaction surfaces specific to each internalin. As with other LRR proteins, the interaction surfaces of the internalins are likely to be formed by the β-strand regions that constitute the concave faces of these molecules. The concave face has been observed to form the major binding surface in complexes of RI and U2LRR with their target proteins (41, 43) and is inferred to form the binding surface for rna1p (44). The sides of RI adjacent to the concave face are also involved in binding. In InlB, the concave face contains separate patches of hydrophobic and negatively charged residues that could constitute protein–protein interaction surfaces (39).
Sequence characteristics of the internalins also point to the concave face as being important to protein–protein interactions. Three-quarters of the hydrophobic residues predicted to be surface exposed in the L. monocytogenes internalins are located on the concave face (positions 3, 4, 6, 8, and 9) (Fig. 1). Likewise, greater than three-quarters of the negatively charged residues predicted to be surface exposed are also located on the concave face (Fig. 1). In general, the concave faces of the internalins possess the hydrophobic and negative charge characteristics observed for InlB. The sides adjacent to the concave face (positions 11 and 13 on one side, and positions 21 and 1 on the other) are highly enriched in positively charged residues, as also observed in InlB. In contrast to the distinctive pattern of residues found on the concave face and sides, the convex face (positions 16, 19, and 20) of the internalins appears to be relatively featureless (Fig. 1). The exception to this is InlC, for which the convex face is predicted to contain a small hydrophobic patch. Although the high sequence conservation of the internalin LRR allows strong predictions about protein–protein interaction sites based on the InlB structure, it does not lead as easily to predictions about target specificity.
The challenge for the future will be in identifying binding partners for individual internalins and in determining how the LRR structural motif is used to generate a variety of specific functional interactions.
M.M. was supported by National Institutes of Health Training Grant GM07240-24. This work was in part supported by a W. M. Keck Distinguished Young Scholar award and an American Heart Association grant (9930135N) to P.G.
1. Finlay, B. B. & Cossart, P. ( 1997) Science 276, 718–725.
2. Lorber, B. ( 1997) Clin. Infect. Dis. 24, 1–9.
3. Cossart, P. & Lecuit, M. ( 1998) EMBO J. 17, 3797–3806.
4. Gaillard, J. L., Berche, P., Frehel, C., Gouin, E. & Cossart, P. ( 1991) Cell 65, 1127–1141.
5. Dramsi, S., Biswas, I., Maguin, E., Braun, L., Mastroeni, P. & Cossart, P. ( 1995) Mol. Microbiol. 16, 251–261.
6. Lingnau, A., Domann, E., Hudel, M., Bock, M., Nichterlein, T., Wehland, J. & Chakraborty, T. ( 1995) Infect. Immun. 63, 3896–3903.
7. Braun, L., Ohayon, H. & Cossart, P. ( 1998) Mol. Microbiol. 27, 1077–1087.
8. Greiffenberg, L., Goebel, W., Kim, K. S., Weiglein, I., Bubert, A., Engelbrecht, F., Stins, M. & Kuhn, M. ( 1998) Infect. Immun. 66, 5260–5267.
9. Müller, S., Hain, T., Pashalidis, P., Lingnau, A., Domann, E., Chakraborty, T. & Wehland, J. ( 1998) Infect. Immun. 66, 3128–3133.
10. Parida, S. K., Domann, E., Rohde, M., Muller, S., Darji, A., Hain, T., Wehland, J. & Chakrabory, T. ( 1998) Mol. Microbiol. 28, 81–93.
11. Mengaud, J., Ohayon, H., Gounon, P., Mege, R. M. & Cossart, P. ( 1996) Cell 84, 923–932.
12. Ireton, K., Payrastre, B., Chap, H., Ogawa, W., Sakaue, H., Kasuga, M. & Cossart, P. ( 1996) Science 274, 780–782.
13. Ireton, K., Payrastre, B. & Cossart, P. ( 1999) J. Biol. Chem. 274, 17025– 17032.
14. Carpenter, C. L. & Cantley, L. C. ( 1996) Curr. Opin. Cell Biol. 8, 153–158.
15. Engelbrecht, F., Chun, S. K., Ochs, C., Hess, J., Lottspeich, F., Goebel, W. & Sokolovic, Z. ( 1996) Mol. Microbiol. 21, 823–837.
16. Domann, E., Zechel, S., Lingnau, A., Hain, T., Darji, A., Nichterlein, T., Wehland, J. & Chakraborty, T. ( 1997) Infect. Immun. 65, 101–109.
17. Dramsi, S., Dehoux, P., Lebrun, M., Goossens, P. L. & Cossart, P. ( 1997) Infect. Immun. 65, 1615–1625.
18. Raffelsbauer, D., Bubert, A., Engelbrecht, F., Scheinpflug, J., Simm, A., Hess, J., Kaufmann, S. H. & Goebel, W. ( 1998) Mol. Gen. Genet. 260, 144–158.
19. Engelbrecht, F., Domainguez-Bernal, G., Hess, J., Dickneite, C., Greiffenberg, L., Lampidis, R., Raffelsbauer, D., Daniels, J. J., Kreft, J., Kaufmann, S. H., et al. ( 1998) Mol. Microbiol. 30, 405–417.
20. Engelbrecht, F., Dickneite, C., Lampidis, R., Götz, M., DasGupta, U. & Goebel, W. ( 1998) Mol. Gen. Genet. 257, 186–197.
21. Gaillard, J. L., Jaubert, F. & Berche, P. ( 1996) J. Exp. Med. 183, 359–369.
22. Gregory, S. H., Sagnimeni, A. J. & Wing, E. J. ( 1997) Infect. Immun. 65, 5137–5141.
23. Jonquières, R., Bierne, H., Mengaud, J. & Cossart, P. ( 1998) Infect. Immun. 66, 3420–3422.
24. Lecuit, M., Dramsi, S., Gottardi, C., Fedor-Chaiken, M., Gumbiner, B. & Cossart, P. ( 1999) EMBO J. 18, 3956–3963.
25. Kobe, B. & Deisenhofer, J. ( 1994) Trends Biochem. Sci. 19, 415–421.
26. Buchanan, S. G. & Gay, N. J. ( 1996) Prog. Biophys. Mol. Biol. 65, 1–44.
27. Kajava, A. V. ( 1998) J. Mol. Biol. 277, 519–527.
28. Leung, K. Y. & Straley, S. C. ( 1989) J. Bacteriol. 171, 4623–4632.
29. Hartman, A. B., Venkatesan, M., Oaks, E. V. & Buysse, J. M. ( 1990) J. Bacteriol. 172, 1905–1915.
30. Miao, E. A., Scherer, C. A., Tsolis, R. M., Kingsley, R. A., Adams, L. G., Bäumler, A. J. & Miller, S. I. ( 1999) Mol. Microbiol. 34, 850–864.
31. Van Gijsegem, F., Gough, C., Zischek, C., Niqueux, E., Arlat, M., Genin, S., Barberis, P., German, S., Castello, P. & Boucher, C. ( 1995) Mol. Microbiol. 15, 1095–1114.
32. Dixon, M. S., Jones, D. A., Keddie, J. S., Thomas, C. M., Harrison, K. & Jones, J. D. ( 1996) Cell 84, 451–459.
33. Boyes, D. C., Nam, J. & Dangl, J. L. ( 1998) Proc. Natl Acad. Sci. USA 95, 15849–15854.
34. Gassmann, W., Hinsch, M. E. & Staskawicz, B. J. ( 1999) Plant J. 20, 265–277.
35. Belvin, M. P. & Anderson, K. V. ( 1996) Annu. Rev. Cell. Dev. Biol. 12, 393–416.
36. Hoffmann, J. A., Kafatos, F. C., Janeway, C. A. & Ezekowitz, R. A. ( 1999) Science 284, 1313–1318.
37. Lecuit, M., Ohayon, H., Braun, L., Mengaud, J. & Cossart, P. ( 1997) Infect. Immun. 65, 5309–5319.
38. Braun, L., Nato, F., Payrastre, B., Mazié, J. C. & Cossart, P. ( 1999) Mol. Microbiol. 34, 10–23.
39. Marino, M., Braun, L., Cossart, P. & Ghosh, P. ( 1999) Mol. Cell 4, 1063–1072.
40. Kobe, B. & Deisenhofer, J. ( 1993) Nature ( London) 366, 751–756.
41. Kobe, B. & Deisenhofer, J. ( 1995) Nature ( London) 374, 183–186.
42. Papageorgiou, A. C., Shapiro, R. & Acharya, K. R. ( 1997) EMBO J. 16, 5162–5177.
43. Price, S. R., Evans, P. R. & Nagai, K. ( 1998) Nature ( London) 394, 645–650.
44. Hillig, R. C., Renault, L., Vetter, I. R., Drell, T., IV, Wittinghofer, A. & Becker, J. ( 1999) Mol. Cell 3, 781–791.
45. Kobe, B. & Deisenhofer, J. ( 1995) Curr. Opin. Struct. Biol. 5, 409–416.
46. Yoder, M. D., Keen, N. T. & Jurnak, F. ( 1993) Science 260, 1503–1507.
47. Heffron, S., Moe, G. R., Sieber, V., Mengaud, J., Cossart, P., Vitali, J. & Jurnak, F. ( 1998) J. Struct. Biol. 122, 223–235.
48. Kraulis, P. ( 1991) J. Appl. Crystallogr. 24, 946–950.
49. Merrit, E. A. & Bacon, D. J. ( 1997) Methods Enzymol. 277, 505–524.





