
Colloquium
Exploitation of host cells by enteropathogenic Escherichia coli
B. A. Vallance and B. B. Finlay*
Biotechnology Laboratory, University of British Columbia, Vancouver, BC, Canada V6T 1Z3
Microbial pathogens have evolved many ingenious ways to infect their hosts and cause disease, including the subversion and exploitation of target host cells. One such subversive microbe is enteropathogenic Escherichia coli (EPEC). A major cause of infantile diarrhea in developing countries, EPEC poses a significant health threat to children worldwide. Central to EPEC-mediated disease is its colonization of the intestinal epithelium. After initial adherence, EPEC causes the localized effacement of microvilli and intimately attaches to the host cell surface, forming characteristic attaching and effacing (A/E) lesions. Considered the prototype for a family of A/E lesion-causing bacteria, recent in vitro studies of EPEC have revolutionized our understanding of how these pathogens infect their hosts and cause disease. Intimate attachment requires the type III-mediated secretion of bacterial proteins, several of which are translocated directly into the infected cell, including the bacteria's own receptor (Tir). Binding to this membrane-bound, pathogen-derived protein permits EPEC to intimately attach to mammalian cells. The translocated EPEC proteins also activate signaling pathways within the underlying cell, causing the reorganization of the host actin cytoskeleton and the formation of pedestal-like structures beneath the adherent bacteria. This review explores what is known about EPEC's subversion of mammalian cell functions and how this knowledge has provided novel insights into bacterial pathogenesis and microbe-host interactions. Future studies of A/E pathogens in animal models should provide further insights into how EPEC exploits not only epithelial cells but other host cells, including those of the immune system, to cause diarrheal disease.
T he study of bacterial pathogenesis has undergone a dramatic resurgence in interest, in part because of the reemergence of old diseases such as tuberculosis, the emergence of new bacterial diseases, and the development of antibiotic resistance in many bacterial pathogens. Much of this recent work has focused on defining the molecular and cellular mechanisms underlying how microbes cause disease and has led to a new appreciation of bacterial pathogenesis. An emerging theme in this field is the ability of many bacteria to exploit host cell signal transduction pathways and cytoskeletal/membrane components to allow colonization and invasion of their hosts. In most cases, the bacteria that take this approach are intracellular pathogens such as Shigella, Listeria, and Salmonella (1). These microbes obtain entry into the host by triggering their own uptake by both phagocytic and nonphagocytic host cells. In contrast, enteropathogenic Escherichia coli (EPEC) is an extracellular pathogen that causes disease by binding to the surface of host cells and directly injecting virulence factors into the underlying cell through its type III secretion system ( 2). These translocated bacterial proteins then interact with host cell components and alter signaling pathways, resulting in disease. EPEC is a serious and widespread cause of infantile diarrhea, particularly in developing countries (3). During infection, EPEC induces a characteristic “attaching and effacing” (A/E) histopathology on gut enterocytes. A/E lesions are characterized by the localized effacement of microvilli and marked cytoskeletal changes, including the accumulation of polymerized actin, directly beneath the adherent bacteria (4, 5). The reorganization of actin forms a pedestal-like structure upon which the bacterium resides (4, 5). A/E lesion formation thus firmly anchors the bacterium to the host cell, and this intimate attachment is thought to be essential for EPEC pathogenicity.
In recent years, our laboratory and others have made significant progress determining the mechanisms by which EPEC attaches to mammalian cells in culture and in defining the role of EPEC's virulence factors in the regulation of host cytoskeletal rearrangements and gene expression during infection. These secreted bacterial proteins are thought to corrupt host cell systems, redirecting the cell's own structural components to support the attachment of EPEC. They also induce changes in host cell signaling pathways that likely act not only in pedestal formation, but also in mediating the diarrheal response to EPEC infection. Perhaps the most significant finding of recent years was the discovery that EPEC does not bind to a host receptor during the process of intimate attachment, but instead inserts its own receptor [translocated intimin receptor (Tir)] into the membrane of the target host cell (6). Since this novel finding, other A/E lesion-causing bacterial pathogens have also been shown to produce Tir homologues (7), suggesting that the process of intimate attachment is conserved amongst many enteric pathogens. These include the hemolytic uremic syndrome causing enterohemorrhagic E. coli (EHEC; O157:H7), as well as the rabbit enteropathogenic E. coli (REPEC) and Citrobacter rodentium. Studies investigating EPEC's virulence factors are the most advanced, with EPEC considered the prototype for the family of A/E inducing pathogens. In this paper, we will review the recent progress made in elucidating the mechanisms of EPEC virulence and its exploitation of host cells. Further, we will discuss the role of these factors in the intact host and how future studies may aid our understanding of the contributions that bacterial, as well as host factors, make to EPEC mediated disease.
Clinical Symptoms and Pathology
As a human pathogen, outlining the symptoms and pathology associated with EPEC infection provides context to the recent advances made defining the molecular basis for EPEC mediated disease. One of several categories of diarrheagenic E. coli, EPEC
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviations: EPEC, enteropathogenic Escherichia coli; REPEC, rabbit enteropathogenic Escherichia coli; EHEC, enterohemorrhagic Escherichia coli; A/E, attaching and effacing; Tir, translocated intimin receptor; LEE, locus of enterocyte effacement; Esp, EPEC-secreted protein; BFP, bundle forming pilus; WAS, Wiskott-Aldrich syndrome; PMN, polymorphonuclear cell.
|
* |
To whom reprint requests should be addressed at: Biotechnology Laboratory, Room 237, Wesbrook Building, 6174 University Boulevard, University of British Columbia, Vancouver, BC, Canada V6T 1Z3. E-mail: bfinlay@interchange.ubc.ca. |
is a well established cause of human diarrhea, particularly in young children. Although outbreaks were still frequent in developed countries until the 1940s and 1950s (8), the incidence of EPEC infection in the United States and United Kingdom has since declined. However, EPEC is still responsible for occasional outbreaks in daycare centers and pediatric wards (9). EPEC has remained an important cause of infant mortality in developing countries, with recent outbreaks reporting a mortality rate of 30% (10). Thus, EPEC infection is estimated to cause the deaths of several hundred thousand children per year (2). The hallmark of EPEC infection is the A/E histopathology often observed in small bowel biopsy specimens from infected patients, and seen after the infection of epithelial cells in tissue culture (2, 3). Infection generally causes acute diarrhea, but severe cases can lead to a protracted disease (3). Aside from profuse watery diarrhea, both vomiting and the development of fever are common symptoms of EPEC infection (3). Based on the morbidity and mortality associated with this microbe, EPEC strains remain a significant health threat to children worldwide.
The Locus of Enterocyte Effacement
Unlike the nonpathogenic strains of E. coli found within the human intestine, EPEC and other pathogenic E. coli strains contain pathogenicity islands within their genome. All of the genes necessary for the formation of A/E lesions and pedestals are contained within a 35-kbp pathogenicity island termed the locus of enterocyte effacement (LEE) (2, 5). The G + C content of the LEE is 38.4%, significantly lower than the 50% composition of the nonpathogenic E. coli K-12 chromosome. This discrepancy suggests that the LEE was originally acquired from a foreign source and was subsequently inserted into EPEC's chromosome. The insertion site for the LEE region in the E. coli K-12 genome is at the site encoding the tRNA for selenocysteine. Interestingly, this location appears to be a hot spot for the insertion of several virulence factor genes, including a large but different pathogenicity island found in uropathogenic E. coli (11). The complete LEE region has been sequenced and contains a type III secretion system (2), as well as other genes necessary for pedestal formation. These include several genes coding for type III-secreted proteins, termed Esps (EPEC-secreted proteins), including EspA, EspB, EspD, and EspF, as well as an adhesin, intimin, and its translocated receptor, Tir. Mutation of any of these bacterial factors, with the exception of EspF (12), prevents A/E lesion formation in epithelial cell culture models (3). As with other type III secretion systems, cytosolic chaperone proteins have been shown to be required for the translocation of secreted effector proteins. Two chaperones have been identified in the LEE, CesD for EspB and EspD (13), and CesT that chaperones Tir (14). DNA sequences with a high degree of homology to the EPEC LEE have been found in the other A/E lesion-causing bacteria, including EHEC, as well as the mouse pathogen C. rodentium, suggesting a common pathway underlying A/E lesion formation (2). This pathway is also self contained because the introduction of the cloned LEE of EPEC into a previously nonpathogenic E. coli strain conferred the ability to form A/E lesions (15).
Localized EPEC Adherence to Epithelial Cells
Interactions between EPEC and host cells entail several distinct steps and have classically been viewed as a three-stage process. The first stage in EPEC pathogenesis involves the initial adherence of the bacterium to the host's intestinal epithelium. In this stage, EPEC form dense microcolonies on the surface of tissue culture cells in a pattern known as localized adherence (3). The bacterium is thought to initially attach to the host cell through a plasmid-encoded bundle forming pilus (BFP). Although mutants lacking this plasmid still attach to host cells, they do not form microcolonies and produce fewer A/E lesions than wild-type EPEC ( 5, 16). Even so, BFP remains an important virulence factor because BFP mutant strains show severe impairment in their ability to cause diarrhea in human volunteers (17). This loss of virulence probably indicates that both initial adherence to host cells as well as microcolony formation are critically involved in the ability of EPEC to successfully infect its host. Curiously, the mediators of initial attachment appear to vary among the family of A/E pathogens. The related pathogen EHEC lacks BFP and, unlike EPEC, infects the human colon rather the ileum (3). Therefore, whether bacterial colonization occurs preferentially in the small or the large bowel may be influenced by the expression of BFP and other adhesins as well as by environmental factors regulating the expression of other virulence factors (18).
EPEC-Secreted Proteins
The second stage of EPEC pathogenesis involves the production of bacterial proteins including EspA, EspB, and EspD. These proteins are translocated from the bacterial cytoplasm to the external environment by a type III secretion system (Fig. 1), encoded by the esc and sep genes, also found within the LEE pathogenicity island. The type III secretion machinery is thought to generate a pore permitting this translocation to occur (2, 5). Type III secretion systems also play an important role during infection by other Gram-negative pathogenic bacteria such as Yersinia and Salmonella, enabling virulence factors to be translocated directly from the bacterial cytoplasm to the host-cell membrane or cytoplasm. Although the majority of the Esps produced by EPEC are necessary for A/E lesion formation, their precise role in EPEC pathogenesis is not well defined. EspA makes filamentous appendages surrounding the bacterium that are transiently present on the bacterial surface (19). These filaments interact with the host cell, possibly forming a translocation tube reaching into the host cell. In support of this theory, EspB is translocated into the host cytosol and membrane by a process dependent on EspA (19). EspD is known to be inserted into the host cell membrane (20). Although the exact functions of EspB and EspD are unknown, their sequence homology to the
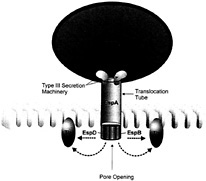
Fig. 1. Translocation of EPEC-secreted proteins (Esps) occurs through a type III secretion system that forms a pore through EPEC's membranes. Once translocated outside the bacteria, EspA forms a filamentous translocation tube whereas EspB and EspD are inserted into the host cell membrane, putatively forming a pore structure, allowing the passage of other effector proteins, such as Tir into the host cell membrane.
YopD and YopB system in Yersinia, and their ability to lyse red blood cells (21), suggests they function as components of the translocation apparatus machinery, forming a pore structure in the host membrane. Thus, the primary function of EspB and EspD may be to deliver other virulence proteins to the host cell, rather than acting as translocated effectors themselves. However, transfection of EspB into HeLa cells also leads to changes in cellular morphology and the reorganization of stress fibers at late time points (22), suggesting that EspB may also act as a cytoskeletal toxin. EspF is also a LEE-encoded EPEC secreted protein, although its role in EPEC pathogenesis is undefined because mutants lacking EspF still form A/E lesions (12).
Intimate Adherence and the Role of Tir
The third stage of EPEC infection is characterized by enterocyte effacement, pedestal formation, and intimate bacterial attachment to the host cell. Intimate attachment requires the outer bacterial membrane protein, intimin. A 94-kDa outer membrane protein encoded by the eae gene, intimin mediates intimate attachment to the host cell by binding to a 90-kDa protein in the host membrane (23). This receptor, now called Tir, was originally thought to be a host protein but was recently shown to be a bacterial protein that is translocated into the host cell membrane (6). The bacterial form of Tir migrates as 78 kDa when analyzed by SDS/PAGE, but, after translocation into the host membrane, Tir undergoes phosphorylation on tyrosine (6) and probably serine and threonine residues (24), and these modifications account for the apparent shift to 90 kDa.
EPEC's use of Tir represents the first example of a pathogen injecting its own receptor into mammalian cells and has revised our concepts of bacterial pathogenesis. By ignoring the usual dependence on the expression of a host-derived receptor, EPEC can, at least in tissue culture, infect cells of most species and tissue origins. Based on its novel role in EPEC pathogenesis, Tir has undergone intense study since its identification. Much like EspB and EspD, the transfer of Tir to the host cell requires the type III secretion system and the Esps critical for the formation of A/E lesions. Tir is required for pedestal formation because its deletion prevents EPEC from forming A/E lesions in tissue culture (6). Tir has two predicted transmembrane domains with a hairpin model proposed for Tir conformation in the host membrane (2). As predicted, the N- and C-terminal regions of Tir are located within the cell whereas the intervening region between the transmembrane domains forms an extracellular loop. Several groups have recently shown that intimin binds to this extracellular loop termed the intimin binding domain, via its C-terminal region (24, 25 and 26). Tir-intimin binding has been shown to be essential for pedestal formation and actin condensation (6). Further, Liu et al. recently used latex beads coated with the C-terminal region of EHEC intimin to trigger A/E lesion formation on HEp-2 cells (27). Pedestals only formed when cells were preinfected with an EPEC intimin mutant, which translocates Tir into the host cell. This confirms the essential role of Tir-intimin interactions in pedestal formation.
Studies examining A/E lesion formation in tissue culture have shown that only a portion of surface intimin is required to interact with translocated Tir. After A/E lesion formation, the expression of surface intimin not bound to Tir is down-regulated (28). Intimin has also been shown to bind to host cells in vitro through its C-terminal region (int280) in a Tir-independent manner (26, 29), suggesting more than one receptor for intimin on epithelial cells. Such binding requires the cysteine 937 residue (26, 29). β1 integrins have been proposed to be the host cell receptor mediating such binding. Although integrins are not present on the apical surface of enterocytes, they are expressed by other cell types, including on the apical surface of M cells located on the luminal surface of Peyer's patches (30). Interestingly, recent studies have shown that, during C. rodentium infection of mice, intimin from C. rodentium or EPEC can induce colonic epithelial hyperplasia concurrent with a strong T helper cell 1 immune response (31). These responses did not depend on bacterial viability because exposure to formalin fixed bacteria still elicited these events ( 32) whereas an intimin substitution mutant for cysteine 937 failed to do so. This suggests that intimin binding to a host cell receptor precipitates the host response. In fact, C. rodentium and EPEC may directly interact with mucosal immune cells because many mucosal T lymphocytes express β1 integrins. Despite these findings, the question of a host-derived receptor for intimin remains controversial, with reports from other laboratories finding that intimin does not bind β1 integrins and that Tir is a necessary requirement for EPEC binding to host cells (23, 33).
Structure of the A/E Lesions
The host cell undergoes a number of alterations during infection by EPEC, but the most striking change is the formation of actin pedestals. In fact, the resulting localized actin accumulation is so distinct that it forms the basis of an in vitro diagnostic test for EPEC (34). The process of pedestal formation begins following the adherence of EPEC to epithelial cells in vitro or in vivo, in concert with host cell microvilli effacement. Within 3 h of tissue culture cell infection by EPEC, pedestals begin to form directly beneath the bacteria (2, 5). The epithelial membrane beneath the adherent organisms is raised to form pedestal-like structures that can extend up to 10 µm away from the cell to form a pseudopodlike structure (35) (Fig. 2). Beyond providing strong attachment of EPEC to the cell surface, presumably to prevent being dislodged during the ensuing diarrheal response by the host, the role of pedestals is unclear. It may, however, represent a strategy by EPEC to remain extracellular, in keeping with the ability of EPEC to block its own phagocytosis by macrophages (36).
Immunofluorescence studies have shown that, in addition to membrane bound Tir, pedestals contain predominantly filamentous (F)-actin (37) (Fig. 3). α-actinin, talin, ezrin, and villin also accumulate along the length of the pedestal, as these cytoskeletal components act in the cross-linking of actin microfilaments. Nonmuscle myosin II and tropomyosin are also found, but at the base of the pedestal (37). Based on its location at the pedestal tip, as well as its predicted structure, Tir is the most likely
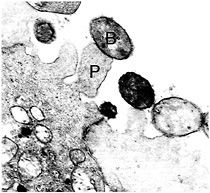
Fig. 2. Transmission electron micrograph of A/E lesions in rabbit intestinal epithelial tissue (Peyer's patch) caused by REPEC O103. Bacteria labeled with “B”; pedestal labeled with “P.” (Photograph is courtesy of Ursula Heczko, Biotechnology Laboratory, University of British Columbia.) (×20,000.)

Fig. 3. Pedestal formation on epithelial cells induced by enteropathogenic Escherichia coli. Fluorescence microscopy of the EPEC pedestal, triple-labeled for actin (green), EPEC (blue), and Tir (red) (courtesy of Danika Goosney, Biotechnology Laboratory, University of British Columbia), (×1,000.)
bacterial candidate to link EPEC to the host cytoskeleton and direct actin accumulation and pedestal formation. To accomplish this, Tir may exploit regulators of actin dynamics to initiate actin polymerization. Members of the Rho family of small GTP-binding proteins were considered potential candidates for this role, but investigations have shown that Rac, Rho, and Cdc42-dependent pathways are not involved in pedestal formation (38). Recent studies have instead implicated members of the Wiskott-Aldrich syndrome (WAS) family of proteins (WASP and N-WASP) as well as an actin nucleating factor, the heptameric Arp2/3 complex. These factors are recruited to the pedestal tip (39) (Fig. 4), and mutation of the GTPase binding domain of WASP prevented the recruitment of the Arp2/3 complex, and pedestal formation. Although host cell cytoskeletal rearrangements are responsible for pedestal formation, they are probably also responsible for microvilli effacement. Although this remains to be proven, the disappearance of microvilli may result from a bacterial-triggered depolymerization of microvilli actin, which is then used to form pedestals. However, some proteins found in pedestals are not derived from the microvilli, suggesting a more complex process than a simple rebuilding of microvilli beneath the adherent bacteria. It is currently unclear whether all of the cytoskeletal proteins identified within pedestals are essential for their formation.
Signal Transduction
Another critical form of cellular exploitation used by EPEC involves the subversion of host signaling pathways to aid in infection. As a result, several signal transduction pathways are stimulated within epithelial cells after EPEC infection. One such pathway results in tyrosine phosphorylation of substrates that co-localize with the accumulated actin beneath adherent bacteria. The major phosphorylation substrate detected in EPEC-infected cells is Tir (6). This phosphorylation event requires both the type III secretion system, as well as the Esps, because their mutation prevents EPEC from localizing tyrosine phosphorylated proteins such as Tir beneath adherent EPEC (2, 6). This suggests that EPEC triggers these responses through bacterial effector molecules. Because phosphorylation occurs after Tir enters the host cell, tyrosine kinase activity must be recruited to the vicinity of Tir within the cell. Although the identity of the involved tyrosine kinase is unknown, it is probably of host origin, presumably recruited or activated by bacterial effector proteins, or by Tir mimicking an endogenous substrate. Alternatively, a bacterial effector with tyrosine kinase activity could be translocated into the cell along with Tir. There is speculation that Tir is also serine/threonine-phosphorylated (24). The tyrosine phosphorylation event during EPEC infection is critical for actin nucleation because, in its absence, A/E lesions do not form (2, 6). Surprisingly, tyrosine phosphorylation of Tir does not occur with the related pathogen EHEC O157:H7 (7), although this pathogen readily forms pedestals. This divergence in signaling requirements suggests that subtle but important differences exist between these two pathogens in their modes of pedestal formation, although this awaits further examination.
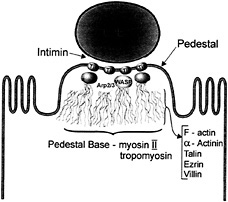
Fig. 4. The structure of the EPEC pedestal. EPEC intimately attaches to the host cell through intimin-Tir binding. N-WASP and the Arp 2/3 complex are recruited to the pedestal tip, nucleating actin. F-actin, α-actinin, talin, ezrin, and villin are found along the length of the pedestal whereas nonmuscle myosin II and tropomyosin are found at the pedestal base.
EPEC also induces other signaling cascades within the host cell, including inositol phosphate fluxes, activation of protein kinase C, phospholipase-Cγ, and NF-κB (2, 5). One consequence of inositol trisphosphate fluxes is the release of Ca2+ from intracellular stores. Several groups have investigated the effect of EPEC infection on changes in intracellular calcium, but the results have been inconclusive. Crane and Oh measured protein kinase C activity and found an enhancement in membrane-associated protein kinase C. However, increasing protein kinase C activity required intimate adherence of the bacterium because intimin mutants did not affect activity (40). EPEC also induces the activation of NF-κB in a T84 epithelial cell culture model, in association with increased interleukin-8 production and the recruitment of polymorphonuclear cells (PMNs) (41, 42). Although the importance of many of these signaling events in infection is demonstrated by their requirement for pedestal formation, much further characterization is required. In fact, it remains unclear whether these responses are specifically triggered by bacterial effector proteins or are a nonspecific consequence of EPEC infection. In either case, changes in cell signaling likely play a major role in the symptomatology of EPEC-mediated disease, and particularly in mediating the resulting diarrhea.
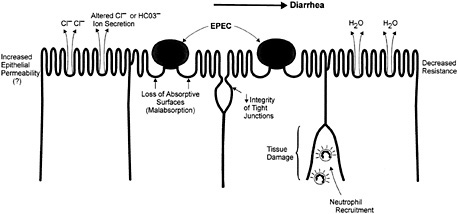
Fig. 5. Putative mechanisms underlying EPEC induced diarrhea include increased epithelial permeability and alterations in Cl− and HCO3− ion secretion. Contributing structural changes include loss of absorptive surfaces, reduced tight junction integrity, and tissue damage.
Mechanisms of EPEC-Mediated Diarrhea
The primary symptom of EPEC infection is diarrhea. Unfortunately, despite the advances made in our understanding of EPEC pathogenesis at the genetic and cellular level, how EPEC triggers diarrhea is still uncertain. In fact, it remains unclear if the resulting diarrhea is triggered by a specific manipulation of the host by EPEC virulence factors, or if EPEC is the recipient of a stereotyped host response to bacterial adhesion. Whatever the cause, diarrhea may prove of benefit to A/E lesion-causing bacteria and particularly those that inhabit the large bowel. As they colonize the intestine, A/E lesion-causing pathogens not only must interact with the cells of their eukaryotic hosts, but they are often in competition with normal intestinal flora and even other pathogens. Mechanisms like diarrhea that disturb the normal host-prokaryote equilibrium presumably provide EPEC with an advantage over competing flora. By intimately binding to the host 's enterocytes, EPEC can remain attached to the host's intestinal surface whereas other less adherent microbes are flushed away.
The diarrhea seen during EPEC infection could be caused by the dramatic loss of absorptive microvilli in the A/E lesion (3). Alternatively, at least one study has shown that EPEC infection reduces the tight junction integrity of epithelial cell monolayers, based on altered distribution of tight junction proteins such as zona occludens (ZO)-1 (43). This observation remains controversial because other studies showed no change in such proteins during infection (44). However, in volunteer studies, the incubation period between EPEC ingestion and the onset of diarrhea is less than 4 h, suggesting that a more active secretory response may be involved (45). Studies examining other diarrheal pathogens have identified changes in chloride ion secretion as one of the most common mechanisms leading to secretory diarrhea (46). Interestingly, reports have shown that EPEC can actively alter ion transport, causing a rapid but transient increase in short circuit current (Isc) in intestinal epithelial cell monolayers mounted in Ussing chambers, with chloride ion secretion implicated in this effect (47, 48). Mutation of espB but not the gene encoding intimin abrogated these ionic changes (47, 48). This agrees with studies of EPEC infection in adult human volunteers. Although EPEC intimin mutants were less virulent, diarrhea still developed in 4 of 11 volunteers who ingested the intimin mutant ( 45), indicating the involvement of other virulence factors in EPEC induced diarrhea. It should be noted that not all studies have supported a role for chloride ion secretion. Hecht and Koutsouris recently implicated changes in bicarbonate (HCO3−) ions rather than chloride ions in the EPEC mediated changes in intestinal ion transport (49).
Finally, other host factors beyond those present in epithelial cell cultures may also contribute to diarrhea. There is substantial recruitment of neutrophils and other PMNs to the site of in vivo infection (50). The inflammatory response may be attributable to bacterial triggered signals from infected cells because EPEC activates both NF-κB and interleukin-8 expression in tissue culture cells (41, 42). These signals were associated with transmigration of PMNs through epithelial cell monolayers. Increased paracellular permeability and stimulation of chloride secretion could be a consequence of this EPEC-induced PMN infiltration (Fig. 5). In vivo, an inflammatory response should take longer than 3 h to develop, suggesting it is not the mechanism that initiates EPEC-mediated diarrhea, although inflammation may contribute to the duration and severity of the diarrheal response. Care must be taken interpreting these responses, however, because many enteric pathogens, including Salmonella (51), activate NF-κB and IL-8 expression simply by adhering to the surface of host cells. This response could therefore be a generalized mechanism used by pathogens to initiate diarrhea, or could reflect a stereotyped innate host response to bacterial adhesion. Whatever the cause, the PMNs and other inflammatory cells recruited to the infected intestine also cause considerable tissue damage through the release of toxic inflammatory mediators (46). Although the resultant pathology probably contributes to changes in epithelial function, many of the cytokines expressed during A/E lesion causing bacterial infections (31), such as γ-interferon and TNF-α, have also been shown to directly alter epithelial cell function in tissue culture (52).
Pathogenesis in Animal Models
Despite the progress made defining the molecular basis for EPEC-host cell interactions, we still know very little about EPEC-mediated disease. Infections affect more than a single cell type in isolation and need to be considered in the context of the complexity of their hosts. Because EPEC is primarily a pediatric pathogen, ethical considerations have generally precluded tak-
ing biopsy samples during infection. As a result, only a few studies of EPEC infection have been carried out using adult human volunteers (45). These findings have been recently reviewed by Nataro and Kaper (3). These studies used very large bacterial doses and, although important, represent an artificial situation. There is now an urgent need to examine the new information regarding EPEC pathogenesis in the context of animal models. Unfortunately, EPEC is a human specific pathogen and does not infect most laboratory animal species. In this case, the family of enteric A/E lesion-causing pathogens can be used to draw conclusions about EPEC, by using microbes that naturally infect other animal species. E. coli capable of forming A/E lesions have been isolated from rabbits (REPEC), as well as calves, sheep, pigs, dogs, and mice. The rabbit pathogen REPEC produces a disease similar to EPEC, infecting the small bowel of weanling rabbits and causing both diarrhea and weight loss (53). Despite these advantages, there are limitations to the model, as genetic and immunological resources are not plentiful for the rabbit. In this respect, C. rodentium (formerly known as Citrobacter freundii biotype 4280) offers an advantage. C. rodentium produces A/E lesions in mice and, like EHEC in humans, colonizes the large rather than the small bowel. Unlike REPEC, this pathogen induces a strong host Th1 immune response as well as epithelial cell hyperplasia rather than diarrhea (31). Although the symptomatology may differ, the basic mechanisms of A/E lesion formation and host response likely remain the same. As a result, the wide array of reagents available for the mouse, including antibodies, recombinant cytokines, and gene knockout strains, makes this model highly suited to study the host response to infection.
Although animal models offer clear opportunities to improve our understanding of how A/E lesion-causing pathogens cause disease, in vivo studies have lagged behind in vitro studies of EPEC. We are only beginning to identify the bacterial factors necessary for disease, but, so far, results from animal models have been encouraging, as they have validated those findings made in tissue culture. Using the REPEC model, Abe et al. demonstrated the crucial nature of the bacterial proteins EspA and EspB in both A/E lesion formation and diarrheal disease (53). REPEC strains lacking either gene lost the ability to form A/E lesions, although they were still able to locally adhere to the small bowel. The essential role of EspB in the formation of A/E lesions has also been shown in the C. rodentium model in mice (54). Interestingly, the recent studies on C. rodentium infection by Higgins et al. identified an important component of the bacterial triggered host response not dependent on A/E lesion formation (32). The discovery that intimin expression was required to induce both the epithelial hyperplasia and T helper cell 1 immune activation confirms the need to assess potential virulence factors of EPEC in animal models.
EPEC Interactions with the Intact Host
As described above, the focus of research on EPEC pathogenesis has been on the microbe. Through the mutation of genes encoding potential bacterial virulence factors, their role in the infectious process has been surmised by the ability to form A/E lesions in tissue culture, or their ability to cause diarrhea in vivo. This has proven successful because most of EPEC's LEE-encoded genes have been assessed in vitro for their importance in A/E lesion formation (2, 3, 5), and several have been tested in rabbit models for their importance in the diarrheal response (53). Unfortunately, these studies only demonstrate the necessity of a bacterial protein for pedestal formation or diarrhea but do not identify its actual role(s) in the infectious process. As a result, only a few of EPEC's arsenal of effector proteins have been well characterized, and, likely, even these factors have as yet unrecognized functions, as exemplified by the discovery of intimin's role in generating the immune response during C. rodentium infection (32). Characterizing the functions of EPEC's secreted and translocated effectors is the next step in the field of EPEC pathogenesis. This will require continued intensive cellular and biochemical analysis of the changes elicited within infected host cells. To fully appreciate EPEC's capacity to exploit mammalian cells, these studies also need to be examined within the complexity of an intact host.
There are a number of areas that need to be assessed, including how EPEC interacts with and manipulates the array of cell types present within the intestine. The mammalian intestinal epithelium is a highly specialized tissue that maintains complex and selective secretory and absorptive functions while interfacing with the external luminal environment. Particularly in the colon, the epithelium exists within a diverse microfloral ecology, which contributes to and regulates the physiology of the lower gut. As a result, intestinal epithelial cells have evolved selective physical, chemical, and immunological barriers that permit this mutually beneficial co-existence (51). Not surprisingly, studies using epithelial cell cultures can only model the intestinal epithelium in a limited fashion. The epithelial lining of the intestine is also a dynamic system, with epithelial cells undergoing rapid turnover every 3–4 days (55). Thus, the epithelial layer of the gut contains cells at varying stages of differentiation, ranging from immature crypt cells to mature enterocytes. As well, more specialized forms of epithelial cells are found interspersed among the columnar epithelium. These include the mucus-secreting goblet cells and the antigen-sampling M cells that overlie the gut-associated lymphoid tissues. Although EPEC is probably capable of interacting with these cells to form pedestals (56), whether EPEC subverts their function by other means has yet to be examined.
Besides interactions with the host's intestinal epithelium, A/E lesion-causing bacteria must also encounter and presumably circumvent innate host defenses. Unfortunately, little is known about innate immunity against EPEC infection, but histological examination of infected tissues has identified both neutrophils and macrophages responding to both EPEC and C. rodentium infection (31, 50). This recruitment of inflammatory cells may be in response to signals sent by the infected epithelium because epithelial cells in culture can produce the neutrophil chemoattractant IL-8 in response to infection by EPEC (41, 42). An active role for intestinal epithelial cells in host defense is not new; many studies have shown that epithelial cells can secrete a number of pro-inflammatory and antimicrobial agents in response to bacterial infection or after immune stimulation (46, 52). Other aspects of innate immunity against EPEC, such as the actions of antimicrobial peptides, have yet to be studied. However, both human colostrum and milk have been shown to strongly inhibit the adhesion of EPEC to HEp-2 cells in vitro with the inhibitory activity found in both the sIgA and oligosaccharide fractions (57). These results suggest that breast-feeding may protect infants from EPEC infection, although a better understanding of the mechanisms involved is required.
Just as the host has developed protective measures, EPEC has evolved measures to counteract and subvert the host's immune response. EPEC can block its own uptake by professional phagocytes like macrophages (36), presumably to inhibit antigen presentation by macrophages. This requires a functioning type III secretion system and the expression of EspA, EspB, and EspD, but not Tir. These requirements suggest that EPEC disrupts the phagocytic process by directly contacting macrophages, rather than through a soluble mediator. EPEC can also inhibit the host's immune response, with Malstrom and James reporting that EPEC lysates inhibited IL-2, IL-4,
and IFN-γ production by both mucosal and splenic lymphocytes (58). This effect did not depend on the expression of either EspA or EspB. However, it is unclear how relevant these interactions may be when taken in the context of the massive immune activation seen in the related C. rodentium model (32).
Conclusions
Research in recent years has made remarkable progress in our understanding of the molecular mechanisms underlying EPEC pathogenesis. Molecular biology, genetics, and cell biology have provided many new insights into how EPEC and related pathogens interact with and exploit host cells during infection. The identification of a type III secretion system within EPEC's genome, as well as the discovery of Tir, has improved our understanding of how EPEC subverts the host cytoskeleton to permit bacterial attachment. This research has identified features common to other enteric pathogens as well as strategies apparently unique to A/E lesion-causing bacteria, such as the translocation of the bacterial receptor Tir into the host membrane.
Although studying the molecular and cellular aspects of bacterial virulence factors has informed us about the mechanisms of bacterial disease, these factors can also function as tools to study various aspects of mammalian cell functions. As such, they have received much interest in the field of microbial pathogenesis, as well as from cell biologists interested in the mechanisms underlying actin dynamics and cytoskeletal rear-rangements (39). These studies also have relevance to unrelated pathogens, such as Helicobacter pylori, which produces A/E-like lesions (59). However, H. pylori-induced pedestals lack the intense actin accumulation observed with the A/E intestinal pathogens, and no homologues of the A/E genes have been found in H. pylori. There are also similarities between the immunopathology associated with A/E lesion-inducing bacteria, and that seen in inflammatory bowel diseases (31). Although no infectious causal agent has yet been identified in inflammatory bowel diseases, the potential contribution of maladaptive microbial-host interactions to the chronicity of these diseases has long been of great interest to gastroenterologists (60).
Despite this progress, more studies are needed characterizing EPEC 's effector molecules as well as their role in causing diarrhea and disease within the intact host. Other gene products that contribute to colonization of the host must be identified, and their role in the infectious process examined in vivo. Although it is true that infections may differ between animals and humans, the similarities will probably prove greater than the differences. Studies integrating host genetics, physiology, and the immune system, all of which are critical determinants to the outcome of infection, should provide a better understanding of EPEC and other A/E pathogens, and together these developments may lead to new therapeutic strategies. With our present knowledge of the factors that mediate bacterial adhesion to the host cell, and the demonstration that preventing bacterial adherence prevents most aspects of the disease, we have already identified potential targets for vaccination. Although the pathogenic effects of A/E lesion formation still need to be separated from other bacterial actions during infection, a successful approach may involve vaccination against the factors involved in bacterial adhesion such as the components and effectors of the type III secretion machinery. Alternatively, with the recent interest in microbe-microbe interactions, and in the use of probiotics, identifying bacterial species that can out-compete EPEC for attachment to host cells may be an attractive option. By continuing to characterize EPEC's effector molecules, their specific effects, and the host's response to infection, we should look forward to new advances in the prevention and treatment of A/E pathogen mediated diarrhea.
We thank Jean Celli, Annick Gauthier, and Carrie Rosenberger for helpful discussions and critical reading of the manuscript and Danika Goosney, Ursula Heczko, and Fern Ness for figures. Work in our laboratory is supported by operating grants from the Medical Research Council of Canada and a Howard Hughes International Scholar award (to B.B.F.). B.B.F. is a Medical Research Council Scientist.
1. Steele-Mortimer, O., Knodler L. A. & Finlay, B. B. ( 2000) Traffic 1, 107–118.
2. DeVinney, R., Gauthier, A., Abe, A. & Finlay, B. B. ( 1999) Cell Mol. Life Sci. 55, 961–976.
3. Nataro, J. P. & Kaper, J. B. ( 1998) Clin. Microbiol. Rev. 11, 142–201.
4. Kaper, J. B. ( 1998) Trends Microbiol. 6, 169–172.
5. Frankel, G., Philipps, A. D., Rosenshine, I., Dougan, G., Kaper, J. B. & Knutton, S. ( 1998) Mol. Microbiol. 30, 911–921.
6. Kenny, B., DeVinney, R. D., Stein, M., Reinscheid, D. J., Frey, E. A. & Finlay, B. B. ( 1997) Cell 91, 511–520.
7. DeVinney, R., Stein, M., Reinscheid, D., Abe, A., Ruschkowski, S. & Finlay, B. B. ( 1999) Infect. Immun. 67, 2389–2398.
8. Robins-Browne, R. M. ( 1987) Rev. Infect. Dis. 9, 28–53.
9. Bower, J. R., Congeni, B. L., Cleary, T. G., Stone, R. T., Wanger, A., Murray, B. E., Mathewson, J. J. & Pickering, L. K. ( 1989) J. Infect. Dis. 160, 243–247.
10. Senerwa, D., Olsvik O., Mutanda L. N., Lindqvist, K. J., Gathuma, J. M., Fossum, K. & Wachsmuth, K. ( 1989) J. Clin. Microbiol. 27, 1307–1311.
11. Blum, G., Ott, M., Lischewski, A., Ritter, A., Imrich, H., Tschape, H. & Hacker, J. ( 1994) Infect. Immun. 62, 606–614.
12. McNamara, B. P. & Donnenberg, M. S. ( 1998) FEMS Microbiol. Lett. 166, 71–78.
13. Wainwright, L. A. & Kaper, J. B. ( 1998) Mol. Microbiol. 27, 1247–1260.
14. Abe, A., de Grado, M., Pfuentzer, R. A., Sanchez-SanMartin, C., DeVinney, R., Puente, J. L., Strynadka, C. J. & Finlay, B. B. ( 1999) Mol. Microbiol. 33, 1162–1175.
15. McDaniel, T. K. & Kaper J. B. ( 1997) Mol. Microbiol. 23, 399–407.
16. Donnenberg, M. S., Kaper, J. B. & Finlay, B. B. ( 1997) Trends Microbiol. 5, 109–114.
17. Levine, M. M., Nataro, J. P., Karch, H., Baldini, M. M., Kaper, J. B., Black, R. E., Clements, M. L. & O'Brien, A. D. ( 1985) J. Infect. Dis. 152, 550–559.
18. Kenny, B., Abe, A., Stein, M. & Finlay, B. B. ( 1997) Infect. Immun. 65, 2606–2612.
19. Knutton, S., Rosenshine, I., Pallen, M. J., Nisan, I., Neves, B. C., Bain, C., Wolff, C., Dougan, G. & Frankel, G. ( 1998) EMBO J. 17, 2166–2176.
20. Kresse, A. U., Rohde, M. & Guzman, C. A. ( 1999) Infect. Immun. 67, 4834–4842.
21. Warawa, J., Finlay, B. B. & Kenny, B. ( 1999) Infect. Immun. 67, 5538–5540.
22. Taylor, K. A., Luther, P. W. & Donnenberg, M. S. ( 1999) Infect. Immun. 67, 120–125.
23. Rosenshine, I., Ruschkowski, S., Stein, M., Reinscheid, D. J., Mills, S. D. & Finlay, B. B. ( 1996) EMBO J. 15, 2613–2624.
24. Kenny, B. ( 1999) Mol. Microbiol. 31, 1229–1241.
25. de Grado, M., Abe. A., Gauthier, A., Steele-Mortimer, O., DeVinney, R. & Finlay B. B. ( 1999) Cell. Microbiol. 1, 7–17.
26. Hartland, E. L., Batchelor, M., Delahay, R. M., Hale, C., Matthews, S., Dougan, G., Knutton, S., Connerton, I. & Frankel, G. ( 1999) Mol. Microbiol. 32, 151–158.
27. Liu, H., Magoun, L., Luperchio, S., Schauer, D. B. & Leong, J. M. ( 1999) Mol. Microbiol. 34, 67–81.
28. Knutton, S., Adu-Bobie, J., Bain, C., Philipps, A. D., Dougan, G. & Frankel, G. ( 1997) Infect. Immun. 65, 1644–1652.
29. Frankel, G., Philips, A. D., Novakova, M., Batchelor, M., Hicks, S. & Dougan, G. ( 1998) Mol. Microbiol. 29, 559–570.
30. Clark, M. A., Hirst, B. H. & Jepson, M. A. ( 1998) Infect. Immun. 66, 1237–1243.
31. Higgins, L. M., Frankel, G., Douce, G., Dougan, G. & MacDonald, T. T. ( 1999) Infect. Immun. 67, 3031–3039.
32. Higgins, L. M., Frankel, G., Connerton, I., Goncalves, N. S., Dougan, G. & MacDonald, T. T. ( 1999) Science 285, 588–591.
33. Liu, H., Magoun, L. & Leong, J. M. ( 1999) Infect. Immun. 67, 2045–2049.
34. Knutton 359 Knutton, S., Baldwin, T, Williams, P. H. & McNeish, A. S. ( 1989) Infect. Immun. 57, 1290–1298.
35. Moon, H. W., Whipp, S. C., Argenzio, R. A., Levine, M. M. & Gianella R. A. ( 1983) Infect. Immun. 41, 1340–1351.
36. Goosney, D. L., Celli, J., Kenny, B. & Finlay, B. B. ( 1999) Infect. Immun. 67, 490–495.
37. Sanger, J. M., Change, R., Ashton, F., Kaper, J. B. & Sanger J. W. ( 1996) Cell Motil. Cytoskeleton 34, 279–287.
38. Ben-Ami, G., Ozeri, V., Hanski, E., Hofmann, F., Aktories, K., Hahn, K. M., Bokoch, G. M. & Rosenshine, I. ( 1998) Infect. Immun. 66, 1755–1758.
39. Kalman, D., Weiner, O. D., Goosney, D. L., Sedat, J. W., Finlay, B. B., Abo, A. & Bishop J. M. ( 1999) Nat. Cell Biol. 1, 389–391.
40. Crane, J. K. & Oh, J. S. ( 1997) Infect. Immun. 65, 3277–3285.
41. Savkovic, S. D., Koutsouris, A. & Hecht, G. ( 1996) Infect. Immun. 64, 4480–4487.
42. Savkovic, S. D., Koutsouris, A. & Hecht, G. ( 1997) Am. J. Physiol. 273, C1160–C1167.
43. Philpott, D. J., McKay, D. M., Sherman, P. M. & Perdue, M. H. ( 1996) Am. J. Physiol. 270, G634–645.
44. Canil, C., Rosenshine, I., Ruschkowski, S., Donnenberg, M. S., Kaper, J. B. & Finlay, B. B. ( 1993) Infect. Immun. 61, 2755–2762.
45. Donnenberg, M. S., Tacket, C. O., James, S. P., Losonsky, G., Nataro, J. P., Wasserman, S. S., Kaper, J. B. & Levine M. M. ( 1993) J. Clin. Invest. 92, 1412–1417.
46. Perdue, M. H. & McKay, D. M. ( 1994) Am. J. Physiol. 267, G151–G156.
47. Collington, G. K., Booth, I. W., Donnenberg, M. S., Kaper, J. B. & Knutton, S. ( 1998) Infect. Immun. 66, 6049–6053.
48. Collington, G. K., Booth, I. W. & Knutton, S. ( 1998) Gut 42, 200–207.
49. Hecht, G. & Koutsouris, A. ( 1999) Am. J. Physiol. 276, G781–G788.
50. Ulshen, M. H. & Rollo, J. L. ( 1980) N. Engl. J. Med. 302, 99–101.
51. Gewirtz, A. T., Rao, A. S., Simon, P. O., Merlin, D., Carnes, D., Madara, J. L. & Neish, A. S. ( 2000) J. Clin. Invest. 105, 79–92.
52. McKay, D. M. & Baird, A. W. ( 1999) Gut 44, 283–289.
53. Abe, A., Heczko, U., Hegele, R. G. & Finlay, B. B. ( 1998) J. Exp. Med. 188, 1907–1916.
54. Newman, J. V., Zabel, B. A., Jha, S. S. & Schauer, D. B. ( 1999) Infect. Immun. 67, 6019–6025.
55. Karam, S. M. ( 1999) Front. Biosci. 15, D286–D298.
56. Van Moll, L. K. & Cantey, J. R. ( 1997) Infect. Immun. 65, 3788–3793.
57. Delneri, M. T., Carbonare, S. B., Silva, M. L., Palmeira, P. & Carneiro-Sampaio, M. M. ( 1997) Eur. J. Pediatr. 156, 493–498.
58. Malstrom, C. & James, S. ( 1998) Infect. Immun. 66, 3120–3127.
59. Hessey, S. J., Spencer, J., Wyatt, J. I., Sabala, G., Rathbone, B. J., Axon, A. T. & Dixon, M. F. ( 1990) Gut 31, 134–138.
60. Sartor, R. B. ( 1997) Ailment Pharmacol. Ther. 11, 17–22.








