
Colloquium
Plants and animals share functionally common bacterial virulence factors
Laurence G. Rahme*†‡, Frederick M. Ausubel§, Hui Cao†‡, Eliana Drenkard§, Boyan C. Goumnerov†‡, Gee W. Lau†‡, Shalina Mahajan-Miklos§¶, Julia Plotnikova§, Man-Wah Tan§||, John Tsongalis‡, Cynthia L Walendziewicz‡, and Ronald G. Tompkins†‡
† Department of Surgery, Harvard Medical School,
‡ Department of Surgery, Massachusetts General Hospital and Shriner 's Burn Institute, Boston, MA 02114;
§ Department of Genetics, Harvard Medical School, and Department of Molecular Biology, Massachusetts General Hospital, Boston, MA 02114; and
|| Society of Fellows, Harvard University, 78 Mount Auburn Street, Cambridge, MA 02138
By exploiting the ability of Pseudomonas aeruginosa to infect a variety of vertebrate and nonvertebrate hosts, we have developed model systems that use plants and nematodes as adjuncts to mammalian models to help elucidate the molecular basis of P. aeruginosa pathogenesis. Our studies reveal a remarkable degree of conservation in the virulence mechanisms used by P. aeruginosa to infect hosts of divergent evolutionary origins.
B acterial pathogens infect a wide variety of evolutionary distinct hosts, including both lower and higher eukaryotes. In all of these cases, the pathogen must have the ability to recognize, become associated with, exploit the nutrient reserves of, and combat the defense responses of its specific host. To accomplish these tasks, pathogens use an extensive arsenal of virulence-related factors.
Many pathogens cause disease in a single or limited number of host species as a consequence of a long coevolutionary history. However, the interactions between host and pathogen that limit host range and determine host resistance or susceptibility are poorly understood. Although many bacterial virulence components are thought to be host-specific, numerous studies have demonstrated the existence of what appear to be universal virulence mechanisms used by diverse bacterial species (1). Similarly, recent work has revealed common features underlying host defense responses against pathogens in plants, insects, and mammals (2). Thus, some of the underlying virulence mechanisms of pathogens, as well as the host defenses against them, are likely to have ancient evolutionary origins preserved across phylogeny.
There remains a great deal to learn about the molecular nature of antagonistic encounters between pathogenic bacteria and their hosts. Despite our limited knowledge, significant advances have been made in developing techniques that facilitate our understanding of virulence mechanisms and the critical role of the host during pathogenesis (3). Our laboratories have developed a technique, which we refer to as multihost pathogenesis, to study pathogens that cause disease in both vertebrate and nonvertebrate hosts (4, 5, 6, 7 and 8).
Epidemiological studies carried out in the 1970s suggested that clinical isolates of Pseudomonas aeruginosa might be capable of causing disease in plants (9, 10). Based on this premise, we developed a model pathogenesis system by using a human clinical isolate of P. aeruginosa, strain UCBPP-PA14 (PA14) that elicits disease in plants, nematodes, insects, and mice (4, 5, 6 and 7, 11). P. aeruginosa is the most common causative organism of sepsis in burned patients and the leading cause of pulmonary infections and mortality in cystic fibrosis patients (12). In addition, this important human opportunistic pathogen infects injured, immunodeficient, or otherwise compromised individuals ( 13). The pathophysiology of infections caused by P. aeruginosa is complex as shown by the clinical diversity of diseases associated with this organism and the multiplicity of virulence factors it produces. Although a natural soil inhabitant, P. aeruginosa is versatile in its metabolic potential, which allows it to survive in a variety of natural and hospital environments. It appears that the combination of environmental persistence, versatility in virulence mechanisms, and multiple virulence factors allows P. aeruginosa to be effective both as an opportunistic human pathogen and as a plant pathogen.
None of the currently available animal models fully mimics all aspects of any single human disease caused by P. aeruginosa. Moreover, it is not practical to use any current vertebrate animal model to systematically screen for pathogenicity-related P. aeruginosa mutants. This latter problem has become acutely apparent as the Pseudomonas Genome Project nears completion, and the relevance of thousands of P. aeruginosa genes to human pathogenesis needs to be determined. In contrast to vertebrate animal models, host-pathogen interaction models that use nonvertebrate hosts permit genomewide screening for P. aeruginosa virulence factors required for pathogenesis. If there is substantial overlap in the virulence factors required for pathogenesis in invertebrate and mammalian hosts, large-scale screening in invertebrate hosts can become an efficient method for identifying previously unknown virulence-related genes relevant to human pathogenesis.
Using P. aeruginosa strain PA14, we provided direct evidence that P. aeruginosa uses a shared subset of virulence factors to elicit disease in both plants (Arabidopsis thaliana) and animals (mice) (4, 11). More recently, we extended the Arabidopsis-P. aeruginosa model to additional nonvertebrate hosts, including the nematode Caenorhabditis elegans (7, 14, 15) and the insects Galleria melonella (8) and Drosophila melanogaster (S.M.-M., G.W.L., L. Perkins, F.M.A., and L.G.R., unpublished data).
This paper summarizes the use of a plant pathogenesis model to identify previously unknown virulence factors and highlights the remarkable conservation in the virulence mechanisms used by P. aeruginosa to infect evolutionary divergent hosts.
P. aeruginosa Pathogenesis
Despite quite detailed knowledge about some of the extracellular proteins and several surface-associated components synthesized by P. aeruginosa, our understanding of P. aeruginosa
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host-Pathogen Interactions: Common Features Between Plants and Animals,” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
|
* |
To whom reprint requests should be addressed. E-mail: rahme@frodo.mgh.harvard.edu. |
|
¶ |
Present address: Micrbobia, Inc., One Kendall Square, Building 1400W, Suite 1418, Cambridge, MA 02139. |
infections is only rudimentary. Much remains to be learned about individual virulence factors essential for pathogenicity and the mechanism by which these factors work together during pathogenesis.
P. aeruginosa virulence factors facilitate tissue invasion and systemic spread and include pili and flagella, endotoxins, exotoxins, vascular permeability factors, and a variety of excreted enzymes (16). P. aeruginosa proteases degrade a variety of host proteins and have a direct destructive effect on skin tissue (17, 18). Elastase (LasB), a potent metalloprotease with broad substrate specificity, degrades host proteins, such as elastin, collagen, transferrin, Ig, and some of the components of complement (19). The LasB elastase acts in concert with both LasA protease and alkaline protease to cause efficient elastolysis, which is required for the tissue damage associated with P. aeruginosa pathogenesis (20). The P. aeruginosa alginate capsule plays an essential role in chronic pulmonary infection in cystic fibrosis patients (21, 22). Finally, the P. aeruginosa lipopolysaccharide also has been shown to be an important virulence factor (23).
The synthesis of several P. aeruginosa-secreted virulence factors is transcriptionally regulated by environmental stimuli (24, 25 and 26). For example, phospholipase C is regulated by the availability of phosphate (27, 28), whereas exotoxin A and elastase are regulated by the concentration of iron in the growth medium (29, 30, 31 and 32). Moreover, the production of a large number of exoproducts, including elastase, alkaline protease, the LasA protease, hemolysin, pyocyanin, and rhamnolipids is cell densitydependent and is regulated by the so-called quorum-sensing cascade (33). The P. aeruginosa quorum-sensing cascade is mediated by low molecular mass homoserine lactones that are synthesized by the products of the lasI and rhlI genes. The concentration of these homoserine lactones is monitored by the products of the lasR and rhlR genes, which serve as global transcriptional activators of a variety of exoproducts relevant for P. aeruginosa pathogenesis. In addition to homoserine lactones, it recently has been shown that 2-heptyl-3-hydroxy-4-quinolone also serves as an intracellular signal molecule involved in the activation of pathogeneicity-related factors (34). This molecule belongs to the 4-quinolone chemical family, best known for the antibiotic activity of many of its members.

Fig. 1. Macroscopic and microscopic symptoms elicited by P. aeruginosa strain UCBPP-PA14 infiltrated into Arabidopsis leaves. (A) The upper part of an Arabidopsis leaf (ecotype LL-O) was infiltrated with bacterial suspensions at a titer of 103 cfu/cm2 and photographed 2 days postinfection. A chlorotic zone (white arrow) surrounds the soft-rot symptoms. (B and C) Arabidopsis leaves infiltrated with bacterial suspension and stained with trypan blue 2 days postinfection. Whole leaves were examined by using a Zeiss Axioskop and photographed. Bacterial movement was observed along leaf veins (arrows indicate vessel parenchyma filled with bacteria). Bacteria are absent in xylem vessels (arrowheads).
In addition to regulating the production of a variety of specific virulence-related factors, the quorum-sensing cascade as well as other cell-to-cell signaling mechanisms are involved in the differentiation of P. aeruginosa biofilms (35, 36), matrix-enclosed bacterial populations that attach to each other and to biotic, or abiotic surfaces (37). P. aeruginosa is protected from adverse environmental conditions and from antibacterial agents while growing in this matrix-enclosed structure that has been implicated as a contributing factor in the persistence and severity of P. aeruginosa infections. It is thought that when nutrient conditions become limiting, bacteria are shed from the biofilm and enter the planktonic (free-living) phase (37). In this manner, cells are able to colonize new habitats and form new biofilms.
Another quorum-sensing regulated virulence factor is pyocyanin, a blue-green-colored phenazine (33, 38). This secondary metabolite has antimicrobial activity against several species of bacteria, fungi, and protozoa, a quality attributed to its redoxactive potential (39). Although little is known about the nature of the enzymes that catalyze the formation of pyocyanin in P. aeruginosa, the conversion of chorismate to anthranilate is thought to be a key step in the pathway that is most likely catalyzed by the anthranilate synthetase encoded by the phnA and phnB genes (40). Even though pyocyanin-induced free radical production appears to be responsible for much of its antimicrobial activity (41), the role of pyocyanin in P. aeruginosa-associated tissue injury is less clear.
Development of the Arabidopsis-P. aeruginosa Model
As described briefly above, we developed the Arabidopsis-P. aeruginosa pathogenesis model based on the P. aeruginosa strain PA14. This was accomplished as follows. A collection of 75 P. aeruginosa strains, of which 30 were human isolates, was screened for its ability to elicit disease on leaves of at least four different Arabidopsis ecotypes (4). Several strains elicited disease characterized by varying degrees of soft-rot symptoms in all four ecotypes (wild varieties) tested. Importantly, two strains, PA14 (a human isolate) and UCBPP-PA29 (a plant isolate), caused severe soft-rot symptom in some, but not all of the ecotypes tested. The finding that these two P. aeruginosa strains exhibit ecotype specificity is typical of plant pathogen interac-
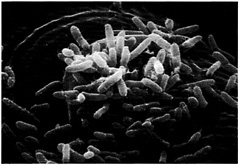
Fig. 2. P. aeruginosa entering the mesophylic compartment of an Arabidopsis leaf through a stomatal opening. Detached Arabidopsis leaves were soaked in a bacterial suspension at a titer of 103 cfu/ml. At 24 h postinfection, the leaves were fixed in osmium tetroxide, followed by dehydration in ethanol. The leaves then were coated in Technics Sputter Coater Hummer II and subsequently examined and photographed by using an Amray (Bedford, MA) 1000 scanning electron microscope. Bacteria are shown to be primarily concentrated on the surface of guard cells, above the stoma.
tions and reflects the selection of plant accessions (ecotypes) that are resistant to particular strains (races) of a pathogen. These observations support the notion that P. aeruginosa is a natural plant pathogen in the wild.
Fig. 1A illustrates the symptomatology observed on a susceptible 6-week-old Arabidopsis plant when the upper part of a leaf is inoculated with P. aeruginosa strain PA14. The severe symptoms elicited by PA14 include a water-soaked reaction zone and chlorosis (yellowing). Soft-rot symptoms begin as small water-soaked lesions, which enlarge rapidly in diameter. Complete maceration and collapse of the infiltrated leaf occurs 4 –5 days after infection. As shown in Fig. 1B, trypan blue staining of infected leaves reveals the presence of P. aeruginosa in the intercellular spaces of the leaf as well as bacterial movement along the veins and bacterial proliferation in vessel parenchyma and companion cells. No bacteria are detected in xylem. Fast propagation of P. aeruginosa in the vessel parenchyma is the prelude to systemic infection and maceration of the entire plant. As illustrated in Fig. 2, detached leaves of Arabidopsis exposed to PA14 culture suspension show that P. aeruginosa cells attach to the leaf epidermis and congregate at the stomal openings where they gain entry to the leaf interior.
Similar to other phytopathogenic bacteria, P. aeruginosa strains that elicit disease symptoms are also able to proliferate in Arabidopsis leaves. Table 1 lists the maximal levels of growth reached by several P. aeruginosa strains and PA14 mutants at the fourth day postinfection. The degree of proliferation was correlated with the severity of disease symptoms. In each case, reduced symptoms were associated with reduced bacterial counts in leaves (Table 1; ref.4).
Table 1. Animal mortality and proliferation studies of P. aeruginosa strains and isogenic UCBPP-PA14 mutants in Arabidopsis and in a mouse burned model
|
P. aeruginosa strain |
cfu/cm2 leaf area* |
Mean titer ± SD in biopsies adjacent to burn† |
% Mouse mortality‡ |
|
UCBPP-PA14 |
2.6 × 107 ± 2.0 × 107 |
6.0 × 107 ± 2.1 × 107 |
77 |
|
UCBPP-PA29 |
2.7 × 107 ± 1.3 × 107 |
8.2 × 107 ± 2.0 × 107 |
5 |
|
PAK |
6.0 × 104 ± 3.0 × 104 |
6.0 × 107 ± 1.2 × 107 |
20 |
|
PAO1 |
8.0 × 105 ± 2.7 × 105 |
4.0 × 107 ± 1.8 × 107 |
25 |
|
UCBPP-PA14 plcS |
1.1 × 105 ± 0.5 × 105 |
6.7 × 104 ± 7.5 × 104 |
40 |
|
UCBPP-PA14 toxA |
1.5 × 106 ± 1.0 × 106 |
1.4 × 108 ± 1.7 × 108 |
40 |
|
UCBPP-PA14 gacA |
6.0 × 103 ± 2.1 × 103 |
NT |
0 |
|
For both quantitation and mortality studies, mice were injected with 5 × 104 cfu/ml. NT, not determined. * Means of four samples ± SD of maximum bacterial counts obtained at 5 days postinfection with 103 cells. † No viable bacterial cells were retrieved from the underlying rectus abdominus muscle immediately after bacterial injection or in animals that received a sham injury in other studies. ‡ The number of animals that died of sepsis was monitored each day for 10 days. |
|||
In our efforts to identify additional plant species susceptible to P. aeruginosa infection, we found that soft-rot symptoms also were observed when PA14 was inoculated into the midrib of lettuce leaf stem (Fig. 3B). In addition to PA14, lettuce is susceptible to the well-characterized P. aeruginosa strains PAK and PA01. In later stages of infection, all three P. aeruginosa strains invade the entire midrib of lettuce leaves, resulting in complete maceration and collapse of the tissue (Fig. 3B).
Pathogenicity of PA14 in a Mouse Burn Model
We primarily have used a mouse full-thickness skin burn model to assess PA14 pathogenicity in a mammalian host (42). Several factors facilitate the colonization and establishment of the bacteria in this model. First, the inoculum is delivered in a scald eschar induced on the abdominal surface of an anesthetized mouse. This method provides the bacteria with necrotic tissue as nutrients and with protection from the active components of the immune system, because the circulation in this area is impeded. Second, it is well known that burn trauma itself induces a generalized decrease in the efficiency of the immune system, which aids in the development of systemic infections and sepsis. The first signs of developing infection and sepsis can be observed clinically and histopathologically 12–24 h postinoculation. Mortality studies were conducted to assess the presence of systemic infection (Table 1). Tissue biopsies from the site of inoculation were obtained and compared to assess local invasiveness. As presented in Table 1, strains PA14 and PA29 as well the P. aeruginosa human isolates PA01 and PAK all proliferated and invaded the muscle underlying the eschar, but strain PA14 resulted in a higher mortality than the other strains. Animals usually succumbed to the infection 36–48 h postinoculation.
We conducted a series of morphological studies to further investigate the dynamics of PA14 infections in the burned mouse model. Specimens from control animals and from burned and inoculated animals were processed by standard histologic techniques. Inoculated mice were killed at varying time intervals and histopathologic examination of parenchymal organs was undertaken. The findings revealed that a local inflammatory response developed during the first 24 h at the site of inoculation and that systemic infection, including multiple organ involvement, occurred between 24 and 36 h. Histologic sections removed from livers and kidneys 36 h postinoculation revealed massive perivascular bacterial invasion of the organs and associated tissue
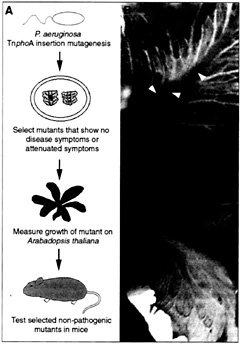
Fig. 3. (A) Strategy used to screen for P. aeruginosa virulence-associated genes. (B) Midribs of lettuce leaves were inoculated with 10 µl of a bacterial suspension at a titer of 103 cfu/ml and photographed 4 days postinfection. Bottom leaf was inoculated with the wild-type strain PA14 (black arrowhead) and top leaf was inoculated with the PA14-toxA mutant. Brown spots (white arrowheads) on the midrib of the top leaf reveal inoculation sites.
necrosis (Fig. 4). These studies demonstrate the ability of PA14 to overcome the immune defense mechanisms in the host and establish a generalized infection.
Shared Virulence Factors in Plant and Animal Pathogenesis
The relevance of the Arabidopsis-P. aeruginosa model to mammalian pathogenesis initially was demonstrated by testing whether two P. aeruginosa virulence factors, exotoxin A and phosopholipase C, thought to influence tissue invasion and systemic spread in mammals (4), also play a role in Arabidopsis pathogenesis. Exotoxin A (toxA) is a potent mammalian protein synthesis inhibitor (43). Phospholipase C (plcS) preferentially degrades phospholipids found in eukaryotic cell membranes (44), which leads to a systemic inflammatory response (45). Conversely, a known virulence determinant important for plant pathogenesis in Pseudomonas syringae, gacA, was tested for its role in mouse pathogenesis. GacA encodes a transcriptional activator of genes encoding extracellular products involved in pathogenicity (46). Isogenic PA14 plcS, toxA, and gacA mutants were constructed by marker exchange, and the resulting mutants were tested for pathogenicity in the Arabidopsis leaf infiltration assay and in the burned mouse model. Decreased pathogenicity was observed in both hosts with all three of the isogenic PA14 mutants. Unlike the wild-type PA14, none of the mutants caused maceration and collapse of Arabidopsis leaves, and the growth of the mutants was significantly reduced (4). Similarly, all three mutants exhibited significantly lower mortality than the wildtype strain in the mouse model. Consistent with its known role in pathogenesis, the plcS mutant exhibited reduced ability to proliferate and invade the tissue adjacent to the burn area (Table 1). Subsequent to these experiments, it was shown that GacA functions upstream of lasI-lasR and rhlI-rhlR quorumsensing regulatory systems (47) to regulate virulence gene expression (33) and type 4 pilus-mediated twitching motility in P. aeruginosa (48).
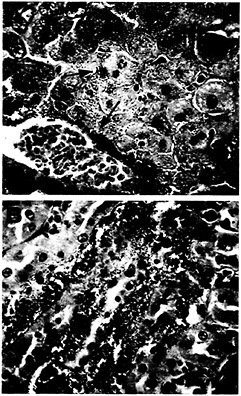
Fig. 4. Histologic section of mouse liver and kidney taken 36 h postinoculation with P. aeruginosa strain PA14. (A) Massive perivascular bacterial infiltration and associated necrosis of the surrounding hepatocytes (arrow) are visible in the liver section. Brown and Hopps (B&H) Gram stain at ×40 magnification, (B) Section of kidney from the same animal showing large bacterial infiltrates within the tubules of the renal medulla (arrow). B& H Gram stain at ×40 magnification.
Use of Nonvertebrate Hosts to Identify Virulence-Associated Genes Involved in Mammalian Pathogenesis
The observation that P. aeruginosa uses common virulence genes to infect both animals and plants led us to theorize that previously unknown virulence determinants required for P. aeruginosa pathogenesis in animals could be identified by screening randomly mutagenized PA14 clones for ones that exhibited
Table 2. P. aeruginosa mutants causing decreased virulence in plants
|
Strain |
Symptoms elicited in Arabidopsis * |
% Mouse mortality † |
Gene identity and comments |
|
PA14 |
Severe |
100 |
Wild type |
|
Group I mutants isolated from plant screens |
|||
|
33C7 |
None |
0 |
Unknown‡ |
|
1D7 |
Weak |
50 |
gacA, quorum-sensing regulator, two-component system lemA-gacA |
|
25A12 |
Weak |
87 |
Unknown |
|
plcS |
Moderate |
40 |
plcS, degrades phospholipids of eukaryotic membranes |
|
toxA |
Moderate |
40 |
toxA, inhibits eukaryotic protein synthesis |
|
33A9 |
Moderate |
0 |
Unknown, absent in PA01, reduced motility, increased surface attachment |
|
25F1 |
Moderate |
20 |
Homologue of Chlorobium tepidum orfT |
|
34H4 |
Moderate |
33 |
Unknown, contains a bipartite nuclear localization signal |
|
pho34B12 |
Moderate |
56 |
Unknown, phenazine regulator, contains a HTH motif |
|
pho15 |
Moderate |
62 |
dsbA, periplasmic S-S forming enzyme |
|
16G12 |
Moderate |
100 |
Unknown |
|
Group II mutants isolated from C. elegans screens |
|||
|
36A4 |
None |
0 |
Homologue of P. syringae hrpM |
|
50E12 |
Weak |
0 |
Homologue of A vinelandii PstP protein, required for accumulation of poly-β-hydroxybutyrate |
|
48D9 |
Weak |
50 |
Homologue of lemA, sensor of the two-component system lemA-gacA |
|
41C1 |
Weak |
81 |
Homologue of putative E. coli integral membrane protein AefA |
|
23A2 |
Weak |
85 |
mexA, non-ATPase membrane efflux pump |
|
41A5 |
Weak |
100 |
Unknown |
|
6A6, 3E8 |
Moderate |
18 |
phzB, phenazine biosynthesis |
|
12A1 |
Moderate |
50 |
lasR, quorum-sensing regulator |
|
8C12 |
Moderate |
63 |
Unknown |
|
* Symptoms observed 4–5 days postinfection. None, no symptoms; weak, localized water soaking and chlorosis of tissue circumscribing the inoculation site; moderate, moderate water-soaking and chlorosis with most of the tissue softened around the inoculation site at 2–3 days postinfection. † Mice were injected with ≈5 × 105 cells. ‡ Unknown, BLASTX analysis yielded no encoded proteins with significant homology. |
|||
decreased virulence in plants. This experimental approach overcomes a major limitation imposed by an animal model, that is, the large number of animals required to undertake a systematic genetic screen for the identification of virulence-related genes.
P. aeruginosa strain PA14 was mutagenized with transposon TnphoA, and 2,500 prototrophic mutants were screened for impaired virulence in a lettuce stem assay. In these highthroughput screens, we substituted Arabidopsis with lettuce because several mutants could be tested on one lettuce midrib by “tooth-picking” single colonies directly from plates that contain the mutant clones (Fig. 3; ref.5). Following the methodology depicted in Fig. 3, the virulence-attenuated mutants identified subsequently were tested for their ability to elicit disease symptoms and proliferate in Arabidopsis leaves. As summarized in Table 2, in addition to toxA and plcS a total of 11 virulencerelated genes were identified among the 2,500 prototrophs screened that elicited null, weak, or moderate rotting symptoms on lettuce and Arabidopsis plants (Table 2, group I). Importantly, the reduced ability of the mutants to cause disease symptoms correlated with reduced bacterial counts in leaves (5).
In addition to demonstrating its ability to cause disease in plants and mice, we have shown that PA14 kills C. elegans when it is presented to the nematodes as a food source (15). In low-salt medium, PA14 kills worms over a period of 2–3 days (slow killing) by an infection-like process that correlates with the accumulation of bacteria in the worm intestine (14). Alternatively, PA14 grown in a high-salt, rich medium kills worms within 4–24 h (fast killing) by a toxin-mediated process (7). Fast and slow killing appear to be mechanistically distinct because most P. aeruginosa mutants that are attenuated in slow killing are not attenuated in fast killing and vice versa. Altogether, 13 P. aeruginosa genes have been identified that when mutated exhibit reduced virulence in the C. elegans slow or fast killing assays. Of these 13 genes, nine also are required for maximum pathogenicity in the Arabidopsis leaf infiltration assay and are listed inTable 2 (group II genes; gacA was identified in both the plant and nematode screens).
Eight of the 20 P. aeruginosa genes listed in Table 2 that were identified by screening in plants and nematodes do not correspond to previously identified proteins from other species ( 5, 6 and 7). Remarkably, at least 15 of these 20 genes are required for pathogenesis in the burned mouse model.
What Types of Virulence Mechanisms Are Conserved Through Evolution?
The “multihost” pathogenesis screens performed in our laboratories identified a variety of virulence-associated genes that encode proteins involved in transcriptional control, posttranscriptional control, efflux systems, biosynthetic enzymes involved in phenazine production, toxins, and proteins of unknown function. Table 2 highlights the striking conservation in the virulence mechanisms used by P. aeruginosa to infect plants, nematodes, and mammals. It is remarkable that at least 15 of the P. aeruginosa mutants isolated in either a plant or a nematode screen were found to be required for full virulence in the burned mouse model.
One virulence-related factor found to play a critical role in pathogenesis in plants, nematodes, and mice is the periplasmic disulfide bond-forming enzyme encoded by the dsbA gene (49), whose function is likely to affect several periplasmic virulencerelated proteins. An important role for dsbA in pathogenesis has been described in several human pathogens, including Shigella flexneri (50) and Vibrio cholera (51), and in the bacterial phytopathogen Erwinia chrysanthemi (52).
Three virulence-related genes identified in our screen, hrpM, gacS, and gacA, previously had been shown by other researchers to be important in phytopathogenesis. However, our studies found that these genes also play an important role in mammalian
pathogenesis. The three genes also are required for effective killing of C. elegans (6, 7). The hrpM gene of P. syringae pv. syringae was identified as a homologue of the Escherichia coli mdoH gene, which is part of an operon involved in the synthesis of membrane-derived oligosaccharides (MDO) (53). Although MDO have been found to have different functions in a variety of Gram-negative bacteria, the role of MDO in bacterial pathogenesis is not well understood. Mutation in the hrpM locus of the plant pathogen P. siryngae pv. syringae abolishes both the development of disease symptoms on host plants as well as the hypersensitivity response in nonhost plants (54). The GacS protein is a sensor kinase of a two-component bacterial family of regulators (55). GacA, the cognate response regulator of GacS, initially was identified as a global regulator of secondary metabolites in P. fluorescens (56, 57). It has been shown that mutations in both the gacA and gacS genes in P. siryngae lead to decreased lesion formation in beans and no production of the toxin syringomycin (46, 55, 58).
The requirement of the lasR, gacS, and gacA gene products for pathogenicity in plants, nematodes, and mice provides evidence that quorum sensing and regulated export of proteins are general features of pathogenesis for all three hosts. The lasR, gacS, and gacA genes are present in numerous plant and animal bacterial pathogens as well as in saprophytes. It is likely that LasR, GacS, and GacA initially served as master regulators, enabling ancestral Gram-negative organisms to adapt to their environment. Subsequently, these proteins evolved to regulate a variety of genes that allowed prokaryotes to invade and establish their presence in eukaryotic hosts.
Another class of conserved virulence factors important in plant and nematode pathogenesis is the genes involved multidrug efflux pumps. The mexA gene corresponding to mutant 23A2 encodes a component of a multidrug efflux pump; it recently has been shown to be involved in active efflux of P. aeruginosa autoinducers that are not freely diffusable (59). However, mutant 23A2 was only marginally compromised in the mouse burn model. Further studies using a lower inoculum need to be conducted to ascertain its role as a virulence factor in mammalian pathogenesis. An additional protein relevant to plant pathogenesis, but not to mammalian pathogenesis, is a homologue of the putative E. coli integral membrane protein AefA (Proposite: PS01246). The function of this protein is yet unknown.
Other known proteins identified in our screens, but not previously shown to be involved in pathogenesis, include a PtsP homologue of A. vinelandii required for poly-β-hydroxybutyrate accumulation. The ptsP gene is predicted to encode enzyme INtr, a presumptive transcriptional regulator of RpoN-dependent operons (60).
One of the novel virulence factors identified in the plant screen (Pho34B12) affects hemolytic and elastolytic activity as well as pyocyanin production (refs.5 and 7; H.C. and L.G.R., unpublished work), all of which are under quorum-sensing regulation. The protein encoded by pho34B12 contains a helixturn–helix DNA binding motif similar to that found in the LysR family of transcriptional regulators (ref.5; H.C. and L.G.R., unpublished work). This class of proteins includes regulators involved in both mammalian and plant pathogenesis (1).
Our multihost pathogenesis study results indicate that phenazines are an important class of virulence-related effector molecules. Despite intensive in vitro analyses of phenazines, the physiological significance of their role in P. aeruginosa pathogenesis in mammals has been controversial (61). Before our studies, there had been no demonstration of their role in vivo. Mutants 3E8 and 6A6 correspond to the previously identified phzB gene in P. fluorescens and phzY gene in P. aureofaciens. Both phzB and phzY are present in operons known to regulate production of phenazine-1-carboxylate (62). Additional effector molecules that play a role in multihost pathogenesis include the ToxA protein and the previously unidentified virulence gene encoding the 34H4 protein. Current work indicates that 34H4 contains a bi-partite nuclear localization signal required for translocation of the protein into the nucleus of mammalian cells (G.W.L. and L.G.R., unpublished work).
A common phenotype of at least two of the mutants, 33A9 and 3E8, is reduced motility and altered surface attachment ability (Table 2). Mutant 3E8 exhibits reduced attachment to abiotic surfaces, such as polyvinilchloride plastic surfaces (ref.63; S.M.-M. and F.M.A., unpublished work) whereas 33A9 exhibited increased surface attachment ability (E.D., G. O'Toole, F.M.A., and L.G.R., unpublished work). Bacterial adhesion is an essential step in biofilm formation and consequently, in bacterial virulence. No significant similarity to any known genes was found in the gene corresponding to mutant 33A9. Interestingly, DNA sequence analysis of the region containing the 33A9 gene revealed that this gene is not present in the recently sequenced genome of the P. aeruginosa strain PAO1.
Conclusions
In summary, we can draw the following conclusions from our studies of P. aeruginosa multihost pathogenesis. First, the variety of virulence-associated genes described in our experiments indicates that the multihost strategy has few limitations with regard to the categories of virulence-related functions that can be identified. Second, the fact that most of the genes identified were not previously known to be involved in pathogenesisrelated functions demonstrates that the multihost strategy is particularly efficient in identifying novel P. aeruginosa virulence factors. Third, and perhaps most importantly, even though many pathogens cause disease in a single or limited number of host species these studies provide strong evidence that there exists several universal bacterial virulence mechanisms highly conserved across phylogeny.
Although some of the virulence-associated genes identified have homologues in other pathogenic bacteria, the exact role of these genes in pathogenesis remains unclear in most cases. The use of genetically tractable host systems, such as plants, nematodes, and insects, will generate important information about host responses and lead to a better understanding of the fundamental molecular mechanisms that underlie bacterial pathogenesis.
1. Finlay, B. B. & Falkow, S. ( 1997) Microbiol. Mol. Biol. Rev. 61, 136–169.
2. Kopp, E. B. & Medzhitov, R. ( 1999) Curr. Opin. Immunol. 11, 13–18.
3. Strauss, E. J. & Falkow, S. ( 1997) Science 276, 707–711.
4. Rahme, L. G., Stevens, E. J., Wolfort, S. F., Shao, J., Tompkins, R. G. & Ausubel, F. M. ( 1995) Science 268, 1899–1902.
5. Rahme, L. G., Tan, M.-W., Le, L., Wong, S. M., Tompkins, R. G., Calderwood, S. B. & Ausubel, F. M. ( 1997) Proc. Natl. Acad. Sci. USA 94, 13245–13250.
6. Tan, M.-W., Rahme, L. G., Sternberg, J. A., Tompkins, R. G. & Ausubel, F. M. ( 1999) Proc. Natl. Acad. Sci. USA 96, 2408–2413.
7. Mahajan-Miklos, S., Tan, M.-W., Rahme, L. G. & Ausubel, F. M. ( 1999) Cell 96, 47–56.
8. Jander, G., Rahme, L. G. & Ausubel, F. M. ( 2000) J. Bacteriol. 182, 3843–3845.
9. Kominos, S. D., Copeland, C. E., Grosiak, B. & Postic, B. ( 1972) Appl. Microbiol. 24, 567–570.
10. Cho, J. J., Schroth, M. N., Kominos, S. D. & Green, S. K. ( 1975) Phytopathology 65, 425–431.
11. Finlay, B. B. ( 1999) Cell 96, 315–318.
12. Doring, D. ( 1993) in Pseudomonas aeruginosa as an Opportunistic Pathogen, ed. Campa, M. (Plenum, New York), pp. 245–273.
13. Wood, R. E. ( 1976) Hosp. Prac. 11, 91–100.
14. Tan, M.-W., Mahajan-Miklos, S. & Ausubel, F. M. ( 1999) Proc. Natl. Acad. Sci. USA 96, 715–720.
15. Tan, M.-W. & Ausubel, F. M. ( 2000) Curr. Opin. Microbiol. 3, 29–34.
16. Fink, R. B., Jr., ed. ( 1993) in Pseudomonas aeruginosa the Opportunist: Pathogenesis and Disease (CRC, Boca Raton, FL), pp. 1–5.
17. Kawaharajo, K., Homma, J. Y., Aoyama, Y., Okada, K. & Morihara, K. ( 1975) Jpn. J. Exp. Med. 45, 79–88.
18. Holder, I. A. & Neely, A. N. ( 1991) Antib. Chemother. 44, 99–105.
19. Galloway, D. R. ( 1991) Mol. Microbiol. 5, 2315–2321.
20. Peters, J. E. & Galloway, D. R. ( 1990) J. Bacteriol. 172, 2236–2240.
21. Ohman, D. E., Cryz, S. J. & Iglewski, B. H. ( 1980) J. Bacteriol. 142, 836–842.
22. Zielinski, N. A., DeVault, J. D., Roychoudhury, S., May, T. B., Kimbara, K., Kato, J., Shinabarger, D., Kitano, K., Berry, A., Misra, T. & Chakrabarty, A. M. ( 1990) in Pseudomonas: Biotransformations, Pathogenesis, and Evolving Biotechnology , eds. Silver, S., Chakrabarty, A. M., Iglewski, B. & Kaplan, S. (Am. Soc. Microbiol., Washington, DC), pp. 15–27.
23. Pitt, T. L. ( 1989) Antibiot. Chemother. 42, 1–7.
24. Nicas, T. I. & Iglewski, B. H. ( 1986) J. Clin. Microbiol. 23, 967–969.
25. Jones, S., Yu, B., Bainton, N. J., Birdsall, M., Bycroft, B. W., Chhabra, S. R., Cox, A. J. R., Golby, P., Reeves, P. J., Stephens, S., et al. ( 1993) EMBO J. 12, 2477–2482.
26. Passador, L., Cook, J. M., Gambello, M. J., Rust, L. & Iglewski, B. H. ( 1993) Science 260, 1127–1130.
27. Pritchard, A. E. & Vasil, M. L. ( 1986) J. Bacteriol. 176, 291–298.
28. Shortridge, V. D, Lazdunski, A. & Vasil, M. L. ( 1992) Mol. Microbiol. 6, 863–871.
29. Grant, C. C. R. & Vasil, M. L. ( 1986) J. Bacteriol. 168, 1112–1119.
30. Lory, S. ( 1986) J. Bacteriol. 168, 1451–1456.
31. Frank, D. W., West, S. E. H. & Iglewski, B. H. ( 1990) in Molecular Basis of Bacterial Pathogenesis, eds. Iglewski, B. H. & Clark, V. L. (Academic, San Diego), Vol. 11, pp. 427–455.
32. Iglewski, B. H., Rust, L. & Bever, R. A. ( 1990) in Pseudomonas: Biotransformations, Pathogenesis, and Evolving Biotechnology , eds. Silver, S., Chakrabarty, A. M., Iglewski, B. & Kaplan, S. (Am. Soc. Microbiol., Washington, DC), pp. 36–43.
33. Pesci, E. C. & Iglewski, B. H. ( 1997) Trends Microbiol. 4, 132–134.
34. Pesci, E. C., Milbank, J. B. J., Pearson, J. P., McKnight, S., Kende, A. S., Greenberg, E. P. & Iglewski, B. H. ( 1999) Proc. Natl. Acad. Sci. USA 96, 11229–11234.
35. McLean, R. J. C., Whiteley, M., Stickler, D. J. & Fuqua, W. C. ( 1997) FEMS Microbiol. Lett. 154, 259–263.
36. Davies, D. G., Parsek, M. R., Pearson, J. P., Iglewski, B. H., Costerton, J. W. & Greenberg, E. P. ( 1998) Science 280, 295–298.
37. Costerton, J. W., Lewandowski, Z., Caldwell, D. E., Korber, D. R. & LappinScott, H. M. ( 1995) Annu. Rev. Microbiol. 49, 711–745.
38. Fuqua, W. C., Winans, S. C. & Greenberg, E. P. ( 1994) J. Bacteriol. 176, 269–275.
39. Sorensen, R. U., Klinger, J. D., Cash, H. A., Chase, P. A. & Dearborn, D. G. ( 1983) Infect. Immun. 41, 321–330.
40. Essar, D. W, Eberly, L., Hadero, A. & Crawford, I. P. ( 1990) J. Bacteriol. 172, 884–900.
41. Hassan, H. M. & Fridovich, I. ( 1980) J. Bacteriol. 141, 1556–1163.
42. Stevens, E. J., Ryan, C. M., Friedberg, J. S., Barnhill, R. L., Yarmush, M. L. & Tompkins, R. G. ( 1994) J. Burn Care Rehabil. 15, 232–235.
43. Iglewski, B. H. & Kabat, D. ( 1975) Proc. Natl. Acad. Sci. USA 72, 2284–2288.
44. Berka, R. M. & Vasil, M. L. ( 1982) J. Bacteriol. 152, 239–245.
45. Besterman, J. M., Duronio, V. & Cuatrecasas, P. ( 1986) Proc. Natl. Acad. Sci. USA 83, 6785–6789.
46. Rich, J. J., Kinscherf, T. G., Kitten, T. & Willis, D. K. ( 1994) J. Bacteriol. 176, 7468–7475.
47. Reimmann, C., Beyeler, M., Latifi, A., Winteler, H., Foglino, M., Lazdunski, A. & Haas, D. ( 1997) Mol. Microbiol. 24, 309–319.
48. Glessner, A., Smith, R. S., Iglewski, B. H. & Robinson, J. B. ( 1999) J. Bacteriol. 181, 1623–1629.
49. Bardwell, J. C. A., McGovern, K. & Beckwith, J. ( 1991) Cell 67, 581–589.
50. Watarai, M., Tobe, T., Yoshikawa, M. & Sasakawa, C. ( 1995) Proc. Natl. Acad. Sci. USA 92, 4927–4931.
51. Peek, J. A. & Taylor, R. K. ( 1992) Proc. Natl. Acad. Sci. USA 89, 6210–6214.
52. Shevchik, V. E., Bortoli-German, I., Robert-Baudouy, J., Robinet, S., Barras, F. & Condemine, G. ( 1995) Mol. Microbiol. 16, 745–753.
53. Loubens, I., Debarbieux, L., Bohin, A., Lacroix, J.-M. & Bohin, J.-P. ( 1993) Mol. Microbiol. 10, 329–340.
54. Anderson, D. M. & Mills, D. ( 1985) Phytopathology 75, 104–108.
55. Hrabak, E. M. & Willis, D. K. ( 1992) J. Bacteriol. 174, 3011–3020.
56. Laville, J., Voisard, C., Keel, C., Maurhofer, M., Défago, G. & Haas, D. ( 1992) Proc. Natl. Acad. Sci. USA 89, 1562–1566.
57. Gaffney, T. D., Lam, S. T., Ligon, J., Gates, K., Frazelle, A., DiMaio, J., Hill, S., Goodwin, S., Torkewitz, N., Allshouse, A. M., et al. ( 1994) Mol. PlantMicrobe Interact. 7, 455–463.
58. Hrabak, E. M. & Willis, D. K. ( 1993) Mol. Plant-Microbe Interact. 6, 368–375.
59. Pearson, J. P., Van Delden, C. & Iglewski, B. H. ( 1999) J. Bacteriol 181, 1203–1210.
60. Reizer, J., Reizer, A., Merrick, M. J., Plunkett, G., Rose, D. J. & Saier, M. H. J. ( 1996) Gene 181, 103–108.
61. Sorensen, R. U. & Joseph, F. J. ( 1993) in Pseudomonas aeruginosa as an Opportunistic Pathogen, eds. Campa, M., Bendenelli, M. & Friedman, H. (Plenum, New York), pp. 43–57.
62. Mavrodi, D. V., Ksenzenko, V. N., Bonsall, R. F., Cook, R. J., Boronin, A. M. & Thomashow, L. S. ( 1998) J. Bacteriol. 180, 2541–2548.
63. O'Toole, G. A. & Kolter, R. ( 1998) Mol. Microbiol. 28, 449–461.







