
Colloquium
Bad bugs and beleaguered bladders: Interplay between uropathogenic Escherichia coli and innate host defenses
Matthew A. Mulvey*, Joel D. Schilling, Juan J. Martinez, and Scott J. Hultgren
Department of Molecular Microbiology and Microbial Pathogenesis, Box 8230, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, MO 63110
Strains of uropathogenic Escherichia coli (UPEC) are the causative agents in the vast majority of all urinary tract infections. Upon entering the urinary tract, UPEC strains face a formidable array of host defenses, including the flow of urine and a panoply of antimicrobial factors. To gain an initial foothold within the bladder, most UPEC strains encode filamentous surface adhesive organelles called type 1 pili that can mediate bacterial attachment to, and invasion of, bladder epithelial cells. Invasion provides UPEC with a protective environment in which bacteria can either replicate or persist in a quiescent state. Infection with type 1-piliated E. coli can trigger a number of host responses, including cytokine production, inflammation, and the exfoliation of infected bladder epithelial cells. Despite numerous host defenses and even antibiotic treatments that can effectively sterilize the urine, recent studies demonstrate that uropathogens can persist within the bladder tissue. These bacteria may serve as a reservoir for recurrent infections, a common problem affecting millions each year.
U rinary tract infections (UTIs), including cystitis and pyelonephritis, affect a large proportion of the world population and account for significant morbidity and high medical costs (1). It is estimated that one-third of American women will have at least one UTI before the age 65 and many will experience more than one infection per year. Uropathogenic Escherichia coli (UPEC) is the causative agent in 70–95% of communityacquired UTIs and about 50% of all cases of nosocomial UTIs (1, 2). The large intestine, along with the vaginal introitus and periurethral tissue, can function as reservoirs for uropathogenic strains of E. coli (3). However, in contrast to resident intestinal strains and other E. coli isolates, UPEC strains encode a number of virulence factors that enable them to colonize the urinary tract and persist in the face of highly effective host defenses. Virulence factors associated with UPEC include toxins such as hemolysin and cytotoxic necrotizing factor, the siderophore aerobactin, capsules, lipopolysaccharide (LPS), and a number of adhesive organelles (4). The ability to adhere to host epithelial cells within the urinary tract is likely the most important determinant of pathogenicity.
Bacterial Adherence in the Urinary Tract
Adhesins expressed on the microbial surface help dictate the tissue tropism of invading bacteria and can modulate host cell responses to infection. Bacteria assemble adhesins on their surface as monomers, as simple oligomers, or as supramolecular fibers called fimbriae or pili (5). The adhesive organelles that are associated with UPEC strains include S pili, Dr family adhesins, P pili, and type 1 pili (4). S pili recognize sialosyloligosaccharide residues on host cells and may help promote colonization of the upper urinary tract by UPEC. Dr family adhesins, including Dr fimbriae and the afimbrial adhesins AFA-I and AFA-II, bind the Dra blood group antigen present on decay accelerating factor and may also facilitate ascending colonization of the urinary tract (6). P pili bind the α-D-galatopyranosyl-(1-4)-β-D-galactopyranoside moiety present in the globoseries of glycolipids that are expressed by erythrocytes and host cells present in the kidney (5). Consistent with this binding specificity, P pili have been shown to be major virulence factors associated with pyelonephritis caused by uropathogenic strains of E. coli. Of the adhesive organelles that are associated with the pathogenesis of UTIs, type 1 pili are the most widely distributed among UPEC isolates (7). The mechanisms and consequences of type 1 pilus-mediated interactions between E. coli and bladder epithelial cells are considered here.
Type 1 pili are composite fibers ranging from a few fractions of a micrometer to more than 3 µm in length (8). They consist of a 7-nm-thick helical rod made up of repeating FimA subunits joined to a 3-nm-wide distal tip fibrillum structure containing two adapter proteins, FimF and FimG, and the adhesin, FimH (8, 9) (Fig. 1 Left). FimH binds mannose-containing glycoprotein receptors and can mediate bacterial attachment to a variety of different host cell types ( 10, 11, 12 and 13). The FimH adhesin consists of two domains: a COOH-terminal pilin domain involved in the incorporation of FimH into type 1 pili and an NH3-terminal adhesin domain that contains a carbohydrate binding pocket capable of accommodating a D-mannose residue (Fig. 1 Right) (14). The adhesin domain of FimH is an 11-stranded elongated β barrel with an overall jelly roll topology. This domain is connected to the pilin domain via an extended linker that may provide maximum flexibility for proper positioning of the carbohydrate binding site so that it can interact with mannose residues presented by host glycoprotein receptors. Interactions between FimH and receptors expressed on the luminal surface of the bladder epithelium are critical to the ability of many UPEC strains to colonize the bladder and cause disease ( 7, 13, 15, 16).
The luminal surface of the bladder, which is first encountered by invading uropathogens, is lined by a stratified transitional epithelium (urothelium) that is usually three to four cell layers deep (17, 18). A thin basement membrane and lamina propria separate the epithelial cells from the smooth muscular and serous layers of the outer wall of the bladder. The urothelium itself is comprised of small, relatively undifferentiated basal and intermediate epithelial cells underlying a single layer of highly differentiated, large, multinucleate superficial facet cells. These
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviations: UTI, urinary tract infection; UPEC, urapathogenic Escherichia coli; LPS, lipopolysaccharide; AUM, asymmetric unit membrane; EM, electron microscopy; PMN, polymorphonucleocyte.
|
* |
To whom reprint requests should be addressed. E-mail: mulvey@borcim.wustl.edu. |
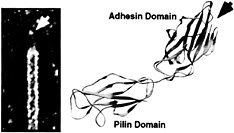
Fig. 1. Type 1 pilus architecture and crystal structure of the FimH adhesin. (Left) High-resolution EM reveals the composite structure of the type 1 pilus. The pilus tip containing the FimH adhesin is indicated. (Right) FimH has two domains, each with Ig-like folds. The adhesin domain has a binding pocket (arrow) that can accommodate D-mannose.
cells, also known as umbrella cells, deposit on their apical surfaces a quasi-crystalline array of hexagonal complexes comprised of four integral membrane proteins known as uroplakins (18). The uroplakins (UPIa, UPIb, UPII, and UPIII), which are highly conserved among all mammalian species, are assembled into 16-nm-wide hexagonal particles that are further organized into plaques 0.3– 0.5 µm in diameter that cover almost the entire luminal surface of the bladder. All of the uroplakins have large luminal domains and, with the exception of UPIII, relatively small cytoplasmic domains. In cross section, the luminal leaflet of the apical plasma membrane appears twice as thick as the cytoplasmic leaflet. This uroplakin-embedded membrane has been termed the AUM (asymmetric unit membrane) and it is proposed to serve as a permeability barrier and to strengthen and stabilize facet cells, preventing rupturing as the bladder distends to accommodate more urine. In vitro binding assays have shown that two of the uroplakins, UPIa and UPIb, derived from diverse mammalian species, including mice and humans, can specifically bind to E. coli expressing type 1 pili (19). Binding is inhibited by the soluble FimH receptor analogue, D-mannose, and by enzymatic deglycosylation of UPIa and UPIb.
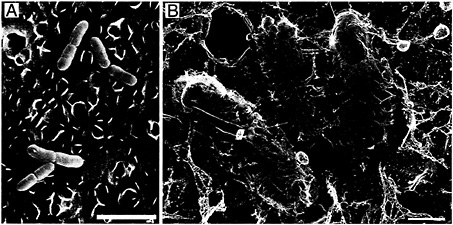
Fig. 2. Type 1 pilus-mediated bacterial attachment to the bladder epithelium. After inoculation of C57BL/6 mice with type 1-piliated UPEC, numerous bacteria (yellow) can be found attached to the luminal surface of the bladder (blue) as detected by scanning EM (A) and high-resolution freeze-dry/deep-etch EM (B). Type 1 pili mediating bacterial attachment were resolved with the high-resolution technique. The scalloped appearance of the bladder surface is attributable to the presence of the uroplakin plaques (≈0.5 µm in diameter). [Bars = 3 µm (A) and 0.5 µm (B).]
Interactions between type 1 pili and host receptor molecules such as UPIa and UPIb allow UPEC to establish an initial foothold within the urinary tract. The ability of type 1 pili to mediate bacterial attachment to the AUM of the facet cells lining the bladder lumen has been investigated by using a mouse cystitis model and microscopic techniques (13). Shortly after inoculation of UPEC into mouse bladders, numerous bacteria can be found attached to the urothelial surface as detected by scanning electron microscopy (EM) (Fig. 2A). Bacteria adhered to the bladder surface both singly and, occasionally, in large biofilmlike colonies. Bacteria were often situated in grooves and niches formed by the AUM of the facet cells (13). Almost no bacteria were found associated with bladders taken from mice infected with mutant E. coli strains that express type 1 pili lacking the FimH adhesin. High-resolution freeze-dry and freeze-fracture/deep-etch EM revealed that the FimH-containing adhesive tips of type 1 pili could interact directly with the uroplakin-embedded AUM, seemingly tethering the bacteria to the bladder surface (Fig. 2B). Type 1 pili also appeared able to interact with other adherent bacteria on the bladder surface. This is an interesting observation considering that type 1 pili have been shown to function in the formation of biofilms in vitro (20).
Type 1 Pilus-Mediated Bacterial Invasion
Uropathogenic strains of E. coli, contrary to prevailing assumptions, are not strictly extracellular pathogens. Over 10 years ago, transmission EM studies of infected rat and mouse bladders demonstrated that bladder epithelial cells can internalize UPEC in vivo (21, 22). Bacteria were observed within membrane-bound vacuoles and free within the cytoplasm of the facet cells that line the luminal surface of the bladder. It was proposed that the bladder epithelial cells internalized bacteria as part of an innate host defense mechanism. A more recent study, however, suggested that bacterial internalization by bladder epithelial cells could benefit the pathogens (13). It was shown by using a murine
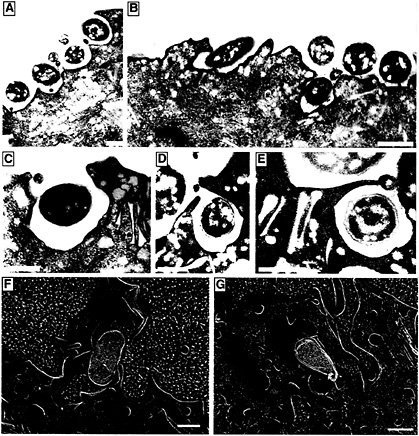
Fig. 3. Internalization of type 1-piliated UPEC by superficial facet cells. (A–F) Transmission EM shows the AUM of mouse superficial facet cells in various stages of enveloping adherent type 1-piliated UPEC. ( F and G) High-resolution freeze-fracture/deep-etch EM also reveals type 1-piliated E. coli (yellow) seemingly being enveloped by facet cells (brown; the AUM and extracellular milieu are colored blue). The cytoplasmic face of three uroplakin plaques, separated by smooth interplaque regions, can be seen in the host membrane that is partially enveloping the bacterium in G. All images were obtained by using C57BL/6 mouse bladders recovered about 1 h after infection with UPEC. [Bars = 1 µm (A and B) and 0.5 µm (C–G).]
cystitis model that a subpopulation of type 1-piliated E. coli inoculated into the bladder could invade the urothelium and that these bacteria appeared to have a distinct survival advantage over their extracellular counterparts.
EM studies of infected mouse bladders have indicated that the AUM of the superficial facet cells can zipper around and envelop attached bacteria, apparently via interactions with type 1 pilus tips (Fig. 3) (13). In vitro assays have shown that cultured human bladder carcinoma epithelial cells can also envelop and internalize type 1-piliated E. coli (23). Standard gentamicin protection assays used to evaluate the ability of a microbe to invade host cells demonstrated that fimH+ type 1-piliated clinical cystitis isolates and laboratory E. coli K12 strains, but not fimH− mutants, could invade cultured bladder epithelial cells. Additional experiments have shown that cultured bladder epithelial cells efficiently internalize latex beads coated with the FimH adhesin in a mannose-inhibitable fashion (Fig. 4). In contrast, BSA-coated latex beads and beads coated with the type 1 pilus periplasmic chaperone FimC were rarely internalized (23).
These observations indicate that FimH is sufficient to mediate bacterial uptake by bladder epithelial cells. This suggests that FimH can function as an invasin, a class of molecules, exemplified by the invasin protein encoded by Yersinia pseudotuberculosis and internalin A expressed by Listeria monocytogenes, that can mediate bacterial internalization into host cells in the absence of other virulence factors (24, 25). Initial investigations have demonstrated that FimH-mediated bacterial invasion of bladder epithelial cells requires the activation of host signal transduction cascades involving protein tyrosine kinases, phosphoinositide-3 kinase, and localized host actin cytoskeletal rearrangements (23). Furthermore, FimH-mediated invasion correlates with the formation of complexes between focal adhesin
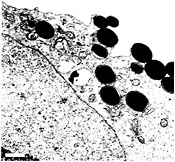
Fig. 4. FimH-mediated internalization of latex beads. Native FimH can be purified from bacteria in complex with the type 1 pilus chaperone FimC (14). Transmission EM (and other data not shown) demonstrate that latex beads coated with FimC-FimH complexes are readily internalized by bladder epithelial cells (5637 cells) grown in culture. Beads coated with FimC alone or with BSA are not internalized (23). The envelopment of FimC-FimH-coated beads by 5637 cells is similar in appearance to the envelopment of type 1-piliated E. coli by bladder epithelial cells. (Bar = 1µm.)
kinase and phosphoinositide-3 kinase and between the cytoskeletal components α-actinin and vinculin. These events likely contribute to the modulation and stabilization of localized actin cytoskeletal alterations that can lead to the envelopment and internalization of E. coli subsequent to FimH-mediated bacterial attachment.
Type 1 pilus-mediated invasion of bladder epithelial cells may provide UPEC refuge from the flow of urine through the bladder and from numerous host defenses present within the urine and bladder tissue. Invasion may also give UPEC strains access to a more nutrient-rich environment. Assays designed to detect and measure intracellular bacterial replication after invasion have demonstrated that type 1-piliated UPEC isolates can survive and replicate within bladder epithelial cells both in vitro and in vivo (M.A.M., J.D.S., and S.J.H., unpublished results). In some cases intracellular bacterial replication can reach very high levels, essentially converting host bladder epithelial cells into bacterial factories. Rather than replicating, we have found that individual uropathogens can also apparently enter a quiescent state within host bladder epithelial cells. External cues, perhaps provided by the host cells, may effect whether intracellular UPEC enters a replicative or quiescent state. Type 1-piliated laboratory E. coli K12 strains, although able to invade bladder cells, are unable to proliferate intracellularly. This suggests that intracellular growth is not a general property of all E. coli strains but may require additional virulence factors unique to uropathogenic isolates.

Fig. 5. Exfoliation of bladder epithelial cells. (A) The luminal surface of the bladder is normally covered by extremely long-lived, highly differentiated superficial facet cells with distinctive pentagonal or hexagonal outlines. These large cells (ranging from 20 to 150 µm in diameter) are often bi- or multinucleate and can be easily identified on the surface of whole-mount bladders stained with Hoechst dye. (B) Within 6 h after inoculation with type 1-piliated E. coli, many infected facet cells in C57BL/6 mice exfoliate and are rinsed away, revealing the smaller, mono-nucleate underlying epithelial cells. (C) A stained paraffin section from an infected mouse bladder shows a facet cell in the process of exfoliating and taking a large mass of adherent E. coli with it. Host cell nuclei were stained with Hoechst dye whereas bacteria were stained red by using anti-E. coli primary and Cy3-labeled secondary antibodies. [Bars = 100 µm (A and B) and 10 µm (C).]
Primary Bladder Defenses
The urinary tract is typically a sterile environment, and bacterial colonization of the bladder epithelium does not go unchallenged. The bulk flow of urine through the bladder and micturition can work to rinse away nonattached or weakly adherent microbes from the bladder surface (3). The low pH and osmolarity of urine can be inhibitory to bacterial growth, and the salts, urea, and organic acids present in urine can reduce bacterial survival within the urinary tract. In addition, lactoferrin within urine can scavenge essential iron away from incoming microbes and a number of soluble and cell associated factors within the bladder, including Tamm-Horsfall protein, low molecular weight sugars, secretory IgA, and uromucoid, can act as anti-adherence factors, competitively inhibiting bacterial attachment to the bladder surface. If a microbe manages to sidestep these constitutive defenses and make contact with the urothelium, its continued presence can trigger the activation of additional host defense mechanisms.
Bladder Cell Exfoliation as a Defense
Normally, the epithelial cells lining the luminal surface of the bladder have an extremely slow turnover rate (17). However, exfoliated bladder cells along with associated bacteria are often found in the urine of patients with UTIs (21, 22, 26). Based on such observations, it has been proposed that the exfoliation and clearance of infected and damaged bladder epithelial cells can function as a host defense (21, 22). Using a mouse cystitis model, we have recently shown that bladder epithelial cells will exfoliate in response to infection by type 1-piliated UPEC strains (13). Like UPEC isolates, recombinant laboratory E. coli K12 strains expressing type 1 pili, but lacking other potential adhesins and virulence factors specifically associated with uropathogens, also trigger massive exfoliation of urothelial cells (Fig. 5 A and Fig. 5 B). Isogenic E. coli mutants that express type 1 pili lacking the FimH adhesin have no obvious effects on the bladder urothelium, suggesting that FimH-mediated bacterial attachment and/or invasion is critical in the induction of bladder cell exfoliation during a UTI.
Bladder cell exfoliation in response to infection with type 1-piliated E. coli occurs via an apoptosis-like mechanism involv-
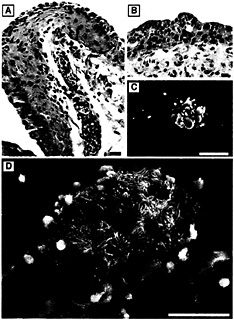
Fig. 6. Neutrophil influx into the urothelium in response to infection. (A) Paraffin sections of C57BL/6 mouse bladders recovered 6 h after infection with type 1-piliated UPEC and stained with hematoxylin and eosin show PMNs (small, darkly stained cells) migrating from blood vessels within the lamina propria and into the urothelium. (B) PMNs appeared to aggregate beneath the luminal surface of the bladder and could occasionally be seen, by scanning EM, emerging in the vicinity of adherent bacteria on the surface of newly exposed immature urothelial cells (C). PMNs were also found associated with infected facet cells in the process of exfoliating (D). [Bars = 20 µm (A), 10 µm (B), 50 µm (C), and 30 µm (D).]
ing host DNA fragmentation and the activation of proteolytic enzymes known as caspases (13). These cysteine proteases are central components in the initiation and execution of apoptotic pathways (27). Exfoliation in C57BL/6 mice can be inhibited by treating mouse bladders with a cell permeable caspase inhibitor, Boc-aspartyl(Ome)-fluormethylketone (13). In addition to inhibiting bladder cell exfoliation, Boc-aspartyl(Ome)-fluormethylketone treatment also significantly reduced the rate of bacterial clearance from the bladder. At 12 h after infection with a type 1-piliated UPEC strain, untreated mouse controls had about 85% fewer bacteria within their bladders in comparison with Boc-aspartyl(Ome)-fluormethylketone-treated mice. These results demonstrate that exfoliation can act as a host defense mechanism. The capacity of exfoliation to facilitate bacterial clearance from the urinary tract is illustrated in Fig. 5C.
Rather than directly triggering the exfoliation response, FimH-mediated bacterial attachment or invasion may serve to deliver other bacterial factors, such as LPS, to host bladder cells. Indeed, it has been reported that soluble LPS can induce the exfoliation of single bladder cells and cell clusters when inoculated into mouse bladders from Albany or ICR mice (28, 29). However, recent studies have shown that bladder epithelial cells in C3H/HeJ mice, which are nonresponsive to LPS, still exfoliate in response to infection with type 1-piliated E. coli (data not shown). It is possible that infection by type 1-piliated E. coli can trigger bladder epithelial cell exfoliation by more than one pathway. The genetic background of the host can also effect the rate and degree of bladder cell exfoliation after infection. For example, massive exfoliation of bladder epithelial cells occurs in C57BL/6 mice within 6 h after infection with type 1-piliated E. coli (Fig. 5 A and Fig. 5 B) whereas similar levels of exfoliation are not observed until 24 –72 h after infection of other mouse strains tested (e.g., C3H/HeN, C3H/HeJ, and FVB/N mice).
Neutrophil Influx and Cytokines
Six hours after transurethral inoculation of C57BL/6 mice with type 1-piliated UPEC, as massive bladder cell exfoliation becomes apparent, neutrophils can be seen infiltrating the bladder and entering the urothelium and bladder lumen (Fig. 6). Neutrophils, or polymorphonucleocytes (PMNs), are phagocytic inflammatory cells that can mediate bacterial killing through the generation of reactive oxygen intermediates and/or the release of preformed anti-microbial peptides. Neutrophil recruitment has been shown to be critical for bacterial clearance from both the bladder and kidney, and the presence of neutrophils in the urine (pyuria) is a hallmark of UTIs (30). Interactions between the neutrophil receptor CD11b/CD18 (Mac-1) and the adhesion molecule ICAM-1 on bladder epithelial cells seem to be critical for neutrophil migration into the urothelium (31). Interestingly, bacterial infection has been shown to induce the expression of ICAM-1 by bladder epithelial cells (31).
Over the past decade, elucidation of the molecular mechanisms involved in neutrophil recruitment into the bladder and kidneys has highlighted the importance of cytokines and chemokines. These soluble molecules, which are produced by a number of host cell types in response to a variety of different stimulatory molecules such as LPS, can regulate and orchestrate inflammatory responses. In patients with cystitis or pyelonephritis, the cytokine interleukin 6 (IL-6) and chemokine interleukin 8 (IL-8) are present in the urine, suggesting a potential role for
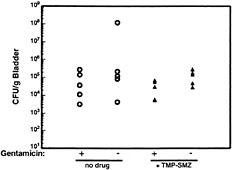
Fig. 7. Resistance of intracellular UPEC to antibiotic treatment. C57BL/6 mice were infected with 1 × 108 colony-forming units of the clinical cystitis isolate NU14. Six hours after infection, mice were given trimethoprim-sulfamethoxazole (54 µg/ml and 270 µg/ml, respectively) in their drinking water or were left untreated. Mice were provided fresh water with or without antibiotics daily, and at 3 days after inoculation bladders were recovered and bisected. Bladder halves were treated with either gentamicin (to kill any extracellular bacteria) or were left untreated before homogenization and determination of bacterial titers. Control experiments (not shown) demonstrated that trimethoprim-sulfamethoxazole completely inhibited the growth of NU14 in vitro.
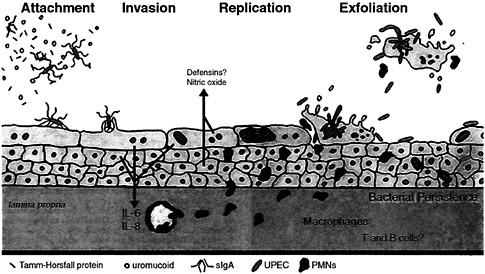
Fig. 8. Interplay between innate host defenses and UPEC within the bladder. The flow of urine in addition to a variety of host factors that can act as soluble receptor analogues for type 1 pili can impede contact between type 1-piliated UPEC and host superficial facet cells. If contact is established, FimH-receptor interactions can trigger the internalization of adherent bacteria into facet cells, in which UPEC can replicate to high levels. However, attachment and/or invasion can result in the activation of apoptotic pathways within facet cells, leading to the eventual exfoliation and clearance of infected host cells. The release of infected bladder cells in urine may facilitate the spread of UPEC strains in the environment. Initial interactions between type 1-piliated E. coli and urothelial cells can also result in the induction of pro-inflammatory cytokines, leading to the influx of PMNs into the urothelium. To avoid clearance by exfoliation, UPEC is able to escape from dying facet cells and can go on to infect surrounding and underlying epithelial cells. These bacteria may eventually be able to enter a niche within the urothelium in which they can persist (at subclinical levels) undetected by immunosurveillance mechanisms.
these molecules in UTI pathogenesis (32, 33, 34 and 35). Bladder and kidney epithelial cells appear to be a major source of IL-6 and IL-8 after infection with UPEC (36, 37).
IL-6 is a pleiotropic cytokine that has a variety of immunoregulatory functions, including the amplification of signals involved in neutrophil recruitment (38, 39). Urine IL-6 levels from patients with UTIs correlate with disease severity, although the exact role of IL-6 in UTI pathogenesis is unclear (34). The function of IL-8, and other related chemokines, during UTIs has been easier to delineate. IL-8 is a member of the CXC chemokine family and a potent neutrophil chemotatic molecule (40). In humans, the induction of IL-8 after infection with UPEC correlates well with the appearance of neutrophils in the urine (32). By using a mouse pyelonephritis model, the appearance of neutrophils in the urine was recently shown to be directly related to the production of MIP-2, a CXC chemokine that is functionally homologous to IL-8 (which mice lack) (41). Mice injected with an antibody to MIP-2 before infection with UPEC have a defect in transepithelial neutrophil migration.
Type 1- and P-piliated E. coli strains have been shown to elicit significantly more cytokines than their nonpiliated isogenic counterparts (36). P-pili can activate kidney epithelial cell cytokine production, independent of LPS, through a ceramide and serine/threonine kinase-dependent signaling pathway (42, 43). In contrast, we have found that type 1 pili indirectly activate IL-6 production by bladder epithelial cells by mediating bacterial internalization, which in turn boosts bladder epithelial responses to LPS (J.D.S., M.A.M., R. G. Lorenz, and S.J.H., unpublished data). Consistent with these results, LPS nonresponsive C3H/HeJ mice have been shown to be severely defective in their inflammatory response to UPEC within the urinary tract (44, 45).
Persistent and Recurrent UTIs
The exfoliation of infected bladder cells and the influx of neutrophils pose a formidable challenge to the survival of UPEC within the urinary tract. In addition, the production of antimicrobial factors such as nitric oxide and defensins by urothelial cells along with the activities of immune cells, such as mast cells and macrophages, may also help control UTIs. However, despite these various host defenses, significant numbers of bacteria can persist within the bladder for days to weeks (refs.45 and 46; M.A.M., J.D.S., and S.J.H., unpublished results). In addition, Hvidberg and coworkers have shown that UPEC strains can persist within mouse bladder tissue virtually undeterred in the face of antibiotic treatments that effectively reduce bacterial titers within the urine (46). This persistence appears to be related to the ability of type 1-piliated E. coli to invade bladder epithelial cells. Recently, we have shown that the treatment of mice with trimethoprim-sulfamethoxazole, an antibiotic combination that is often used to treat UTIs (1), fails to clear bacteria from bladder tissue, although the drugs can effectively reduce bacterial titers within the urine (Fig. 7). The bacteria persisting after trimethoprim-sulfamethoxazole treatment are also protected, for the most part, from the ex vivo treatment of bladders with gentamicin, suggesting that these bacteria are intracellular.
It is not entirely clear how invasion can facilitate the survival of UPEC within the bladder, especially considering that host
urothelial cells can exfoliate in response to infection. Recent observations, however, suggest that UPEC is able to flux into and out of bladder epithelial cells (M.A.M., J.D.S., and S.J.H., unpublished data). This may allow invading uropathogens to multiply and subsequently escape from their host bladder cells before the host cells can complete the exfoliation process. This could also facilitate the dissemination of UPEC within the urinary tract and help bacteria evade both innate and adaptive host defenses. Furthermore, exfoliation of bladder epithelial cells could leave lower layers of the urothelium more susceptible to infection. It is in the underlying epithelial layers that UPEC may be able to persist for long intervals, sequestered within host bladder epithelial cells, possibly in a quiescent state and undetectable within the urine. These bacteria could potentially serve as a source for recurrent infections, one of the more vexing problems associated with UTIs. A model depicting some of the key factors and events that appear to contribute to the pathogenesis of bladder infections is shown in Fig. 8. Future research is aimed at identifying bacterial and host factors that can exacerbate or attenuate the severity of UTIs. It is hoped that such studies will eventually lead to more efficacious antibacterial therapeutics.
We thank R. Roth and J. Heuser for their help with the high-resolution EM; M. Veith for assistance with scanning EM; and L. LaRose and M. Levy for help with transmission EM. This work was supported by National Institutes of Health Grants RO1AI29549 and RO1DK51406. M.A.M. was supported by National Institutes of Health Fellowship AI09787.
1. Hooton, T. M. & Stamm, W. E. ( 1997) Infect. Dis. Clin. North Am. 11, 551–581.
2. Svanborg, C. & Godaly, G. ( 1997) Infect. Dis. Clin. North Am. 11, 513–529.
3. Sobel, J. D. ( 1997) Infect. Dis. Clin. North Am. 11, 531–549.
4. Johnson, J. R. ( 1991) Clin. Microbiol. Rev. 4, 80–128.
5. Hultgren, S. J., Jones, C. H. & Normark, S. N. ( 1996) in Escherichia Coli and Salmonella; Cellular and Molecular Biology, ed. Neidhardt, F. C. (Am. Soc. Microbiol., Washington, DC), pp. 2730–2756.
6. Goldhar, J. ( 1996) Adv. Exp. Med. Biol. 408, 63–72.
7. Langermann, S., Palaszynski, S., Barnhart, M., Auguste, G., Pinkner, J. S., Burlein, J., Barren, P., Koenig, S., Leath, S., Jones, C. H. & Hultgren, S. J. ( 1997) Science 276, 607–611.
8. Jones, C. H., Pinkner, J. S., Roth, R., Heuser, J., Nicholes, A. V., Abraham, S. N. & Hultgren, S. J. ( 1995) Proc. Natl. Acad. Sci. USA 92, 2081–2085.
9. Russell, P. W. & Orndorff, P. E. ( 1992) J. Bacteriol. 174, 5923–5935.
10. Tewari, R., MacGregor, J. I., Ikeda, T., Little, J. R. 1., Hultgren, S. J. & Abraham, S. N. ( 1993) J. Biol. Chem. 268, 3009–3015.
11. Baorto, D. M., Gao, Z., Malaviya, R., Dustin, M. L., van der Merwe, A., Lublin, D. M. & Abraham, S. N. ( 1997) Nature ( London) 389, 636–639.
12. Malaviya, R. & Abraham, S. N. ( 1998) J. Mol. Med. 76, 617–623.
13. Mulvey, M. A., Lopez-Boado, Y. S., Wilson, C. L., Roth, R., Parks, W. C., Heuser, J. & Hultgren, S. J. ( 1998) Science 282, 1494–1497.
14. Choudhury, D., Thompson, A., Stojanoff, V., Langermann, S., Pinkner, J., Hultgren, S. J. & Knight, S. D. ( 1999) Science 285, 1061–1066.
15. Connell, H., Agace, W., Klemm, P., Schembri, M., Marild, S. & Svanborg, C. ( 1996) Proc. Natl. Acad. Sci. USA 93, 9827–9832.
16. Thankavel, K., Madison, B., Ikeda, T., Malaviya, R., Shah, A. H., Arumugam, P. M. & Abraham, S. N. ( 1997) J. Clin. Invest. 100, 1123–1136.
17. Hicks, R. M. ( 1975) Biol. Rev. 50, 215–246.
18. Sun, T. T., Zhao, H., Provet, J., Aebi, U. & Wu, X. R. ( 1996) Mol. Biol. Rep. 23, 3–11.
19. Wu, X. R., Sun, T. T. & Medina, J. J. ( 1996) Proc. Natl. Acad. Sci. USA 93, 9630–9635.
20. Pratt, L. A. & Kolter, R. ( 1998) Mol. Microbiol. 30, 285–293.
21. Fukushi, Y., Orikasa, S. & Kagayama, M. ( 1979) Invest. Urol. 17, 61–68.
22. McTaggart, L. A., Rigby, R. C. & Elliott, T. S. ( 1990) J. Med. Microbiol. 32, 135–141.
23. Martinez, J. J., Mulvey, M. A., Schilling, J. D., Pinkner, J. & Hultgren, S. J. ( 2000) EMBO J. 19, 2803–2812.
24. Isberg, R. R., Voorhis, D. L. & Falkow, S. ( 1987) Cell 50, 769–778.
25. Mengaud, J., Ohayon, H., Gounon, P., Mege, R. M. & Cossart, P. ( 1996) Cell 84, 923–932.
26. Elliott, T. S., Reed, L., Slack, R. C. & Bishop, M. C. ( 1985) J. Infect. 11, 191–199.
27. Cohen, G. M. ( 1997) Biochem. J. 326, 1–16.
28. Aronson, M., Medalia, O., Amichay, D. & Nativ, O. ( 1988) Infect. Immun. 56, 1615–1617.
29. Jezernik, K., Medalia, O. & Aronson, M. ( 1995) Cell Biol. Int. 19, 887–893.
30. Haraoka, M., Hang, L., Frendeus, B., Godaly, G., Burdick, M., Stricter, R. & Svanborg, C. ( 1999) J. Infect. Dis. 180, 1220–1229.
31. Agace, W. W., Patarroyo, M., Svensson, M., Carlemalm, E. & Svanborg, C. ( 1995) Infect. Immun. 63, 4054–4062.
32. Agace, W., Hedges, S., Ceska, M. & Svanborg, C. ( 1993) J. Clin. Invest. 92, 780–785.
33. Ko, Y. C., Mukaida, N., Ishiyama, S., Tokue, A., Kawai, T., Matsushima, K. & Kasahara, T. ( 1993) Infect. Immun. 61, 1307–1314.
34. Otto, G., Braconier, J., Andreasson, A. & Svanborg, C. ( 1999) J. Infect. Dis. 179, 172–179.
35. Hedges, S., Anderson, P., Lidin-Janson, G., de Man, P. & Svanborg, C. ( 1991) Infect. Immun. 59, 421–427.
36. Hedges, S., Svensson, M. & Svanborg, C. ( 1992) Infect. Immun. 60, 1295–1301.
37. Hedges, S., Agace, W., Svensson, M., Sjogren, A. C., Ceska, M. & Svanborg, C. ( 1994) Infect. Immun. 62, 2315–2321.
38. Kopf, M., Ramsay, A., Brombacher, F., Baumann, H., Freer, G., Galanos, C., Gutierrez-Ramos, J. C. & Kohler, G. ( 1995) Ann. N.Y. Acad. Sci. 762, 308–318.
39. Romano, M., Sironi, M., Toniatti, C., Polentarutti, N., Fruscella, P., Ghezzi, P., Faggioni, R., Luini, W., van Hinsbergh, V., Sozzani, S., et al. ( 1997) Immunity 6, 315–325.
40. Murphy, P. M. ( 1997) Semin. Hematol. 34, 311–318.
41. Hang, L., Haraoka, M., Agace, W. W., Leffler, H., Burdick, M., Strieter, R. & Svanborg, C. ( 1999) J. Immunol. 162, 3037–3044.
42. Hedlund, M., Svensson, M., Nilsson, A., Duan, R. D. & Svanborg, C. ( 1996) J. Exp. Med. 183, 1037–1044.
43. Hedlund, M., Wachtler, C., Johansson, E., Hang, L., Somerville, J. E., Darveau, R. P. & Svanborg, C. ( 1999) Mol. Microbiol. 33, 693–703.
44. Hagberg, L., Hull, R., Hull, S., McGhee, J. R., Michalek, S. M. & Svanborg Eden, C. ( 1984) Infect. Immun. 46, 839–844.
45. Hopkins, W. J., Gendron-Fitzpatrick, A., Balish, E. & Uehling, D. T. ( 1998) Infect. Immun. 66, 2798–2802.
46. Hvidberg, H., Struve, C., Krogfelt, K. A., Christensen, N., Rasmussen, S. N. & Frimodt-Moller, N. ( 2000) Antimicrob. Agents Chemother. 44, 156–163.







