
Colloquium
Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens
Carl Nathan* and Michael U. Shiloh
Department of Microbiology and Immunology and Department of Medicine, Weill Cornell Medical College and Program in Immunology, Weill Graduate School of Medical Sciences of Cornell University, New York, NY 10021
This review summarizes recent evidence from knock-out mice on the role of reactive oxygen intermediates and reactive nitrogen intermediates (RNI) in mammalian immunity. Reflections on redundancy in immunity help explain an apparent paradox: the phagocyte oxidase and inducible nitric oxide synthase are each nonredundant, and yet also mutually redundant, in host defense. In combination, the contribution of these two enzymes appears to be greater than previously appreciated. The remainder of this review focuses on a relatively new field, the basis of microbial resistance to RNI. Experimental tuberculosis provides an important example of an extended, dynamic balance between host and pathogen in which RNI play a major role. In diseases such as tuberculosis, a molecular understanding of host–pathogen interactions requires characterization of the defenses used by microbes against RNI, analogous to our understanding of defenses against reactive oxygen intermediates. Genetic and biochemical approaches have identified candidates for RNI-resistance genes in Mycobacterium tuberculosis and other pathogens.
T his overview begins by rationalizing the prominence in immunity of chemically reactive micromolecules whose widespread interactions contrast with the exquisite specificity of B and T cell receptors, the signature proteins of the immune system. Next we consider the limitations of asking knock-out mice to tell us what is important in host defense. Interpretation of knock-outs is often based on the premise that the importance of a gene is defined by its nonredundancy. The shortcomings of this premise can be particularly problematic in immunity. Recent evidence from knock-out mice is weighed, then measured against a disease of commanding interest, tuberculosis. The foregoing analysis prompts a key question: Is there an enzymatic basis of resistance to reactive nitrogen intermediates (RNI), as there is to reactive oxygen intermediates (ROI)? [The term “RNI” refers to oxidation states and adducts of the nitrogenous products of nitric oxide synthases, ranging from nitric oxide (·NO) to nitrate (NO3−), that arise in physiological environments, including NO−, ·NO2, NO2−, N2O3, N2O4, S-nitrosothiols, peroxynitrite (OONO−), and dinitrosyl-iron complexes. The term “ROI” refers to intermediate reduction products of O2 en route to water, namely, superoxide ![]() hydrogen peroxide (H2O2), and hydroxyl radical (OH·), and reactive products of these with halides and amines.] Answers are beginning to emerge for both microbes and mammalian cells. Here we survey findings in microbes, in an effort to help lay the groundwork for molecular control over one important facet of host–pathogen relationships.
hydrogen peroxide (H2O2), and hydroxyl radical (OH·), and reactive products of these with halides and amines.] Answers are beginning to emerge for both microbes and mammalian cells. Here we survey findings in microbes, in an effort to help lay the groundwork for molecular control over one important facet of host–pathogen relationships.
Chemically Reactive Micromolecules in Host Defense
The Uses of Nonspecificity. Immunity is the ability of the largest inherited genome in a body to restrict the replication of the smaller, noninherited genomes in the same body. Immune recognition, the process by which one genome identifies another, affords some of the most impressive examples of specificity in biology. When it comes to actually inactivating DNA, however, specificity is hard to preserve, because the reactions by which DNA is built, copied, guarded, and repaired are extensively shared among organisms.
Perhaps the most striking example of nonspecificity in the immune system is its reliance on the production of chemically reactive micromolecules that do not discriminate the genomic source of their chemical targets (Fig. 1). Reactive oxygen intermediates (ROI) (1) and reactive nitrogen intermediates (RNI) (2) can damage DNA and several chemical moieties on which its propagation and protection depend, including Fe-S clusters, tyrosyl radicals, hemes, sulfhydryls, thioethers, and alkenes. ROI are produced by all aerobic cells and RNI by many. High output production of ROI is the specialty of mammalian phagocytes, with polymorphonuclear leukocytes in the lead and immunologically activated macrophages capable of generating about 1/3–1/2 as much. High output production of RNI is attainable by many mammalian cells in response to appropriate inflammatory stimuli, macrophages considerably outpacing polymorphonuclear leukocytes (2). Macrophages have the opportunity to produce superoxide ![]() and nitric oxide (˙NO) in nearly equimolar amounts and thus can be prolific generators of their joint and particularly destructive product, peroxynitrite (OONO−) (see Fig. 1 legend) (3, 4 and 5). RNI of dietary origin are put to use as antimicrobial agents in gastric juice, a key component of the innate immune system of epithelium (6).
and nitric oxide (˙NO) in nearly equimolar amounts and thus can be prolific generators of their joint and particularly destructive product, peroxynitrite (OONO−) (see Fig. 1 legend) (3, 4 and 5). RNI of dietary origin are put to use as antimicrobial agents in gastric juice, a key component of the innate immune system of epithelium (6).
It seems puzzling that evolution has entrenched defenses with the potential to damage uninfected cells. However, from the perspective of the whole organism, it is both a privilege and a necessity of multicellularity to lose parts to save the whole. As abscess formation illustrates in mammals, phagocytes are quick to sacrifice themselves and any volume of host tissue to prevent microbial metastasis. Plants meet the same need with the hypersensitivity reaction, a phagocyte-free version of abscess formation that depends on production of ROI and RNI (7, 8, 9 and 10).
A downside of highly specific recognition as a pillar of the immune response is that a microbe can sometimes escape recognition by altering a molecular feature that flags it, such as the order of monomers in its polymers. Conversely, the advantage of using ROI and RNI for defense is that a microbe cannot
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviations: NOS, nitric oxide synthase; phox, phagocyte oxidase; ROI, reactive oxygen intermediates; RNI, reactive nitrogen intermediates; SOD, superoxide dismutase; STM, signature-tagged transposon mutagenesis.
|
* |
To whom reprint requests should be addressed. E-mail: cnathan@med.cornell.edu. |
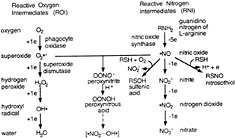
Fig. 1. ROI and RNI production in mammalian cells via phox and NOS: parallel but connecting paths. Nitroxyl anion (NO−), a one-electron reduction product of nitric oxide (˙NO), is unlikely to arise from ˙NO under physiologic conditions, but is considered by some investigators to be a primary and more toxic product of NOS (91). Reaction of RNI with cysteine sulfhydryls can lead either to S-nitrosylation or to oxidation to the sulfenic acid, as well as to disulfide bond formation (not shown), all of which are potentially reversible. Peroxynitrite anion (OONO−) and peroxynitrous acid (OONOH) have distinct patterns of reactivity (92), but for simplicity, the text refers to both as peroxynitrite. OONOH spontaneously decomposes via species resembling the reactive radicals, hydroxyl (OH˙) and/or nitrogen dioxide (˙NO2). When L-arginine is limiting, NOS can produce superoxide ![]() along with ˙NO, favoring the formation of peroxynitrite (5).
along with ˙NO, favoring the formation of peroxynitrite (5).
readily evade them by dispensing with their targets, because the targets are atomic rather than macromolecular, and confer essential chemical functions (2). Instead, pathogens interfer with host cell production of ROI or RNI, catabolize them, or repair their damage (11).
The Limits of Nonredundancy. A cardinal tenet of contemporary research holds that the role and importance of a biochemical pathway are established by the phenotype of its mutant. In effect, the function of a gene product is described by its nonredundancy. However, it does not necessarily follow that apparent redundancy indicates dispensability. A great many null mutations do not yield phenotypes, most likely because the range of conditions tested is narrow (12, 13). Interpretation of redundancy as dispensability is particularly inappropriate for genes engaged in competition with other genomes. To the extent that a well designed defense backs up its most important elements, redundancy can argue for utility. For example, if pathogens are challenged to inhibit production of a given class of host antimicrobial compounds, then the host is challenged to evolve diverse approaches to their synthesis. Moreover, the few dozen pathogens favored by investigators for experiments are a tiny fraction of the hundreds swarming at a host's gates. A given antimicrobial mechanism may be redundant against the pathogens tested but nonredundant against others. Phagocyte antimicrobial mechanisms often work synergistically (14). A phenotype indicating that a given gene product is important for resistance to a pathogen does not imply that other gene products are unimportant in defense against the same pathogen. In short, redundancy and synergy are essential features of the immune system that complicate the interpretation of knock-outs. Recourses include generation of compound knock-outs and diversification of settings in which they are tested. There remains the problem that species with induced deficiencies do not recapitulate key aspects of human immunity. For example, in contrast to humans, mouse neutrophils lack defensins and bacterial permeability increasing factor and respond feebly to tumor necrosis factor and formylated peptides. Mice lack type I CD1 molecules, which are specialized to present particularly hydrophobic microbial antigens, and have not been reported to express granulysin, the only known antibacterial protein of T cells.
Table 1. Antimicrobial products of human phagocytes that are delivered to phagosomes
|
Product |
Neutrophils* |
Macrophages† |
|
ROI |
+ |
+ |
|
RNI |
+ |
+ |
|
Myeloperoxidase |
+ |
− |
|
Lactoferrin |
+ |
− |
|
Bacterial permeability increasing factor |
+ |
− |
|
Serprocidins (elastase, cathepsin G, protease 3, azurocidin) |
+ |
− |
|
Phospholipase A2 |
+ |
− |
|
Cathelicidin |
+ |
− |
|
Lysozyme |
+ |
− |
|
Defensins (HNPs) 1, 2, 3, 4 |
+ |
− |
|
* Antibacterial proteins specific to eosinophils are not included. † Monocytes contain some of the antimicrobial proteins of neutrophils until they differentiate into macrophages. |
||
With these reflections in mind, one approaches the lessons from knock-out mice anticipating that a major effector mechanism in host defense could be found to be both nonredundant and redundant. This proves to be the case for ROI and RNI.
Nonredundant Roles of Phagocyte Oxidase (phox) and Nitric Oxide Synthase 2 (NOS2). Table 1 lists phagocyte products that are microbicidal in vitro and are delivered to phagosomes. From these features it is reasonable to presume that the physiological roles of these products include antimicrobial action. Table 2 lists the five such products that have been shown to play a nonredundant role in mice. Mice whose phagocytes are deficient in elastase or cathepsin G, two of four antimicrobial serprocidins (15), are susceptible to experimental infection with Klebsiella pneumoniae (6), Escherichia coli (16), and Aspergillus fumigatis (75). Myeloperoxidase converts H2O2 into more toxic hypohalites (1); mice lacking myeloperoxidase have increased susceptibility to Candida albicans (17). Mice deficient in the phagocyte oxidase (phox), the major source of pathogen-triggered ROI production (18, 19), are susceptible to several inoculated pathogens. Finally, mice deficient in the high output pathway of nitric oxide production, catalyzed by ˙NO synthase type 2 (NOS2 or iNOS), have a worse course of infection than wild-type mice after inoculation with diverse organisms (20, 21). However, the autotoxic potential of RNI is illustrated by the greater severity of influenza virus pneumonitis (22) and Mycobacterium avium infection (23) in wild-type mice than in NOS2-deficient mice.
An experimental alternative to knock-outs is administration of inhibitors. The major problem is specificity. No phox inhibitors have been reported that are effective and nontoxic in experimental animals. The most potent known inhibitor of phox in vitro, diphenylene iodonium, is 20-fold more potent as an inhibitor of NOS2 (24). In contrast, L-arginine analogs serve as nontoxic, phox-sparing NOS2 inhibitors (25). DeGroote and Fang list reports in which NOS2 inhibitors have exacerbated infection by 80 species of viruses, bacteria, fungi, and protozoa (26).
As for ROI, RNI are critical in host defense not only because they can damage pathogens but also because they are immunoregulatory ( 21). For example, RNI can inhibit G proteins (27), activate or inhibit kinases (28), caspases (29), metalloproteases (106), transcription factors (30), and DNA methyltransferase (31), inhibit lymphocyte proliferation, alter cytokine and prostaglandin (107)
Table 2. Antimicrobial mechanisms of phagocytes that are substantially nonredundant in host defense
|
Phenotype of knock-out mice |
||||
|
Gene encoding |
Gene essential for host defense |
Gene contributory to host defense |
Gene detrimental to host defense |
Gene dispensable for host defense |
|
Klebsiella pneumonia Escherichia coli Aspergillus fumigatus |
||||
|
Cathepsin G (75) |
Aspergillus fumigatus |
|||
|
Myeloperoxidase (17) |
Candida albicans |
Staphylococcus aureus |
||
|
Salmonella typhimurium Aspergillus fumigatus Staphylococcus aureus |
Listeria monocytogenes Mycobacterium tuberculosis |
|||
|
Mycobacterium tuberculosis Leishmania major Leishmania donovani Ectromelia virus Coxsackie B3 virus |
Listeria monocytogenes Toxoplasma gondii Salmonella typhimurium Staphylococcus aureus Chlamydia pneumoniae Mycoplasma pneumoniae Entamoeba histolytica Murine cytomegalovirus Hepatitis B |
Influenza A virus Mycobacterium avium |
Plasmodium chabaudi Plasmodium yoelii Plasmodium berghei Trypanosoma cruzi Pseudomonas aeruginosa Legionella pneumophila Chlamydia trachomatis Borrelia burgdorferi |
|
|
All pathogens were experimentally inoculated. Spontaneous infectionsarose from wounds in gp47phox–/– mice (18) but have not been reported in gp91phox-deficient mice. |
||||
production, and either cause or prevent apoptosis of host cells (32). The immune response of plants also depends on ROI and RNI for signaling and apoptosis, and possibly for necrosis and direct antimicrobial actions (7, 8, 10).
Redundant Roles of phox and NOS2. Investigators are now beginning to breed mice with compound genetic deficiencies to study interactions of host defense pathways (75). Perhaps the most dramatic phenotype to emerge so far from this approach has resulted from combined deficiency of NOS2 and the gp91 component of phox (33). Nearly all gp91phox–/–NOS2–/– mice died of spontaneous infections with commensal organisms, unless reared under specific pathogen-free conditions with life-long antibacterial and antifungal medication (33). Even with medication, during nearly 2 years of observation of this colony, about one-fifth of young adult mice developed massive, leukocyte-filled abscesses of internal organs caused by commensal bacteria or fungi of the gastrointestinal or respiratory tracts [a recent change in antibiotics may have lowered this incidence (M.U.S., unpublished observations)]. In contrast, mice with single deficiencies in gp91phox or NOS2 rarely develop spontaneous infections, even when the conditions of husbandry are routine and no medication is given. Thus, phox and NOS2 each appear to compensate in large part for isolated deficiency of the other.
The relative futility of host defense against commensal organisms in gp91phox–/–NOS2–/– mice makes the point that epithelia with their antimicrobial peptides, plasma with its antibody, complement, and mannose binding protein, granulocytes with their serprocidins, phospholipases, lysosomal hydrolases, and phagosomal acid, and T cells and natural killer cells with their perform, granzymes, and Fas ligand are apparently incapable of ensuring the survival of the species in the combined absence of phox, NOS2, and veterinary intervention. The phenotype of marked susceptibility to spontaneous infections has apparently not been reported for any other genetically altered mouse that can mobilize normal numbers of phagocytes to inflammatory sites.
Relevance for Humans. Of the five induced genetic deficiencies listed in Table 2, a clear-cut human counterpart is known for only one: genetic deficiency of phox components gives rise to chronic granulomatous disease, a syndrome of recurrent infections that establishes the nonredundance and importance of phox in human biology (34). Isolated deficiencies of serprocidins have apparently not been identified in humans. Myeloperoxidase deficiency is the most common known genetic abnormality of human leukocytes but appears to confer no phenotype in vivo, except perhaps in combination with diabetes mellitus (35). No spontaneously arising, primary genetic deficiency of NOS2 has been identified.
The lack of any known primary deficiency state of human NOS2 has intensified interest in experiments with isolated human macrophages. Infections, microbial products, and cytokines readily induce expression of NOS2 in tissue macrophages from rodents, but the same stimuli do not consistently or extensively induce NOS2 when applied to mononuclear phagocytes cultured from the blood of healthy humans. It has often been overlooked that few if any have reported the induction of NOS2 in mononuclear phagocytes cultured from the blood of mice. Most important, human mononuclear phagocytes from inflamed or infected subjects commonly express NOS2, generally at a higher level in tissue macrophages than in blood monocytes (25, 36, 37). In sum, if one takes into account the anatomic source and differentiation state of the macrophages and the health status of the donor, what some authors declare to be a species difference between mice and humans strikes others as a conserved pattern of gene expression. Likewise, human neutrophils from the blood of normal donors lack NOS2, but cytokines sometimes induce it (38) and neutrophils from some infected sites express it (39). When human macrophages express NOS2 at a high level in vitro, they can use it to kill leishmania (40) and mycobacteria (41). Expression of NOS2 at a low level usually does not confer antimicrobial activity. This may help explain the limited extent to which human monocytes from the blood of healthy donors kill leishmania and mycobacteria in vitro. In sum, with respect to the ability of human mononuclear phagocytes to express NOS2, the deficiency lies not in the cells but in our understanding and control of their differentiation (25).
Tuberculosis as a Paradigm for a Host–Pathogen Relationship in Which Host-Derived RNI Play a Major Role. Mycobacterium tuberculosis is one of the most successful pathogens of humankind in terms of
Table 3. Evidence for a role of host-derived RNI in control of tuberculosis
|
Evidence |
Ref. |
|
In vitro and in mice |
|
|
Sensitivity of mycobacteria to RNI in vitro |
|
|
Expression of NOS2 at sites of disease in immunocompetent mice |
|
|
Lack of NOS2 in immunocompromised mice with progressive disease: malnutrition; knock-outs of β2-microglobulin, T cell receptors, interferon-γ or its receptor, TNF receptor 1 |
|
|
Exacerbation of infection in macrophages with NOS inhibitors |
|
|
Exacerbation of disease in vivo with NOS inhibitors |
|
|
Exacerbation of disease in mice with disrupted NOS2 alleles |
|
|
In human cells |
|
|
Expression of NOS2 in macrophages from lungs of patients with tuberculosis |
|
|
Production of potentially mycobactericidal amounts of RNI by macrophages from lungs of patients with TB |
|
|
Production of RNI by M. tuberculosis-infected macrophages from lungs of normal donors |
|
|
Production of RNI by M. tuberculosis-infected blood mononuclear cells from both normal and tuberculous donors |
|
|
Killing of mycobacteria by pulmonary macrophages only if they express NOS2, and prevention of killing with a NOS inhibitor |
the proportion of the population infected (about one-third), the duration of infection (often lifelong), and the number of resulting deaths (2–3 million a year, among the most for any single infectious agent). Most infected individuals remain disease-free despite harboring viable organisms within or surrounded by macrophages. Thus, it is of paramount interest to understand what chemistry is used by macrophages to hold the tubercle bacillus in check, and what permits the bacillus to escape host control in about 5–10% of immunocompetent individuals and a higher proportion of those who are immunocompromised.
Table 3 summarizes evidence regarding the role of NOS2 in experimental and human tuberculosis. The evidence is of three types, (i) RNI have long been recognized as toxic to many microorganisms in culture (25, 26). Consistent with this, RNI in liquid and gaseous form can kill M. tuberculosis. For example, 2 days' exposure to 90 ppm of ˙NO gas kills more than 99% of M. tuberculosis in culture (42). As a point of reference, concentrations up to 80 ppm have been administered to patients for days or weeks to dilate the pulmonary vasculature. M. tuberculosis is also sensitive to nitrogen dioxide, but is much more resistant than other mycobacteria to peroxynitrite (43). The biochemical basis of selective, species-specific resistance to certain forms of RNI is a question of considerable interest, ( ii) NOS2 inhibitors exacerbate the infection in macrophages and in mice treated during either the acute or the chronic phases of the disease (44). (iii) For M. tuberculosis infection of mice, expression of NOS2 at sites of disease in well nourished, wild type mice is associated with control of infection for a substantial period of the host's normal lifespan. Conversely, failure to express NOS2 correlates with proliferation of tubercle bacilli and early death of the mice. Exacerbation of disease in association with reduced expression of NOS2 has been observed in malnourished or glucocorticoidtreated wild-type mice, and in well nourished mice with genetic deficiencies in immunologic pathways that, among other things, control the induction of NOS2 (44). The capstone of this evidence is that M. tuberculosis grows rapidly in and quickly kills mice that have been rendered selectively NOS2-deficient through homologous recombination (45).
Follow-up studies have added a startling observation: chemotherapy that appears to cure M. tuberculosis infection in immunocompetent mice fails to do so in NOS2-deficient mice (J. McKinney, personal communication). This is diametrically opposed to what one might have predicted from the observation that NOS2 helps prevent M. tuberculosis from replicating (44) and the expectation that chemotherapy will be more effective against dividing than dormant mycobacteria. Thus, some widely used drugs that are tuberculocidal in vitro are comparably effective in vivo only with help from NOS2. This has important implications: (i) The physiology of tubercle bacilli grown in vitro may differ so fundamentally from that of the bacilli in the host as to make drug screening in pure culture seriously incomplete. ( ii) Chemotherapy might be more effective in immunocompromised hosts if accompanied by delivery of RNI. (iii) Sensitization of tubercle bacilli to RNI might shorten the time to chemotherapeutic effect in immunocompetent hosts. The requirement for prolonged chemotherapy is a major reason why patients interrupt treatment, fostering the emergence of drug resistant strains.
What is the human relevance? Macrophages from the lungs of patients with tuberculosis express NOS2 (46, 47) in potentially mycobactericidal amounts (47) and can use it to kill mycobacteria in vitro (41). In some studies, expression of NOS2 has not conferred mycobactericidal activity (see, e.g., ref.48). Perhaps the activity of NOS2 was not high enough; a synergizing pathway was not operative; or the bacteria were resistant. Finally, chemotherapy is generally less successful in immunodeficient than in immunocompetent patients, suggesting that the human immune system contributes a biochemistry that synergizes with chemotherapy, as in the mouse.
Interest in the candidacy of RNI in helping to control tuberculosis is heightened by the lack of evidence for a major role of phox-derived ROI in this disease. Some chronic granulomatous disease patients have contracted tuberculosis, but it is not clear that they are unusually susceptible to the disease or that they experience an atypical course. Phox-deficient mice allowed increased bacillary proliferation in one of three organs examined (49), but the increase was only evident between weeks 2 and 4 (50), and, in another study, no difference in viable organisms was observed (R. North, personal communication). Finally, phoxdeficient mice have not been reported to succumb to experimental tuberculosis more rapidly than wild-type mice.
Microbial Resistance to RNI
The biochemical basis of resistance to ROI has been under study for 100 years, ever since the characterization of catalase launched the field of enzymology. Resistance to RNI, in contrast, is a relatively new concept. As such, RNI resistance takes center-stage for the rest of this review, beginning with a biological perspective.
Infection as a Dynamic Balance. Tuberculosis teaches that a balance can teeter for years between host immunity and a pathogen's resistance. Malnutrition, HIV infection, and immunosuppressive medication predispose toward progression of primary infection or reactivation of latent infection. The balance can tip in favor of the pathogen after decades of well being, even when no defect in host immunity is evident. In short, immunity to tuberculosis is often static, not sterilizing. Thus, the concept of microbial resistance is central not just to the pharmacology but also to the immunology of tuberculosis. Because RNI are essential for the control of murine tuberculosis and are produced in human tuberculosis, and control of the pathogen is imperfect in both species, tubercle bacilli may express mechanisms for RNI resistance.
Table 4. Parallels between ROI and RNI
|
Feature |
ROI |
RNI |
|
Primary catalyst |
Multisubunit flavocytochrome |
Multisubunit flavocytochrome |
|
Substrates |
O2, NADPH |
O2, NADPH, L-arginine |
|
Primary product |
Inorganic radical |
Inorganic radical (˙NO) |
|
Actions at low levels |
Activate or inhibit receptors, enzymes, transcription factors |
Activate or inhibit receptors, enzymes, transcription factors |
|
Actions at high levels |
Cause mutagenesis, apoptosis, necrosis |
Cause mutagenesis, apoptosis, necrosis |
|
Basis of cellular resistance |
SODs; catalase; peroxiredoxins; redox cycles involving glutathione, thioredoxin, glutaredoxin, trypanothione, ovothione, mycothione; methionine sulfoxide reductase; ascorbate; γ-tocopherol; urate; α-keto acids |
Under study |
Parallels Between ROI and RNI: Extension to RNI Catabolizing Pathways. With evidence from both animals and plants pointing to an important role for ROI and RNI in host defense, it is instructive to compare these two sets of compounds in broad terms. From the perspective presented in Table 4, it seems inescapable that metabolic pathways must exist to detoxify RNI. Because RNI are prevalent in soil as well as hosts, it seems likely that such pathways will be present in microbes, including pathogens.
Having postulated the existence of RNI-resistance mechanisms, how many should one expect? Again, it seems reasonable to turn to ROI for an indication. The panoply of enzymatic defense mechanisms against ROI includes catalases, superoxide dismutases (SODs), peroxiredoxins (see below), peptidyl methionine sulfoxide reductase and enzymes that make, oxidize, and reduce glutathione, glutaredoxin, thioredoxin, tryparedoxin, trypanothione, and probably mycothione (51), along with nonenzymatic reactants such as ascorbate, tocopherol, urate, and α-keto acids such as pyruvate (52).
Differences in species, cell types, subcellular localization, and regulated expression help account for this multiplicity, but there is a more instructive explanation: chemical specificity. The earlier discussion of nonspecificity of chemically reactive micromolecules referred to the species of origin of their submolecular targets. The enzymology by which ROI are catabolized or their damage repaired is a different matter. Here, the specificity can be as exquisite as for any other enzymatic reaction. For example, superoxide ![]() and hydrogen peroxide (H2O2) differ by merely two electrons and protons. Yet, each induces a separate stress regulon in some enterobacteria (soxRS and oxyR, respectively), and each is catabolized by enzymes (SODs and catalases, respectively) that are inactive against the other's substrate. Likewise, different enzymes reduce oxidized sulfur when it is part of either cysteine or methionine. At the level of survival, it is clear that some microbes can distinguish one form of RNI from another. For example, as noted above, all species of mycobacteria tested, both virulent and nonvirulent, were sensitive to being killed by ˙NO or ˙NO2, but only the virulent species were resistant to being killed by OONO− (43). Thus, it seems reasonable to anticipate that different enzymatic defenses may exist to protect microbes against different forms of RNI.
and hydrogen peroxide (H2O2) differ by merely two electrons and protons. Yet, each induces a separate stress regulon in some enterobacteria (soxRS and oxyR, respectively), and each is catabolized by enzymes (SODs and catalases, respectively) that are inactive against the other's substrate. Likewise, different enzymes reduce oxidized sulfur when it is part of either cysteine or methionine. At the level of survival, it is clear that some microbes can distinguish one form of RNI from another. For example, as noted above, all species of mycobacteria tested, both virulent and nonvirulent, were sensitive to being killed by ˙NO or ˙NO2, but only the virulent species were resistant to being killed by OONO− (43). Thus, it seems reasonable to anticipate that different enzymatic defenses may exist to protect microbes against different forms of RNI.
Table 5. Methods for identification of candidate microbial RNI resistance genes
|
Method |
Examples |
Ref. |
|
Loss of function with in vitro selection (mutagenesis; selection for hypersusceptibility to RNI; complementation of mutant with library from wild type of same species) |
metL |
|
|
Loss of function with in vivo selection (differential signature tagged mutagenesis: comparison of recovery of transposonmutagenized bacteria from wild-type mice, mice deficient in phox, mice deficient in NOS2 and mice deficient in both) |
Underway |
|
|
Gain of function (survival of recombinants expressing library from species of interest under selection pressure from RNI in vitro) |
NOXR1, NOXR3 |
|
|
Promoter trap (differential fluorescence induction or other in vivo expression technology to identify genes whose transcription is increased upon exposure to RNI) |
Underway |
|
|
Induction (detection of RNI catabolizing activity after sublethal exposure to RNI; purification monitored by bioactivity) |
hmp (flavohemoglobin) |
|
|
Biochemical hypothesis |
SODC; ahpC |
Redundancy is another instructive explanation for a multiplicity of microbial RNI resistance mechanisms, for the same reasons discussed earlier in reference to the host. As with knock-outs of immune system components, redundancy of RNI resistance mechanisms complicates interpretation of the phenotypes of their isolated deficiencies. For example, Salmonella typhimurium can express either or both of two periplasmic SODs (SODC1 and 2). Only the more virulent serovars expressed both, and such a strain was only attenuated in wild-type mice when both were disrupted ( 53). Similarly, nearly all nonviral genomes sequenced to date encode at least one peroxiredoxin, but many bacteria have more than one, and some as many as four (54). In bacteria with up to three peroxiredoxins, knock-out of a single one has produced a phenotype of increased in vitro sensitivity to ROI and/or RNI in some cases [E. coli (55, 56); S. typhimurium (55, 57)] but not in others [Streptococcus mutans (56)].
Routes to the Identification of Candidate RNI Resistance Genes. Table 5 summarizes approaches by which investigators have begun to identify candidate RNI resistance genes or gene products.
Mutagenesis followed by selection for loss of function has been carried out with chemical sources of RNI in vitro (58) and is now being undertaken with biological sources of RNI in vivo. Loss of function screens have the advantage that mutations are introduced directly in the species of interest. Signature-tagged transposon mutagenesis (STM) (59) offers the further advantage that the primary read-out (failure to grow in mice) is a preliminary
report of pathogenic relevance. STM generates a library in the pathogen of interest through transposon insertion. Panels of individual clones are pooled for injection into mice. DNA is prepared from a sample of the input pool as well as from the pool of bacteria recovered from the mice. Sequences unique to each transposon are used to Southern-blot filters that display the clones comprising the input pool. Comparison of the hybridization patterns of the input and recovered pools helps identify genes dispensable for growth in vitro but indispensable in vivo. An adaptation termed “differential” STM collects all of the bacterial clones that were negatively selected in wild-type mice and places them into new panels, together with a suitable number of control bacterial clones that had not displayed a survival disadvantage in vivo. These second-generation panels are used to repeat the selection in wild-type mice and simultaneously to extend the selection to mice genetically disrupted in a host defense pathway of interest: for example, gp91phox or NOS2. Bacterial clones that are once again negatively selected in wild-type mice but are recovered (“attenuation reversal”) in phox- or NOS2-deficient mice are candidates for carrying mutant genes whose wild-type alleles may act selectively to confer resistance to phox or to NOS2, respectively (M.U.S., K. Hisert, D. Holden, and C.N., unpublished work). STM, first applied to S. typhimurium (59), has recently been extended to M. tuberculosis (60, 61). Differential STM as defined above is underway using both organisms. Because STM is a competitive assay, defects in growth in vivo may only be apparent during competition with bacteria that are wild-type with respect to the gene in question; conversely, defects in growth in vivo may be obscured by the presence of bacteria that supply a missing function in trans.
Selection for a gain of function (resistance to nitrite) by expressing a library from M. tuberculosis in E. coli (62) was used to bypass what at the time were substantial technical difficulties in the mutagenesis of M. tuberculosis. A disadvantage of this approach is the inability to detect members of biochemical pathways under multigenic control whose other members are absent from the host in which the library is expressed. Another shortcoming is the lack of direct insight into the physiologic relevance of a gene expressed in a surrogate host. The same method was applied with another library prepared from the same strain of M. tuberculosis, using a different form of RNI (S-nitrosoglutathione) for selection in a different host (S. typhimurium) (63). The two unannotated mycobacterial genes identified in these two experiments conferred different patterns of resistance to RNI (62, 63).
A third approach is based on the premise that RNI- or ROI-resistance mechanisms will not be expressed under standard growth conditions in vitro, but will be induced by a sublethal level of the stress itself. This has led to the biochemical demonstration of induction by RNI of eight proteins in M. tuberculosis (64). At least one of these, α-crystallin, is also induced by growth in mouse and human macrophages and is essential for the normal growth of M. tuberculosis in wild-type mice (65). A variant of this approach has used catabolism of RNI as an assay with which to monitor purification of a gene product induced by RNI (66). Yet another variant is to use pathophysiologically relevant production of ROI and RNI by host cells in vitro or in vivo and detect transcriptionally induced bacterial genes by promoter traps (67). These methods may miss genes that are regulated posttranscriptionally, as well as genes that are important for RNI resistance but expressed under basal conditions. These methods may also identify genes that are induced by RNI but not involved in RNI resistance.
Other candidate RNI resistance genes have been identified based on biochemical hypotheses (108). These encode proteins with known functions that have been hypothesized to have an additional function of conferring resistance to RNI.
This field is young, and few of the candidates have been reported to meet all of the following criteria: the head-to-head comparison of the wild-type pathogen, the pathogen disrupted selectively in the gene of interest, and the latter pathogen complemented with the wild-type allele, tested for resistance not only to RNI delivered as chemical reagents, but also to RNI-producing phagocytes and experimental animals.
Classification of Candidate RNI Resistance Gene Products by Presumptive Mechanism of Action. (See Table 6, published as supplemental data on the PNAS web site, www.pnas.org.)
(i) Interference with production or uptake of RNI. By reducing the level of superoxide, SODs decrease the extent to which superoxide combines with nitric oxide to form a product, peroxynitrite, that is more bactericidal than either of its precursors. In this indirect sense, periplasmic Cu, Zn-SODs help confer RNI resistance (68). In E. coli (69) but not in S. typhimurium (70), ˙NO can activate the SoxR transcription factor that induces the soxS regulon controlling SOD expression (71). The soxRS system is not operative in Mycobacteria. The ability of S-nitrosoglutathione to halt the replication of S. typhimurium is markedly reduced by a mutation in dpp, which encodes a dipeptide permease (72). Apparently, Salmonella prepare this RNI for import by relieving it of its γ-glutamyl residue through the extracellular action of γ-glutamyltranspeptidase. Remnant S-nitrosocysteinylglycine must then be taken up by the bacterium to exert its cytostatic action, implicating an intracellular target. In this sense, a defective allele of dpp or a suppressor of dpp could be considered an RNI resistance gene. Although no such genes are known to contribute to salmonellosis, the phenotype of the dpp null strain has been informative.
(ii) Mechanisms likely to involve conversion of RNI to less toxic forms. S. typhimurium's OxyR, a cysteine-dependent transcription factor, is activated by intramolecular disulfide bond formation upon oxidation by hydrogen peroxide. Oxidized OxyR induces a stress regulon that includes catalase and alkylhydroperoxide reductase (73). When glutathione is depleted (73), S-nitrosothiols can also activate OxyR in association with its S-nitrosylation (69).
OxyR contributes to RNI resistance in E. coli (69), most likely because it represents one route to the induction of alkylhydroperoxide reductase (Ahp). However, the small subunit of Ahp (AhpC) is expressed in many organisms that lack OxyR, such as M. tuberculosis (57). Indeed, AhpC homologs appear to be more widely distributed than SOD and catalase, and much more widely distributed than the large subunit of Ahp encoded by ahpF (74). ahpC is now regarded as the founding member of the ubiquitous peroxiredoxin family (74). Several members of this family have been shown to reduce hydrogen peroxide or alkylperoxides. It was recently discovered that peroxiredoxins serve also in protection against RNI. A strain of S. typhimurium selectively deficient in ahpC was markedly more susceptible than the wild type to killing by RNI (57). The ahpC homolog from M. tuberculosis complemented the defect as well as ahpC from S. typhimurium itself (57). Expression of ahpC from M. tuberculosis conferred marked resistance to S-nitrosoglutathione, nitrite, and products of NOS2, and even protected transfected mammalian cells from RNI generated by NOS2 (57). Peroxiredoxins purified from S. typhimurium, M. tuberculosis, and H. pylori converted peroxynitrite to nitrite fast enough (second order rate constant, ≈1.5 × 10−6·M·s−1) to forestall oxidative destruction of bystander DNA. The catalytic mechanism involved reversible oxidation of an active-site cysteine to the sulfenic acid, followed by intermonomeric disulfide bond formation and reduction back to the sulfhydryl by a flavoprotein and NADH or by thioredoxin, thioredoxin reductase, and NADPH (108).
In E. coli, S. typhimurium, and M. tuberculosis, RNI induce expression of a flavohemoglobin encoded by hmp (66, 77, 78, 79, 80 and 81). Deletion mutants of hmp in S. typhimurium and E. coli were more susceptible than the wild type in vitro to ˙NO and S-nitrosothiols (77, 81). A mechanism for hmp's protective role against
S-nitrosothiols was suggested by the demonstration of an ˙NO dioxygenase activity of the E. coli flavohemoglobin that required NADPH, FAD, and O2 (66, 79). Expression of hmp also protected E. coli from nitrosative stress under anaerobic conditions. In this case, the mechanism appears to involve formation of a nitrosyl-heme intermediate that oxidizes NADH, converting ˙NO to N2O (82). Special interest attaches to the flavohemoglobin: like AhpC, it is widely expressed and enzymatically catabolizes preformed RNI; and like SOD, it may help prevent formation of peroxynitrite by drawing off a precursor.
Glucose 6-phosphate dehydrogenase, encoded by zwf, initiates the hexose monophosphate shunt to generate NADPH. S. typhimurium disrupted in zwf were hypersusceptible to S-nitrosoglutathione. The mutant bacteria regained partial virulence in mice whose NOS2 was inhibited (83). Dependence of salmonella on zwf for resistance to S-nitrosoglutathione may reflect the role of NADPH in the redox cycling of glutathione, thioredoxin, AhpC, and/or flavohemoglobin.
Low molecular weight thiols, such as glutathione and homocysteine, react with ˙NO. Mutation of gshB (26), which controls glutathione synthesis, and of metL (58), which controls the biosynthesis of homocysteine, each rendered S. typhimurium hypersusceptible to RNI. Virulence of the metL-disrupted organisms was reduced in mice and restored by inhibition of NOS (58). It is usually assumed that low molecular weight thiols protect cells from RNI by scavenging them. It is possible that low molecular weight thiols function additionally or alternatively to help reverse the oxidation or S-nitrosylation of other targets by RNI. Both mechanisms (scavenging and repairing) must be reconciled with the fact that the resulting S-nitrosothiols, such as S-nitrosoglutathione, can themselves be bacteriostatic (72) or bactericidal (57, 76). Presumably, formation of S-nitrosothiols in the course of scavenging RNI or repairing their lesions is only protective if the rate of formation of S-nitrosothiols does not exceed the pathogens' capacity to catabolize S-nitrosothiols. The rate-limiting step in the latter process remains to be identified (84). Thioredoxin can accelerate the decomposition of S-nitrosothiols (85), but whether this reaction protects bacteria from RNI is untested. Glutathione peroxidase can catabolize OONO−in vitro (86), and many small biological molecules can react with OONO− or its toxic products, including glutathione, cysteine, methionine, and tyrosine, but their contributions to microbial defense against OONO− are undefined.
(iii) Mechanisms likely to involve repair of RNI-dependent lesions. RNI acting in the service of host defense have DNA as their ultimate target. OONO−, ˙NO2, and higher oxides of nitrogen are mutagenic (87, 88 and 89). The DNA repair proteins RecB and RecC are important for survival of S. typhimurium in mice and macrophages (90). A recBC mutant of S. typhimurium was attenuated in both wild-type and NOS2-deficient mice, but regained partial virulence in phox-deficient mice and complete virulence in mice deficient in both NOS2 and phox (33). Thus, it appears that RNI help control S. typhimurium in part by promoting DNA damage, although to a lesser extent than ROI. Repair of oxidized proteins in RNI-stressed bacteria also has an enzymatic basis (G. St. John, J. Ruan, N. Brot, H. Weissbach, and C.N., unpublished work).
(iv) Mechanisms unknown. NOXR1 and NOXR3, each cloned from M. tuberculosis, confer enhanced resistance to RNI upon S. typhimurium, E. coli, and Mycobacterium smegmatis, but their mechanism of action is unknown (62, 63). Transformed M. smegmatis expressing NOXR1 were markedly more resistant than vector-transformed controls to being killed by macrophages, whether the macrophages were wild-type, NOS2-deficient, or phox-deficient. This was consistent with the evidence that NOXR1 afforded resistance to both RNI and ROI (62).
Perspective
Thirteen years ago conventional wisdom held that mammals could not produce RNI because RNI would be toxic. Now it is accepted that RNI production is widespread among eukaryotes in both nontoxic (signaling) and toxic formats. The toxicity is harnessed for host defense at a cost to the host. This state of affairs parallels the past history and current view of mammalian production of ROI. As for ROI, there are enzymes with the capacity to protect both pathogens and host cells from RNI. The genes encoding some RNI resistance enzymes are widely distributed, and a given cell may express several. The distribution and diversity of RNI resistance genes suggest that RNI exert evolutionary pressure and that different RNI pose distinct biochemical challenges. Agents that inhibit pathogens' RNI resistance mechanisms may improve immunity in those diseases for which RNI represent an important but imperfect element of host control.
Critical comments by R. Bryk, A. Ding, S. Ehrt, M. Fuortes, K. Hisert, D. Marciano, J. McKinney, and G. St. John are much appreciated. We thank C. Bogdan for sharing preprints. The National Heart, Lung, and Blood Institute supported this work.
1. Klebanoff, S. J. ( 1999), in Inflammation, eds. Gallin, J. I., Snyderman, R., Fearon, D. T., Haynes, B. F. & Nathan, C. (Lippincott, Philadelphia), pp. 721–768.
2. Nathan, C. ( 1992) FASEB J. 6, 3051–3064.
3. Ischiropoulos, H., Zhu, L. & Beckman, J. S. ( 1992) Arch. Biochem. Biophys. 298, 446–451.
4. Zingarelli, B., O'Connor, M., Wong, H., Salzman, A. L. & Szabo, C. ( 1996) J. Immunol. 156, 350–358.
5. Xia, Y. & Zweier, J. L. ( 1997) Proc. Natl. Acad. Sci. USA 94, 6954–6958.
6. Benjamin, N. & Dykhuizen, R. ( 1999)in Nitric Oxide and Infection, ed. Fang, F. C. (Kluwer/Plenum, New York), pp. 215–230.
7. Delledonne, M., Xia, Y., Dixon, R. A. & Lamb, C. ( 1998) Nature ( London) 394, 585–588.
8. Durner, J., Wendehenne, D. & Klessig, D. F. ( 1998) Proc. Natl. Acad. Sci. USA 95, 10328–10333.
9. Bolwell, G. ( 1999) Curr. Opin. Plant Biol. 2, 287–294.
10. Durner, J. & Klessig, D. F. ( 1999) Curr. Opin. Plant Biol. 2, 369–374.
11. Shiloh, M. U. & Nathan, C. ( 1999) in Phagocytosis: The Host, ed. Gordon, S. (JAI Press, Greenwich, CT), Vol. 5, pp. 407–439.
12. Miklos, G. L. & Rubin, G. M. ( 1996) Cell 86, 521–529.
13. Ashburner, M., Misra, S., Roote, J., Lewis, S. E., Blazej, R., Davis, T., Doyle, C., Galle, R., George, R., Harris, N., et al. ( 1999) Genetics 153, 179–219.
14. Elsbach, P., Weiss, J. & Levy, O. ( 1999) in Inflammation, ed. Gallin, J. I., Snyderman, R., Fearon, D. T., Haynes, B. F. & Nathan, C. (Lippincott, Philadelphia), pp. 801–818.
15. Campanelli, D., Melchior, M., Fu, Y., Nakata, M., Shuman, H., Nathan, C. & Gabay, J. E. ( 1990) J. Exp. Med. 172, 1709–1715.
16. Belaaouaj, A., McCarthy, R., Baumann, M., Gao, Z., Ley, T. J., Abraham, S. N. & Shapiro, S. D. ( 1998) Nat. Med. 4, 615–618.
17. Aratani, Y., Koyama, H., Nyui, S., Suzuki, K., Kura, F. & Maeda, N. ( 1999) Infect. Immun. 67, 1828–1836.
18. Jackson, S. H., Gallin, J. I. & Holland, S. M. ( 1995) J. Exp. Med. 182, 751–758.
19. Pollock, J. D., Williams, D. A., Gifford, M. A., Li, L. L., Du, X., Fisherman, J., Orkin, S. H., Doerschuk, C. M. & Dinauer, M. C. ( 1995) Nat. Genet. 9, 202–209.
20. Nathan, C. ( 1997) J. Clin. Invest. 100, 2417–2423.
21. Bogdan, C., Rollinghoff, M. & Diefenbach, A. ( 2000) Curr. Opin. Immunol. 12, 64–76.
22. Karupiah, G., Chen, J. H., Mahalingam, S., Nathan, C. F. & MacMicking, J. D. ( 1998) J. Exp. Med. 188, 1541–1546.
23. Gomes, M. S., Florido, M., Pais, T. F. & Appelberg, R. ( 1999) J. Immunol. 162, 6734–6739.
24. Stuehr, D. J., Fasehun, O. A., Kwon, N. S., Gross, S. S., Gonzalez, J. A., Levi, R. & Nathan, C. F. ( 1991) FASEB J. 5, 98–103.
25. MacMicking, J., Xie, Q. W. & Nathan, C. ( 1997) Annu. Rev. Immunol. 15, 323–350.
26. DeGroote, M. A. & Fang, F. C. ( 1999) in Nitric Oxide and Infection, ed. Fang, F. C. (Kluwer/Plenum, New York), pp. 231–264.
27. Lander, H. M., Hajjar, D. P., Hempstead, B. L., Mirza, U. A., Chait, B. T., Campbell, S. & Quilliam, L. A. ( 1997) J. Biol. Chem. 272, 4323–4326.
28. Diefenbach, A., Schindler, H., Rollinghoff, M., Yokoyama, W. M. & Bogdan, C. ( 1999) Science 284, 951–955.
29. Mannick, J. B., Hausladen, A., Liu, L., Hess, D. T., Zeng, M., Miao, Q. X., Kane, L. S., Gow, A. J. & Stamler, J. S. ( 1999) Science 284, 651–654.
30. Hierholzer, C., Harbrecht, B., Menezes, J. M., Kane, J., MacMicking, J., Nathan, C. F., Peitzman, A. B., Billiar, T. R. & Tweardy, D. J. ( 1998) J. Exp. Med. 187, 917–928.
31. Hmadcha, A., Bedoya, F. J., Sobrino, F. & Pintado, E. ( 1999) J. Exp. Med. 190, 1595–1603.
32. Brune, B., von Knethen, A. & Sandau, K. B. ( 1998) Eur. J. Pharmacol. 351, 261–272.
33. Shiloh, M. U., MacMicking, J. D., Nicholson, S., Brause, J. E., Potter, S., Marino, M., Fang, F., Dinauer, M. & Nathan, C. ( 1999) Immunity 10, 29–38.
34. Rotrosen, D. & Gallin, J. I. ( 1987) Annu. Rev. Immunol. 5, 127–150.
35. Lehrer, R. I. & Cline, M. J. ( 1969) J. Clin. Invest. 48, 1478–1488.
36. Weinberg, J. B. ( 1998) Mol. Med. 4, 557–591.
37. Facchetti, F., Vermi, W., Fiorentini, S., Chilosi, M., Caruso, A., Duse, M., Notarangelo, L. D. & Badolato, R. ( 1999) Am. J. Pathol. 154, 145–152.
38. Evans, T. J., Buttery, L. D., Carpenter, A., Springall, D. R., Polak, J. M. & Cohen, J. ( 1996) Proc. Natl. Acad. Sci. USA 93, 9553–9558.
39. Wheeler, M. A., Smith, S. D., Garcia-Cardena, G., Nathan, C. F., Weiss, R. M. & Sessa, W. C. ( 1997) J. Clin. Invest. 99, 110–116.
40. Vouldoukis, I., Riveros-Moreno, V., Dugas, B., Ouaaz, F., Becherel, P., Debre, P., Moncada, S. & Mossalayi, M. D. ( 1995) Proc. Natl. Acad. Sci. USA 92, 7804–7808.
41. Nozaki, Y., Hasegawa, Y., Ichiyama, S., Nakashima, I. & Shimokata, K. ( 1997) Infect. Immun. 65, 3644–3647.
42. Long, R., Light, B. & Talbot, J. A. ( 1999) Antimicrob. Agents Chemother. 43, 403–405.
43. Yu, K., Mitchell, C., Xing, Y., Magliozzo, R. S., Bloom, B. R. & Chan, J. ( 1999) Tuber. Lung Dis. 79, 191–198.
44. Chan, J. & Flynn, J. ( 1999) in Nitric Oxide and Infection, ed. Fang, F. C. (Kluwer/Plenum, New York), pp. 281–310.
45. MacMicking, J. D., North, R. J., La Course, R., Mudgett, J. S., Shah, S. K. & Nathan, C. F. ( 1997) Proc. Natl. Acad. Sci. USA 94, 5243–5248.
46. Nicholson, S., Bonecini-Almeida, M. d. G., Lapa e Silva, J. R., Nathan, C., Xie, Q. W., Mumford, R., Weidner, J. R., Calaycay, J., Geng, J., Boechat, N., et al. ( 1996) J. Exp. Med. 183, 2293–2302.
47. Wang, C. H., Liu, C. Y., Lin, H. C., Yu, C. T., Chung, K. F. & Kuo, H. P. ( 1998) Eur. Respir. J. 11, 809–815.
48. Rich, E. A., Torres, M., Sada, E., Finegan, C. K., Hamilton, B. D. & Toossi, Z. ( 1997) Tuber. Lung Dis. 78, 247–255.
49. Adams, L. B., Dinauer, M. C., Morgenstern, D. E. & Krahenbuhl, J. L. ( 1997) Tuber. Lung Dis. 78, 237–246.
50. Cooper, A. M., Segal, B. H., Frank, A. A., Holland, S. M. & Orme, I. M. ( 2000) Infect. Immun. 68, 1231–1234.
51. Patel, M. P. & Blanchard, J. S. ( 1999) Biochemistry 38, 11827–11833.
52. O' Donnell-Tormey, J., Nathan, C. F., Lanks, K., DeBoer, C. J. & de la Harpe, J. ( 1987) J. Exp. Med. 165, 500–514.
53. Fang, F. C., DeGroote, M. A., Foster, J. W., Baumler, A. J., Ochsner, U., Testerman, T., Bearson, S., Giard, J., Xu, Y., Campbell, G. & Laessig, T. ( 1999) Proc. Natl. Acac. Sci. USA 96, 7502–7507.
54. Jin, D.-Y. & Jeang, K-T. ( 2000) in Antioxidation and Redox Regulation of Genes, eds. Sen, C. K., Sies, H. & Baeuerle, P. A. (Academic, New York), pp. 381–407.
55. Storz, G., Jacobson, F. S., Tartaglia, L. A., Morgan, R. W., Silveira, L. A. & Ames, B. N. ( 1989) J. Bacteriol. 171, 2049–2055.
56. Higuchi, M., Yamamoto, Y., Poole, L. B., Shimada, M., Sato, Y., Takahashi, N. & Kamio, Y. ( 1999) J. Bacteriol. 181, 5940–5947.
57. Chen, L., Xie, Q. W. & Nathan, C. ( 1998) Mol. Cell 1, 795–805.
58. DeGroote, M. A., Testerman, T., Xu, Y., Stauffer, G. & Fang, F. C. ( 1996) Science 272, 414–417.
59. Hensel, M., Shea, J. E., Gleeson, C., Jones, M. D., Dalton, E. & Holden, D. W. ( 1995) Science 269, 400–403.
60. Camacho, L. R., Ensergueix, D., Perez, E., Gicquel, B. & Guilhot, C. ( 1999) Mol. Microbiol. 34, 257–267.
61. Cox, J. S., Chen, B., McNeil, M. & Jacobs Jr., W. R. ( 1999) Nature ( London) 402, 79–83.
62. Ehrt, S., Shiloh, M. U., Ruan, J., Choi, M., Gunzburg, S., Nathan, C., Xie, Q. & Riley, L. W. ( 1997) J. Exp. Med. 186, 1885–1896.
63. Ruan, J., St John, G., Ehrt, S., Riley, L. & Nathan, C. ( 1999) Infect. Immun. 67, 3276–3283.
64. Garbe, T. R., Hibler, N. S. & Deretic, V. ( 1999) Infect. Immun. 67, 460–465.
65. Yuan, Y., Crane, D., Simpson, R. M., Zhu, Y., Hickey, M. J., Sherman, D. R. & Barry, C. E., III ( 1998) Proc. Natl. Acad. Sci. USA 95, 9578–9583.
66. Hausladen, A., Gow, A. J. & Stamler, J. S. ( 1998) Proc. Natl. Acad. Sci. USA 95, 14100–14105.
67. Chiang, S. L., Mekalanos, J. J. & Holden, D. W. ( 1999) Annu. Rev. Microbiol. 53, 129–154.
68. DeGroote, M. A., Ochsner, U. A., Shiloh, M. U., Nathan, C., McCord, J. M., Dinauer, M. C., Libby, S. J., Vazquez-Torres, A., Xu, Y. & Fang, F. C. ( 1997) Proc. Natl. Acad. Sci. USA 94, 13997–4001.
69. Hausladen, A., Privalle, C. T., Keng, T., De Angelo, J. & Stamler, J. S. ( 1996) Cell 86, 719–729.
70. Fang, F. C., Vazquez-Torres, A. & Xu, Y. ( 1997) Infect. Immun. 65, 5371–5375.
71. Hidalgo, E., Ding, H. & Demple, B. ( 1997) Cell 88, 121–129.
72. DeGroote, M. A., Granger, D., Xu, Y., Campbell, G., Prince, R. & Fang, F. C. ( 1995) Proc. Natl. Acad. Sci. USA 92, 6399–6403.
73. Zheng, M., Aslund, F. & Storz, G. ( 1998) Science 279, 1718–1721.
74. Chae, H. Z., Robison, K., Poole, L. B., Church, G., Storz, G. & Rhee, S. G. ( 1994) Proc. Natl. Acad. Sci. USA 91, 7017–7021.
75. Tkalcevic, J., Novelli, M., Phylactides, M., Iredale, J. P., Segal, A. W. & Roes, J. ( 2000) Immunity 12, 201–210.
76. Shiloh, M. U., Ruan, J. & Nathan, C. ( 1997) Infect. Immun. 65, 3193–3198.
77. Crawford, M. J. & Goldberg, D. E. ( 1998) J. Biol. Chem. 273, 12543–12547.
78. Crawford, M. J. & Goldberg, D. E. ( 1998) J. Biol. Chem. 273, 34028–34032.
79. Gardner, P. R., Gardner, A. M., Martin, L. A. & Salzman, A. L. ( 1998) Proc. Natl. Acad. Sci. USA 95, 10378–10383.
80. Hu, Y., Butcher, P. D., Mangan, J. A., Rajandream, M. A. & Coates, A. R. ( 1999) J. Bacteriol. 181, 3486–3493.
81. Membrillo-Hernandez, J., Coopamah, M. D., Anjum, M. F., Stevanin, T. M., Kelly, A., Hughes, M. N. & Poole, R. K. ( 1999) J. Biol. Chem. 274, 748–754.
82. Kim, S. O., Orii, Y., Lloyd, D., Hughes, M. N. & Poole, R. K. ( 1999) FEBS Lett 445, 389–394.
83. Lundberg, B. E., Wolf, R. E., Jr., Dinauer, M. C., Xu, Y. & Fang, F. C. ( 1999) Infect. Immun. 67, 436–438.
84. Gaston, B. & Stamler, J. S. ( 1999) in Nitric Oxide and Infection, ed. Fang, F. C. (Kluwer/Plenum, New York), pp. 37–55.
85. Nikitovic, D. & Holmgren, A. ( 1996) J. Biol. Chem. 271, 19180–19185.
86. Briviba, K., Kissner, R., Koppenol, W. H. & Sies, H. ( 1998) Chem. Res. Toxicol. 11, 1398–1401.
87. Wink, D. A., Kasprzak, K. S., Maragos, C. M., Elespuru, R. K., Misra, M., Dunams, T. M., Cebula, T. A., Koch, W. H., Andrews, A. W., Allen, J. S., et al. ( 1991) Science 254, 1001–1003.
88. Juedes, M. J. & Wogan, G. N. ( 1996) Mutat. Res. 349, 51–61.
89. Tamir, S., Burney, S. & Tannenbaum, S. R. ( 1996) Chem. Res. Toxicol. 9, 821–827.
90. Buchmeier, N. A., Lipps, C. J., So, M. Y. & Heffron, F. ( 1993) Mol. Microbiol. 7, 933–936.
91. Ma, X. L., Gao, F., Liu, G.-L., Lopez, B. L., Christopher, T. A., Fukuto, J. M., Wink, D. A. & Feelisch, M. ( 1999) Proc. Nat. Acad. Sci. USA. 96, 14617–14622.
92. Radi, R., Beckman, J. S., Bush, K. M. & Freeman, B. A. ( 1991) J. Biol. Chem. 266, 4244–4250.
93. Doi, T., Ando, M., Akaike, T., Suga, M., Sato, K. & Maeda, H. ( 1993) Infect. Immun. 61, 1980–1989.
94. O'Brien, L., Carmichael, J., Lowrie, D. B. & Andrew, P. W. ( 1994) Infect. Immun. 62, 5187–5190.
95. Dalton, D. K., Pitts-Meek, S., Keshav, S., Figari, I. S., Bradley, A. & Stewart, T. A. ( 1993) Science 259, 1739–1742.
96. Flynn, J. L., Chan, J., Triebold, K. J., Dalton, D. K., Stewart, T. A. & Bloom, B. R. ( 1993) J. Exp. Med. 178, 2249–2254.
97. Huang, S., Hendriks, W., Althage, A., Hemmi, S., Bluethmann, H., Kamijo, R., Vilcek, J., Zinkernagel, R. M. & Aguet, M. ( 1993) Science 259, 1742–1745.
98. Flynn, J. L., Goldstein, M. M., Chan, J., Triebold, K. J., Pfeffer, K., Lowenstein, C. J., Schreiber, R., Mak, T. W. & Bloom, B. R. ( 1995) Immunity 2, 561–572.
99. Chan, J., Tian, Y., Tanaka, K. E., Tsang, M. S., Yu, K., Salgame, P., Carroll, D., Kress, Y., Teitelbaum, R. & Bloom, B. R. ( 1996) Proc. Natl. Acad. Sci. USA 93, 14857–14861.
100. Denis, M. ( 1991) Cell. Immunol. 132, 150–157.
101. Flesch, I. E. & Kaufmann, S. H. ( 1991) Infect. Immun. 59, 3213–3218.
102. Chan, J., Xing, Y., Magliozzo, R. S. & Bloom, B. R. ( 1992) J. Exp. Med. 175, 1111–1122.
103. Chan, J., Tanaka, K., Carroll, D., Flynn, J. & Bloom, B. R. ( 1995) Infect. Immun. 63, 736–740.
104. Flynn, J. L., Scanga, C. A., Tanaka, K. E. & Chan, J. ( 1998) J. Immunol. 160, 1796–1803.
105. Kwon, O. J. ( 1997) J. Korean Med. Sci. 12, 481–487.
106. Zhang, Z., Kolls, J. K., Oliver, P., Good, D., Schwarzenberger, P. O., Joshi, M. S., Ponthier, J. L. & Lancaster, J. R., Jr. ( 2000) J. Biol. Chem. 275, 15839–15844.
107. Marnett, L. J., Wright, T. L., Crews, B. C., Tannenbaum, S.R. & Morrow, J. D. ( 2000) J. Biol. Chem. 275, 13427–13430.
108. Bryk, R., Griffin, P. & Nathan, C. ( 2000) Nature ( London), in press.








