
Colloquium
Nitric oxide and salicylic acid signaling in plant defense
Daniel F. Klessig*†, Jörg Durner*‡, Robert Noad*§, Duroy A. Navarre*, David Wendehenne*¶, Dhirendra Kumar*, Jun Ma Zhou*, Jyoti Shah*∥, Shuqun Zhang*,**, Pradeep Kachroo*, Youssef Trifa*, Dominique Pontier††, Eric Lam††, and Herman Silva*‡‡
* Waksman Institute and Department of Molecular Biology and Biochemistry, Rutgers, The State University of New Jersey, 190 Frelinghuysen Road, Piscataway, NJ 08854-8020; and
†† Plant Science Department/AgBiotech Center, Rutgers, The State University of New Jersey, 59 Dudley Road, New Brunswick, NJ, 08901
Salicylic acid (SA) plays a critical signaling role in the activation of plant defense responses after pathogen attack. We have identified several potential components of the SA signaling pathway, including (i) the H2O2-scavenging enzymes catalase and ascorbate peroxidase, (ii) a high affinity SA-binding protein (SABP2), (iii) a SA-inducible protein kinase (SIPK), (iv) NPR1, an ankyrin repeat-containing protein that exhibits limited homology to IκBα and is required for SA signaling, and (v) members of the TGA/OBF family of bZIP transcription factors. These bZIP factors physically interact with NPR1 and bind the SA-responsive element in promoters of several defense genes, such as the pathogenesis-related 1 gene (PR-1). Recent studies have demonstrated that nitric oxide (NO) is another signal that activates defense responses after pathogen attack. NO has been shown to play a critical role in the activation of innate immune and inflammatory responses in animals. Increases in NO synthase (NOS)-like activity occurred in resistant but not susceptible tobacco after infection with tobacco mosaic virus. Here we demonstrate that this increase in activity participates in PR-1 gene induction. Two signaling molecules, cGMP and cyclic ADP ribose (cADPR), which function downstream of NO in animals, also appear to mediate plant defense gene activation (e.g., PR-1). Additionally, NO may activate PR-1 expression via an NO-dependent, cADPR-independent pathway. Several targets of NO in animals, including guanylate cyclase, aconitase, and mitogen-activated protein kinases (e.g., SIPK), are also modulated by NO in plants. Thus, at least portions of NO signaling pathways appear to be shared between plants and animals.
MAP kinases | Ca+2 cADP ribose | cGMP | disease resistance
D isease resistance in plants is usually associated with the activation of a wide variety of defense responses that serve to prevent pathogen replication and/or movement. In some plant-pathogen interactions, the ability of the host plant to recognize the pathogen and activate these responses is regulated in a gene-for-gene-specific manner by the direct or indirect interaction between the products of a plant resistance (R) gene and a pathogen avirulence (Avr) gene (1, 2). When either the plant or the pathogen lacks its cognate gene, activation of the plant's defense responses either fails to occur or is delayed sufficiently so that pathogen colonization ensues. In contrast to this race/cultivar-specific form of resistance, which is relatively rare, many plant species exhibit non-host resistance. Non-host resistance is characterized by the activation of many of the same defense responses as are associated with race/cultivar-specific resistance; however, it occurs in the absence of any known R/Avr gene combination.
One of the earliest responses activated after host plant recognition of an Avr protein or a non-host specific elicitor is the oxidative burst, in which levels of reactive oxygen species (ROS) rapidly increase (3, 4). Other rapid responses include the cross-linking of cell wall proteins, the activation of protein kinases, and the increased expression of various plant protectant and defense genes (5, 6). Some of these genes encode peroxidases, glutathione S-transferases, proteinase inhibitors, and various biosynthetic enzymes, such as phenylalanine ammonia lyase (PAL). PAL is the first enzyme in the phenylpropanoid pathway, which is involved in the synthesis of low molecular weight, antimicrobial compounds known as phytoalexins.
At somewhat later times, resistant plants often develop a hypersensitive response (HR), in which necrotic lesions form at the site(s) of pathogen or elicitor entry (5, 7). The localized cell death associated with the HR resembles animal programmed cell death, and it may help prevent the pathogen from spreading to uninfected tissues. Just before or concomitant with the appearance of a HR is the increased synthesis of several families of pathogenesis-related (PR) proteins in the inoculated leaves ( 8). Many of these proteins have been shown to exhibit antimicrobial activity either in vitro or in vivo. PR proteins are subsequently expressed in the uninoculated portions of the plant, concurrent with the development of a long lasting, broad-based resistance known as systemic acquired resistance (SAR) (9). Because of this correlation, increased PR gene expression is frequently used as a marker for SAR. Interestingly, although plants lack a circula-
This paper was presented at the National Academy of Sciences colloquium “Virulence and Defense in Host–Pathogen Interactions: Common Features Between Plants and Animals, ” held December 9–11, 1999, at the Arnold and Mabel Beckman Center in Irvine, CA.
Abbreviations: ADPRC, ADP-ribosyl cyclase; BTH, benzothiadiazole; cADPR, cyclic ADP ribose; CWD, cell wall-derived; GC, guanylate cyclase; HR, hypersensitive response; INA, 2,6-dichloroisonicotinic acid; IRP-1, iron regulatory protein 1; JA, jasmonic acid; MAP, mitogen-activated protein; NO, nitric oxide; NOS, NO synthase; NtACO1, Nicotiana tabacum aconitase 1; PAL, phenylalanine ammonia lyase; PR, pathogenesis-related; ROS, reactive oxygen species; SA, salicylic acid; SAR, systemic acquired resistance; SIPK, salicylic acid-induced protein kinase; TMV, tobacco mosaic virus; WIPK, wounding-induced protein kinase.
|
† |
To whom reprint requests should be addressed. E-mail: klessig@mbcl.rutgers.edu. |
|
‡ |
Present address: Institute of Biochemical Plant Pathology, GSF–National Research Center for Environment and Health, D-85764 Oberschleissheim, Germany. |
|
§ |
Present address: Natural Environment Research Council Institute of Virology and Environmental Microbiology, University of Oxford, Mansfield Road, Oxford OX1 3SR, U.K. |
|
¶ |
Present address: Biochimie, Biologie Cellulaire et Moleculaire des Interactions-Plantes/Microorganismes, Unite Mixte Institut National de la Recherche Agronomique-Universite de Bourgogne, Institut National de la Recherche Agronomique, BV 1540, 17 rue Sully, 21034 Dijon, France. |
|
∥ |
Present address: Division of Biology, Kansas State University, 303 Ackert Hall, Manhattan, KS, 66506. |
|
** |
Department of Biochemistry, University of Missouri, 117 Schweitzer Hall, Columbia, MO 65211. |
|
‡‡ |
Department of Biology, Faculty of Sciences, University of Chile, Casilla 653, Santiago, Chile. |
tory system and do not produce antibodies, SAR shares several common characteristics with the innate immune system of animals.
A substantial body of evidence indicates that salicylic acid (SA) is a critical signaling molecule in the pathway(s) leading to local and systemic disease resistance, as well as PR expression (10, 11). In addition, recent studies have demonstrated that ethylene and jasmonic acid (JA) mediate the activation of various defense responses and resistance to certain pathogens (12, 13). The relationship between the SA, ethylene, and JA signaling pathways is not well defined. SA has been shown to work synergistically with ethylene or the JA derivative methyl jasmonate to activate PR expression in tobacco and Arabidopsis (14, 15); however, other defense responses appear to be regulated by ethylene-and/or JA-dependent pathways that are independent of SA (16, 17).
Another signaling molecule that has been implicated in the activation of plant defenses is nitric oxide (NO). This compound has previously been shown to serve as a key redox-active signal for the activation of various mammalian defense responses, including the inflammatory and innate immune responses (18, 19). In contrast to the extensive studies during the past decade or more on NO's role in animal defense, only recently has NO's involvement in the plant defense response to pathogens been addressed (20). To date, NO's participation has been documented in at least three different plant-pathogen systems (21, 22). Current evidence suggests that there are many parallels between NO action in plants and animals.
In this paper, we will review some of our findings concerning NO-and SA-mediated signaling in plants. Particular emphasis will be placed on the results generated over the past few years. In addition, new evidence implicating NO and its second messenger cyclic ADP ribose (cADPR) in tobacco PR-1 gene activation is presented.
Results and Discussion
SA-Interacting Proteins in Tobacco. To clarify the mechanism(s) through which SA activates plant defense responses, we have sought to identify the effector(s) with which SA interacts. The first protein shown to reversibly bind SA was a catalase from tobacco, originally termed SABP, for SA binding protein (23). Catalases convert H2O2 to H2O and O2; this activity was inhibited by SA both in vitro and in vivo. Catalase activity was also inhibited by the synthetic SA functional analogs 2,6-dichloroisonicotinic acid (INA) and benzothiadiazole (BTH), as well as by various biologically active SA analogs, which are capable of inducing PR expression and enhancing disease resistance (23, 24 and 25). By contrast, biologically inactive analogs of SA and INA failed to inhibit catalase activity (23, 24). Interestingly, H2O2 and various prooxidants induced PR-1 expression in tobacco whereas several antioxidants suppressed the SA-, INA-, or BTH-mediated activation of this gene (23, 25). Thus, SA was proposed to activate PR expression by increasing the levels of H2O2 and other ROS, which could then serve as second messengers in the defense signaling pathway.
Analyses of ascorbate peroxidase (26), the other major H2O2-degrading enzyme in plant cells, provided additional support for this hypothesis. SA, INA, and BTH were all found to inhibit ascorbate peroxidase activity whereas inactive analogs of SA did not (25, 26). However, different studies from both our lab (27, 28) and others (29, 30 and 31) have suggested that H2O2 functions upstream rather than, or in addition to, downstream of SA in the defense signaling pathway. Thus, the role played by SA-mediated catalase inhibition and elevated ROS levels in the induction of defense responses is currently unclear.
Another mechanism through which SA-mediated inhibition of catalase and ascorbate peroxidase might activate defenses is via the generation of SA free radicals. SA has been shown to inhibit catalase by serving as a one-electron donating substrate (32). In this process, SA is converted into a free radical, which could then initiate lipid peroxidation. SA and its biologically active analogs have been shown to induce lipid peroxidation in tobacco suspension cells (33). Moreover, exogenously applied lipid peroxides (the products of lipid peroxidation) induce PR expression in suspension cells.
In addition to catalase, a second SA-binding protein (SABP2) has been identified. This soluble, low molecular weight protein exhibits an affinity for SA (Kd = 90 nM) that is approximately 150 times higher than that of catalase (34). Strikingly, SABP2's affinity for BTH is 15-fold higher than its affinity for SA; this is consistent with BTH's greater efficacy at inducing PR expression and SAR.
Mitogen-Activated Protein (MAP) Kinases and the Activation of Defense Responses in Tobacco. Protein phosphorylation and/or dephosphorylation are known to play important roles in many signal transduction pathways. Based on studies using protein kinase and phosphatase inhibitors, at least two phosphoproteins were implicated in the SA signaling pathway of tobacco (35). One of these proteins appears to work upstream of SA whereas the other works downstream. A 48-kDa kinase that is rapidly and transiently activated by SA treatment was subsequently identified in tobacco suspension cells (36). Sequence analysis of a cDNA clone encoding this SA-induced protein kinase (SIPK) revealed that it is a member of the MAP kinase family. In addition to SA, SIPK was shown to be activated at the enzyme level by various pathogen-associated stimuli, including two elicitins and a cell wall-derived (CWD) carbohydrate elicitor from Phytophthora spp. (37), and bacterial harpin (ref.38; Table 1). SIPK also was activated in a gene-for-gene specific manner by the Avr9 peptide from Cladosporium fulvum in transgenic tobacco expressing the cognate resistance gene Cf-9 (39) and in tobacco mosaic virus (TMV)-infected Xanthi nc carrying the cognate resistance gene N (40). Additionally, SIPK was activated to high levels by wounding (41). Thus, this kinase appears to be involved in multiple signaling pathways.
The discovery that SIPK encodes a 48-kDa kinase that is strongly activated by wounding raised questions as to the identity of the wounding-induced protein kinase (WIPK) gene product. Transcripts of WIPK were shown to accumulate after wounding stress (42). This observation led to the hypothesis that WIPK encoded a wounding-activated kinase of approximately 46 kDa. However, through the use of a WIPK-specific antibody, it was demonstrated that wounding induced little, if any, increases in WIPK protein and enzymatic activity levels, in contrast to a significant rise in its transcript level (41). Rather, WIPK was found to be activated by most of the pathogen-associated stimuli that activate SIPK, including the CWD elicitor, both elicitins, TMV infection, and the presence of Avr9 peptide in transgenic tobacco expressing Cf-9 (Table 1). These studies also revealed that WIPK activation is regulated at multiple levels. Some stimuli, such as the CWD elicitor, rapidly and transiently induced WIPK enzymatic activity to only low levels whereas the slower and more dramatic increase in its mRNA and protein levels did not result in higher enzymatic activity (43). On the other hand, the dramatic rise in WIPK activity after TMV infection or elicitin treatment was preceded by and required both increases in WIPK transcription and translation, as well as posttranslational phosphorylation (40, 43). By contrast, activation of SIPK, like that for MAP kinases in yeast and animals, was regulated strictly at the posttranslational level by dual phosphorylation of threonine and tyrosine residues, presumably in the conserved TEY motif located between subdomains VII and VIII of the kinase catalytic domain.
Table 1. Activation of SIPK and WIPK at multiple levels by various stimuli
|
SIPK |
WIPK |
|||||
|
Stimuli |
mRNA ⇑ |
Protein ⇑ |
Activity ⇑* |
mRNA ⇑ |
Protein ⇑ |
Activity ⇑* |
|
−∥ |
− |
++ |
− |
− |
− |
|
|
− |
− |
++ |
++ |
++ |
−/+** |
|
|
− |
− |
++ |
++ |
++ |
++ |
|
|
− |
− |
++ |
NT |
NT |
NT |
|
|
TMV†† |
− |
− |
++ |
++ |
++ |
++ |
|
NT |
− |
++ |
++ |
− |
++/+ |
|
|
Wounding†† |
− |
− |
++ |
+ |
−/+ |
−/+** |
|
* Kinase activity was measured by immunoselecting WIPK or SIPK with the WIPK-specific antibody Ab-p44N or the SIPK-specific antibody Ab-p48N and then performing a kinase assay with MBP 0.1 mg/ml and 10 micromolar ATP with 1 µCi of [γ-32P]ATP as substrates. † The cell wall-derived (CWD) carbohydrate elicitor was from Phytophthora parasitica. ‡ Purified elicitins were from Phytophthora parasitica (parasiticein) and Phytophthora cryptogea (cryptogein). § Purified harpin from Erwinia amylovora (38); or infection with Pseudomonas syringae pv springae strain 61 with wild-type harpin but not with strain Pss61 hrpH−. ¶ Recipient was transgenic tobacco expressing the Cf-9 gene from tomato that confers resistance to Cladosporium fulvum expressing Avr9 (39). ∥ ++, high level activation; +, low level activation; ±, little or no activation; −, no activation; NT, not tested. ** Weak, transient activation that does not correlate with increases in WIPK mRNA and protein. †† Tested in planta. ‡‡ Tested in suspension cell culture. |
||||||
Arabidopsis Mutants in the SA Signal Transduction Pathway. To identify components involved in the SA signaling pathway, Arabidopsis thaliana mutants that exhibit altered disease resistance have been generated and characterized by several laboratories. Many of these mutants can be divided into two classes. Those in the first class constitutively express PR genes and accumulate elevated levels of SA in the absence of either pathogen infection or an elicitor and exhibit enhanced disease resistance (6, 10). A subset of these mutants, such as cep (constitutive expression of PR-1 gene) (44), also form spontaneous HR-like lesions. Because all of the constitutive SAR mutants contain high levels of SA, the corresponding genes likely encode components that function upstream of SA. Alternatively, they may encode components of a separate pathway(s) that impinges on the SA pathway at or upstream of SA.
In comparison, mutants belonging to the second class fail to express PR genes and/or exhibit enhanced disease susceptibility despite treatment with SA or its analogs. Several allelic mutants belonging to this class have been identified, including npr1 (non-expresser of PR genes) (45, 46), nim1 (noninducible immunity) (47), and sai1 (SA-insensitive) (48). The inability of these mutants to respond to SA and its synthetic functional analogs suggests that the wild-type protein functions between SA and PR expression in the defense pathway. Recently, NPR1 was cloned and shown to encode a 65-kDa protein that bears limited homology to IκBα and contains ankyrin repeats (49, 50). These repeats, which often are involved in protein-protein interactions, appear to be critical for NPR1 function; they are altered in many of the npr1 mutants (48, 49 and 50).
Seven suppressors of the SA-insensitive (ssi) phenotype of sai1 (renamed npr1-5) have been identified and placed into four complementation groups. Mutations belonging to each of these groups have been mapped to distinct chromosomal locations. For example, ssi1 maps to a 68-kb region on the long arm of chromosome 4, ssi2 maps to a 40-kb segment of chromosome 2, and ssi3 and ssi4 map to chromosome 5. Analysis of the ssi1 mutant suggests that the wild-type protein functions as a switch that modulates cross-talk between the SA/NPR1-mediated pathway for PR-1 induction and the JA-/ethylene-mediated pathway leading to defensin gene (PDF1.2) expression (51).
NPR1-Interacting Proteins. As a complementary approach to identifying SA signaling components, a yeast two-hybrid screen was used to identify proteins that directly interact with NPR1. Several NPR1-interacting proteins (NIPs) were identified; two of them are members of the TGA/OBF family of bZIP transcription factors (52). This family of proteins has previously been implicated in the induction of PR-1 and other SA-responsive genes. To extend these findings, the ability of six TGA/OBF family members to bind NPR1 was assessed. TGA2 and TGA3 exhibited the greatest affinity for NPR1 whereas TGA5 and TGA6 showed weaker affinity and that of TGA1 and TGA4 was very low to undetectable (52). The ankyrin repeat region of NPR1 appears to be critical for binding TGA2 and TGA3 because four distinct point mutations in this region disrupted the interaction between these proteins. These mutations have also been shown to abolish SA signaling in planta. Combined with the discovery that TGA2 and TGA3 bind the TGACG element of the PR-1 promoter, which is required for SA and INA inducibility in planta (53), these results strongly argue that members of the TGA/OBF transcription family transduce the SA signal from NPR1 to the activation of SA-inducible genes. Recent reports from two other laboratories have also shown that several members of this transcription factor family can bind NPR1 (54, 55).
NO Signaling in Plant Defense. ROS are believed to perform multiple roles during plant defense responses to microbial attack by acting directly in the initial defense and, possibly, as cellular signaling molecules. Because ROS-associated plant defense responses have similarities to the inflammatory and immune responses of animals, where NO is an important signaling molecule, we examined whether NO serves a similar function in plants.
To address whether a mammalian type nitric oxide synthase (NOS)-like activity was elevated during plant-pathogen interactions, we took advantage of the reversible, high temperature inhibition of TMV-induced defense responses in tobacco. When Xanthi nc (NN) plants are inoculated with TMV and incubated
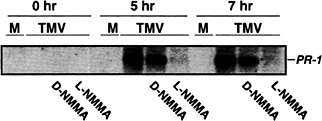
Fig. 1. The effect of a NOS inhibitor on TMV induction of PR-1 gene expression. Xanthi nc (NN) plants were mock inoculated (M) or inoculated with the U1 strain of TMV (1 µg/ml) and then were maintained for 48 h at 32°C in a growth chamber. One hour before shifting plants from 32°C to 22°C, the intercellular spaces of the inoculated leaves were infiltrated with either H2O, the active form (5 mM NG-monomethyl-L-arginine monoacetate, L -NMMA), or inactive form (5 mM D -NMMA) of an NOS inhibitor. Samples were taken for RNA preparation at 0, 5, 7 h after the shift to 22°C. A portion (10 µg) of total RNA was fractionated on a formaldehyde-agarose gel and was subjected to Northern blot analysis using standard protocols (74). The tobacco acidic PR-1a cDNA clone was used as a probe.
at temperatures >28°C, they fail to (i) produce elevated levels of SA and PR proteins, (ii) develop an HR, and (iii) restrict virus replication and spread. However, when the infected plants are moved to lower temperatures (22°C), all of the above defense responses are rapidly and strongly induced (56). We found that by 6 h after shifting TMV-infected Xanthi nc (NN) to 22°C, NOS-like activity rose ≈5-fold whereas little or no change in activity was detected in mock-infected Xanthi nc (NN) or in TMV-infected susceptible Xanthi (nn) (22).
To determine whether this increase in NOS-like activity was required for TMV-induced PR-1 gene expression, similar temperature shift experiments were carried out, except that some of the TMV-inoculated leaves were injected with the NOS inhibitor, NG-monomethyl-L -arginine monoacetate, 1 h before the plants were shifted from 32 °C to 22°C. NG-monomethyl-L -arginine monoacetate repressed expression of PR-1 (Fig. 1). In contrast, the inactive D form of this inhibitor had little or no effect, thus establishing the specificity of the inhibition. Expression of PAL, an early defense gene, was also monitored. Suppression of its expression was more variable and less complete than that observed for PR-1 (data not shown). This result suggests that PAL activation by TMV may use a NO-independent pathway as well as a NO-dependent pathway.
The suppression of TMV-induced PR-1 (and partial suppression of PAL) expression by NG-monomethyl-L -arginine monoacetate argues that NO participates in activating these genes after infection. To further establish NO's involvement, the levels of NO were artificially elevated, and the expression of PR-1 and PAL genes was monitored. Injection of 1 or 5 units of recombinant rat neuronal NOS, together with its cofactors and substrate, into the intercellular spaces of tobacco leaves induced PR-1 expression (Fig. 2; ref.22). Tobacco suspension cells also responded to exogenous NO, provided as an NO donor (e.g., S-nitroso-N-acetyl-DL -penicillamine or S-nitroso-L-glutathione), by activating PR-1 and PAL (22).
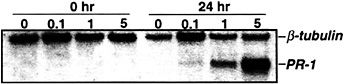
Fig. 2. Induction of PR-1 gene expression with various concentrations of NOS. Five- to eight-week-old Xanthi nc (NN) plants were injected with 40 mM Hepes (pH 7.4) containing all of the cofactors and substrate of NOS (22) and different concentrations of recombinant rat neuronal NOS (0–5 units per ml; Alexis Biochem, San Diego). Samples were taken for RNA preparation immediately after (0 h) or 24 h after injection; the RNA was subjected to Northern blot analysis as described in Fig. 1, with the addition that β-tubulin was used as an internal control for gel loading and transfer.
NO's Targets and Downstream Messengers. The NO signaling pathway leading to defense gene expression in tobacco appears to be similar to that defined in animals. In animals, NO frequently acts through a cGMP-dependent pathway (57). In this pathway, NO posttranslationally activates guanylate cyclase (GC), which leads to a transient increase in the second messenger cGMP (Fig. 3). In some types of animal cells, cGMP in turn activates ADPribosyl cyclase (ADPRC), through a cGMP-dependent protein kinase. The resulting elevated levels of another second messenger, cyclic ADP ribose (cADPR), then stimulate Ca+2 release into the cytoplasm through ruthenium red-sensitive ion channels called ryanodine receptors. In tobacco, NO induced a transient but dramatic increase in cGMP levels (22). In addition, a membrane-permeable analogue of cGMP activated PAL expression whereas NO induction of PAL was suppressed by GC inhibitors, 6-anilino-5,8-quinolinedione (LY83583), and 1H- (1, 2, 4)-oxadiazole[4,3-α]quinoxalin-1-one (ODQ). cADPR also activated PR-1 and PAL expression. This activation was suppressed by the Ca2+ channel inhibitor ruthenium red, arguing that Ca2+ participates downstream of cADPR in the signal transduction pathway.
Although the above results demonstrate that cADPR can activate PR-1 and PAL expression, they did not establish whether NO acts through cADPR to induce these genes. To address this issue, expression of PR-1 was monitored after injecting tobacco leaves with recombinant NOS or recombinant NOS plus the cADPR antagonist 8-bromo-cADPR. 8-bromo-cADPR suppressed PR-1 activation (Fig. 4), suggesting that cADPR mediates NO induction of PR-1. However, because this suppression was variable and never complete, we cannot conclude that NO

Fig. 3. Proposed SA- and NO-mediated pathway for activation of certain defense genes and elevation of intracellular free Fe2+. Key signaling molecules in the cascade include NO, cGMP, cADPR, Ca2+, and SA. Important enzymes are NOS, GC, cGMP-dependent protein kinase, and ADPRC. PAL and PR-1 are two important defense genes activated by pathogens. Activation of NOS by TMV infection increases NO levels, which activate GC and lead to elevated cGMP levels. cGMP activates ADPRC, via a cGMP-dependent protein kinase, which results in rising cADPR levels. cADPR activates ruthenium red (RR)-sensitive Ca2+ ion channels, which leads to higher cytosolic Ca2+ levels. Ca2+ induces SA biosynthesis, perhaps by activation of PAL gene expression. The SA-induced MAP kinase, SIPK, may play a role in SA signaling through NPR1. NPR1 transmits the SA signal to the PR-1 gene via its interaction with members of the TGA/OBF family of transcription factors, which bind the SA-responsive TGACG element of the PR-1 promoter. In a separate branch of the pathway, NO inhibits cytosolic aconitase and may convert it into a iron regulatory protein (IRP), thereby facilitating an increase in intracellular free Fe2+. GSNO and SNAP are NO donors whereas PTIO is an NO scavenger.
activation of PR-1 is exclusively via a cGMP/cADPR-dependent pathway. The variable and less than complete nature of this inhibition by 8-bromo-cADPR may in fact suggest that NO activation of PR-1 occurs through more than one pathway. For example, in mammalian systems there is evidence that NO directly regulates Ca2+ channels via S-nitrosylation (58), which would be independent of cGMP and cADPR. Furthermore, cGMP can also act independently of cADPR via activation of certain cGMP-dependent kinases not involved in cADPR signaling (59).
Because PR-1 expression is often regulated by SA, the relationship between the NO and SA signaling pathways was assessed. NO treatment was shown to induce substantial increases in the level of total SA (22). Moreover, SA was critical for the NO- and cADPR-mediated activation of PR-1 expression; neither signaling molecule induced this gene in SA-deficient transgenic tobacco expressing the bacterial nahG gene. By contrast, the NO- and cADPR-mediated induction of PAL was unaffected in these plants. Thus, NO and cADPR appear to regulate the expression of various defense genes through either SA-dependent or SA-independent pathways.
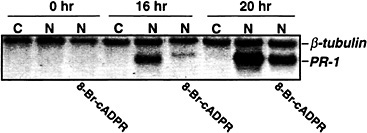
Fig. 4. A cADPR antagonist suppresses the NO induction of PR-1 gene expression. Five- to eight-week-old Xanthi nc (NN) plants were injected with 40 mM Hepes (pH 7.4) containing all of the cofactors and substrate of NOS (22) either without NOS as a negative control (C) or with NOS (1 unit/ml) as a positive control (N). A second set of plants were co-injected with NOS, cofactors and substrate plus 200 µM of the cADPR antagonist 8-bromo-cADPR. Samples were taken for RNA preparation at 0, 16, and 20 h after injection; the RNA was subjected to Northern blot analysis as described in Fig. 2.
In addition to GC, the citric acid cycle enzyme aconitase is a major direct target of NO in animals (60). NO inhibits mammalian mitochondrial and cytosolic aconitases. In addition, it converts the cytosolic aconitase into an mRNA binding protein known as IRP-1 (iron regulatory protein) (61). IRP-1 binds specific sites, termed iron-responsive elements, on the transcripts of genes involved in iron metabolism and/or energy metabolism, such as the transferrin receptor. Binding of IRP-1 to these sites, which have hairpin-like structures, alters the stability or translatability of the corresponding transcripts. Thus, IRP-1 plays a major role in regulating free iron concentrations in animal cells. It is through this mechanism that NO stimulates increased levels of intracellular free iron, but the physiological relevance of the NO-mediated IRP-1 activation remains unclear.
We have recently demonstrated that tobacco cytosolic and mitochondrial aconitase activities also are inhibited by NO (62). In addition, we have cloned a tobacco cytosolic aconitase gene (NtACO1). Sequence analysis revealed that the encoded protein shares 76% similarity with the human IRP-1. Moreover, several residues that are critical for IRP-1's ability to bind mRNA are conserved in NtACO1. These results suggest that NtACO1 might perform an analogous function to IRP-1 in plants. The potential of NtACO1 to increase free iron levels in response to NO could provide a defensive function after pathogen attack. In the presence of ROS, free iron promotes oxidative damage via the Fenton reaction (63, 64). Thus, NO-mediated increases in iron may help create a lethal environment that would contribute to the HR, as well as kill the pathogen (62).
NO has also been shown to indirectly modulate the activity of MAP kinases in mammalian tumor cells and neurons (65, 66). Thus, NO's effect on the pathogen-activated tobacco MAP kinases SIPK and WIPK was assessed (67). Two NO donors and recombinant NOS were shown to cause a transient activation of SIPK. This activation appears to be mediated via a SA-dependent pathway because SIPK enzymatic activity was not induced in NOS-treated transgenic tobacco plants expressing the nahG gene. By contrast, neither recombinant NOS nor the NO donors caused substantial activation of WIPK. Taken together,
our results suggest that plants contain a functional NO signaling system whose components and targets are highly analogous to those identified in animals (Fig. 3).
Conclusions
The results presented in this paper strongly argue that both SA and NO play important roles in the activation of plant defense responses after pathogen attack. However, the interrelationship between their respective signaling pathways is currently unclear. NO, as well as other ROS, have been shown to stimulate the accumulation of SA, and SA induces the production of ROS, such as H2O2 and NO (20, 68). Thus, these signals appear to be self-amplifying. One likely role for NO, SA, and ROS is to promote the HR and pathogen killing. Both ROS and SA (or a factor downstream of SA) have been shown to synergize with NO to enhance host cell death in soybean suspension cells ( 21). This situation may parallel that in animals, in which NO collaborates with ROS to regulate apoptosis and produce peroxynitrite, a highly toxic compound that is thought to directly kill the pathogen (20). In addition, SA may mediate and/or potentiate NO's effects by altering the activity of various NO-regulated enzymes, such as aconitase, ascorbate peroxidase, and catalase (ref.62 and D. Clark, J.D., and D.F.K., unpublished results). SA also induces the synthesis of a pathogen-inducible oxygenase in plants that has strong homology to a mammalian cyclooxygenase (69). This enzyme is posttranslationally activated by NO in mammals (70). Thus, SA and NO may work synergistically to transduce the defense signal by targeting the same effector proteins and/or their genes.
In addition to acting synergistically with NO to activate various defense responses, SA may also antagonize the NO signaling pathway. In mammalian cells, salicylates are potent scavengers of NO and its derivatives (71). Moreover, salicylates inhibit the activity and transcription of iNOS (72). In plants, SA may also antagonize NO's ability to inhibit respiration (and thereby cause oxidative stress) by activating the NO-insensitive alternative oxidase (73). Considering the many interactions that are currently emerging between the pleiotropic effectors SA, NO, and other ROS, it is apparent that we are at a very early stage in understanding the complexity of their action in disease resistance.
We thank D'Maris Dempsey for assistance in preparation of this manuscript. This work was supported by Grants MCB 9723952 and MCB 9904660 from the National Science Foundation and 9802200 from the U.S. Department of Agriculture.
1. Flor, H. ( 1971) Annu. Rev. Phytopathol. 9, 275–296.
2. Keen, N. T. ( 1990) Annu. Rev. Genet. 24, 447–463.
3. Baker, C. J. & Orlandi, E. W. ( 1995) Annu. Rev. Phytopathol. 33, 299–321.
4. Lamb, C. & Dixon, R. A. ( 1997) Annu. Rev. Plant Physiol. Plant Mol. Biol. 48, 251–275.
5. Hammond-Kosack, K. E. & Jones, J. D. G. ( 1996) Plant Cell 8, 1773–1791.
6. Yang, Y., Shah, J. & Klessig, D. F. ( 1997) Genes Dev. 11, 1621–1639.
7. Dangl, J. L., Dietrich, R. A. & Richberg, M. H. ( 1996) Plant Cell 8, 1793–1807.
8. Kombrink, E. & Somssich, I. E. ( 1997) in Plant Relationships, eds. Carroll, G. C. & Tudzynski, P. (Springer-Verlag, Berlin), pp. 107–128.
9. Ryals, J. A., Neuenschwander, U. H., Willits, M. G., Molina, A., Steiner, H.-Y. & Hunt, M. D. ( 1996) Plant Cell 8, 1809–1819.
10. Dempsey, D., Shah, J. & Klessig, D. F. ( 1999) Crit. Rev. Plant Sci. 18, 547–575.
11. Shah, J. & Klessig, D. F. ( 1999) in Biochemistry and Molecular Biology of Plant Hormones, eds. Hooykaas, P. P. J., Hall, M. A. & Libbenga, K. R. (Elsevier, Amsterdam), pp. 513–541.
12. Dong, X. ( 1998) Curr. Opin. Plant Biol. 1, 316–323.
13. Pieterse, C. M. J. & van Loon, L. C. ( 1999) Trends Plant Sci. 4, 52–58.
14. Xu, Y., Chang, P. F. L., Liu, D., Narasimhan, M. L., Raghothanma, K. G., Gasegawa, P. M. & Bressan, R. A. ( 1994) Plant Cell 6, 1077–1085.
15. Lawton, K. A., Potter, S. L., Uknes, S. & Ryals, J. ( 1994) Plant Cell 6, 581–588.
16. Pieterse, C. M. J., Wees, S. C. M., van Hoffland, E., Pelt, J. A. & van Loon, L. C. ( 1996) Plant Cell 8, 1225–1237.
17. Penninckx, I. A. M. A., Eggermont, K., Terras, F. R. G., Thomma, B. P. H. J., De Samblanx, G. W., Buchala, A., Metraux, J.-P., Manners, J. M. & Broekaert, W. F. ( 1996) Plant Cell 8, 2309–2323.
18. Schmidt, H. H. & Walter, U. ( 1994) Cell 78, 919–925.
19. Stamler, J. S. ( 1994) Cell 78, 931–936.
20. Durner, J. & Klessig, D. F. ( 1999) Curr. Opin. Plant Sci. 2, 369–374.
21. Delledonne, M., Xia, Y., Dixon, R. A. & Lamb, C. ( 1998) Nature ( London) 394, 585–588.
22. Durner, J., Wendehenne, D. & Klessig, D. F. ( 1998) Proc. Natl. Acad. Sci. USA 95, 10328–10333.
23. Chen, Z., Silva, H. & Klessig, D. F. ( 1993) Science 262, 1883–1886.
24. Conrath, U., Chen, Z., Ricigliano, J. W. & Klessig, D. F. ( 1995) Proc. Natl. Acad. Sci. USA 92, 7143–7147.
25. Wendehenne, D., Durner, J., Chen, Z. & Klessig, D. F. ( 1998) Phytochemistry 47, 651–657.
26. Durner, J. & Klessig, D. F. ( 1995) Proc. Natl. Acad. Sci. USA 92, 11312–11316.
27. Du, H. & Klessig, D. F. ( 1997) Mol. Plant–Microbe Interact. 10, 922–925.
28. Takahashi, H., Chen, Z., Du, H., Liu, Y. & Klessig, D. F. ( 1997) Plant J. 11, 993–1005.
29. Bi, Y. M., Kenton, P., Mur, L., Darby, R. & Draper, J. ( 1995) Plant J. 8, 235–245.
30. Neuenschwander, U., Vernooij, B., Friedrich, L., Uknes, S., Kessmann, H. & Ryals, J. ( 1995) Plant J. 8, 227–233.
31. Chamnongpol, S., Willekens, H., Moeder, W., Langebartels, C., Sandermann Jr., H., Van Montagu, M., Inzé, D. & Van Camp, W. ( 1998) Proc. Natl. Acad. Sci. USA 95, 5818–5823.
32. Durner, J. & Klessig, D. F. ( 1996) J. Biol. Chem. 271, 28492–28501.
33. Anderson, M. D., Chen, Z. & Klessig, D. F. ( 1998) Phytochemistry 47, 555–566.
34. Du, H. & Klessig, D. F. ( 1997) Plant Physiol. 113, 1319–1327.
35. Conrath, U., Silva, H. & Klessig, D. F. ( 1997) Plant J. 11, 747–757.
36. Zhang, S. & Klessig, D. F. ( 1997) Plant Cell 9, 809–824.
37. Zhang, S., Du, H. & Klessig, D. F. ( 1998) Plant Cell 10, 435–449.
38. Hoyos, M. E., Zhang, S., Johal, G. S., Klessig, D. F., Hirt, H., Pike, S. M. & Novacky, A. J. ( 1998) Plant Physiol., Suppl., 46–47.
39. Romeis, T., Piedras, P., Zhang, S., Klessig, D. F., Hirt, H. & Jones, J. D. G. ( 1999) Plant Cell 11, 273–287.
40. Zhang, S. & Klessig, D. F. ( 1998) Proc. Natl. Acad. Sci. USA 95, 7433–7438.
41. Zhang, S. & Klessig, D. F. ( 1998) Proc. Natl. Acad. Sci. USA 95, 7225–7230.
42. Seo, S., Okamoto, M., Seto, H., Ishizuka, K., Sano, H. & Ohashi, Y. ( 1995) Science 270, 1988–1992.
43. Zhang, S., Liu, Y. & Klessig, D. F. ( 2000) Plant J., in press.
44. Silva, H., Yoshioka, K., Dooner, H. K. & Klessig, D. F. ( 1999) Mol. PlantMicrobe Interact. 12, 1053–1063.
45. Cao, H., Bowling, S. A., Gordon, A. S. & Dong, X. ( 1994) Plant Cell 6, 1583–1592.
46. Glazebrook, J., Rogers, E. E. & Ausubel, F. M. ( 1996) Genetics 143, 973–982.
47. Delaney, T. P., Friedrich, L. & Ryals, J. A. ( 1995) Proc. Natl. Acad. Sci. USA 92, 6602–6606.
48. Shah, J., Tsui, F. & Klessig, D. F. ( 1997) Mol. Plant–Microbe Interact. 10, 69–78.
49. Cao, H., Glazebrook, J., Clarke, J. D., Volko, S. & Dong, X. ( 1997) Cell 88, 57–63.
50. Ryals, J., Weymann, K., Lawton, K., Friedrich, L., Ellis, D., Steiner, H.-Y., Johnson, J., Delaney, T. P., Jesse, T., Vos, P. & Uknes, S. ( 1997) Plant Cell 9, 425–439.
51. Shah, J., Kachroo, P. & Klessig, D. F. ( 1999) Plant Cell 11, 191–206.
52. Zhou, J.-M., Trifa, Y., Silva, H., Pontier, D., Lam, E., Shah, J. & Klessig, D. F. ( 2000) Mol. Plant–Microbe Interact. 13, 191–202.
53. Lebel, E., Heifetz, P., Thorne, L., Uknes, S., Ryals, J. & Ward, E. ( 1998) Plant J. 16, 223–233.
54. Zhang, Y., Fan, W., Kinkema, M., Li, X. & Dong, X. ( 1999) Proc. Natl. Acad. Sci. USA 96, 6523–6528.
55. Depres, C., DeLong, C., Glaze, S., Liu, E. & Fobert, P. R. ( 2000) Plant Cell 12, 279–290.
56. Malamy, J., Hennig, J. & Klessig, D. F. ( 1992) Plant Cell 4, 359–366.
57. McDonald, L. J. & Murad, F. ( 1995) Adv. Pharmacol. 34, 263–276.
58. Xu, L., Eu, J. P., Meissner, G. & Stamler, J. S. ( 1998) Science 279, 234–237.
59. Murad, F. ( 1994) Adv. Pharmacol. 26, 19–33.
60. Gardner, P. R., Costantino, G., Szabo, C. & Salzman, A. L. ( 1997) J. Biol. Chem. 272, 25071–25076.
61. Domachowske, J. B. ( 1997) Biochem. Mol. Med. 60, 1–7.
62. Navarre, D. A., Wendehenne, D., Durner, J., Noad, R. & Klessig, D. F. ( 2000) Plant Physiol. 122, 573–582.
63. Liochev, S. L. ( 1996) Free Radical Res. 25, 369–384.
64. Rouault, T. & Klausner, R. ( 1997) Curr. Top. Cell. Regul. 35, 1–19.
65. Mott, H. R., Carpenter, J. W. & Campbell, S. L. ( 1997) Biochemistry 36, 3640–3644.
66. Yun, H. Y., Gonzalez-Zulueta, M., Dawson, V. L. & Dawson, T. M. ( 1998) Proc. Natl. Acad. Sci. USA 95, 5773–5778.
67. Kumar, D. & Klessig, D. F. ( 2000) Mol. Plant–Microbe Interact. 13, 347–351.
68. Van Camp, W., Van Montagu, M. & Inzé, D. ( 1998) Trends Plant Sci. 3, 330–334.
69. Sanz, A., Moreno, J. I. & Castresana, C. ( 1998) Plant Cell 10, 1523–1537.
70. Nogawa, S., Forstner, C., Zhang, F., Nagayama, M., Ross, M. E. & Ladecola, C. ( 1998) Proc. Natl. Acad. Sci. USA 95, 10966–10971.
71. Hermann, M., Kapiotis, S., Hofbauer, R., Exner, M., Seelos, C, Held, I. & Gmeiner, B. ( 1999) FEES Lett. 45, 212–214.
72. Farivar, R. S. & Brecher, P. ( 1996) J. Biol Chem. 271, 31585–31592.
73. Millar, A. H. & Day, D. A. ( 1997) Trends Plant Sci. 2, 289–290.
74. Sambrook, J., Fritsch, E. F. & Maniatis, T. ( 1989) Molecular Cloning: A Laboratory Manual (Cold Spring Harbor Lab. Press, Plainview, NY).
75. Zhang, S. & Klessig, D. F. ( 1999) in MAP Kinases in Plant Signal Transduction, ed. Hirt, H. (Springer-Verlag, Heidelberg), pp. 65–84.







