
Page 147
9
Managing Carbon Losses for Selective Oxidation Catalysis
Leo E. Manzer
DuPont Central Research and Development
Catalytic oxidations are among the least selective of all catalytic reactions and the source of much of the carbon dioxide from chemical processes. These processes are often carried out at high temperatures, resulting in selectivity for the desired oxygenated products of less than 90%. The major by-product is usually carbon dioxide. Table 9.1 shows typical selectivity for the major product in these large-scale commercial operations. Since most of these processes have production capacities of several hundred million pounds per year, the amount of CO2 generated is quite significant. For example, for a malefic anhydride plant operating at a capacity of 200 million pounds per year, with 60% selectivity, more than 500 million pounds per year of carbon dioxide is produced per year. Therefore, there is a large incentive to improve yield of the desired hydrocarbon product in these processes. This can be done by improving the catalyst in these existing processes or by completely changing the chemistry and engineering of existing processes.
ANAEROBIC VERSUS AEROBIC OXIDATIONS
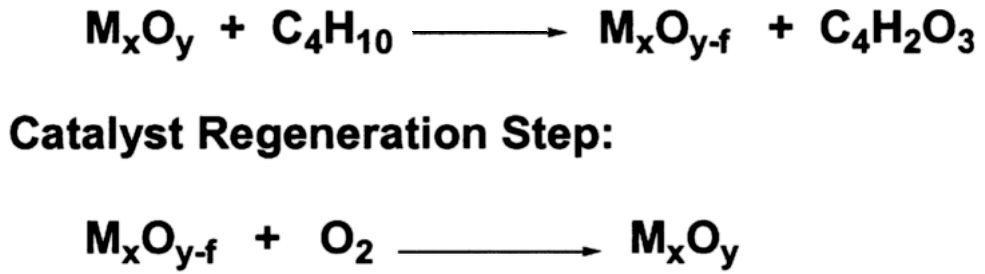
It has been known for many years that in certain oxidation reactions the most selective catalysis occurs when the oxygen in the final product is derived directly from the lattice of the oxide catalyst. However, most large-scale commercial processes are carried out in the nonflammable region, which usually requires a feed of less than 3% organic and the balance air. This large excess of gas-phase oxygen results in the low selectivities shown in Table 9.1. The Mars-van Krevelen mechanism ( Figure 9.1) suggests that a gain in selectivity is possible by keeping gas-phase oxygen from the process. The lattice oxygen of the catalyst is used in a stoichiometric reaction with a hydrocarbon to yield the oxygenated product. The reduced oxide catalyst is then transported to a separate zone and reoxidized by air. The process is referred to as anaerobic oxidation.
For an anaerobic oxidation process to be successful and economically viable, several requirements must be met. First, there must be a selectivity improvement for the desired product by operating in this mode. Next, the catalyst must have a high oxygen-carrying capacity per unit weight, to minimize the amount of catalyst circulated through the reactor. This is very important because the catalyst is often
Page 148
|
Oxidation Process |
Major Product |
Selectivity (%) |
|
Butane oxidation |
Malefic anhydride |
60 |
|
Propylene oxidation |
Acrolein or acrylic Acid |
75 |
|
Propylene ammoxidation |
Acrylonitrile |
80 |
|
Ethylene oxidation |
Ethylene oxide |
88 |
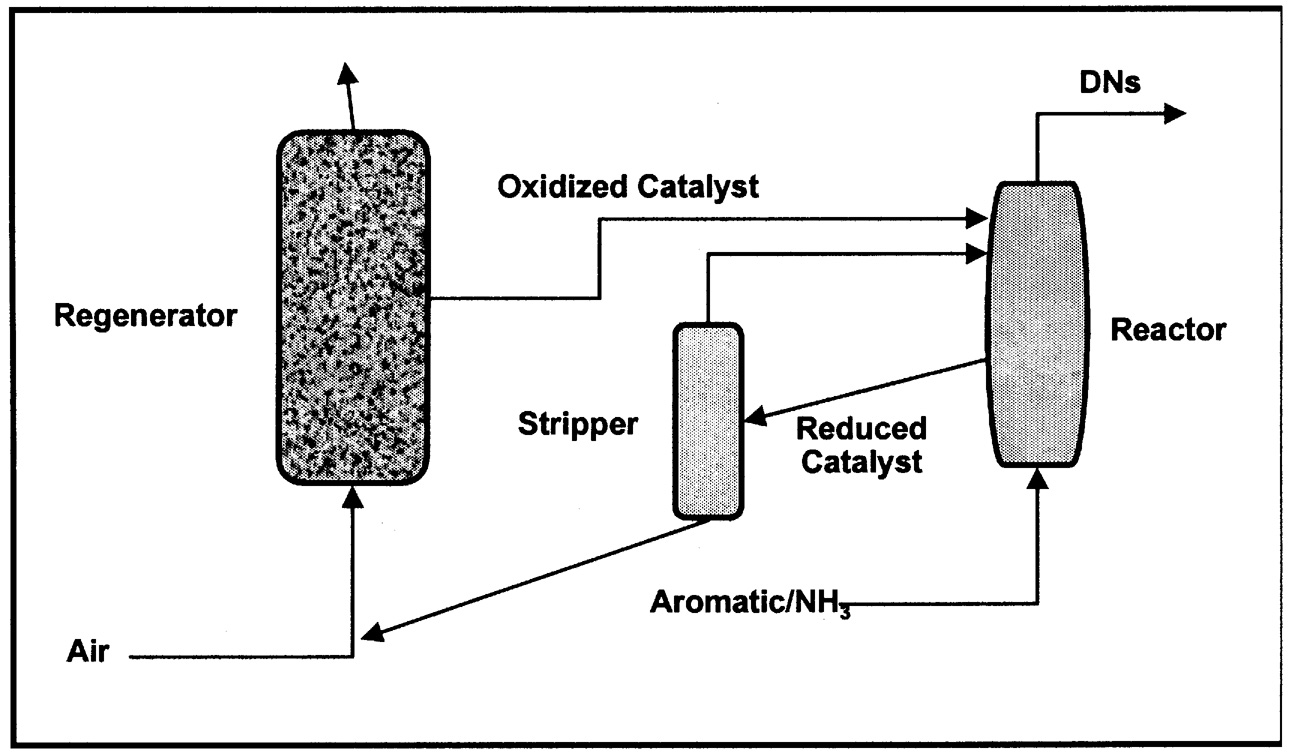
more expensive than the oxygenated product. Reoxidation of the reduced catalyst in the regenerator should occur at a temperature similar to that of the oxidation step to minimize the need to heat or cool the catalyst solids (energy minimization). Finally, since the reaction is essentially stoichiometric between catalyst and organic, a large amount of solid must be circulated around the reaction system. The catalyst must therefore be very resistant to attrition and must maintain structural integrity through many redox cycles. An excellent, early example of a two-step, anaerobic oxidation is the Lummus process for ammoxidation of o-xylene to o-phthalonitrile (dinitriles, DNs).2 A simplified schematic is shown in Figure 9.2. A higher selectivity is claimed for the two-step process relative to the single-stage, aerobic oxidation.
Another large development effort was carried out by ARCO Chemical during the 1970s to convert methane to ethylene.3 The reaction occurs at a very high temperature of 850-900°C. Patent and literature references, which illustrate the use of a Li0.5B0.5MnMg2.8Ox/SiO2 catalyst, show that at a conversion of 22%, the selectivity to two-carbon compounds was about 60% under both aerobic and anaerobic conditions. However, the yield of CO2 was reduced from 34% to 22% when the reaction was conducted in a cyclic mode, by carrying out the oxidation under anaerobic conditions. A major development effort was terminated when the price of oil decreased.
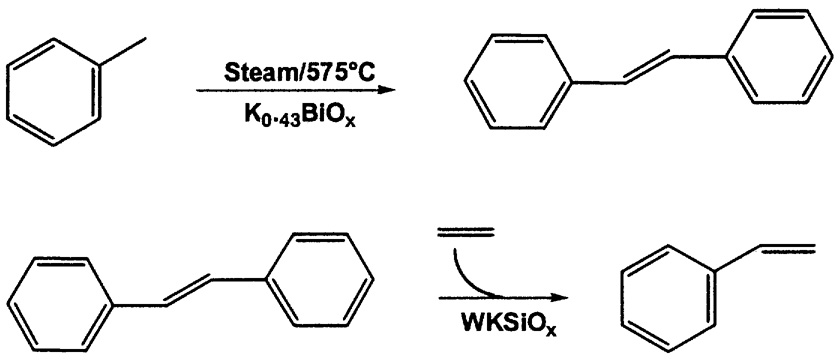
Scientists and engineers at Monsanto studied the oxidative dimerization of toluene to stilbene,4 as part of a new styrene process ( Figure 9.3). In the first step, using a K0.43BiOx catalyst at 575°C, the anaerobic process showed higher conversion (46 vs. 38%) and higher selectivity to stilbene (81.3 vs. 72.7%).
Emig has recently studied the oxidative dimerization of isobutylene to 2,5-dimethylhexadiene (DMH).5 Under aerobic conditions, a conversion of 24% was obtained with a selectivity of 38% to DMH giving a single-pass yield of 9.1%. Under anaerobic oxidation, the conversion dropped to 11%, the yield remained constant at 9.9%, and the selectivity to DMH increased from 38 to 90%. This is a remarkable example of CO2 reduction using a two-step process.
A recent patent has described the oxidation of propylene to acrolein and acrylic acid using a multicomponent metal oxide catalyst in a circulating solids reactor (CSR).6 Under anaerobic conditions
~ enlarge ~
FIGURE 9.1 Mars-van Krevelen oxidation of butane to maleic anhydride
Page 149
~ enlarge ~
FIGURE 9.2 Simplified schematic for the two-stage oxidation of o-xylene.
at 350°C, propylene conversion was 16% and selectivity was 95.5%. When air was introduced into the CSR, the conversion increased to 21% and selectivity dropped to 82%, once again showing the substantial advantage of keeping gas-phase oxygen out of the catalytic oxidation zone.
DuPont recently commercialized a new process for the oxidation of butane to maleic anhydride using a CSR.7 The maleic anhydride is scrubbed from the reaction zone as maleic acid and then hydrogenated to tetrahydrofuran. The advantages are well documented in the references. A key to this process was the development of an attrition-resistant catalyst obtained by spray-drying a solution of micronized vanadium-phosphorus-oxygen (VPO) catalysts in polysilicic acid.8 In the spray dryer, a porous shell of very hard silica is formed to protect the soft VPO catalyst.
These few examples show an advantage of anaerobic oxidations for selected reactions, to minimize CO2 formation. A few other opportunities for further study should include the oxidation of o-xylene to phthalic anhydride, oxidative dehydrogenation of ethylbenzene to styrene, oxidation of isobutylene to methacrolein and methacrylic acid, and oxidative dehydrogenation of paraffins to olefins.
PARAFFIN OXIDATIONS
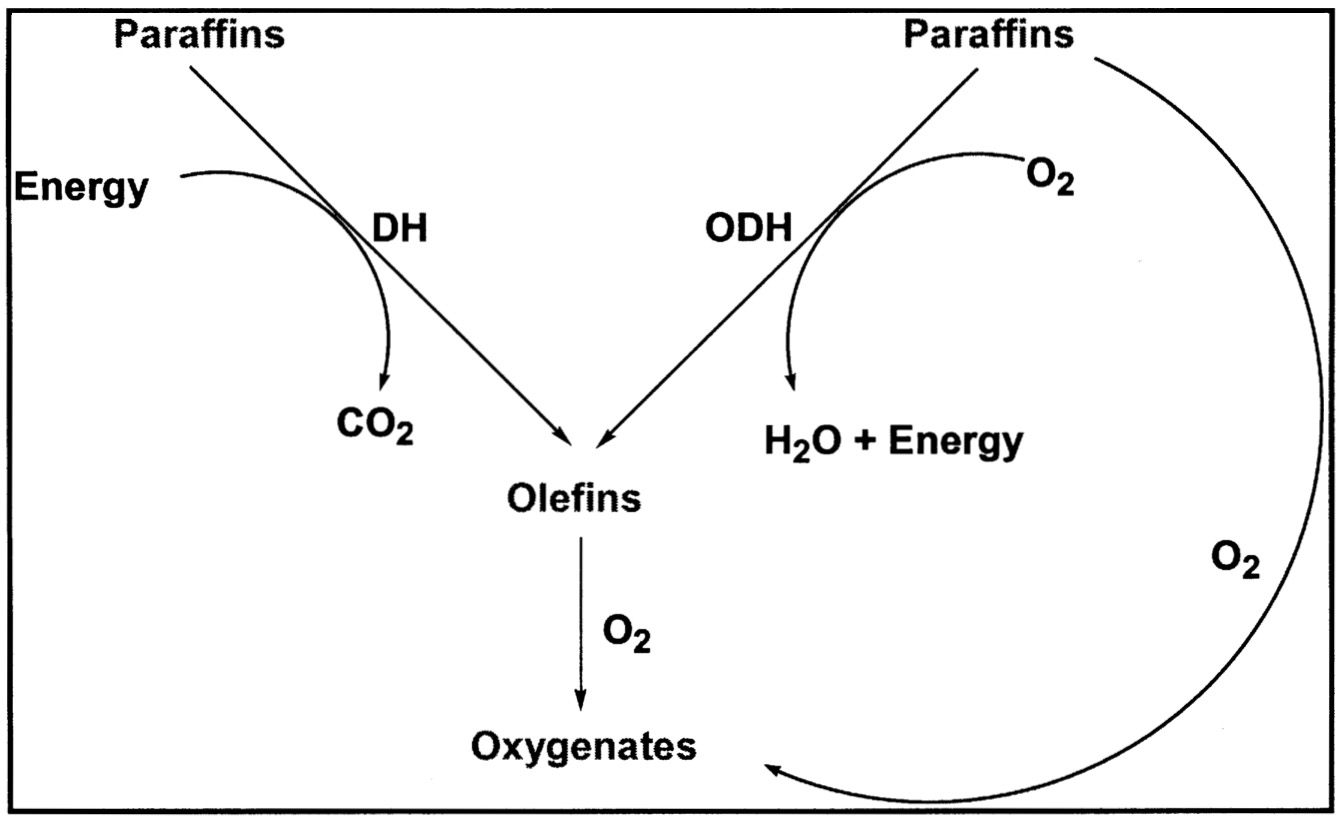
Currently, there are no commercial processes involving the direct gas-phase oxidation of paraffins to an oxygenated product or olefin. The conventional approach involves endothermic dehydrogenation
~ enlarge ~
FIGURE 9.3 Monsanto anaerobic oxidative coupling of toluene.
Page 150
of paraffin to the desired olefin followed by oxidation to the desired product. This process generates CO2 in the endothermic dehydrogenation step because heat must be provided to the reaction. By comparison, if direct oxidation of the paraffin to the desired oxygenates could be achieved at high selectivity, there would be a net reduction in CO2 and the process, in fact, would export energy ( Figure 9.4). A number of companies have been very active in this area for many years.9
The results from selected patents for the ammoxidation of propane to acrylonitrile are shown in Table 9.2. Significant advances have been made over the past 10 years. High conversions and selectivities approaching 64% have been obtained. BP has announced that a pilot plant is operational to collect basic data for commercial design. The stake is high for this development because of the lower cost of propane compared to propylene and the reduction in CO2 emissions.
Another well-studied process, in which significant progress has been made, involves the oxidation of propane to acrylic acid.10 (See Table 9.3.) These results are quite impressive, with selectivity reported in excess of 80%. By contrast, direct catalytic oxidation of isobutane to methacrylic acid has been less developed.11 Sumitomo has reported that 42% methacrylic acid can be obtained at 25% conversion.
NEW PROCESS CHEMISTRY OR CONDITIONS
Up to this point, the focus has been on improving the yield of the catalytic reaction to reduce CO2 emission. However it is important to consider entirely new process chemistry that might reduce the number of steps, lower the temperature, and as a result, also lower CO2 production. An excellent illustration of this point involves the production of methyl methacrylate (MMH). Current commercial catalytic routes use C4 feedstocks and involve two high-temperature gas-phase catalytic steps followed by esterification. The first two steps occur above 350°C with an overall yield of about 75%. The main by-product is carbon dioxide. A new process to methyl methacrylate is under development by Asahi Chemical.12 This process combines the second and third steps into a single oxidative esterification step
~ enlarge ~
FIGURE 9.4 Incentive for direct oxidation of paraffins.
Page 151
|
Company |
Catalyst |
Conversion (%) |
Selectivity (%) |
|
BP |
VSb1.4Sn0.2Ti0.1Ox |
30.5 |
58.3 |
|
Asahi |
NbSbaCrbXyOn |
29.1 |
30.7 |
|
Mitsui Toatsu |
V1Li0.1P1.1Ox |
54.8 |
58.8 |
|
Mitsubishi |
MoVxTe0.2Nb0.1O4.25 |
79.4 |
63.5 |
(Figure 9.5). Using a Pd/Pb/Mg-Al2O3 catalyst, Asahi reports better than 98% conversion of the methacrolein to methacrylic acid with a selectivity greater than 95%. The reaction occurs at a mild 80°C in a slurry-phase reactor. The overall yield is significantly higher than in the conventional process, less CO2 is generated, and capital investment is lower. A plant with a capacity of 135 million pounds per year is currently under construction. Mitsubishi Rayon13 has also been active in this area with a Pd5Bi2Fe/CaCO3 catalyst giving better than 97% selectivity for MMA at 76% conversion.
ALTERNATIVE OXIDANTS
Oxygen or air will likely be the preferred source of oxygen from an economic standpoint for many years. However, a growing number of developmental applications with hydrogen peroxide (H2O2), nitrous oxide (N2O), and alkylhydroperoxides as the oxygen source are appearing in the literature. A relative comparison of costs of various oxygen sources is shown in Table 9.4. Clearly, from a cost standpoint, it will be difficult to justify new commodity chemical processes using on-purpose production of N2O and H2O2 as oxygen source. However for fine chemical applications such as pharmaceuticals and agrochemicals, the cost may well be justified. Hydrogen and oxygen mixtures may be economically justified, although safety issues will likely require a greater investment.
Panov has extensively studied the use of N2O as a selective oxidant for aromatics.14 Using zeolites that contain only small amounts of iron, he has shown that benzene can be oxidized to phenol with selectivity of over 99% at around 300°C. Emig has studied the mechanism of this interesting reaction.15 He proposes that at temperatures of less than 300°C in the absence of benzene, all the N2O reacts with the surface to give an α-oxygen site that is very stable ( Figure 9.6.) Cooling the solid below room temperature and introducing benzene give phenol in high selectivity. The lifetime of the site is 0.5 seconds at 500°C and 1.75 seconds at 420°C.
|
Company |
Catalyst |
Conversion (%) |
Selectivity for Acrylic Acid (%) |
|
Wang (Fudan U) |
V1Zr0.5P1.5Ox |
18.3 |
81.0 |
|
Ce0.01VPO |
27.2 |
68.3 |
|
|
Toa Gosei |
V-Sb-Mo-NbOx |
30.9 |
29.4 |
|
Mitsubishi |
V0.3Te0.23Nb0.12Bi0.017MoOx |
56.2 |
42.6 |
|
Rohm and Haas |
V/Te/Nb/MoOx |
71.0 |
59.0 |
Page 152
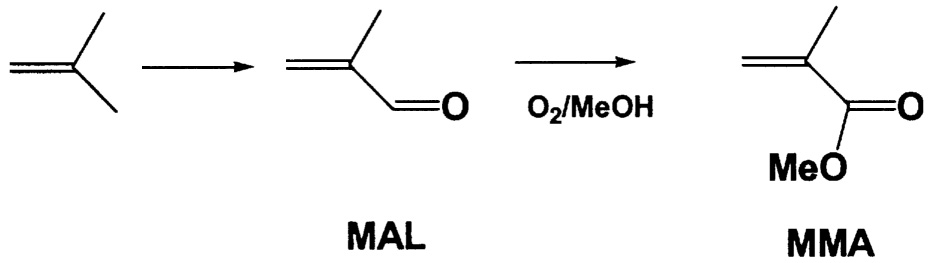
~ enlarge ~
FIGURE 9.5 New route to methyl methacrylate. NOTE: MAL = methacrolein.
Through a very extensive collaboration with Solutia, this technology has been integrated into nylon intermediates manufacturing. Conventional technology for the production of adipic acid from cyclohexane provides cyclohexanone (K) and cylcohexanol (A) as intermediates. Nitric acid is used to oxidize K/A to adipic acid and in that step produces significant amounts of N2O. Historically, the N2O-containing gas stream has been vented to the atmosphere, but due to ozone depletion issues, most producers now abate the N2O. Solutia decided to separate the N2O after the nitric acid oxidation of K/A and to react it with benzene to produce phenol, which can be hydrogenated to K and oxidized to adipic acid. This provides an opportunity to use the N2O for expansion purposes and to provide a higher incremental yield of adipic acid. The process is reported to be in pilot plant production.16 Application of this technology for uses other than retrofit options will be highly dependent on the cost of N2O. A recent patent to Solutia17 involves the use of Bi/Mn/AlOx as a catalyst for the oxidation of ammonia to nitrous oxide. N2O selectivity is reported to be about 92% at 99.2% conversion of NH3. The cost of N2O is projected to be about 25% that of H2O2, so if this new process is commercialized, it might be a serious alternative to O2 oxidation.
Titanosilicalites18 are now well known to oxidize a wide variety of organics with hydrogen peroxide or alkylhydroperoxides as the oxygen source ( Figure 9.7.) Selectivity is generally very high, although utilization of the peroxide is often low. Because of the high cost of hydrogen peroxide ( Table 9.4), commercial use will likely be restricted to fine chemical applications. However the use of hydrogen-oxygen mixtures is currently showing great promise.
For many years, DuPont studied the direct catalytic combination of hydrogen and oxygen to hydrogen peroxide.19 The use of platinum and palladium bimetallic catalysts on silica was studied extensively. Selectivity for hydrogen peroxide was highly dependent on the weight ratio of platinum to total metal loading on silica. Optimum ratios were 0.02-0.2, which yielded selectivity for H2O2 of about 70%. Very high pressures were reported to be used and concentrations of hydrogen peroxide exceeded 20%. A key to achieving the high selectivity was the addition of a promoter such as Cl− or Br−.
Hoelderich has been studying the epoxidation of propylene to propylene oxide using titano- or vanadiosilicalite (VS) catalysts.20 Using a titanosilicalite (TS) support with Pt/Pd and an NaBr promoter ( Figure 9.8), with a mixture of hydrogen and oxygen, he was able to achieve selectivity for propylene
|
Oxygen Source |
Cost ($/lb-mole) |
Cost ($/lb) |
|
O2 |
0.64 |
0.02 |
|
N2O |
7.5 |
0.17 |
|
H2O2 |
17 |
0.50 |
|
H2 + O2 |
0.65 |
0.02 |
Page 153
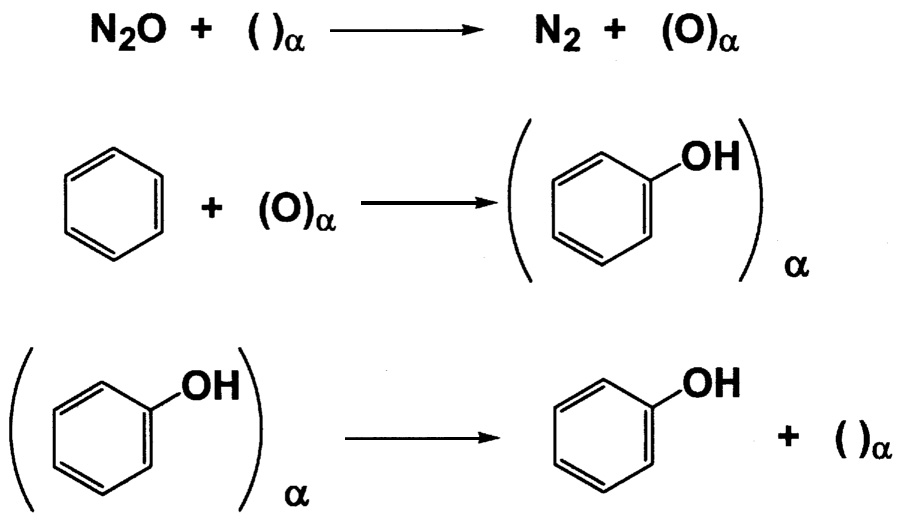
~ enlarge ~
FIGURE 9.6 Emig's proposed mechanism for the selective oxidation of aromatics by N2O.
oxide of 87.3% at 19.4% conversion. In the absence of the NaBr promoter, selectivity dropped to 34.1%. The result is an exciting illustration of the potential for in situ production of a peroxo species as a selective oxidant.
Recently, carbon dioxide has been reported to act as a mild oxidant with chromium-based catalysts at very high temperatures. Longya et al. have studied the oxidative dehydrogenation of ethane using modified chromium catalysts that are support on silicate-2.21 At 1073°K, good selectivity for ethylene is seen ( Table 9.5.) An evaluation of the data suggested that the overall chemistry is very complex ( Figure 9.9). Two successive coupling reactions occur. The first involves the dehydrogenation of ethane to ethylene and hydrogen. The second reaction involves the reverse water-gas shift reaction, to form CO and water, thus allowing for continuous removal of hydrogen during the dehydrogenation step. The process is reported to be commercial for the conversion of ethane to ethylene in an FCC (fluid catalytic cracking) tail-gas stream. The ethylene produced is acceptable for the formation of ethylbenzene.22
Recently Wang et al. have studied the effect of the support on the oxidative dehydrogenation of ethane with ethylene using carbon dioxide.23 The data are shown in Table 9.6. All data are at 600°C. Silica appears to be the optimum support in this study as well. Very high selectivity for ethylene is seen.
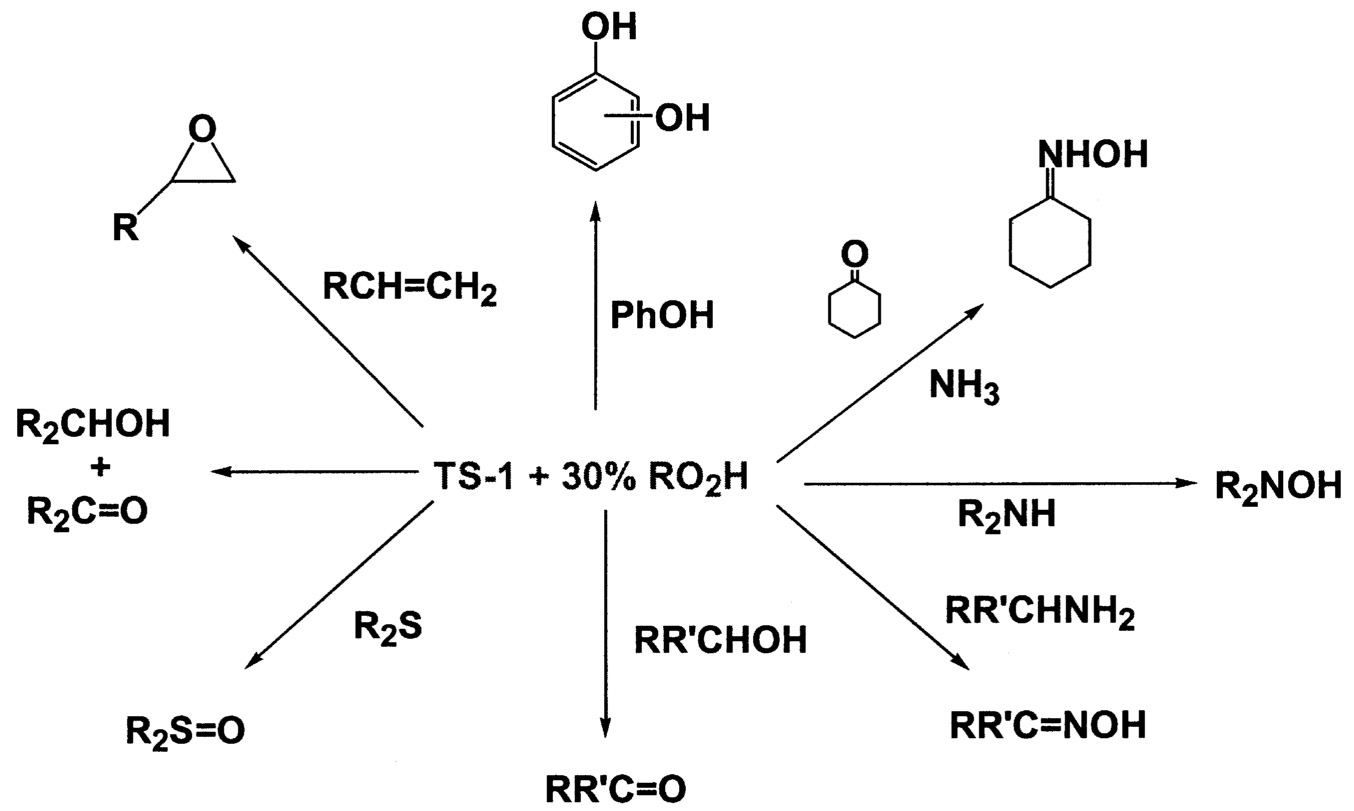
~ enlarge ~
FIGURE 9.7 Range of products made by using titanosilicalites.
Page 154
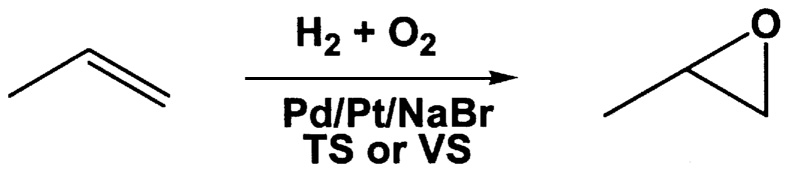
~ enlarge ~
FIGURE 9.8 Preparation of propylene oxide from hydrogen-oxygen mixtures.
CONCLUSIONS
A significant reduction in the amount of CO2 that is emitted to the atmosphere is a desirable global goal. This workshop has suggested a number of opportunities to convert CO2 into useful products. However, for chemistry, the ultimate goal is to eliminate the production of carbon dioxide completely in new processes. Clearly, it will be uneconomical to replace existing plant investment, so end-of-pipe treatment is important. The purpose of this chapter is to show that indeed there are a number of emerging new catalysts and catalytic processes that have higher selectivity for products and thus result in reduced CO2 production. While most of these developments are only in the discovery or early development phase, there is sufficient progress to indicate that commercialization is a real possibility in the future. The following are the key points of this chapter:
1. Anaerobic oxidation of hydrocarbons can offer significant reduction of CO2 in several cases. However, several criteria must be met for economical viability.
2. Alternative oxidants such as H2O2, RO2H, and N2O can provide higher selectivity in many reactions, but economics are currently attractive in only a few isolated cases.
|
Catalyst |
Conversion (%) CO2 |
Conversion (%) C2H6 |
Selectivity (wt%) CH4 |
Selectivity (wt%) C2H4 |
Selectivity (wt%) H2/CO |
|
Cr/Si-2 |
18.6 |
58.9 |
19.6 |
80.4 |
1.4 |
|
Cr-Mn/Si-2 |
22.2 |
62.4 |
18.4 |
81.6 |
1.4 |
|
Cr-Mn-Ni/Si-2 |
24.2 |
67.9 |
18.7 |
81.3 |
1.6 |
|
Cr-Mn-Ni-La/Si-2 |
20.5 |
64.2 |
13.8 |
86.2 |
1.4 |
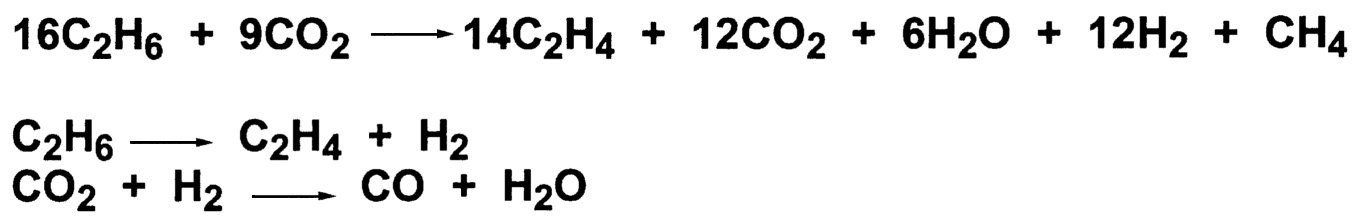
~ enlarge ~
FIGURE 9.9 Oxidative dehydrogenation of ethane with CO2.
Page 155
|
Catalyst |
Conversion (%) CO2 |
Conversion (%) C2H6 |
Selectivity (wt%) CH4 |
Selectivity (wt%) C2H4 |
Selectivity (wt%) H2/CO |
|
Cr2O3/TiO2 |
0.8 |
0.9 |
6.3 |
93.7 |
4.2 |
|
Cr2O3 |
16.8 |
12.1 |
4.6 |
95.4 |
4.8 |
|
Cr2O3/Al2O3 |
4.9 |
12.6 |
2.3 |
97.6 |
5.6 |
|
Cr2O3/ZrO2 |
19.2 |
37.9 |
25.3 |
74.6 |
3.2 |
|
Cr2O3/SiO2 |
9.6 |
38.8 |
4.2 |
95.7 |
1.4 |
3. The use of H2 and O2 mixtures is beginning to show promise as a replacement for H2O2, but safety issues will have to be seriously addressed.
4. Creative new catalytic technology can significantly reduce investment and carbon dioxide production through higher yields and few steps.
5. The direct use of CO2 as a mild oxidant is an interesting new development that should be pursued aggressively.
6. Higher selectivity can also result in less processing, which is an unrecognized benefit that also reduces energy consumption and indirectly CO2 emissions.
REFERENCES
1. , and . Spec. Suppl. Chem. Eng. Sci. 3: 41 ( 1954 ).
2. , and . Hydrocarbon Processing. 103-106 ( February 1976 ).
3. , , , and . J. Catal. 114(2): 422-432 ( 1988 ).
4. , and . U.S. Patent 4, 254, 293 ( 1981 ).
5. and . Oxidative coupling of isobutene in a two step process, pp. 593-602 in 3rd World Congress on Oxidation Catalysis. Elsevier Science B.V. ( 1997 ).
6. , , , , and , WO 99/03809 ( 1999 ).
7. , Chemical and Engineering News, ( April 3, 1995 ); , , , , , , , and , Catal. Today 1(1-2): 49-58. , , , , , and . Stud. Sur. Sci. Catal. 38 (Catalysis 1987): 645-654 ( 1988 ).
8. , ACS Syp. Ser. 411: 55-64. ( 1989 ) CODEN:ACSMC8 ISBN:0097-6156.
9. and . ( Mitsubishi Chemical Industries Ltd.), JP 98-294795.
10. , , , and . ( Misubishi Kasei Corp.), U.S. Patent 5, 380, 933 ( 1995 ); , , , and . ( BASF) WO 9920590 ( 1999 ); , , and ( Toa Gosei Chemical Industry Co., Ltd.). JP 09316023 ( 1997 ); , and . ( Rohm and Haas Ltd.), EP 0962253A2 ( 1999 ).
11. , , and . ( Sumitomo Chemical Co.) JP 95-171855 ( 1997 ).
12. , , , and ( Asahi Chemical Industry Co., Ltd.), JP 10263399 ( 1998 ).
13. , , and , JP 09216850A2 ( 1997 ); , , and , DE 19734242A1 ( 1999 ).
14. , , , , , , and . U.S. Patent 5, 110, 995 ( 1992 ).
15. , , , and . Pp. 847-856 in 3rd World Congress on Oxidation Catalysis, Vol. 110, R.K. Grasselli, S.T. Oyama, A.M. Gaffney and J.JE. Lyons (Editors), Elsevier Science B.V. ( 1997 ).
Page 156
16. , , , , and . Pp. 857-864 in 3rd World Congress on Oxidation Catalysis, Vol. 110, R.K. Grasselli, S.T. Oyama, A.M. Gaffney and J.JE. Lyons (Editors), Elsevier Science B.V. ( 1997 ).
17. WO 9825698.
18. Pp 21-23 in Heterogeneous Catalysis and Fine Chemicals III, M. Guisnet et al. (Editors), Elsevier Science Publishers B.V. ( 1993 ).
19. U.S. Patent 4, 832, 938 ( 1989 ).
20. , German Patent DE 98-19845975.
21. , , , , , and . Natural Gas Conversion V. Pp. 605-610 in Studies in Surface Science and Catalysis, Vol. 119, A. Parmaliana et al. (Editors), Elsevier Science B.V. ( 1998 ).
22. , , , , CN 87105054.4 ( 1987 ).
23. , , , , and , Applied Catalysis A: General 196: I-8 ( 2000 ).
DISCUSSION
Dave Cole, Oak Ridge National Laboratory: I would like to ask two questions. First, would you comment on the discussion yesterday that pointed to the fact that mitigating CO2 through commercial ventures such as this is a very small part of the CO2 mitigation problem?
Second, as I recall, some years ago in one of the multilab reports on carbon management, there was a suggestion that one might generate carbon and hydrogen from methane in decarbonation-type reactions. Do you have anyone working on that? What might the catalytic and thermal requirements be to do that?
Leo Manzer: I agree with yesterday's discussion. For us, 500 million pounds a year of CO2 is a lot of material, but still much smaller than carbon dioxide from electricity facilities. Large plants in the chemical industry generate a few hundred million pounds a year.
I can't deal with those other issues. We are working on oxidation catalysis, and we make a small improvement there. So it is a step in the right direction. I don't follow research in the conversion of methane into hydrogen and carbon. There has been work in that area, but the problem is that you still have to do something with the carbon. If you can use the carbon, fine. If it grows out of catalyst, the carbon will most likely coke up the catalyst; you would have to burn it off, and you would get CO2 again. If you can use the carbon somewhere else, then this might be a way to make hydrogen.
Dave Cole: I have heard some discussion about actually trying to engineer the carbon to get it to hold onto the hydrogen in the form of either the NO2 structures or even buckyballs. I don't know if anyone here has heard of this or done any work on it. If so, I would like to hear about it. In the recovery, is the problem that whatever surfaces you have get coated with the carbon?
Leo Manzer: Probably something like that is needed to generate low-cost buckyballs and buckytubes.
Chandrakant Panchal, Argonne National Laboratory: I think you can take more credit for CO2 reduction. When you improve the selectivity of the chemical reactions, the energy cost for separation of products is reduced significantly. Most of the energy in the chemical industry goes to these operations. I think you can claim much more CO2 reduction by improving selectivity. The Department of Energy's Office of Industrial Technologies program Vision 2020 has identified selectivity improvements as a way of reducing the need for separation—hence a way to reduce CO2.
Leo Manzer: That is a very good point. I make this point in my report as well. If you have 100% selectivity, you can eliminate all refining and save a lot of energy that way.
Page 157
Tobin Marks, Northwestern University: Let me follow up even more on that. If we are going into an era legislatively where there are punitive fines against the industry for CO2 generation, then the things that you were talking about are going to be doubly valuable.
Leo Manzer: I think that is right, Tobin. I think this provides an opportunity for further research that may not be done as much in industry as it used to be. The opportunity is there for folks in academics to develop this technology. It takes a while, and this is tough chemistry, but it is an opportunity.
Tobin Marks: In Europe, is taxation levied against CO2 producers? I have heard that it is in Norway. Can anyone tell us whether that is an issue in other countries?
David Keith, Carnegie Mellon University: There is no other systematic tax. Even the Norwegian tax is only on offshore emissions and is quite limited. There is a lot of talk and some targeted sectoral effects to put an effective price on carbon.
Leo Manzer: That is a good point. I think you have to do this globally to make it work.
John Frost, Michigan State University: I ask this question knowing that DuPont has a curious history in this area. One of the organisms that came out of the national labs—AFP-111, when grown on glucose, has yields in excess of 150% because it actively fixes CO2 at atmospheric pressure during the fermentation. In what potential scenario could you see this type of process becoming competitive with what you are practicing now?
Leo Manzer: You may be aware that we have a fermentation process that we are developing to take glucose to 1,3-propane diol, which is in the pilot plant stage right now.
This is an example of how a biochemical route, starting with something like glucose, can be cost-effective. It can be even better than chemical routes to make propane diol. Does that answer your question? We are not starting with methane.
John Frost: I think it is a question of issues of displacement technology. Where is the crossover point in this game? You have a facility that is obviously well established.
Leo Manzer: Replacement economics are very tough, almost impossible. It is just the game we are playing these days. This is why it is nice to work in an area where there is decent growth, so that every few years or so when you have to build another plant, you can include any new technology in it.
Dave Thomas, BP Amoco: As was pointed out yesterday, the amount of carbon dioxide produced by the chemical industry is relatively small compared to many of the other processes. Every time you take something up and boil it and then cool it off through some mechanical or chemical process, you consume a great deal of energy.
I think the contribution that the chemical industry can make in the area of CO2 mitigation is in high-efficiency processes that reduce the amount of heat input and enhance the energy consumed. As you point out, highly selective reactions could be very valuable.
As an aside, one of the people I talked to in our chemical business said that if we have to go to full-scale sequestration from chemical plant CO2 effluents, this would essentially cause its margins to evaporate. In other words, the business would not be economically viable unless everybody else had to do the same and the price was just raised.
Page 158
The cost of CO2 mitigation for the chemical industry is not small. I was struck by your earlier comments about some of the oxidative reactions using air. You pointed out that in reaction with butane, you have to avoid the explosive limit, and you can produce a lot of CO2 by accident if you don't avoid the combustion limits. Have you thought about using carbon dioxide-oxygen mixtures rather than air in these kinds of reactions?
Leo Manzer: CO2 often is recycled in some of these processes. It is not specifically mixing CO2 and oxygen. That will change the flammability envelope significantly, I think. We have not done this. It may be an opportunity for somebody to look at.
Olaf Walter, Forschungszentrum Karlsruhe (Germany): Oxidations are always a target for increasing selectivity. In recent years, there also have been investigations using microreactors for heterogeneous oxidations, which may increase selectivity, often by avoiding hot spots on the catalysts.
A second technique, also in the same direction, might be the use of supercooling carbon dioxide as a solvent, where you have a one-phase system in the oxidation. This is then a homogeneous reaction, and a catalyst may not be needed. I think we should point out that there is the potential of still further optimization of these processes.
Richard Alkire, University of Illinois: It is very exciting to hear of the need for new chemistry and also new processing. In chemical engineering at least, that has dropped away from the academic side over the years. In my opinion, in the earlier part of the century, chemical engineers in academics actually participated in the invention of new processes. When I think of the deemphasis over the years on the academic side coupled with the reduced industrial research investment, it seems to me that there is the potential for a serious disconnect which may already exist. If I think of trying to do chemical engineering as running across the street at any other university, I can imagine having difficulty engaging in conversation that might be helpful in the way you suggest is needed.
Yet you have mentioned certain research areas in which academics could make contributions. Could you say exactly what the interesting chemistry or engineering is that is needed here? Also, do you have any thoughts on how to engage that part in a useful way?
Leo Manzer: First, you have to have funding available in both chemistry and chemical engineering so that you have enough people available to discuss things with each other. This is the most important part of the process. Consider DuPont's work on the oxidation of butane to maleic anhydride. We worked on this for 15 years. In that situation, chemistry and chemical engineering were completely integrated with folks constantly talking together.
The engineering aspects of running an oxidation reactor in an anaerobic mode like that with lots of butane around, was very complicated. This is one of the biggest issues. If people actually do look at developing some of these other processes, it is scale-up that will be most difficult. The scale issues are incredibly complicated. There is a real opportunity where the funding is available for engineers and chemists to get together to work jointly on these projects. I don't know how to make this happen except to have money available to support your research programs and those of others. I mean, without the people there to do the research, it will never happen.












