
Page 83
5
Carbon Dioxide as a Feedstock
Carol Creutz and Etsuko Fujita
Brookhaven National Laboratory
This chapter is an overview on the subject of carbon dioxide as a starting material for organic syntheses of potential commercial interest and the utilization of carbon dioxide as a substrate for fuel production. It draws extensively on literature sources,1,2 and 3 particularly the report of a 1999 workshop on the subject of catalysis in CO2 utilization,1 but with emphasis on systems of most interest to us.
Atmospheric carbon dioxide is an abundant (750 billion tons of carbon in the atmosphere) but dilute source of carbon (only 0.036% by volume),3 so technologies for utilization at the production source are crucial for both sequestration and utilization. Sequestration—such as pumping CO2 into the seas or the earth—is beyond the scope of this chapter, except where it overlaps utilization—for example, in converting CO2 to polymers. Yet sequestration dominates current thinking on short term solutions to global warming, as should be clear from reports of this and other workshops.4,5 The net anthropogenic increase of 13,000 million tons of carbon dioxide estimated to be added to the atmosphere annually at present can be compared to the 110 million tons of CO2 used to produce chemicals, chiefly urea (75 million tons of CO2), salicylic acid, cyclic carbonates, and polycarbonates.1 Increased utilization of CO2 as a starting material is, however, highly desirable, because it is an inexpensive, nontoxic starting material. There are ongoing efforts to replace phosgene as a starting material.6 Creation of new materials and markets for them will increase this utilization, producing an increasingly positive, albeit relatively small, impact on global CO2 levels. The other uses of interest are utilization as a solvent and for fuel production, and these are discussed in turn.
PRINCIPAL CURRENT USES OF CARBON DIOXIDE
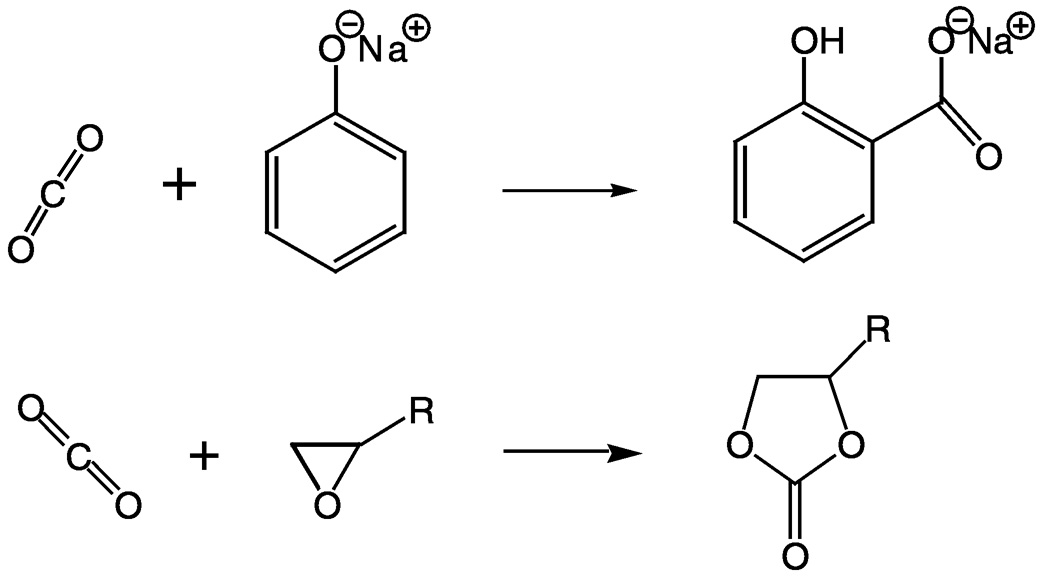
Urea synthesis is currently the largest use of carbon dioxide in organic synthesis. Urea, C(O)(NH2)2, is the most important nitrogen fertilizer in the world. Urea is also an intermediate in organic syntheses such as the production of melamine and urea resins, which are used as adhesives and bonding agents. Carbon dioxide is also used to produce salicylic acid, which is found in pharmaceuticals, and cyclic organic carbonates, high melting, but extremely high boiling solvents for natural and synthetic polymers such as lignin, cellulose, nylon, and poly vinyl chloride ( Figure 5.1). The latter are used extensively in
Page 84
~ enlarge ~
FIGURE 5.1 Examples of reactions involving carbon dioxide and leading to intermediates in industrial synthesis. Top: The reaction of carbon dioxide with sodium phenolate to produce sodium salicylate. Bottom: The reaction of carbon dioxide with an epoxide to make a cyclic organic carbonate.
the production of polyacrylic fibers and paints. Ethylene and propylene carbonates have many uses in chemical synthesis—among them reactions with ammonia and amines to form carbamates and subsequent reactions with diamines to yield di(hydroxyethyl) carbamates, which can react further with urea to form polyurethanes.
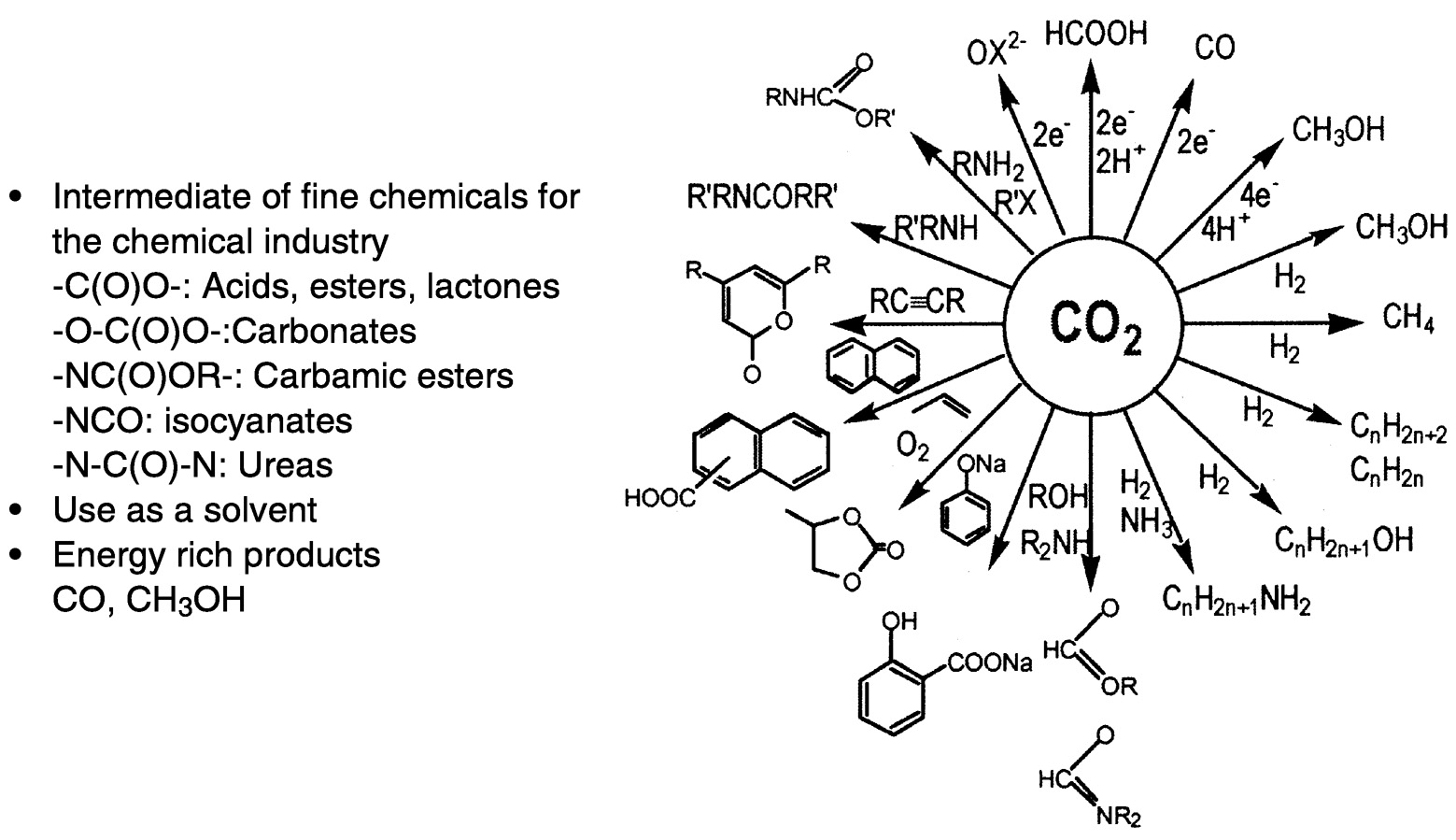
Figure 5.2 provides a broad summary of current and projected utilization of carbon dioxide. Reactions that use CO2 to produce organic chemicals or intermediates for the chemical industry are summarized from 6 to 11:00 o'clock on the diagram. The first examples, such as salicylic acid, are in current practice. Also included in Figure 5.2 are reactions that hold promise for extensive utilization in the future. Many of these involve insertion of carbon dioxide into Y—X bonds, often the C—H bond. The products of interest include esters, carbamic esters, salicylic acid, and cyclic carbonates. These reactions
~ enlarge ~
FIGURE 5.2 Utilization of CO2 in synthetic chemistry. SOURCE: Aresta (1998). 6
Page 85
commonly involve formal insertion of carbon dioxide into an X—H bond, as into the N—H bond in urea formation. Novel insertions under active investigation involve the incorporation of CO2 into polymers—polycarbonates, polypyrones, lactone intermediates, and polyurethanes. Of particular interest is the incorporation of carbon dioxide into polymers, an active area of research and one that is very promising for future applications.1,7 However, the impact of new materials and processes in this area will ultimately depend on market forces, a factor than can be frustrating to the researchers. Other reactions shown in Figure 5.2 are, from 2 to 6 o'clock, hydrogenation reactions and, from 12 to 2 o'clock, hydrogenations accomplished by electrons and protons—both directed toward fuel formation.
Carbon Dioxide as a Solvent
Supercritical carbon dioxide is a hydrophobic solvent that can replace organic solvents in a number of applications. Its critical temperature is 31 °C, and it has very low viscosity. When carbon dioxide is substituted for an organic solvent, solvent costs and emission of toxic organics can be reduced. Furthermore, separation of the products and catalyst can be controlled easily by changing the carbon dioxide pressure. Currently, supercritical carbon dioxide is used in caffeine extraction, dry cleaning, and parts degreasing. These processes can involve high-capacity plants of more than 22.5 × 106 kg per year in the case of decaffeination processes.8 Potential future or developing applications include utilization in food and pharmaceutical processing to defray future liability costs, production of pharmaceutical nanoparticles for injection, polymerizations,9 emulsion polymerization of water-soluble monomers, enhanced oil recovery, and homogeneous10 and phase-separable catalysis, including that based on ionic liquid solvents.11
REACTIVITY OF CARBON DIOXIDE
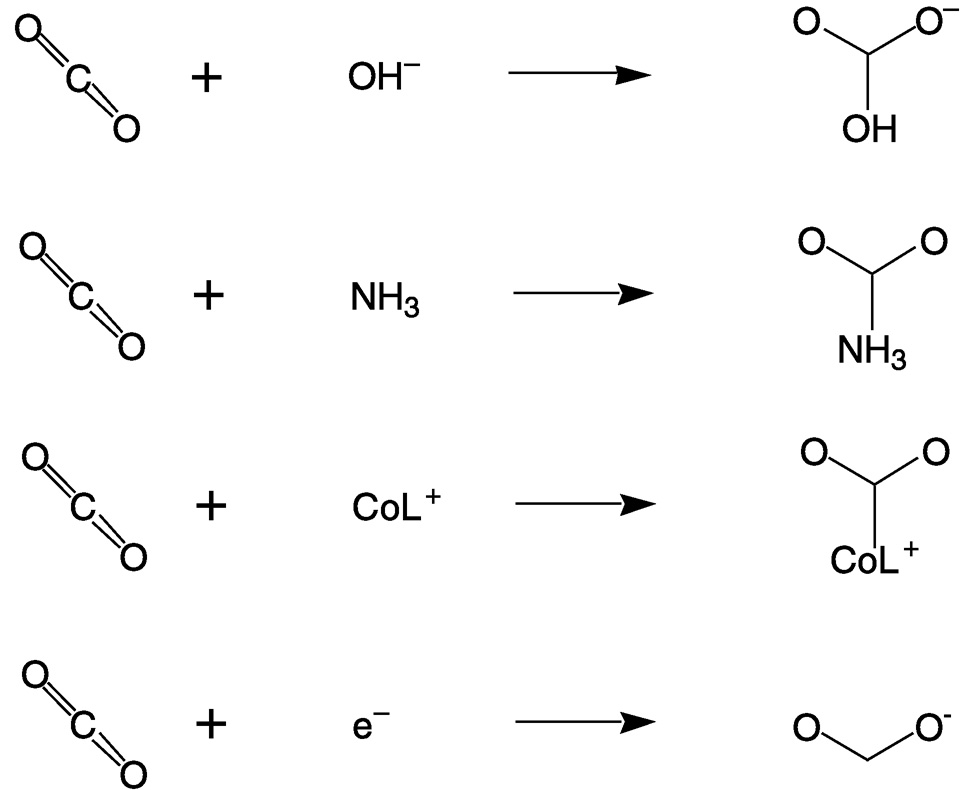
Carbon dioxide is a linear molecule in which the oxygen atoms are weak Lewis (and Brønsted) bases and the carbon is electrophilic. Reactions of carbon dioxide are dominated by nucleophilic attacks at the carbon, which result in bending of the O—C—O angle to about 120°. Figure 5.3 illustrates four very different nucleophilic reactions: hydroxide attack on CO2 to form bicarbonate; the initial addition of ammonia to CO2, which ultimately produces urea; the binding of CO2 to a macrocyclic cobalt(I) complex, which catalyzes CO2 reduction;12,13 and the addition of an electron to CO2 to yield the carbon dioxide radical ion. The first three also exemplify the reactivity of CO2 with respect to nucleophilic attack on the carbon.
Thermodynamic Barriers to CO2 Utilization
Carbon dioxide is a very stable molecule, and accordingly, energy must generally be supplied to drive the desired transformation. High temperatures, extremely reactive reagents, electricity, or the energy from photons may be exploited to carry out carbon dioxide reactions.
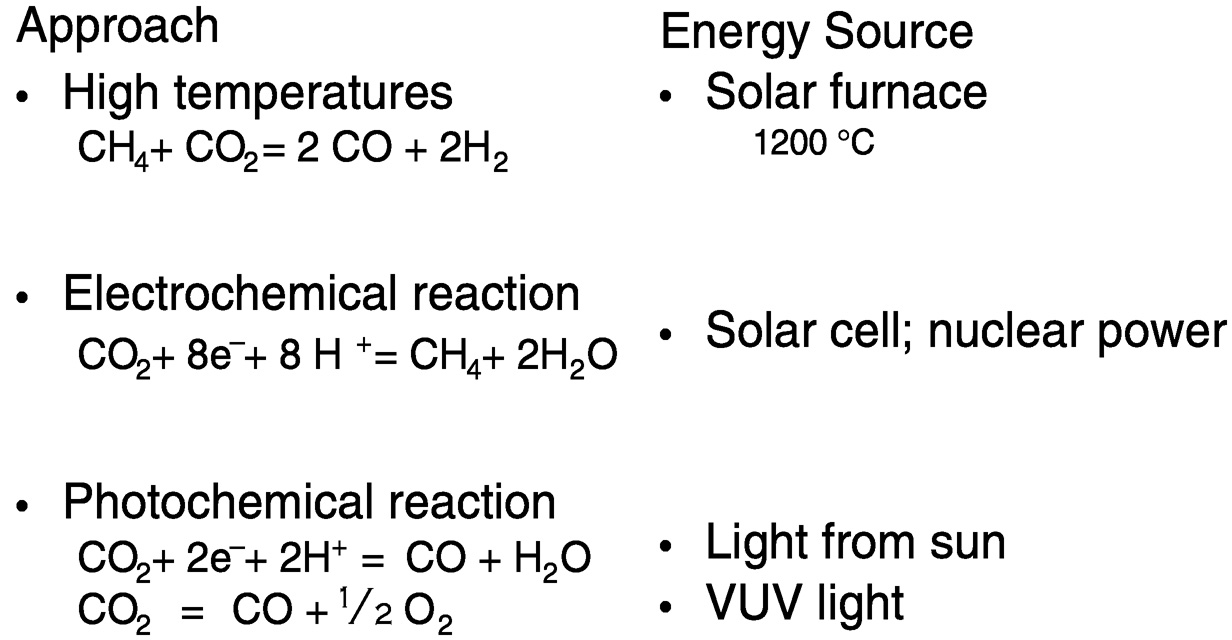
Figure 5.4 depicts reactions in which the energy source is renewable or nuclear. (“Renewable” refers to solar electric, wind hydroelectric, geothermal, solar thermal, and biomass-based energy sources.) The reaction CH4 + CO2 = 2 CO + 2 H2 is called the carbon dioxide reforming of methane. This reaction, if combined with metal production in situ, using solar furnaces to achieve the high temperatures needed (e.g., 1200 °C), could be used to significantly mitigate CO2 produced in cement, lime, and metal (iron, aluminum) production, which amounts to about 10% of total CO2 released annually.3 For electrochemical reduction of CO2 to methane, energy may be derived from a solar cell or
Page 86
~ enlarge ~
FIGURE 5.3 Reactions of carbon dioxide are dominated by nucleophilic attacks at the carbon atom.
nuclear power. Reduction may also be accomplished photochemically by utilizing a dye to absorb visible light, since CO2 itself does not. Interestingly, vacuum ultraviolet (VUV) irradiation of carbon dioxide yields oxygen and carbon monoxide.
Conversion of Carbon Dioxide to Fuels
Direct Hydrogenation
With abundant renewable energy sources, carbon dioxide can be converted to fuels by reduction to methanol or methane. The high energy density of carbon-based fuels and their availability as either gases, liquids, or solids are important reasons for the dominant position of fossil fuels in the current marketplace. Because the value of a fuel is based on its energy content and its ease of transport and
~ enlarge ~
FIGURE 5.4 Overcoming the thermodynamic barriers to CO2 utilization.
Page 87
storage, methane is less desirable than methanol because of its low fuel density and high cost of transport. Today, carbon dioxide is a by-product of fuel use, not a feedstock for fuel production. Conversion of CO2 to fuels using renewable or nuclear power produces no net emission of carbon dioxide (excluding CO2 produced by energy consumption in the reduction process), and it would complement the renewable production of fuels from biomass, which is likely to be insufficient to meet future world demands. Catalysis can play an important role in this area. The objective of such fuel production is to develop strategies for reduction of CO2 that can be adapted for use at different sources and yield fuel products widely utilizable with current and future technologies.
Hydrogenation of carbon dioxide to methanol is slightly exergonic, and to methane to a greater extent, because of the favorable thermodynamics of water formation (see Figure 5.5).
Catalysis of hydrogenations leading to N, N-dimethylformamide (DMF), formate, and hydrocarbons is being addressed successfully.10,14 Reduction to carbon monoxide is also useful when the CO hydrogen mixtures can be used to augment feeds in industrial processes such as ethylene and methanol production. Methanol, lower hydrocarbons (methane, ethane, ethylene, etc.), CO, and formic acid (HCOOH) have been prepared from CO2 and H2 using several different metal and metal oxide catalysts at elevated temperature and pressure.15 Selectivity and catalytic activity depend on the catalyst used (i.e., metal or metal oxide), its size, additives, support, temperature, CO2 to H2 ratio, and pressure. Copper on zinc oxide (ZnO) seems to be the most active catalyst for methanol production.15,16 A small-scale test plant with a production capacity of 50 kg per day of methanol was constructed at the Research Institute of Innovation Technology of the Earth to examine the performance of a Cu/ZnO-based multicomponent catalyst (Cu/ZnO/ZrO2/Al2O3/Ga2O3) under practical conditions.17 Selectivity for methanol production was found to be very high, and direct methanol production from CO2 may be commercially feasible with an inexpensive source of hydrogen.17 Use of hydrogen for this reduction chemistry is not, however, economically attractive at present because of its cost; inexpensive production of hydrogen by solar or nuclear power sources could radically alter this scenario.
Indirect Hydrogenation
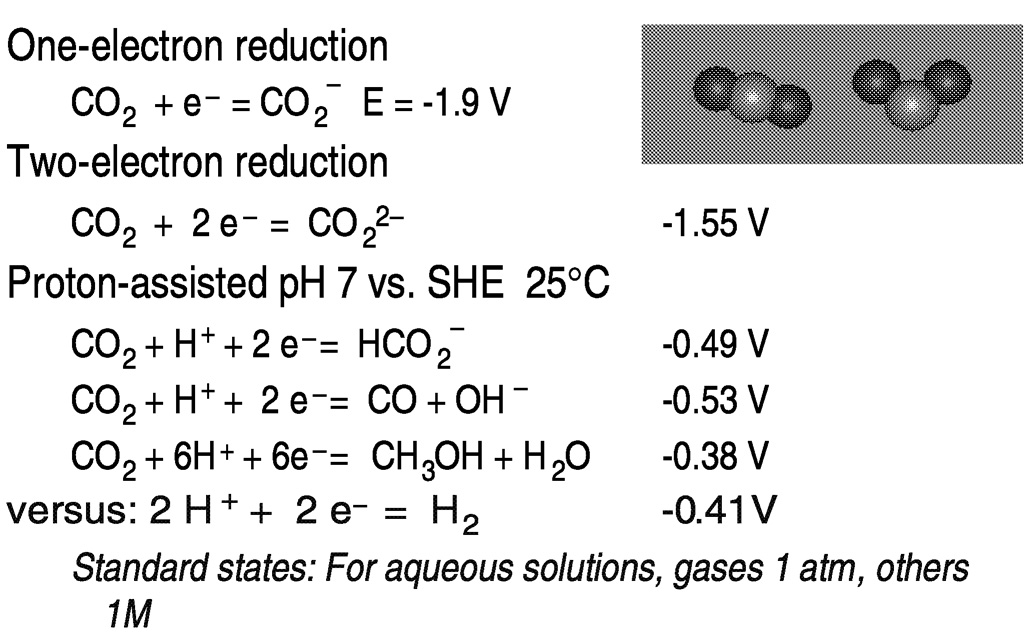
Hydrogen may be replaced by electrons and protons, available, for example, in electrochemical reduction in aqueous media ( Figure 5.6). One-electron reduction of carbon dioxide to the radical anion CO2−presents thermodynamic and kinetic barriers. In aqueous solution, the reduction potential is −1.9 V versus the standard hydrogen electrode (SHE).18 The barrier to outer-sphere electron transfer for the couple is large because of the very different geometries of the linear, neutral carbon dioxide and the bent radical anion.19 Thus, direct (uncatalyzed) electroreduction requires a significant overvoltage. As shown in Figure 5.5, the thermodynamic barriers are reduced by protonating the reduction product.13,20 However, because of the near equivalence of the hydrogen potential to that for the proton-assisted hydrogenation of CO2, reduction of H+ or H2O to H2 may also occur, depending on the system.
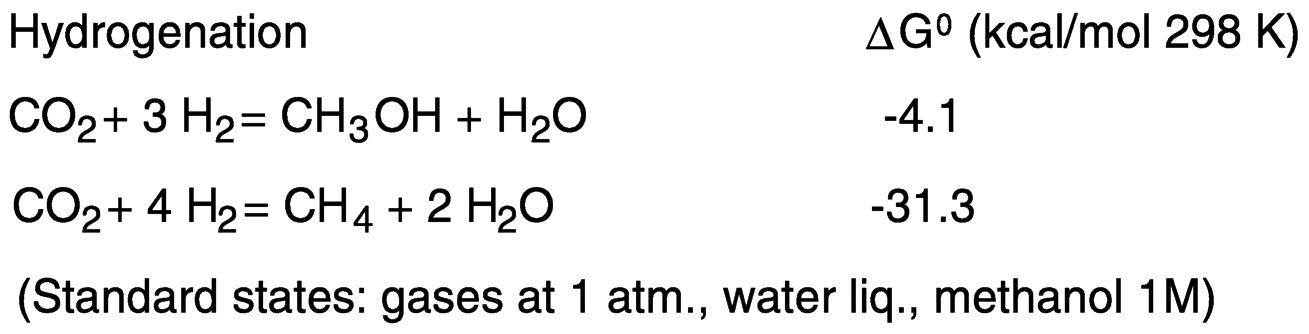
~ enlarge ~
FIGURE 5.5 Hydrogenation of carbon dioxide to methanol and methane.
Page 88
~ enlarge ~
FIGURE 5.6 Thermodynamic and kinetic aspects of CO2 reduction. SOURCE: Sutin et al. (1997),20 Fujita (1999).13
Electrochemical Reduction
As noted earlier, direct electroreduction is achieved at high overvoltage. An unreactive metal or carbon electrode produces carbon dioxide radical anion, which may undergo dimerization to oxalate or disproportionation to carbon monoxide and carbonate.21 By contrast, non-innocent metals, through active sites on their surfaces, can direct CO2 reduction to hydrogenated products at a much lower applied voltage because of the high efficiency of the heterogeneous catalysis. Particularly noteworthy is the work of Hori,22 which showed that copper produces high yields of methane from aqueous bicarbonate at 0 °C and high yields of ethylene at 45 °C. In these systems, the metal serves a dual role, both delivering electrons and stabilizing the reduced fragments. In the case of copper, the reduction is believed to involve the sequence shown in Figure 5.7. The metals ruthenium, cadmium, mercury, indium, tin, and lead yield formate; gold, silver, and zinc yield CO; while aluminum, gallium, platinum, iron, nickel, and titanium exhibit little activity.3 Other important areas of electrochemical reduction are homogeneous catalysis, surface modified by a “molecular” catalyst, and photoelectrochemical systems.
Homogeneous Catalysis of Carbon Dioxide Reduction
Homogeneous catalysts may fulfill the role of the surface metal catalytic sites in the above systems (for example, copper). Homogeneous catalysis is important in electrochemical reductions systems, as well as photochemical systems. Indeed the two approaches share many features, as discussed later.
Catalysts lower the overpotential for CO2 reduction by undergoing reduction at a potential (Ecat) less negative than that for direct CO2 reduction, binding CO2, or undergoing a second reduction at (or more positive than) Ecat. For homogeneous catalysis,21 two-electron reduction to give CO, HCO2−, or C2O42− is most frequently observed, and added proton sources may be required.
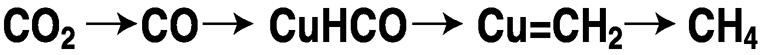
~ enlarge ~
FIGURE 5.7 Sequence of the electrochemical reduction of CO2 with copper to form methane.
Page 89
~ enlarge ~
FIGURE 5.8 Modes of binding carbon dioxide to transition metal centers.
Transition Metal Catalysis
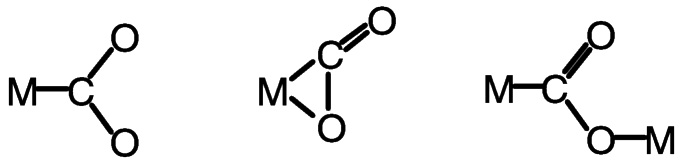
Binding of carbon dioxide to transition metal centers has been reviewed.23,24 Among the several modes in which the metal may bind the CO2 are the three shown in Figure 5.8, end-on, C η1; bridging, C, O η2 ; and bimetallic motifs in which one metal binds C η1 and another metal binds O η1.
Electrochemical Systems
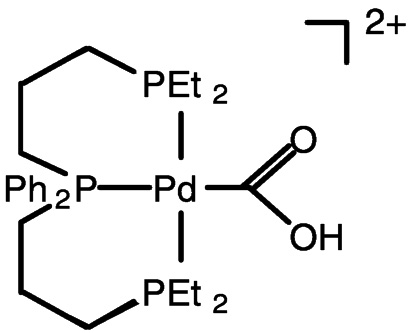
Electrocatalysis by transition metal complexes is elegantly illustrated by the work of DuBois and colleagues.25 In the absence of CO2, the palladium (II) complex undergoes two-electron reduction, but when CO2 is present, the one-electron reduction product binds CO2 (DMF as solvent) ( Figure 5.9). With added acid, carbon monoxide is produced catalytically from carbon dioxide. This chemistry likely involves the protonated carbon dioxide adduct (hydroxycarbonyl complex) shown in Figure 5.9 and Figure 5.10.26 Catalytic turnover numbers greater than 100 have been reported for this and related compounds. Some hydrogen is produced in parallel, evidently via a hydride complex.
Other systems for electrochemical CO2 reduction utilize transition metal complexes of nitrogen-containing (nickel and cobalt) macrocycles (including porphyrins and phthalocyanines) and (ruthenium, cobalt, and rhenium) complexes of 2,2'-bipyridine.27
Photochemical Systems28
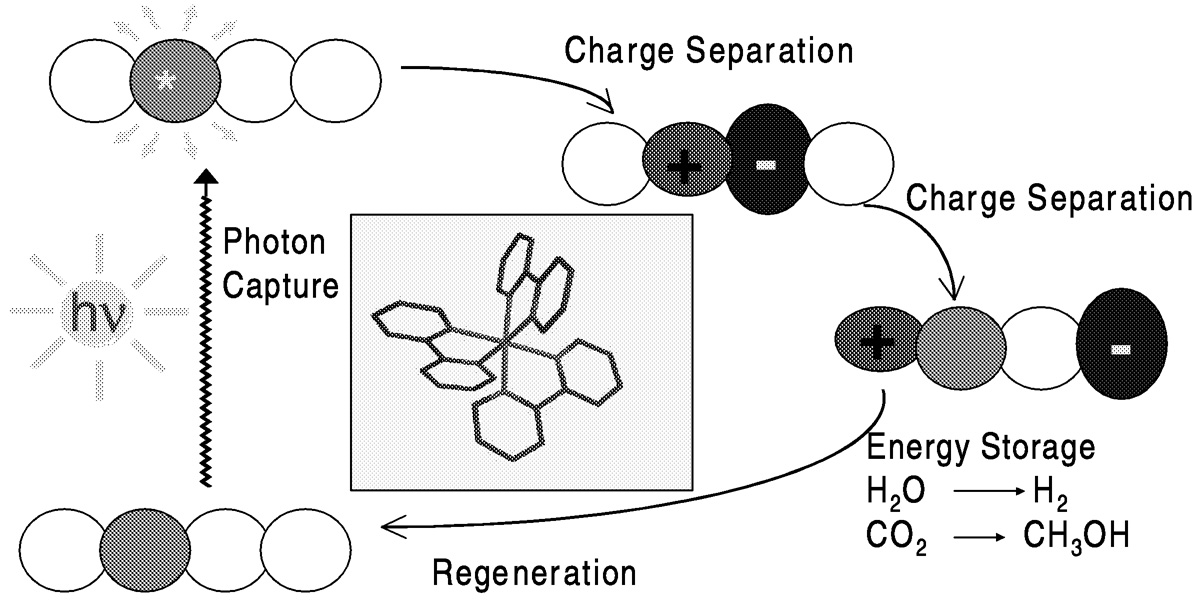
Photochemical reduction systems ( Figure 5.11) require efficient light harvesting, usually by a so-called dye or sensitizer, and efficient charge separation and energy utilization. Transition metal complexes, particularly tris(2,2'-bipyridine)ruthenium(II), serve as sensitizers. The overall reaction carried out must be a useful one. That is, in addition to carbon dioxide reduction, the complementary oxidation process (which provides the electrons) should be a desirable one. Both reduction and oxidation processes generally require catalysis. For carbon dioxide reduction, a number of the catalysts used in electrochemical systems are also effective in photochemical systems, as outlined below.
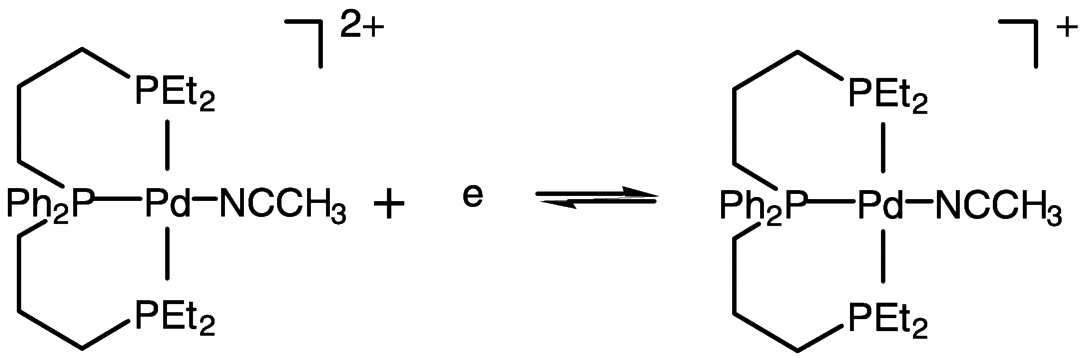
~ enlarge ~
FIGURE 5.9 Reduction of palladium (II) to palladium (I) in the presence of CO2
Page 90
~ enlarge ~
FIGURE 5.10 The protonated carbon dioxide adduct involved in reduction chemistry.
Comparison of Electrochemical and Photochemical Systems
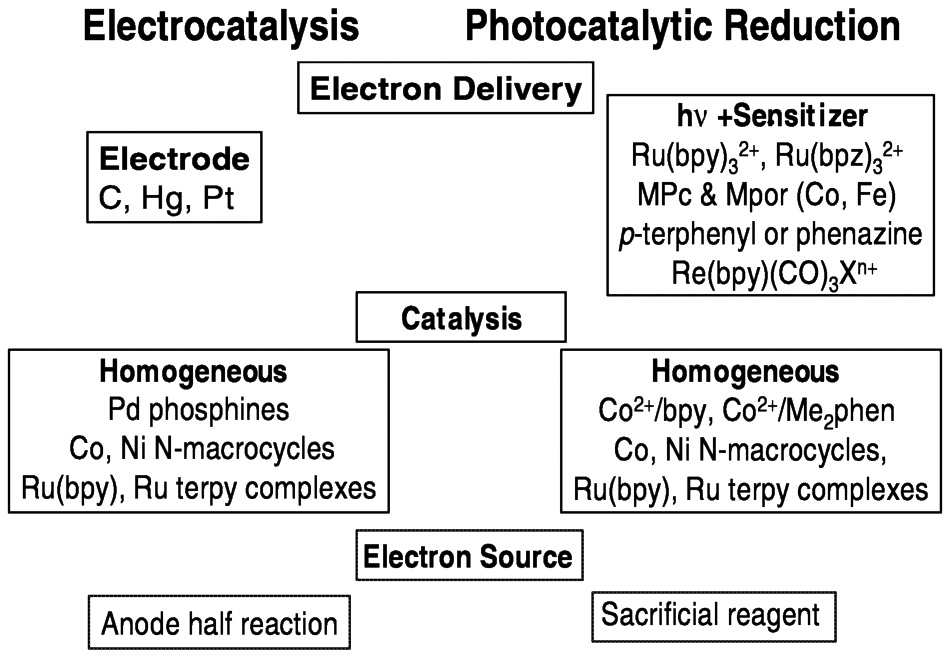
When the catalytic reduction of carbon dioxide is truly homogeneous (occurs in the solution), electrochemical and photochemical systems may have much in common. The means of electron delivery differs, of course, with photoinduced electron transfer processes serving the role of the electrode in the photochemical system. Many of the catalyst systems studied so far—cobalt and nickel macrocycle systems, for example—work in both electrochemical and photochemical systems. In both approaches, the ultimate source of these electrons is an issue ( Figure 5.12). Sacrificial reagents (generally organic compounds that become oxidized) are commonly used, and one of the challenges is to replace these reactions with processes that are less costly and wasteful. For aqueous systems, it would be highly desirable to use the water oxidation half-reaction, that is,
H2O = 2 e− + 1/2 O2 + 2 H+
for this purpose, so that the overall reaction would be
CO2 + 2 H2O → CH3OH + 3/2 O2.
The challenge remains the effective development and deployment of water oxidation catalysts.
At present, electrochemical reduction of CO2 yields carbon monoxide, formate, methane, and so forth, with good current efficiencies and, in photochemical systems, quantum yields of carbon monoxide (or formate or both) up to 40%.
~ enlarge ~
FIGURE 5.11 Photoinduced charge transfer with transition metal complexes.
Page 91
~ enlarge ~
FIGURE 5.12 Comparison of photochemical and electrochemical systems for reducing CO2
OPPORTUNITIES
There are many areas in which ongoing and future research can lead to new modes of carbon dioxide utilization. These include the following:
-
Utilization of CO2 in new polymers
Development and understanding of both homo- and heterogeneous catalysts for the following:
Electrochemical and photochemical electron sources in the presence of proton sources can avoid use of expensive H2, but both need:
1. Polymerization, hydrogenation, electrochemical and photochemical processes 2. Utilization of soluble and surface-anchored nanoparticles of metal and semiconductor clusters 3. Reactions or catalysis in supercritical CO2
1. Faster catalytic processes and more stable catalytic systems 2. Development of useful second half reaction (i.e., elimination of sacrificial reagent or useful anode reaction).
ACKNOWLEDGMENT
We thank D.L. DuBois for helpful comments. This research was carried out at Brookhaven National Laboratory under contract DE-AC02-98CH10886 with the U.S. Department of Energy and was supported by its Division of Chemical Sciences, Office of Basic Energy Sciences.
REFERENCES
1. , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , , and . Chem. Rev. 2001 , in press.
2. 1988 . Carbon Dioxide Activation by Metal Complexes. VCH : Cambridge .
3. , and . 1999 . Greenhouse Gas Carbon Dioxide Mitigation. CRC Press : Boca Raton, Fla.
4. , , , , , , , , , , , and . 1998 . Carbon Management: Assessment of Fundamental Research Needs. U.S. Department of Energy : Washington, D.C.
Page 92
5. , , , , , , , , , , , , , , , , , , , and . 1999 . Carbon Sequestration Research and Development. U. S. Department of Energy : Washington, D.C.
6. 1998 . Advances in Chemical Conversions for Mitigating Carbon Dioxide; Studies in Surface Science and Catalysis 114, 65-76.
7. , and . 1996 . Coord. Chem. Rev. 153, 155-174.
8. , and . 1998 . “Carbon Dioxide” www.AccessScience.com.
9. , , , and . 1999 . Chem. Rev. 99, 543-563.
10. , , and . 1999 . Chem. Rev. 99, 475-493.
11. , , , and . 1999 . Nature 399, 28-29.
12. ; , , and . J. Am. Chem. Soc. 113, 343-353, 1991 .
13. . 1999 . Coord. Chem. Rev. 185–186, 373–384.
14. , , and . 1995 . Chem. Rev. 99, 259-271.
15. . 1998 . Advances in Chemical Conversions for Mitigating Carbon Dioxide; Studies in Surface Science and Catalysis 114, 19-30.
16. , and . 1998 . Advances in Chemical Conversions for Mitigating Carbon Dioxide; Studies in Surface Science and Catalysis 114, 87-96.
17. , , , , and . 1998 . Advances in Chemical Conversions for Mitigating Carbon Dioxide, Delmon, B., and J. T. Yates., Eds. Elsevier : Amsterdam .
18. , and . 1989 . J. Phys. Chem. 93, 409-414.
19. , , and . 1985 . Inorg. Chem. 24, 433-439.
20. , , and . 1997 . Commts. Inorg. Chem. 19, 67-92.
21. , and . 1993 . Pp. 118-140 in Mechanisms of the Electrochemical Reduction of Carbon Dioxide Catalyzed by Transition metal Complexes, Sullivan, B.P., K. Krist, and H.E. Guard, Eds.; Elsevier : New York.
22. , , and . 1989 . J. Chem. Soc., Faraday Trans. 185, 2309-2326.
23. 1993 . Carbon Dioxide Binding to Transition-Metal Center. Pp. 19-67 in Electrochemical and Electrocatalytic Reactions of Carbon Dioxide, B.P. Anllivan, Amsterdam : Elsevier .
24. 1996 . Chem. Rev. 2063-2095.
25. , , , , and . 1996 . Organometallics 3360-3373.
26. , , and . 1991 . J. Am. Chem. Soc. 113, 8753.
27. , and . 1989 . Coord. Chem. Rev. 93, 245-268.
28. and . 2001 . Pp 88-126 in Homogeneous Redox Catalysis of CO2 Fixation, Balzani, V. Ed.; Wiley-VCH , Weinheim Vol IV.
DISCUSSION
Glenn Crosby, Washington State University: Carol, I have not been involved with the Department of Energy for several years in this kind of research, but what has happened to the level of funding for the utilization of, say, protons for promoting photocatalysis and photoelectron over the last few years?
Carol Creutz: I am going to defer to Bill Millman from Chemical Sciences.
Bill Millman, Department of Energy: Well, in flat-budget scenarios, it has essentially gone down approximately 1.5% per year over the last about six years. In real terms—in constant dollars—it is a significant percentage. If you look at staffing at the labs, it means about 25%. This is one of the effects of the constant budgets.
It is safe to say then that the effort in photocatalysis and photoelectric chemistry I observed and was involved in four or five years ago has not kept pace with inflation but has actually decreased significantly in absolute terms.










