
2
Keynote Session
This chapter contains summaries of the individual presentations from the keynote session of the Workshop. The objective was to hear from key stakeholders from research, clinical practice, regulatory agencies, and industry in order to provide their perspectives on innovation and invention in the medical device industry.
HISTORICAL PERSPECTIVE ON IOM’S ROLE IN PROVIDING A FORUM FOR DISCUSSION
Robert W.Mann, Sc.D.
Whitaker Professor Emeritus of Biomedical Engineering Massachusetts Institute of Technology
The inaugural national effort addressing the issues this Workshop is considering was the Committee on the Interplay of Engineering with Biology and Medicine (CIEBM),1 established in 1967 by the National Academy of Engineering (NAE) “to delineate the means by which the national engineering capability can be effectively applied to biology, medicine, and health services.” Collaboration between practitioners of medicine and engineering in the Institute of Medicine (IOM) was insured at its inception in 1970 by charter provisions2 de-
|
1 |
“An Assessment of Industrial Activity in the Field of Biomedical Engineering,” Appendix A, National Academy of Engineering, Washington, D.C., 1971. |
|
2 |
Charter section “II Membership 1. The membership of the Institute shall consist of persons selected from the fields of health and medicine…and from such other fields related to health and medicine as the…medical and biological sciences…and engineering.” The physical sciences are not specifically mentioned. |
fining the membership. In the early 1970s, the IOM Committee on Science Policy for Medicine and Health established as a priority interdisciplinary research collaboration between the life sciences and medicine and the physical sciences and engineering, including the innovation of medical devices. In the early 1980s, further activities facilitated interdisciplinary interactions between the physical sciences and medicine, including the formation of a Working Group on Interdisciplinary Collaboration, also supported by the Whitaker Foundation, and a Committee on Promoting Research Collaboration. Topics addressed by these groups included federal policy, the academic-industrial interface, the role of private foundations, and the role of university and teaching hospital structures in facilitating interdisciplinary research. Interdisciplinary Research: Promoting Collaboration Between the Life Sciences and Medicine and the Physical Sciences and Engineering was published in 1990, stating that, “The committee recognized two different motivations for collaborative research: (1) the desire to increase understanding of natural phenomena, and (2) the need to provide practical benefits.”
To address directly “practical benefits” in terms of new medical devices and explore important issues and interrelationships of engineering, medicine, invention, and public policy, the NAE and IOM, in their first major collaborative effort, jointly convened the symposium, New Medical Devices: Factors Influencing Invention, Development, and Use, in March 1987. The symposium brought physicians, engineers, and scientists together with industry executives, lawyers, ethicists, economists, and government officials to explore key factors that would influence development and use of innovative medical devices during the next decade. Symposium participants identified current trends in federal and private support of technological innovation, medical device regulation, product liability, and health care reimbursement. In addition, participants addressed important general issues, such as how to sustain technological innovation and health care quality in a rapidly changing health care environment, and how to encourage and support inventors.
After the highly successful symposium, in 1988 the National Academy Press published New Medical Devices: Invention, Development and Use, which addressed the three major themes: (1) innovation and use of new medical devices; (2) current trends in federal and private support of technological innovation, medical device regulation, product liability, and health care reimbursement; and (3) several perspectives on how these trends interact to influence the availability and appropriate use of new medical devices.
At the 1988 symposium, five inventors reported that basic science advances were of little direct relevance in their innovation of Technion’s Auto Analyzer, plasmapheresis, the pneumatic extradural intracranial pressure monitor, the electronic retinoscope, the first successful implantable cardiac pacemaker, and wheelchairs for the third world. Edward B.Roberts of the Massachusetts Institute of Technology (MIT) Sloan School, said “innovation in medical devices is usually based on engineering problem-solving by individuals or small firms, is often incremental rather than radical, seldom depends on the results of long-term
research in the basic sciences, and generally does not reflect the recent generation of fundamental new knowledge.” Medical device innovation was reported to be quite different from that of the pharmaceutical industry, where basic research is carried out in large organizations, generating fundamental knowledge in order to create radical drug innovations.
Has the intervening decade changed the earlier assessment, specifically with respect to traditional medical devices, those employing mechano-electrical-electronic-magnetic technologies? To get a sense of what the answer might be, Dr. Mann polled colleagues involved in devising new medical devices and consistently was told that once demand is recognized or anticipated and concept conceived, the process is intrinsically engineering problem solving, evaluation, improvement, and practical and economic manufacture, not to mention finding funding and addressing marketing issues. Clinical trials are expensive and take a long time. Certainly this process is true for medical devices widely deployed, for example, single-use endoscopic instruments, artificial hip and knee joints, stents, and intraocular lenses. The same process applies to even less common instruments, such as the left-ventricular assist device,3 and cochlear implants, and those under development, such as visual prostheses.4
Given the existing cornucopia of physics- and chemistry-based engineering science knowledge, and so many powerful processes and techniques—such as computer-aided design/computer-aided manufacturing, robotics, VLSI chips, and microfab—there is no need for designers of traditional devices to mount or seek basic research.
Dr. Mann offered two contemporary examples of what he calls traditional medical devices. The first is an epiretinal implant to stimulate the ganglion cells of the eye to reverse the progress of blindness in macular degeneration.5 Major problems cited by the innovators were the insertion into, and mechanical compatibility of the stimulating implant with the retina, which they describe as “like a sheet of wet Kleenex.” These are both basically design problems, and concern long-term biocompatibility. Dr. Mann’s second example involves a cardiac surgeon seated at a computer 3-D display of the patient’s heart, moving a manipulandum as he would during open-heart surgery to repair a defective heart valve. This complex employs systems integration and ergonomic design of stereoscopic imaging with optical magnification and tiny robots, combined with an endoscope inserted through centimeter-sized slits in the patient’s chest. The device, manipulated by the surgeon and aided by multi-sensory feedback, is minimally invasive.6
In the 1988 report, Dr. Mann’s foreword stated, “In my opinion, the research areas grievously underserved are interdisciplinary questions undergirding future medical devices. We have run the string of devices nostalgically described by our inventors. Future medical technology will increasingly require more fundamental understanding at the organ, cell, and subcellular levels, and it will be based on collaborative biological and physical science research.” At that time, a number of new areas were emerging, including biomaterials, biosensors, artificial organs, and functional neurostimulation. To today’s list can be added tissue engineering,7 developing biological substitutes for natural tissues—skin or cartilage for example—and ultimately organ transplants, interdigitation of molecular biology and engineering systems analysis through computational modeling of biological systems at the molecular level to understand metabolism, adhesion, mechanical contraction proliferation, differentiation, and molecule-to-cell and cell-to-cell signaling.8 The more holistic tissue engineering and more reductionist modeling will in time converge, leading to a more fundamentally based realization of medical devices, to have a profound positive capability to promote, regain, and extend human health.
The intervening decade has seen a dramatic increase in university programs, departments, and curricula in bioengineering,9 driven partly by the emergence of biology as a subject common to undergraduate education and partly by the generous and dedicated funding contributed to bioengineering and biomedical engineering programs by the Whitaker Foundation.10 The more than 70 biomedical engineering departments and programs in the United States have benefited greatly from the $540 million in grants from Whitaker in the past two decades, but how this large enterprise will be sustained when Whitaker spends itself out in 2006 as planned remains to be seen. Dr. Mann added that federal support for the area has always been modest and peer review committees have not been broadly cognizant of the merits of generous support of the field. The American Institute of Medical and Biological Engineering, composed of academic, governmental, and industrial practitioners with academic, society, and industrial councils, was inaugurated in 1992 to enhance the visibility of the field and lobby for more federal funding, especially from NIH.11 In April 2001, the National Institute of Biomedical Imaging and Bioengineering was established at NIH.
Interorganizational cooperation and coordination among the various professionals engaged in medical device development will certainly advance device realization. The Center for Innovative Minimally Invasive Therapy (CIMIT), a consortium of the Massachusetts General Hospital, Brigham and Women’s Hospital, Draper Laboratories, and Massachusetts Institute of Technology is one such example. CIMIT’s goal is to combine clinical and technological resources in order to generate, develop, and reduce-to-practice innovative and high-impact concepts in minimally invasive therapy that improve the quality and lower the cost of health care delivery. A west coast counterpart is Stanford’s Medical Device Network (MDN), which brings together physicians, engineers and scientists in the San Francisco Bay Area to encourage and facilitate invention, patenting, and early development of biomedical devices and instruments. A $150 million grant to Stanford from the founder of Netscape is intended to support MDN.
Biomedical device innovation—in terms of skilled and committed people, organization, and resources—has advanced significantly since the 1960s NAE-CIEBM efforts and the 1987 NAE-IOM study.
A REGULATORY PERSPECTIVE
David W.Feigal, Jr., M.D., M.P.H.
Director, Center for Devices and Radiological Health Food and Drug Administration
Devices really span the entire culture of FDA because there are devices that are combined with drugs or biologics and there are biologics that are devices. The FDA Center for Drug Evaluation and Research (CDER) is driven by efficacy and exclusivity in drug development, as well as safety. The Center for Biologics Evaluation and Research (CBER), with more of a biotechnology focus than CDER, differs from the latter in that it has limited ability to confer exclusivity. All of FDA grapples with the definition of “biologics.”
The Public Health Service Act has not been substantially modified since 1944; the existing definition does not even mention bacterial products. The Act says, “products analogous to viruses,” and bacterial products are covered on that basis. With biological products, there is a tremendous sensitivity and fear of infectious diseases. Fear of infection is one of the challenges facing xenotransplantation, for example. However, the actual disease transmission rate with blood products today is lower than it has ever been.
One of the things that shapes the device industry is the fact that there is a wide diversity in risk. In addition, devices face the most detailed laws for therapeutics, down to minute specifications of some of the post-marketing features. Device law has authorities that do not exist for other products, such as true recall and product tracking.
The Center for Devices and Radiologic Health (CDRH), which regulates devices, has its cultural origins in responses to unsafe manufacturing practices of the past, false and misleading advertising, and fraud. To address manufacturing
fraud, FDA developed good manufacturing practices, and for laboratory fraud it created good laboratory practices. Good clinical practice is one of the set of the regulations that involve Institutional Review Boards (IRBs), informed consent, record keeping, and ethical treatment of patients and research subjects. In addition, FDA has guidance for tissue screening, and even poor regulatory practice, turning the remedy upon itself to develop good review practices. In the recent past, FDA has made efforts to speed the approval process, responding to criticism from industry, Congress, and the public that it took too long. User fees placed stringent performance criteria not just on product review times but also on the review process itself.
An issue that reviewers and FDA take very seriously is the Food, Drug and Cosmetics Act requirement that a minimum level of quality of evidence is necessary to make some decisions. What is not at all intuitive to physicians is that the advertising standard for manufacturers having an approved claim is higher than that for the practice of medicine. That is why off-label medicine is allowed and physicians have to make choices.
FDA standards for biologics refer to safety, purity, and potency. Potency is a form of efficacy. Other standards require products to be unadulterated and not misbranded. Prior to 1962, devices were regulated as a drug. One device standard requires well-controlled investigations and other valid scientific evidence sufficient to determine effectiveness. That provides a lot of flexibility in the device area, more than exists in the drug standard. Devices explicitly face a risk-based standard, where the type of evidence depends on the classification of the devices. Humanitarian device exemptions are an example where the standard is changed for a specific area. Changes in technology have influenced FDA’s view of product development. Sometimes the technology is embedded in the products themselves, for example, high-throughput screening, rational drug design, bioengineering, and miniaturization.
The culture of industry-FDA interactions has changed; there are more modular and agreement meetings and increased emphasis on determining least burdensome regulatory paths. Another aspect of regulatory change has been to put more emphasis on special populations, meeting the needs of children and the elderly, and making sure that women are adequately studied. Other changes in the process that have occurred include more transparency, harmonization with Europe and the Health Care Financing Administration (HCFA),12 and responding to new laws. Increasingly complex communications exist among FDA, sponsors, manufacturers, research institutions, IRBs, trade associations, professional societies, third-party payers, and, in some cases, the Federal Trade Commission, depending on the nature of the product.
FDA is also seeing more complex conflict-of-interest situations, in which there may be an investigator-manufacturer-innovator with responsibilities to his or her university, or who may have Cooperative Research and Development
Agreement with NIH. This person might also be the health professional who is taking care of the patient at the same time.
Another complex scenario for FDA occurs when a health facility becomes the manufacturer. In the setting of in vitro diagnostics, the local clinical laboratory might decide to develop a unique test (the regulatory nickname for this is “home brew”). FDA traditionally has stepped back from oversight in this area, in part because it could overwhelm its resources, and in part because the Clinical Laboratory Improvement Act provides some supervision of these laboratories. This is an especially hot topic in the area of genetic testing, where laboratories that have specialized in parts of the genome do not intend to become a manufacturer in the usual commercial sense. They will offer the test at one site. FDA still has jurisdiction over these facilities, but it challenges the old-fashioned paradigm in which a manufacturer is making a relatively limited number of products and shipping them nationwide. Another area receiving a lot of attention is the health facility that refurbishes single-use devices. They are manufacturers and thus are subject to FDA oversight.
It is crucial to remember that FDA was established at the time of Henry Ford’s vision of the mass manufacturing of products, which assumed a standardized unit would be sold to everyone. Today, FDA increasingly faces individual products that are customized for the individual, just as customers get personalized coupons at the supermarket checkout based on a computer list of previous purchases.
Every business day, the device industry introduces 50 new products into the marketplace. Of those 50 new products, half are exempt from any type of premarket application. In 1998, CDRH reviewed over 4,500 applications. New drug applications and biologics license applications average between 2.5 and 5 man-years of review time. The amount of time and the resources that FDA dedicates to device approval is already quite low. There are about 900 different device types that FDA could write guidance about, given the proper resources.
One hundred years after the creation of FDA, the challenges are both the same and different. The Internet has replaced magazine ads as the home of the “patent nostrums.” Interstate commerce has become international commerce. New products are still developed for mass markets by large corporations, but increasingly new products are tailored for small markets, sometimes even the individual patient. Eight thousand United States device manufacturers are joined by thousands of clinical laboratories and hospitals in developing custom diagnostics, implantables, and crafting new devices from tissue. Surgery and clinical pathology are blending into manufacturing. Instead of large manufacturers with few products, there are small manufacturers with thousands of variations on custom products. Mechanization, the great hope of the last century, has been replaced by information and the promise of genomics. Innovation and consumer protection are allies. Rapid change requires confidence and assurance of the integrity of the regulatory process.
AN INDUSTRY PERSPECTIVE: CHALLENGES IN THE DEVELOPMENT AND REGULATION OF DRUG-DEVICE COMBINATION PRODUCTS
Tobias Massa, Ph.D., DABT
Executive Director, Global Regulatory Affairs
Eli Lilly and Company
Pharmaceutical manufacturers have traditionally used devices for delivery of parenteral administration of drugs. In the United States, devices such as syringes and infusion pumps are approved by the FDA Center for Devices and Radiological Health (CDRH) as 510(k) applications. In more recent years, disposable and reusable devices for administration of drugs have been approved. These have offered the benefit of convenience and ease of use for patients faced with chronic (and in many cases lifetime) multiple daily injections. When sold together, these drug-device combination products are most often regulated and approved by the Center for Drug Evaluation and Research (CDER).
Drugs for the treatment of asthma, chronic obstructive pulmonary disease, and seasonal allergic rhinitis have made extensive use of metered dose inhalers (MDIs) and dry powder inhalers (DPIs). These represent true drug-device combination products that cannot be developed independently of one another because the dose administered to the patient is dependent on the drug and functional characteristics of the device. The use of MDIs and DPIs currently is expanding beyond the treatment of disorders limited to the respiratory system. Combination products for the systemic distribution of drugs such as insulin, parathyroid hormone (PTH), growth hormone, and other proteins utilizing pulmonary administration are under development. The advantages of inhalation as a route of administration, compared with parenteral administration, are obvious. It is anticipated that the use of the pulmonary route of administration will increase as the technology associated with these dosage forms improves and the issues surrounding their development and approval are addressed. Such drug-device combination products challenge the regulatory system’s approach to review and approval.
Industry and health authorities readily admit that these products pose unique challenges. The dose given to the patient, and therefore the safety and efficacy of the product, is dependent not only on the formulation of the drug product, but also on the performance characteristics of the device. Together, these determine the emitted dose and particle size distribution, and hence the respirable dose given to the patient. One must therefore consider the “product” to be the formulation and the device, which would include the container, valve, actuator, and any associated protective packaging. Development and regulation of these products is complicated further by the variety of devices available. Each is unique and raises specific issues that must be addressed to regulate these products adequately.
Until recently, regulation of MDIs and DPIs occurred exclusively in CDER, as only products intended to treat respiratory disorders utilized these products. CDER acted as the primary reviewer of the investigational new drugs (IND) and new drug applications (NDA) associated with these products, with consultation
from CDRH to insure that the device portion of the combination product was developed properly and met appropriate regulatory standards. Thus, a manufacturer had to address the concerns of more than one office within FDA.
Although MDIs were first introduced in the late 1950s, there still is no comprehensive, approved guidance for their development. Prior to November 1998, manufacturers obtained advice from FDA on a case-by-case basis for each product under development. Although not considered by industry to be directly applicable, manufacturers have been asked to comply with certain aspects of the Reviewer Guidance for Premarket Notification for Anesthesiology and Respiratory Therapy issued in 1993. This process for obtaining guidance was considered highly unsatisfactory, as manufacturers perceived that it resulted in inconsistent, arbitrary, and unnecessarily conservative regulation of these products. Furthermore, United States regulations seemed inconsistent with those of other regions, such as Europe. Thus, the challenge faced by regulators and industry is agreement on a harmonized guidance that provides for adequate and sufficient control to assure safety and efficacy while not being overly conservative.
In November 1998, FDA published “Guidance for Industry on Metered Dose Inhalers (MDI) and Dry Powder Inhaler (DPI) Drug Products: Chemistry, Manufacturing and Controls Documentation,” the first comprehensive guidance for these complex dosage forms. The guidance represents a compilation of the advice given by the FDA Pulmonary Division over the past 10 years and is based on the agency’s experience in reviewing numerous applications for these products.
Industry agrees with many of the points made in the draft guidance. MDIs and DPIs are complex products, and they require special controls not used for the development and manufacture of other dosage forms. There are a number of issues, however, about which there is significant disagreement. There is general industry consensus that a number of the specifications proposed in the draft appear to be overly restrictive and do not offer sufficient flexibility to cover the variety of devices currently approved or under development. Limits for some specifications appear to be inappropriately tight without sufficient scientific justification. Some of these requirements are unique to FDA for these products.
The draft guidance suggests a “one size fits all” approach to several significant specifications, notably particle size distribution and content uniformity (referred to as “dose uniformity” in the European Union [EU]). The United States requirements for content uniformity are highly prescriptive and apply to both MDIs and DPIs. They are based on assumptions about manufacturing and analytical capabilities rather than demonstration of safety and effectiveness. The dosage form must meet the criteria in the guidance relative to the dose claimed on the label, and there is no provision for analysis of outlier test results. The EU guidance takes a more rational approach and suggests that the specification should be based on the data from material that was found to be safe and effective in clinical studies and on a reasonable assessment of potential variability in manufacturing capability and analytical methods. The United States requirements punish manufacturers who have the ability to control and monitor the
manufacturing process carefully. The approach proposed in the draft FDA guidance is inconsistent with the principles for setting specifications in the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) guidance Q6A.
The FDA guidance recommends specifications for spray pattern and extractable profiles. Neither of these is required in the EU guidance for DPIs. The FDA has suggested that spray pattern is an essential quality control parameter to assess valve and actuator function of MDIs. Specifications for size and shape of the spray pattern are recommended. It is acknowledged that one would not want the product emitted from the device to be presented to the patient in a narrow stream that would not allow proper inhalation of the dose. However, it has not been demonstrated that changes or differences in spray pattern are medically relevant; therefore, setting a tight specification for this parameter appears inappropriate. It has been suggested that this be addressed in development and correlated to dimensions of the device (valve and or actuator) that can be measured as a quality control test. If a specification cannot be avoided, a qualitative limit test might be more appropriate unless a quantitative limit can be scientifically justified by FDA.
The requirement for extractable profiles is also of concern. FDA has recommended that the elastomeric components of MDIs and DPIs undergo extraction in a number of different solvents to determine whether any constituents of the components can be extracted or leached by the solvents. The “fingerprint” of the extractables is expected to serve as a QC release test to insure that the supplier of the components has not made any changes in the composition of the constituent materials. It has also been stated that this test is required to insure patient safety. There are many objections to this line of thinking. First, many of the components are not in contact with the formulated drug, or are in contact with the formulation for very short periods of time upon actuation of the device during dosing. The amount of any potential extractables would be small relative to what could be found in a simulated extraction study. This test has little relevance for DPIs, which do not contain any solvents. Lastly, the amount of material extracted in such studies is rarely found to be at toxicologically relevant levels. Industry has suggested that this test is appropriate for use in development of MDIs to determine whether any potentially toxic extractables are present at levels that might be of concern. There appears to be lack of sufficient scientific justification for this requirement as a routine quality control test. The draft guidance also requires determination of stability under a number of conditions that go beyond the ICH stability guidance Q1A.
Companies developing pulmonary dosage forms are frequently asked to comply with certain requirements in the CDRH Guidance for Premarket Notification Submission. This guidance is written to regulate devices intended for use in operating rooms and hospital settings. It was not written with consideration of drug device combination products. For example, these products are expected to pass a fluid spill resistance test, also known as drip testing. The test involves
pouring a large quantity of water over the device, after which the device is expected to meet its functional release testing. It is hard to imagine why this test should apply to dry powder inhalers. It seems much more appropriate to label these products with a warning to keep them away from wet environments. The CDRH guidance also prescribes temperature, pressure, and humidity extremes under which mechanical performance must be demonstrated. Such extremes are not specified in EU guidance for DPIs.
The draft FDA guidance also prescribes extremely tight specification for impurities in propellants used in MDIs. These limits were developed based on process and analytical capabilities of the propellant suppliers, not on toxicological assessment. In many cases, the limits are thousands of times lower than the no-effect toxicological limits for the impurities in question and are much tighter than current compendial limits. Given that these propellants have been used for many years in MDIs, one must ask why the limits have been set so low. FDA has stated that the tight limits are necessary due to the issues surrounding the short supply of CFC propellants (due to the phase out of these materials per the Montreal Protocol) and their concerns with stockpiling, recycling, and illegal importation. While these concerns may be valid, the tight limits appear to penalize manufacturers who adhere to regulations and guidance. A more appropriate method to deal with such concerns might be to provide severe penalties for violation of such regulations if they are indeed such a threat to public health.
The most difficult provision in the FDA draft guidance is the requirement that the commercial drug device combination be used in pivotal clinical trials to determine safety and efficacy. FDA strongly advises against changes to the device once the pivotal clinical and stability studies are initiated. When combined with the requirement for 2-year safety studies of products such as insulin and PTH, the use of the final commercial product in these trials is a significant issue for manufacturers. It requires that commercial development be completed long before the product has been shown to be safe and effective and therefore commercially viable. This policy also creates a system in which device optimization may not occur until after product approval due to severely compressed time lines to have a commercial product by Phase 3. Unlike drugs given by other routes of administration, there is no provision to conduct bridging bioequivalence studies for pulmonary combination products. FDA has stated that it is difficult, if not impossible, to demonstrate the bioequivalence of products delivered using pulmonary administration. This position has resulted from their experience with devices used to treat asthma and COPD. The low doses of the steroids and beta-blockers used in these products make determination of blood levels virtually impossible. The sponsors must rely on clinical trials using pulmonary function tests to demonstrate equivalence. This is difficult at best, as evidenced by the paucity of generic oral and nasal inhalation products. However, this situation might be different for some of the protein products currently under development, such as insulin, PTH, and growth hormone, which may have adequate surrogate markers to allow for determination of bioequivalence. The guidance should allow sufficient flexibility to accommodate this possibility.
It is clearly recognized by industry that these products are unique, in that the dose seen by the patient is dependent on the formulation and the performance characteristics of the device. They require equally unique specifications to ensure that a defined dose can be administered reproducibly to the patient and that the combination product is safe and effective in patient use. There is a need to balance setting reasonable product specifications with protection of public health and safety. The current system will continue to result in specifications being set unreasonably, resulting in batches of product being rejected at release or failing on stability unnecessarily. Harmonization of United States requirements with those of other regions, particularly the EU, is also necessary to insure adequate utilization of resources and avoid unnecessary clinical and in vitro testing.
AN EVALUATOR’S PERSPECTIVE
Joel J.Nobel, M.D.
President, ECRI
and
Jeffrey C.Lerner, Ph.D.
Vice President for Strategic Planning, ECRI
Clinical acceptance of new medical devices, drugs, or biotechnologies and overcoming the hurdles of coverage and payment reimbursements depends heavily on the results of clinical trials and technology assessment. ECRI (formerly the Emergency Care Research Institute) is a non-profit health services research organization that for 30 years has evaluated a wide range of medical products, ranging from complex imaging systems to anti-needle stick devices and blood chemistry analyzers and critical care monitors to surgical gloves. ECRI’s technology assessment program focuses more broadly on drugs, devices, biotechnologies, and medical and surgical procedures, examining efficacy, safety, cost-effectiveness, and, in some cases, ethical and legal issues associated with a specific technology. While ECRI operates at financial arm’s length from industry, it also works closely with industry to resolve differences in views, resolve hazards, and improve products, and for the most part, in a climate of mutual respect.
Clinical trials data is a critical element in evaluation or assessment, regulation, or reimbursement, and so ECRI has about 30 years of experience in examining peer review clinical studies, most of which have emerged from academic institutions, and ECRI examines these studies in a way that is far tougher than the original peer review process. Most peer-reviewed journal articles emerging from the academic medical community simply will not support evidence-based medicine. They do not provide evidence to support decisions about what works, how well it works, and whether or not it is worth doing.
The first step in technology assessment is to collect information on all that is known about a technology. Typically, less than 5% of peer-reviewed journal articles in oncology or surgery can withstand serious scrutiny and stand up well over the years. It is a bit better in the cardiovascular literature. Financial, psychologi-
cal, career, or other incentives can distort the process. In addition, responsibility for clinical research is too diffuse. While this has certain values, it also dilutes responsibility and in doing so makes accountability hard to pinpoint. Even when clinical studies do get it right, the results are often ignored. Electronic fetal monitoring has been repeatedly shown to have little benefit, yet it is ubiquitous.
Another factor is that research in academic centers is not adequately managed. An academic institution produces many intellectual products and some patented physical products, but it lacks any analogue of a quality control manager typical of industry or research laboratories. Academic institutions lack requirements for basic elements like retention time for research records, control of laboratory notebooks, back-up requirements for computer data, data auditing, or any core quality measures typical of most other types of institutions doing research or producing goods.
Research funding sources and funding relationships distort motivation, method, thought processes, results, and presentation. And, whatever its virtues, maintaining the value of academic freedom takes precedence over any type of institutional oversight and responsibility for the quality of research.
Clinical research today deserves examination and reform at a fundamental structural level. Disclosure of research funding sources is essential. The disclosure certification has to ask questions in a very thoughtful way because many types of remuneration have been designed to be concealed. Clinical research needs a code of ethics to which investigators should make a contractual commitment, preferably one with penalties for violation. Clinical research in academic institutions desperately needs ongoing quality management to improve the quality of the studies undertaken by the majority of researchers who are honest.
INNOVATION AND INVENTION IN MEDICAL DEVICES: IMPLANTABLE DEFIBRILLATORS
Glen D.Nelson, M.D.
Vice Chair, Medtronic, Inc.
Approximately 25 million Americans today benefit from therapeutic implants. Because of the site-specific nature of device therapies and method of action, there are few, if any, metabolic side effects or interactions. In contrast to pharmaceuticals, implants are typically immune from patient compliance problems. This holds implications for the overall quality of outcomes for individual patients and patient populations and more easily allows researchers to measure the results.
Implants are indicated only when simpler therapeutic alternatives do not exist or are markedly less effective. For example, an NIH-sponsored, multicenter, unsustained tachycardia trial clarified the risk of sudden cardiac death in certain patients who had suffered a previous myocardial infarction. The risk of sudden death was found to be 32% at 5 years for patients who had a history of coronary heart disease, decreased heart function, and short episodes of non-
sustained ventricular tachycardia, even if frontline medications were administered.13 In contrast, susceptible patients who received an implanted cardio-defibrillator exhibited a 74% reduction in sudden cardiac death compared with those patients receiving medications only. According to the study, if the findings were applied in clinical practice, up to 65,000 lives would be saved.14
Reimbursement for such devices continues to be a significant barrier. Provisional coverage following FDA approval or clearance for PMA devices will speed the availability of technology when it has real value. A secondary effect of reimbursement pressures is a reduction in the amount of public financing available to small start-up companies. This has a restrictive effect on researchers’ ability to develop leading-edge technology. Cost-containment pressure focuses on event costs, and fails to assess the relative comprehensive costs of therapeutic alternatives over the entire course of the disease and over a patient’s life.
Economic models are not well developed, so focusing on the event costs leads to the view that technology is the culprit in health care cost escalation. Economic models tend to ignore broader social and economic benefits as patients return to self-reliance and family members or caregivers are relieved of their support roles. In addition, fear of litigation has often limited researchers’ ability as manufacturers to produce new technology because material manufacturers are afraid of the litigious circumstances.
Medical devices are often mistakenly perceived to follow the pharmaceutical model. Unlike pharmaceuticals—where therapeutic formulation remains essentially unchanged for the commercial lifetime of the agent—medical device technologies undergo continual and progressive evolutionary improvement. For example, defibrillators were developed over a period of 10 years at Medtronic, yielding a series of improvements in the basic device. The most striking improvement is significant size reduction, a result of breakthroughs in power sources, microelectronics, new capacitors, and packaging technology. Size is important to patient comfort. Achieving smaller size without sacrificing capability also reduces the potential for certain complications, including erosion of the implant, which is particularly important in slender people or children. What is even more clinically significant is that device longevity has been extended by years from those first generations from 2 or 3, to 10 or 12 years. Breakthroughs in defibrillation stimulation patterns and lead technology have removed the requirement for opening the chest to implant the leads to use these devices, resulting in a five-fold reduction in the mortality risk of the procedure. Arrhythmia detection algorithms have been enhanced to minimize false positives and negatives. Detection and stimulation advances have led to therapies that can block
the emergence of the most serious arrhythmias and ventricular afibrillation and literally overpace the arrhythmias before you get to the end stage of having to deliver a shock. Finally, onboard EKG storage capabilities provide cardiologists with important information on rhythm detection, therapy effectiveness, and help to guide the ongoing therapy. Figure 1 illustrates how cost-effectiveness for medical devices typically increases with subsequent device generation.
The first generations of implantable defibrillators were marginally cost-effective. Within a few years, cost-effectiveness improved substantially as a result of lower power microelectronics and advances in power source energy capacity. The advances extended the lifetime service of the defibrillators and delayed markedly the need for replacement devices and the attendant costs. Advances in lead technology and stimulation patterns in the 1990s made practical the implantation of these devices via a transvenous route.
Interestingly, although the event costs of implantation are substantial (estimated at around $45,000, including the cost of the devices and the hospitalization), some studies suggest that the current technology generations represent cost savings versus the alternatives, and that is a difficult analysis because often the endpoint with this particular device is death. This cost reduction is the result of continual improvements in efficacy brought about by progressive advances in
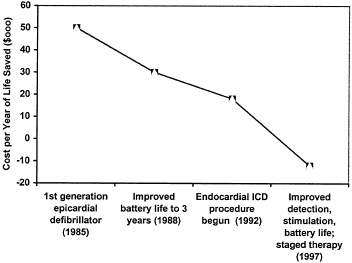
FIGURE 1 The impact of technological evolution on cost-effectiveness of implantable cardioverter-defibrillator.
arrhythmia detection and stimulation algorithms and further extensions of battery life, and also because of the multifunctional capabilities of today’s devices that permit them to adapt automatically to changes in the patient’s clinical and physiologic status.
These technological advances, in combination, have markedly reduced unplanned hospitalizations as well as dependence on costly concomitant medical treatments. While this example relates specifically to implantable pulse defibrillators, the pattern of progressive cost-effectiveness and improvement resulting from next generational technological and technique advances is a hallmark for essentially all medical device technology. Thus, each new generation is almost invariably more cost-effective than its predecessor.
Patients derive demonstrable benefit from a process that encourages the prompt introduction of the next generations of medical devices. These benefits include substantial reductions in morbidity and mortality, enhanced efficacy, fewer complications, and greater transparency between the normal condition and the treated circumstance.
Researchers will continue to see value-added advances in medical device therapies that are currently serving patients. These devices will approximate lost natural function and exhibit intrinsic capabilities that will allow them to automatically adapt to changes in the underlying condition of the patient. Devices will become more biological. Prosthetic heart valves for instance, will probably possess physical and mechanical characteristics that will permit them to be implanted when they are needed to replace natural valves that have become incompetent. In addition to acting as valves, they will serve as substrates for the attraction, differentiation, and ingrowth of living cells that over a period of time will replace the synthetic structures implanted. In time, these tissue-engineered structures will be indistinguishable from normal natural tissue. Such approaches are already entering the clinical setting in the form of skin substitutes and replacement for bone, ligaments, and cartilage.
Combination devices that deliver gene therapies to specific sites of interest and for required periods of time are another way that devices will progressively become more biological. Such approaches offer the distinct possibility of disease cures rather than palliative treatments, as is now the case. Although researchers are naturally attracted to the exciting possibilities of emerging and future therapeutic medical devices, the area of implanted diagnostics and monitoring holds enormous potential.
Next generations of technology will literally close the loop on therapy. The implantable monitor, for example, will continually be vigilant to the patient’s circumstances, and when something serious occurs, appropriate words can be sent by telemetry or the device itself can automatically respond just as the defibrillator does when it detects some abnormal rhythm that will lead to ventricular fibrillation. In some cases, as with a low battery indicator, the alert will be sent only to the patient, so he or she can have the device checked. This scenario presents a dramatic application of monitoring capabilities, and the most clinically relevant contributions will be in quality improvement aspects.
Patient compliance, however, remains a serious limitation in chronic disease treatment. Implantable monitoring will likely provide much better guidance as to when treatments are suboptimal due to the regimen selected or due to a failure to follow prescribed drug administration protocols. Chronic monitoring will permit physicians to match treatment options more closely to the patient’s abilities, thereby improving overall quality. The convergence of traditional medical technology with communication and Internet technology presents a remarkable opportunity to change the face of health care.
Dramatic quality improvements can result from consistent application of statistically based medical care algorithms based on broad and deep databases. Real-time access of these large databases will identify trends so that the health care system can be more dynamic and at the same time knowledge-based, and perhaps far less dependent on small clinical trials. Using databases that are automatically generated by automatic acquisition from devices, combined with caregivers’ databases, will allow researchers to apply deep computing approaches to identify trends early and to create optimal treatment algorithms.
GENERAL DISCUSSION OF THE KEYNOTE SESSION
John Watson of NIH began the discussion by inquiring about the Product Quality Research Institute (PQRI). Tobias Massa from Eli Lilly described the new institute as the brainchild of Roger Williams, whose grand design has industry, academia, and government coming together to try to address common quality issues associated with pharmaceutical products. Funded by pharmaceutical corporations and trade organizations, PQRI currently sponsors four technical committees dealing in the areas of drug substance, drug product, biopharmaceutics (issues related to bioequivalence), and science management, which is seeking to map the regulatory review process. According to Dr. Massa, working groups are looking at specific questions related to those four areas, for example, blend uniformity analysis. That is, what is the best way to assure content uniformity of products? Some of these projects will be carried out at FDA, and some will be carried out at individual companies volunteering to do this work in their own laboratories.
Clifford Goodman then returned to Glen Nelson’s remarks on the challenge of demonstrating cost-effectiveness, pointing out that many devices that Nelson’s company makes call for a substantial initial expense, between hardware and implant surgery, and do not start saving money until later. So, there is a time factor in considering cost-effectiveness. Furthermore, he noted, Nelson’s cost-effectiveness data did not even mention productivity, that is the averted costs of lost productivity, something else difficult to quantify because it does not start saving money immediately, nor do researchers generally capture the financial savings from letting people stay at work. Goodman brought up the case of the HMO that loses 15 percent of its enrollees every year and therefore might not care about long-range savings from use of an expensive device because the pa-
tient will probably not be enrolled long enough to recoup the investment. He then asked the group for their thoughts on whether the inability or unwillingness of such payers is in fact affecting decisions about which technologies to undertake and build.
Dr. Nelson replied that it definitely affects innovation, and drew on his experience as a managed care company director to describe a proposal to reduce the sort of disincentive that Dr. Goodman cited, namely a surcharge that would make funding available for long-term interventions that do not have to be driven by a 1-year actuarial mentality. Unanswered questions include who is given responsibility for administering that fund and whether it would allow for earlier reimbursement for therapies that not only are promising now, but are bound to become dramatically cost-effective in the societal sense after a couple of revisions. Nelson also reinforced Goodman’s complaint that researchers need economic models that include a method of assigning a value to the fact that not only patients but also spouses or other caregivers can go back to work after an expensive intervention like an implant. These outcomes, he asserted, are not only heartwarming, but also have huge economic impacts that vastly outweigh the cost of the device. Where do you recognize those savings, he asked, and how do you channel them back then into the further development of that technology?
Ron Geigle, from the consulting firm Polidais, offered the observation that in the policy world of Washington there is a growing disconnect between the nature of evidence that is appropriate for devices and the nature of evidence that is appropriate for drugs. That is, he said, one can see a growing tendency to insist upon randomized controlled clinical trials as the gold standard, and yet this morning a lot was said about exciting technologies, technologies that are changing rapidly, nanotechnologies, information technologies, Internet-connected-to-patient monitoring, closed-loop systems, quick half-lives. Geigle’s questions for Dr. Nelson and the group were whether there are assessment models that work best for those sorts of technologies, and secondly, what is the effect of the imposition of randomized controlled clinical trials on that kind of device-innovation process and the capacity of companies to innovate and attract venture capital? How do researchers create an understanding in Washington that devices are different from drugs and ought to be evaluated with a different model of evidence?
Dr. Nelson agreed with Dr. Geigle’s articulation of the problem, pointing out that the major R&D costs for companies like his are in the clinical trial phase, where it is extremely difficult to randomize and use sham procedures. He noted that trials seem to have scientific rigor, but are costly, and in his view are not beneficial. Instead, he argued, postmarket surveillance is one means of reducing those costs and maybe at the same time improving the outcomes. Devices or pharmaceuticals or surgical procedures will never be perfect; researchers and practitioners must learn as quickly as possible whether there are imperfections that can be corrected. Nelson pointed out that no one wants to go back to the era where irradiating the thymus gland of children resulted in thyroid
cancer 20 years later, but, by the same token, open heart surgery in the 50s led to a mortality rate of 50 percent for ventricular septal defects. There are no reimbursers in the United States or the world today that would pay for a procedure with those statistics. Yet the current mortality rate for a ventricular septal defect repair is probably about 0.3 percent. The point, according to Dr. Nelson, is that researchers have to move through these periods of potential poor medical results to get to better medical results. He offered the example of Charles Bailey, who lost five patients in a row fracturing mitral valves and lost his privileges at every hospital in Philadelphia, but when he did the sixth, and it was successful, it opened a whole new field of surgery. Nelson confessed he does not know how to draw the line, but feels it is important to accept some lesser level of performance knowing that later iterations will not only be cost-effective but will be enormously valuable in terms of people’s quality of life.
Dr. Nobel replied that he certainly agrees with the proposition that randomized controlled trials are not appropriate for certain types of medical products, and that presents an education problem for the people who provide reimbursement. It is not a reasonable gold standard, he said, and is not even applicable in many cases. It is thoroughly impractical and in some cases plainly unethical to do certain types of studies, but he noted that it is natural for a large government reimburser, or even a large private one, to want to have a simplified rule book. It makes life easy, but the diversity of medical devices requires a diversity of proving methods, and that is an intellectual and educational challenge for the people in this group and people who pay for patient care.
David Feigal from the FDA offered some thoughts on the reimbursement decisions facing HCFA, noting that they have different criteria for reimbursement, one of which is value added, something that is not required for drug approval or device approval by FDA. There are countries in Europe that have a comparative claims requirement. That is, they require comparison of the new product relative to the existing products that they resemble, but that has never been the standard in the United States. Third-party payers often want just that sort of data. This is an area where knowing the rules may help the process because of the opportunity during development to consider whether collecting that comparative data is desirable, even if it is not necessary for FDA approval.
Dr. Feigal also felt that the rapid evolution of devices often makes it unclear what it is that HCFA is agreeing to reimburse. That is, if they approve a pacemaker today, and 3 or 4 years later the manufacturer has a new-generation product, a totally different device, will they revisit that decision or are accept the fact that they already thought through this general category? If the former, does that create incentives not to innovate and to leave things frozen so that people do not lose the reimbursement approval? From FDA’s standpoint it might have been an incremental change that would have been quite acceptable, but given the limited data set available to the third party reimburser, it may well trigger the response, “We need to have some clinical evidence.”
Feigal’s final comment was to remind the group that even for drugs the definition of adequate and well-controlled trials lists five examples, only one of which is the randomized double-blind placebo-controlled trial. The historical control is another, at the other extreme. In between are dose-response, no-treatment control, and active control, and you can find approvals that use each of the different types of evidence. So, he averred, researchers need to ask in every case, “What are we trying to learn, and what are the difficulties with the evidence in this field?” He gave an example from a recent review of cardiac therapy during which an FDA reviewer pointed to the restenosis rates of the control groups of five randomized controlled trials and said, “They have to do more randomized controlled trials because the control groups vary between 5 percent and 30 percent, which is exactly the same range, maybe a bit higher, as the benefit groups.” But whenever there was a direct comparison there always was a benefit! There is not going to be a single answer for this, he concluded, but these are science- and evidence-based decisions, and they require exactly the kind of information clinicians want in making decisions for their patients. Neither they nor FDA want or need overkill.
Jim Benson with the Health Industry Manufacturers Association (HIMA) [Editor’s note: now Advanced Medical Technology Association (AdvaMed)] then returned to an earlier point and asked the group to think about who pays the bills: in the case of HCFA reimbursement, the taxpayers, and in the case of HMOs and other private insurers, the employers. All of it comes out of the worker’s salary in one form or another, yet, he said, researchers do not seem to target their studies and their research toward the benefit that goes to those two communities, the taxpayers and the employers that are paying the HMOs. Maybe the industrial community and others really need to include that in some of their cost-effectiveness work and really aim at convincing not HCFA, which does not really have any choice, but the taxpayers and employers, that some of this innovation can actually save money.
Dr. Nelson agreed that consumers are going to have a lot larger role in determining their health care, and their view of value may be a lot different than the view of their managed care organizations. He pointed out that the problem with just appealing to the individual consumer is that the natural response is to abandon the original spending accounts and let everybody just spend their own money on health care. But, he said, researchers really do need an insurance mentality that takes care of the individual whose costs will be 100 times the average. People have accepted as a society that they are going to amalgamate their risk, although there are lots of things working against it. People cannot pull the bottom can out of a grocery store display and do away with insurance.
Dr. Mann extended the discussion by drawing on his experience chairing a National Academy of Sciences conference on technology to aid the blind that made two points. One was that the federal government at that time was spending on the order of $10 to $100 per cancer and cardiac victim and $.02 per blind person, and the other part was that if one made a blind person a taxpayer through
reading access and mobility one was, in fact, being very cost-effective. The second point was that a lot of the work at MIT on the “Boston Arm” prosthesis was funded in part by the Liberty Mutual Insurance Company. As an insurance company writing worker’s compensation policies it was to their financial advantage to be able not only to advertise that they were doing great things for humanity but also to show that they were reducing their rehabilitation costs by providing a better prosthesis.
Tom Loarie, CEO of Kera Vision Inc., had the last word in the morning’s discussion, using personal experience to illustrate the point that assessing new technologies at the very early stages of development is very difficult, even for sophisticated people in the business. As a young engineer at a company called American Hospital Supply Corporation, he witnessed the company looking into acquiring a small firm making a pacemaker. In those days it was probably a $30 million to $40 million business, and they came back and said, “You know, that is really not going to amount to much.” American Hospital Supply does not exist anymore, but Medtronic does.
Loarie’s second point was his belief that the crux of the problem is that in this and other western countries researchers like to believe that everyone should have equal access to new technology. He asked the group to imagine if researchers had decided in the early 1980s that the car phone was something every citizen of this country should be required to have, or simply have a right to, at $3,000 per unit. A car phone is very much like a medical device, he asserted. It is engineering-based. It goes through the iterative process and as researchers learn, as they improve it, they reduce costs, and the market eventually expands. It is, however, very different from a drug, in that it is introduced into the market at the very highest point in its cost. Researchers face this dilemma of providing it to the masses, while government steadily adds to the cost. He argued that these conflicting demands build in a tremendous prejudice against the approval of new technologies, and it is the only part of this economy where technology is looked at as a scapegoat.






















