
7
Anthrax Vaccine Manufacture
Anthrax Vaccine Adsorbed (AVA) is licensed for manufacture only by the firm BioPort, which acquired the vaccine production facility from the Michigan Biologic Products Institute (MBPI) in September 1998. Production of the vaccine had been suspended in early 1998 when the facility was closed for renovations. Production resumed in 1999. However, vaccine was not released for routine use until the Food and Drug Administration (FDA) approved the license supplement for the renovations, change in labels and package insert, and change to an outside contractor for filling on January 31, 2002. Therefore during the period that the committee held its meetings and most of its deliberations, the vaccine had not yet been released for use following the renovations.
Among the issues that the committee was charged with addressing is “validation of the manufacturing process focusing on, but not limited to, discrepancies identified by the Food and Drug Administration in February 1998.” This chapter provides some background to clarify this portion of the charge, including a description of the role of the FDA in regulating the manufacture and marketing of vaccines, the history of the manufacturing process, and problems particular to the currently licensed anthrax vaccine. The committee’s findings regarding the manufacture of the anthrax vaccine are also presented. The committee’s first step was to interpret the charge.
COMMITTEE’S INTERPRETATION OF THE CHARGE
“Manufacturing process validation” is the formal and detailed defini-
tion of each step in a controlled manufacturing process. The 1987 FDA Guideline on General Principles of Process Validation defines process validation as “establishing documented evidence which provides a high degree of assurance that a specific process will consistently produce a product meeting its pre-determined specifications and quality attributes” (FDA, 1987, p. 4). A manufacturer carries out this validation through careful documentation of all aspects of the manufacturing process, and it is overseen and deemed acceptable or unacceptable by FDA. The Institute of Medicine (IOM) committee could not itself validate the manufacturing process, nor was the committee in a position to second-guess FDA’s inspection and determination of validity. The committee could, however, review and evaluate the process by which BioPort was working to validate the manufacturing process for AVA.
The committee took the following approach to evaluating BioPort’s validation process. The committee requested from BioPort copies of the Form FDA 483s (Form FDA 483 is a list of observations provided by an FDA investigator at the conclusion of an inspection) from FDA inspections conducted since 1998, as well as BioPort’s responses to these inspection reports. The committee was specifically interested in the subset of materials focusing on product characterization and process validation.
BioPort provided these documents, as well as copies of the MBPI strategic plan with updates. The documents provide detailed information about the responsiveness of the company to the FDA’s findings and the FDA’s evaluations of the manufacturer’s progress.
REGULATORY OVERSIGHT OF VACCINE MANUFACTURE
All vaccines are regulated by FDA’s Center for Biologics Evaluation and Research (CBER). As with other biologics, the development of a new vaccine involves preclinical research (before administration of a vaccine to humans), research in studies with humans in which the product is administered under limited study conditions, an application for licensure, and continued monitoring after licensure and marketing.
The application for licensure must be approved by FDA, which reviews the results of clinical trials and other data and information on safety and efficacy. FDA also reviews detailed information on the manufacturing process and the facility where the product will be produced and tests the products. Previously, sponsors of new biological product applications were required to apply separately for approval of the product and approval of the manufacturing facility (Product License Application and Establishment License Application, respectively), but in 1999 the regulations were modified to combine the two into a single Biologics License Application (BLA) for all products licensed by CBER.
A vaccine product must be produced in compliance with good manufacturing practices (GMPs), as specified in the Code of Federal Regulations.1 To ascertain compliance, FDA conducts periodic GMP surveillance inspections. In the case of vaccines, the inspections are carried out by a team of experts in GMPs and experts in the product in question, an approach adopted in 1999 under the Team Biologics program. The Team Biologics program is a partnership between CBER and FDA’s Office of Regulatory Affairs, designed to increase both consistency and focus in FDA’s inspections of biologics manufacturers. When a manufacturer makes a change in the facility or the production process that could have a moderate to substantial potential to affect adversely the quality of the product, the manufacturer must submit a supplemental application for FDA approval of the change.
Because vaccines are produced through complex procedures that depend upon living organisms, there are many points in the process where variance could be introduced into the final product. To ensure consistency, each lot of the product must be individually tested and approved before its release for marketing. The vaccine manufacturer must submit a sample of the lot along with a lot release protocol to FDA, which then reviews the lot testing data and, if necessary, performs additional tests.
In the case of AVA, BioPort resumed manufacture of the vaccine in 1999 after a pause for plant renovation. The BLA supplement for the renovations has been approved, and release of lots manufactured after renovation of the facility was approved in January 2002. One of the sources of difficulty in the regulatory history of AVA may be that the standard regulatory expectations for vaccines in general changed between the licensing of AVA in 1970 and 2001. Vaccines may, in some sense, have been victims of their own success: for many vaccines the decline in the disease burden from communicable diseases was so clear and the effectiveness of the vaccine was so great that there was little incentive to modify the production process. However, as part of its continuing quality improvement effort, FDA instituted more explicitly defined process validation requirements for the manufacture of vaccines and other products licensed before these requirements were codified. As a result, manufacturers of vaccines, including AVA, must now ensure that their production processes are validated.
ANTHRAX VACCINE DEVELOPMENT
Research was conducted at Camp Detrick (later Fort Detrick), Maryland, to develop an anthrax vaccine based on Bacillus anthracis cultures grown in synthetic medium without proteins or other macromolecules (Turnbull, 2000). In 1954, Wright and colleagues described a chemically defined medium that could be used to grow the bacteria and provided evidence that protective antigen in culture filtrates could be concentrated, stabilized, and partially purified by precipitation with alum to make a vaccine. Further refinements to simplify large-scale production included microaerophilic incubation (Wright and Puziss, 1957; Wright et al., 1962), use of a different growth medium (Puziss et al., 1963; Wright et al., 1962), and adsorption onto aluminum hydroxide gel (Alhydrogel) instead of precipitation with alum (Puziss and Wright, 1963). Different strains of B. anthracis were evaluated for use as the vaccine strain (Auerbach and Wright, 1955; Puziss and Wright, 1963), and ultimately, Vollum strain V770-NP1-R was adopted and used for the licensed anthrax vaccine (see Table 7-1 for a list of significant events in AVA development and manufacture).
Anthrax Vaccine Licensure
It is noteworthy that the landmark study evaluating the efficacy of the anthrax vaccine was carried out not with the vaccine that was ultimately licensed but with the earlier vaccine described above. The previously discussed study by Brachman and colleagues (1962), which evaluated the efficacy of a protective antigen-based anthrax vaccine in wool mill workers, was carried out over a 4-year period between 1955 and 1959. The vaccine they evaluated was made by Merck Sharpe & Dohme (hereafter referred to as the Merck vaccine) using a nonencapsulated, nonpro-teolytic mutant of the Vollum strain of B. anthracis called R1-NP that was grown in 599 medium (Puziss and Wright, 1954) to produce protective antigen that was precipitated and concentrated with aluminum potassium sulfate (alum). It differed from the currently licensed vaccine in terms of the B. anthracis strain used to generate protective antigen, in the medium in which it was grown, in the mode of growth (aerobic rather than microaerophilic [fermentation]), and in the mode of antigen concentration and purification (alum precipitation rather than adsorption to aluminum hydroxide gel). In addition, the Merck vaccine used 0.01 percent thimerosal as a preservative, whereas the licensed vaccine uses 0.0025 percent benzethonium chloride (see Table 7-2).
The package insert calls for subcutaneous administration of a basic series of six doses of 0.5 ml each. After the initial dose, subsequent doses are administered at 2 weeks, 4 weeks, 6 months, 12 months, and 18 months.
TABLE 7-1 Events in AVA Development and Manufacture
|
1955–1959 |
Brachman et al. conduct a field evaluation of the anthrax vaccine manufactured by Merck and publish their results in 1962. |
|
1966 |
CDC submits an Investigational New Drug (IND) application for Anthrax Vaccine Adsorbed (AVA). |
|
Nov. 10, 1970 |
Division of Biologics Standards issues a product license to Michigan Department of Public Health (MDPH) to manufacture anthrax vaccine. |
|
1973–1975 |
FDA convenes an external review of AVA using safety data from CDC trials and efficacy data from the field evaluation of Brachman et al. (1962). The panel finds sufficient evidence that AVA is safe and effective “under the limited circumstances for which the vaccine is employed.” |
|
Dec. 13, 1985 |
Findings from an FDA external review are published in the Federal Register. |
|
1993 |
FDA inspects the MDPH anthrax vaccine manufacturing facilities in January; FDA approves the renovations in July. |
|
1996 |
MDPH becomes known as MBPI, an entity controlled by the Michigan State government. |
|
Nov. 1996 |
FDA conducts a surveillance inspection of MBPI (not including the anthrax vaccine manufacturing facilities) in which it finds numerous deviations from regulations. |
|
March 1997 |
FDA issues MBPI a Notice of Intent to Revoke (NOIR) letter based on the Nov. 1996 inspection. |
|
April 1997 |
MBPI responds to the NOIR letter with its Strategic Plan for Compliance. |
|
Jan. 1998 |
MBPI halts production of AVA sublots to begin comprehensive renovation. |
|
Feb. 1998 |
FDA conducts a comprehensive inspection of the MBPI facility to evaluate MBPI’s compliance with its strategic plan. |
|
May 18, 1998 |
Secretary of Defense William Cohen implements the Anthrax Vaccine Immunization Program for all U.S. armed forces. |
|
Sept. 1998 |
MBPI transfers ownership to BioPort Corporation. |
|
Oct. 1998 |
FDA’s GMP inspection of BioPort Corporation notes continuing improvement. |
|
Sept. 1999 |
Submission of BLA supplement for renovation of BioPort’s AVA manufacturing facility. |
|
Nov. 1999 |
Preapproval inspection and complete review letter from FDA. |
|
Oct. 2000 |
FDA inspects BioPort and observes numerous deviations from regulations, including problems with the filling suite. |
|
April 2001 |
BioPort responds to FDA with modifications. |
|
2001 |
BioPort decommissions the filling suite and contracts with Hollister-Stier Laboratories to perform the AVA filling operation. |
|
Dec. 2001 |
Preapproval inspection of BioPort; FDA approval of the BLA supplement for the facility renovations. |
|
Jan. 2002 |
Inspection of the Hollister-Stier Laboratories filling operation facility. |
|
Jan. 31, 2002 |
FDA approval of BLA supplement for the filling operations and labeling change. |
TABLE 7-2 Comparison of AVA and Merck Vaccine
|
|
Vaccine Product |
|
|
Characteristic |
AVA |
Merck |
|
Strain |
Nonproteolytic, nonencapsulated strain V770-NP1-R |
Nonproteolytic, nonencapsulated strain R1-NP |
|
Medium |
1095, chemically defined |
599, chemically defined |
|
Growth conditions |
Microaerophilic (fermentation) |
Aerobic (static culture) |
|
Means of removal of bacteria |
Filtration (hydrophobic, low protein binding) |
Filtration (sintered glass) |
|
Purification and concentration procedure |
Adsorption (aluminum hydroxide) |
Precipitation (alum and acid) |
|
Recovery |
Decantation, centrifugation |
Decantation, centrifugation |
|
Concentration factor from culture filtrate |
10× |
10× |
|
Aqueous vehicle |
Normal saline |
Normal saline |
|
Adjuvant |
Aluminum hydroxide (0.65 mg of aluminum/dose) |
Aluminum potassium sulfate (0.52 mg of aluminum/dose) |
|
Preservative |
0.0025% Benzethonium chloride |
0.01% Thimerosal |
|
Stabilizer |
0.01% Formaldehyde |
None |
|
Schedule (0.5 ml/dose) |
0, 2, 4 weeks; 6, 12, 18 months; annual booster |
0, 2, 4 weeks; 6, 12, 18 months; annual booster |
|
Route |
Subcutaneous |
Subcutaneous |
|
Amount of protein per dose |
Approximately 50 µg |
Unknown |
|
SOURCES: Giri et al. (2001), Myers et al. (2001). |
||
Annual boosters are required. The evidence to justify this dosing schedule is limited. An alum-precipitated predecessor of AVA was given to 55 volunteers in two 0.5-milliliter (ml) injections given subcutaneously 2 weeks apart and found to be acceptable (Wright et al., 1954). A group of 660 people were then given three subcutaneous injections of this same vaccine at 2-week intervals, followed by a booster dose of 0.25 ml after 6 months. No significant local reactions were reported after the first dose, but 0.6
percent of 650 people receiving the second dose and 2.2 percent of 537 people receiving the third dose reported significant local reactions. Of 445 people receiving a booster dose, 2.6 percent reported significant local reactions. Systemic reactions were reported after 0.7 percent of doses, but information was not provided by dose (Wright et al., 1954). Brachman and colleagues (1962) used the same vaccine with a schedule of three 0.5-ml subcutaneous injections given at 2-week intervals, followed by three 0.5-ml booster doses given at 6-month intervals (see Chapters 3 and 6 for additional discussion of this study). Thereafter booster doses were given at yearly intervals. This schedule was then used for the studies leading to licensure of AVA. As mentioned in Chapters 3 and 6, a pilot study has been conducted to evaluate changes in both the route of administration and the dosing schedule (Pittman, 2001; Pittman et al., 2002). A clinical trial will soon begin to further evaluate these modifications.
The Centers for Disease Control and Prevention (CDC) submitted an Investigational New Drug (IND) application for anthrax vaccine to the Division of Biologics Standards (DBS) of the National Institutes of Health on April 14, 1966 (Elengold, 2000). Although Merck Sharp & Dohme had produced one of the vaccine lots evaluated early in the study, the Michigan Department of Public Health (MDPH) made the remainder of the lots evaluated. In 1968, MDPH was awarded a 3-year contract to produce the vaccine. The manufacturing process was that described by Puziss and colleagues in 1963. The process was also described in the materials provided with the progress reports to DBS under the IND application (CDC, 1967–1971). (Safety and efficacy data from the five annual progress reports submitted as part of the IND application are presented elsewhere in this report.) Additional data indicating that the vaccine protected guinea pigs from challenge were also submitted (Pittman, 1969). An ad hoc committee involved with review of the vaccine expressed the desire for additional efficacy data as well as for comparisons of sera from human recipients of the Merck vaccine with sera from recipients of the MDPH vaccine but recommended that licensure be granted (Feeley et al., 1969; Pittman, 1969). On the basis of the information submitted, DBS issued a product license to MDPH to manufacture AVA on November 10, 1970 (Elengold, 2000).
Rereview of Anthrax Vaccine
In 1972, DBS was moved from NIH to become part of FDA, where it was called the Bureau of Biologics (Parkman and Hardegree, 1999). FDA began a process of reexamining the vaccines that had already been licensed to evaluate their safety and efficacy. FDA assigned initial review of each category of biological products to a separate independent advisory panel that was charged with preparing a report for the FDA commissioner to
-
Evaluate the safety and effectiveness of the biological products,
-
Review labeling of the biological products, and
-
Identify the biological products under review that are safe, effective, and not misbranded (FDA, 1985, p. 51002).
The advisory report includes recommendations classifying products into one of three categories:
-
Category I designates those biological products determined by the panel to be safe, effective, and not misbranded;
-
Category II designates those biological products determined by the panel to be unsafe, ineffective, or misbranded; and
-
Category III designates those biological products determined by the panel not to fall within either Category I or II on the basis of the panel’s conclusion that the available data are insufficient to classify such biological products and for which further testing is therefore required (p. 51002).
The panel appointed to review data on bacterial vaccines, toxoids, related antitoxins, and immune globulins evaluated AVA. The panel met numerous times from 1973 to 1975. Its final report on all the products that it reviewed was published in the Federal Register in 1985. In that report the panel recommended that AVA be placed in Category I, the category of products determined to be safe, effective, and not misbranded “because there is substantial evidence of safety and effectiveness for this product” (p. 51058). The panel found the vaccine to be “fairly well tolerated with the majority of reactions consisting of local erythema and edema. Severe local reactions and systemic reactions are relatively rare” (p. 51058). The panel found that the safety of the product was “not a major concern, especially considering its very limited distribution and the benefit-to-risk aspects of occupational exposure in those individuals for whom it is indicated” (p. 51058). The panel noted that the product was intended solely for use for immunization of high-risk individuals such as industrial populations working with animal hides and other products and laboratory workers handling the organism. In considering efficacy, it found protection against cutaneous anthrax in fully immunized subjects to be adequately established by the study by Brachman and colleagues (1962), who used “the very similar Merck . . . vaccine” (p. 51058). The panel reported that “no meaningful assessment of its value against inhalation anthrax is possible due to its low incidence” (p. 51058).
Anthrax Vaccine Manufacturing Process and Vaccine Constituents
Briefly, the process for manufacturing the licensed vaccine is as follows
(Giri et al., 2001; CDC, 1967–1971; Puziss et al., 1963): an inoculum of nonencapsulated, nonproteolytic, avirulent B. anthracis strain V770-NP1-R is placed into sterile growth medium 1095. This chemically defined growth medium, described by Wright and colleagues in 1962, contains amino acids, minerals, glucose, and other specified ingredients found to be optimal for growth of B. anthracis. Bacterial growth takes place through fermentation under microaerophilic (limited oxygen) conditions. The inoculated medium is slowly agitated and held at 37 ± 0.5ºC for 23 ± 1 hours in each of two fermentors (volumes of 10 and 100 liters, respectively) while pH and glucose levels are monitored (Puziss et al., 1963; Myers, 2001).
After incubation, the mixture is filtered by use of hydrophobic, low-protein binding filters. On completion of filtration, sterile aluminum hydroxide gel is added and the mixture is stirred for 17 ± 1 hours. The supernatant (fluid) is removed after a period of settling (73 ± 1 hours) and again after a centrifugation step. The aluminum hydroxide-adsorbed antigen is resuspended in a physiological saline solution that contains formaldehyde (final concentration: 0.01 percent) as a stabilizer and benzethonium chloride (final concentration: 0.0025 percent) as a preservative. Tests for stability, purity, and potency are carried out, and the vaccine is filled in 10-dose vials, stoppered, and sealed before it is labeled and packaged for use.
The time required to manufacture a lot of AVA is approximately 22 weeks from the initiation of sublot production to release for distribution by FDA. The approximate timeline is shown in Table 7-3, but actual times for any given lot may be longer or shorter.
Vaccines are licensed with defined dating periods, but the regulations2 provide that FDA may grant an extension of the expiration dates if the lot in question passes potency and sterility tests. AVA was previously licensed for up to 36 months from the date of manufacture when stored at 2 to 8ºC, including both time in manufacturer’s storage and time in distribution (Elengold, 2001).
One example of the evolution in vaccine standards that has taken place over the last 30 years lies in the characterization of vaccine constituents. In contrast to the development of AVA and other vaccines of an earlier era, consider the analytical data assembled for recombinant hepatitis B vaccines for adults, which were licensed in the late 1980s. In the prelicensing phase of research, physicochemical, immunological, and molecular biological test methods were all used to provide evidence of the identity, purity, and genetic stability of the protein product (Parkman and Hardegree, 1999). Protein characterization techniques included sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), peptide mapping, amino acid
TABLE 7-3 Timeline for Production of AVA
|
Activity |
Week |
|
Initiate sublot production |
1 |
|
Formulate bulk lot |
7 |
|
Fill into vials |
9 |
|
Complete testing |
15 |
|
Submit release protocol to FDA |
17 |
|
Release for distribution by FDA |
22 |
|
SOURCE: Myers (2002b). |
|
composition analysis, amino-terminal sequencing, and high-performance size-exclusion chromatography. Immunological techniques included Western blotting for hepatitis B surface antigen and contaminating yeast proteins, radioimmunoassay, enzyme-linked immunosorbent assay (ELISA), and immunogenicity in mice.
Few protein characterization and immunological techniques were available, however, to describe or specify the constituents of the anthrax vaccine when it was developed and refined in the 1950s and 1960s. Evaluations of the product relied heavily on comparison of relative potency in animals. For example, the relative success of various vaccine formulations was assessed on the basis of the production of effective immunity in rabbits, guinea pigs, and monkeys. Puziss and Wright’s assay of protective antigen (1954) consisted of estimating “protective antigen activity” by immunization and challenge of guinea pigs. In their study described in a 1962 paper, Wright and colleagues also used complement fixation titrations (McGann et al., 1961) to estimate levels of protective antigen.
Detailed characterization of vaccine constituents was not routine in the 1960s. The anthrax vaccine was licensed in 1970, and the specifications of vaccine constituents consisted only of the amounts of stabilizer, preservative, and adjuvant added. No required amount of protective antigen was originally specified in the vaccine license, nor are the maximum (or minimum) amounts of other components that might be of interest or concern, such as edema factor (EF) or lethal factor (LF). The criteria for lot release were simply that the vaccine pass the necessary potency, safety, and purity tests.
Today, with detailed characterization of vaccine constituents clearly feasible, BioPort has undertaken analyses to characterize AVA in support of ongoing process validation studies (Winberry et al., 2001). One challenge is the development of an easy and reliable desorption procedure for the separation of vaccine constituents from Alhydrogel. Because of the difficulty of conducting desorption from Alhydrogel, BioPort investigators analyzed
aliquots of the fermentation filtrate before adsorption. By use of the Bradford (1976) method to determine protein concentration, SDS-PAGE separation, Western blotting, and ELISA, a typical fermentation filtrate was determined to contain 10 micrograms (µg) of Bradford protein per milliliter; over 40 percent protective antigen, based on SDS-PAGE and Western blotting; and 2 to 4 µg of protective antigen per milliliter, based on ELISA. No detectable EF was found by Western blotting, but LF was determined by ELISA to be present in the range of 10 to 30 nanograms per milliliter of filtrate (Winberry et al., 2001).
To explore the biological activity of the LF in the vaccine, the mouse macrophage cytotoxicity assay was performed on 11 vaccine lots. Initial results indicated that the small-molecular-weight additives Phemerol (benzethonium chloride) and formaldehyde have minor toxic effects on macrophage cells. After their removal by dialysis, no toxic effect on the cells was evident. BioPort investigators concluded that the small amount of LF present in the vaccine is inactive and noted that further studies on characterization of the filtrate and the final formulated vaccine were ongoing (Winberry et al., 2001).
In agreement with FDA, BioPort has changed several manufacturing parameters for the production of AVA to provide greater assurance of product consistency. As depicted in Figure 7-1, these changes apply to aspects of production from temperature and time of fermentation to characteristics of the final product. Manufactured lots of vaccine must contain between 5 and 20 µg of total protein per milliliter, with no less than 35 percent of this protein consisting of the 83-kilodalton (kDa) protective antigen (precursor), to be acceptable (Giri et al., 2001). Data from 36 sublots combined as groups of 12 to obtain three consistent lots of filtrate found the Bradford protein to be present at a mean concentration of 7.7 µg/ ml, with a concentration range of 5.1 to 15.8 µg/ml. The mean percentage of protective antigen, based on densitometric scans of the 83-kDa band on SDS-polyacrylamide gels, was 65.6 percent (range, 48.2 to 84.6 percent) for the 36 sublots (Myers, 2002a).
REGULATORY ACTIONS CONCERNING AVA MANUFACTURE
MDPH, a state-owned and -run facility, received the license to manufacture AVA in 1970 and produced vaccine until the facility was transferred to MBPI in 1995. In September 1998 the facility was sold to the private company BioPort, but the existing MBPI management team was retained (see Table 7-1).
FDA conducted an inspection of the MDPH anthrax vaccine manufacturing facility in January 1993, which was followed in July 1993 by FDA approval of amendments to the license application for new equipment and
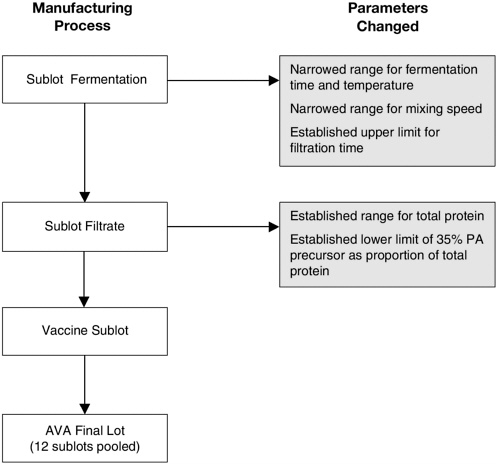
FIGURE 7-1 Changes to parameters of the Anthrax Vaccine Adsorbed manufacturing process.
SOURCES: Giri et al. (2001), Myers (2002a,b).
facilities (Donlon, 1993; FDA, 1993). Meanwhile, FDA inspections of MDPH product lines other than AVA in 1993 and 1995 resulted in findings of significant deviations from GMP and an official warning letter.
During a follow-up inspection of MBPI conducted in November 1996 that did not include the facilities for manufacture of AVA, FDA documented many deviations from the Food, Drug, and Cosmetic Act, FDA regulations, and current GMPs for the manufacture of blood-derived products and bacterial vaccines other than AVA (Elengold, 2000, 2001; Zoon, 1997). FDA issued a Notice of Intent to Revoke (NOIR) letter to MBPI in March 1997, stating that if MBPI’s corrective actions proved to be inadequate, FDA might revoke its licenses. The letter did not mandate closure of
the facility or involve seizure of finished product (Zoon, 1997). MBPI’s response to the NOIR letter was a Strategic Plan for Compliance, provided to FDA in April 1997. Periodic updates to the strategic plan that reported on MBPI’s progress were also provided to FDA (Michigan Biologic Products Institute, 1997–1998). In January 1998, after completion of renovation planning, MBPI shut down the anthrax vaccine production facility for planned renovations.
In February 1998, FDA conducted a comprehensive inspection covering all product lines of the facility to evaluate MBPI’s compliance with the strategic plan. The inspection, which covered the manufacture and testing of all lots of AVA as well as other products, found that MBPI had made progress in achieving its compliance goals but that there were “significant deviations from FDA’s regulations” (Elengold, 2000, p. 2; FDA, 1998a). The FDA inspection report noted that the manufacturing process for anthrax vaccine was not validated and included detailed lists of equipment or processes that had not been adequately described, specified, or documented. It reported a lack of written procedures or specifications for examination, rejection, and disposition of sublots of vaccine and problems with sample selection for potency testing, assignment of expiration dates, and documentation of justification for redating of expired lots. Many problems were also identified in the stability program (FDA, 1999a). As a result of this inspection, MBPI voluntarily quarantined several lots of vaccine after consultation with FDA (FDA, 1999a, p. 14). The facilities were transferred to BioPort in September 1998.
Another FDA inspection of the facility (now BioPort’s facility) took place in October 1998. The inspection covered AVA and other product lines. Regarding AVA, the inspection found problems in the program for monitoring the stability of the vaccine (FDA, 1998b), but it also noted progress in many of the areas of observations made in the February 1998 Form FDA 483 (FDA, 1999a, pp. 20–23).
In September 1999, BioPort submitted a supplement to its BLA to FDA covering the renovations to the manufacturing facility. As part of its review of that application, FDA carried out a preapproval inspection of the BioPort plant in November 1999. The Establishment Inspection Report from that inspection identified observations and possible deviations in the areas of “validation, failure to investigate, manufacturing deviations, deviation reporting, aseptic processing, filling operations, standard operating procedures, stability testing, and environmental monitoring” (Elengold, 2000, p. 3; FDA, 1999b). The next inspection was in October 2000. Again, the inspection noted several items in need of attention, including the filling suite, which was among the categories listed as needing attention in the 1999 inspection (FDA, 2000). In April 2001, BioPort submitted a response detailing the company’s modifications and improvements. In regard to the
filling suite, BioPort decided to decommission the area and, subject to CBER approval, outsource the filling operation, at least temporarily.
The most recent inspection of the BioPort anthrax vaccine production facilities took place from December 12 to 19, 2001. The inspection report (FDA, 2001a) noted seven observations that needed attention, many of which BioPort successfully addressed during the inspection (FDA, 2001b). FDA approved the supplement to BLA on December 27, 2001, with acknowledgment that BioPort has made three commitments for additional postapproval work (Masiello, 2001).3 Before lots of new AVA could be released and become available for shipment, however, FDA also had to approve a supplemental BLA for Hollister-Stier Laboratories, which is performing the filling operation under contract to BioPort. A preapproval inspection of that facility took place in January 2002, and FDA approved the supplement to the BLA for the contract filler on January 31, 2002, with the specification that additional stability data be collected (Maseillo, 2002). Under the approval, lots of AVA filled at Hollister-Stier will have an expiration date 18 months from the date of manufacture when the vaccine is stored at 2 to 8 degrees C, but as before the dating period may be extended with submission of supporting data to FDA.
For purposes of clarity, the committee has categorized the AVA product according to whether it was produced before or after the plant renovation. The available remaining vaccine was produced before January 1998, when MBPI halted production to carry out a comprehensive renovation. The committee heard testimony that FDA believes that the previously manufactured and CBER-released lots of AVA, not presently quarantined by BioPort, are safe and effective for the labeled indications (Elengold, 2000). While FDA has now released postrenovation lots for use, they had not been shipped as of late February 2002. Therefore, all AVA currently or previously used is prerenovation product, regardless of when the particular lot in question may have been released, and any vaccine subsequently manufactured by BioPort will be referred to as “postrenovation vaccine.”4
The General Accounting Office recently reported on changes to the manufacturing facilities not separately reported to FDA (GAO, 2001). In 1990 the manufacturer (then MDPH) changed its filter type from ceramic to nylon and thereafter used several different nylon filters (Elengold, 2001). In 1997 it changed filter types to match the industry standard, a polyvinylidene (nonshedding) filter. According to BioPort, these changes were within the specifications for filters in the original approved license. FDA learned of the changes and contacted BioPort in February 2001. The agency requested data and then accepted the changes in July 2001. On the basis of the data regarding the filter changes that BioPort submitted to FDA, which the company also provided to the IOM committee, all the lots of vaccine used by the Department of Defense not only as part of the Anthrax Vaccine Immunization Program but also, importantly, for safety studies were manufactured after the change from ceramic to nylon filters. Thus, several examinations of the safety of the product produced since the change to nylon filters have, in effect, been conducted. (The safety studies were discussed in Chapter 6.)
FINDINGS AND RECOMMENDATIONS
The committee assembled and reviewed the evidence it received in the course of its study. This evidence includes numerous Form FDA 483s and responses to those reports between FDA and BioPort, as well as statements and explanations to the IOM committee from officials of both FDA and BioPort on July 11, 2001. Furthermore, the committee took into consideration the recent and increasing BioPort and Department of Defense investments in facility renovations and process improvements, including the major action of shutting down the inadequate filling operations and transferal of those operations, with CBER approval, to a contractor meeting GMP standards. Finally, the committee noted the evident availability of technical support and assistance from CBER and Department of Defense research and development resources and the results that have been achieved in the form of progress in correcting the deficiencies noted in the Form FDA 483, as reported by BioPort and confirmed by FDA at the committee’s meeting of July 10 and 11, 2001. As noted at the start of the chapter, FDA has now approved BioPort’s BLA supplements for facility renovation, the changed package insert and label, and the contract filler (Goldenthal, 2002; Masiello, 2001, 2002).
The committee deliberated about the historical evidence concerning regulatory practice with regard to biologics and the special case of the AVA manufacturing facility. The committee took special note of the changes, modernizations, and improvements that FDA has undertaken, including normalization of inspections and creation of the Team Biologics program
(the partnership between CBER and FDA’s Office of Regulatory Affairs). In addition, the committee took special note of the continuing effort at constructive criticism and response between the agency and the manufacturer. The committee also considered the history of the AVA manufacturer, in particular, the switch from a state-owned to a privately owned and operated interstate commercial venture and the coincident changes in FDA oversight and validation requirements. Finally, the committee was most mindful of the changes in scientific and technical knowledge in the process of vaccine manufacture and characterization that have occurred since the original licensure of the AVA product.
Finding: FDA’s process of plant inspection and FDA’s validation of the vaccine manufacturing process have changed and have become more stringent with time.
Finding: With high-priority efforts by the manufacturer and FDA, the manufacturing process for AVA has been validated so that vaccine manufactured postrenovation has been approved for release and distribution.
The manufacturing facility licensed to produce AVA has been the subject of numerous specific citations regarding the manufacturing process and equipment on FDA inspection reports. The manufacturer also responded, however, and worked toward full compliance with FDA requirements and lot release. As a result of the regulatory changes mentioned in the finding above and changes in the myriad important details of materials and equipment and in scientific knowledge, the committee believes that greater consistency will be assured in the postrenovation AVA product.
Finding: AVA will now be produced by a newly validated manufacturing process under strict controls, according to current FDA requirements. As a result the postrenovation product has greater assurance of consistency than that produced at the time of original licensure.
REFERENCES
Auerbach BA, Wright GG. 1955. Studies on immunity in anthrax. VI. Immunizing activity of protective antigen against various strains of Bacillus anthracis. Journal of Immunology 75:129–133.
Brachman PS, Gold H, Plotkin S, Fekety FR, Werrin M, Ingraham NR. 1962. Field evaluation of a human anthrax vaccine. American Journal of Public Health 52:632–645.
Bradford MM. 1976. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry 72:248–254.
CDC (Centers for Disease Control and Prevention). 1967–1971. Application and Report on Manufacture of Anthrax Protective Antigen, Aluminum Hydroxide Adsorbed (DBS-IND 180). Observational Study. Atlanta, Ga.: Centers for Disease Control and Prevention.
Donlon JA. 1993. FDA approval of Michigan Department of Public Health ammendment to establishment license application. Letter to Myers RC, Michigan Department of Public Health, Lansing, Mich.
Elengold, M, Deputy Director, Operations, Center for Biologics Evaluation and Research, FDA. 2000. Anthrax Vaccine Immunization Program—What Have We Learned? Statement at the October 3, 2000, hearing of the Committee on Government Reform, U.S. House of Representatives, Washington, D.C.
Elengold M. 2001. Technical review. E-mail to Joellenbeck L, Institute of Medicine, Washington, D.C., December 12.
FDA (Food and Drug Administration). 1985. Biological products: bacterial vaccines and toxoids: implementation of efficacy review. Proposed rule. Federal Register 50(240): 51002–51117.
FDA. 1987. Guideline on General Principles of Process Validation. Rockville, Md.: Food and Drug Administration.
FDA. 1993. Form FDA 483 issued to Michigan Department of Public Health. Period of inspection: January 14–15, 1993. Letter to Myers R, Michigan Department of Public Health, Lansing, Mich.
FDA. 1998a. Form FDA 483 issued to Michigan Biologic Products Institute. Period of inspection: February 4–20, 1998. Letter to Myers RC, Michigan Biologic Products Institute, Lansing, Mich.
FDA. 1998b. Form FDA 483 issued to BioPort Corporation. Period of inspection: October 19–23, 1998. Letter to El-Hibri F, BioPort Corporation, Lansing, Mich.
FDA. 1999a. Establishment Inspection Report of BioPort Corporation, October 19–23, 1998. Rockville, Md.: FDA.
FDA. 1999b. Form FDA 483 issued to BioPort Corporation. Period of inspection: November 15–23, 1999. Letter to El-Hibri F, BioPort Corporation, Lansing, Mich.
FDA. 2000. Form FDA 483 issued to BioPort Corporation. Period of inspection: October 10–26, 2000. Letter to Kramer RG, BioPort Corporation, Lansing, Mich.
FDA. 2001a. Form FDA 483 issued to BioPort Corporation. Period of inspection: December 12–19, 2000. Letter to Kramer RG, BioPort Corporation, Lansing, Mich.
FDA. 2001b (December 19). Press Release: Pre-approval Inspection of the BioPort Facility. Rockville, Md.: FDA.
Feeley JC, Manclark CR, O’Malley JP, Kolb RW. 1969. Michigan Department of Public Health anthrax vaccine, evaluation of clinical data submitted under IND-180 on January 22, 1969. Memorandum to Pittman M, U.S. Department of Health, Education, and Welfare, Washington, D.C.
GAO (General Accounting Office). 2001. Anthrax Vaccine: Changes to the Manufacturing Process. GAO-02-181T. Washington, DC: GAO.
Goldenthal K. 2002. The labeling supplement to your license application for Anthrax Vaccine Adsorbed has been approved. Letter to Giri L, BioPort Corporation, Lansing, Mich.
Giri L, Kramer R, Myers R, Brennan-Root K, Waytes T, Winberry L. 2001. BioPort response to committee questions. Presentation to the Institute of Medicine Committee to Assess the Safety and Efficacy of the Anthrax Vaccine, Meeting IV, Washington, D.C.
Masiello SA. 2001. FDA approval of a BioPort Corporation biologics license application supplement. Letter to Giri L, BioPort Corporation, Lansing, Mich.
Masiello SA. 2002. Your request to supplement your BLA to include Hollister-Stier Laboratories LLC, Spokane, Washington, as a contract manufacturer has been approved. Letter to Giri L, BioPort Corporation, Lansing, Mich.
McGann VG, Stearman RL, Wright GG. 1961. Studies on immunity in anthrax. VIII. Relationship of complement-fixing activity to protective activity of culture filtrates. Journal of Immunology 86:458–464.
Michigan Biologic Products Institute. 1997–1998. Strategic Plans and Updates. Lansing, Mich.: BioPort Corporation.
Myers R. 2001. Technical review. E-mail to Joellenbeck L, Institute of Medicine, Washington, D.C., December 15.
Myers R. 2002a. Follow-up questions. E-mail to Joellenbeck L, Institute of Medicine, Washington, D.C., January 9.
Myers R. 2002b. Timeline for vaccine manufacture. E-mail to Joellenbeck L, Institute of Medicine, Washington, D.C., February 19.
Myers R, Winberry L, Park S. 2001. Components of the Anthrax Vaccine Adsorbed and contrast with Merck vaccine. Presentation to the Institute of Medicine Committee to Assess the Safety and Efficacy of the Anthrax Vaccine, Meeting II, Washington, D.C.
Parkman PD, Hardegree MC. 1999. Regulation and testing of vaccines. In: Plotkin SA, Orenstein WA, eds. Vaccines, 3rd ed. Philadelphia, Pa.: W.B. Saunders Company. Pp. 1131–1143.
Pittman M. 1969. Michigan Department of Health: application for license for anthrax vaccine. Memorandum to Gibson ST, Department of Health, Education, and Welfare, Washington, D.C.
Pittman PR. 2001. Anthrax vaccine: dose reduction/route change pilot study. Presentation to the Institute of Medicine Committee to Assess the Safety and Efficacy of the Anthrax Vaccine, Meeting II, Washington, D.C.
Pittman PR, Kim-Ahn G, Pifat DY, Coon K, Gibbs P, Little S, Pace-Templeton J, Myers R, Parker GW, Friedlander AM. 2002. Anthrax vaccine: safety and immunogenicity of a dose-reduction, route comparison study in humans. Vaccine 20(9–10):1412–1420.
Puziss M, Wright GG. 1954. Studies on immunity in anthrax. IV. Factors influencing elaboration of the protective antigen of Bacillus anthracis in chemically defined media. Journal of Bacteriology 68:474–482.
Puziss M, Wright GG. 1963. Studies on immunity in anthrax. X. Gel adsorbed protective antigen for immunization of man. Journal of Bacteriology 85:230–236.
Puziss M, Manning LC, Lynch JW, Barclay E, Abelow I, Wright GG. 1963. Large-scale production of protective antigen of Bacillus anthracis in anaerobic cultures. Applied Microbiology 11(4):330–334.
Turnbull PCB. 2000. Current status of immunization against anthrax: old vaccines may be here to stay for a while. Current Opinion in Infectious Diseases 13(2):113–120.
Winberry LK, Bondoc L, Park S, Simon L, Shih CN, Giri L. 2001. Characterization of the US-licensed anthrax vaccine. In: Program and Abstracts Book of the Fourth Annual Conference on Anthrax. Washington, D.C.: American Society for Microbiology.
Wright GG, Puziss M. 1957. Elaboration of protective antigen of Bacillus anthracis under anaerobic conditions. Nature 179:916–917.
Wright GG, Hedberg MA, Slein JB. 1954. Studies on immunity in anthrax. III. Elaboration of protective antigen in a chemically-defined, non-protein medium. Journal of Immunology 72:263–269.
Wright GG, Puziss M, Neely WB. 1962. Studies on immunity in anthrax. IX. Effect of variations in cultural conditions on elaboration of protective antigen by strains of Bacillus anthracis. Journal of Immunology 83:515–522.
Zoon KC. 1997. Inspections of Michigan Biologic Products Institute, November 18 and 27, 1996. Letter to Myers R, Michigan Biologic Products Institute, Lansing, Mich.


















