
Synaptic regulation of protein synthesis and the fragile X protein
William T.Greenough*†‡§¶||, Anna Y.Klintsova*§¶, Scott A.Irwin§¶, Roberto Galvez§¶, Kathy E.Bates*§, and Ivan Jeanne Weiler*§¶
Departments of *Psychology, †Psychiatry, and ‡Cell and Structural Biology, §Neuroscience Program, and ¶Beckman Institute, University of Illinois, 405 North Mathews, Urbana, IL 61801
Protein synthesis occurs in neuronal dendrites, often near synapses. Polyribosomal aggregates often appear in dendritic spines, particularly during development. Polyribosomal aggregates in spines increase during experience-dependent synaptogenesis, e.g., in rats in a complex environment. Some protein synthesis appears to be regulated directly by synaptic activity. We use “synaptoneurosomes,” a preparation highly enriched in pinched-off, resealed presynaptic processes attached to resealed postsynaptic processes that retain normal functions of neurotransmitter release, receptor activation, and various postsynaptic responses including signaling pathways and protein synthesis. We have found that, when synaptoneurosomes are stimulated with glutamate or group I metabotropic glutamate receptor agonists such as dihydroxyphenylglycine, mRNA is rapidly taken up into polyribosomal aggregates, and labeled methionine is incorporated into protein. One of the proteins synthesized is FMRP, the protein that is reduced or absent in fragile X mental retardation syndrome. FMRP has three RNA-binding domains and reportedly binds to a significant number of mRNAs. We have found that dihydroxyphenylglycine-activated protein synthesis in synaptoneurosomes is dramatically reduced in a knockout mouse model of fragile X syndrome, which cannot produce full-length FMRP, suggesting that FMRP is involved in or required for this process. Studies of autopsy samples from patients with fragile X syndrome have indicated that dendritic spines may fail to assume a normal mature size and shape and that there are more spines per unit dendrite length in the patient samples. Similar findings on spine size and shape have come from studies of the knockout mouse. Study of the development of the somatosensory cortical region containing the barrel-like cell arrangements that process whisker information suggests that normal dendritic regression is impaired in the knockout mouse. This finding suggests that FMRP may be required for the normal processes of maturation and elimination to occur in cerebral cortical development.
This paper describes synaptically triggered, synaptically localized protein synthesis discovered initially through electron microscopic studies of synaptic responses to experience and to deafferentation. We have used a “synaptoneurosome” preparation, a relative purification of synapses, to investigate this process. Using this preparation, we discovered that FMRP, the protein that is absent in fragile X mental retardation syndrome, is synthesized in synaptoneurosomes in response to application of glutamate or metabotropic glutamate receptor (mGluR) agonists. We have also discovered that a knockout mouse lacking the ability to produce complete FMRP exhibits a very substantial reduction in the ability to translate mRNA in response to activation in the synaptoneurosome preparation as well as a reduction in the presence of postsynaptic polyribosomal aggregates in vivo.
Evidence for a Role of Synaptic Protein Synthesis in Synaptogenesis.
The studies that led us to investigate the synthesis of protein at synapses and to determine that at least some synthesis was regulated by the neurotransmitter glutamate began with the finding that synapses formed in response to experience. The context for this finding is the evidence that the effects of experience on synapse number in brain regions such as cerebral cortical sensory regions involve two processes. In the first, termed “experience-expectant” synaptogenesis (1), synapses appear to form in numbers in excess of what will survive, in the apparent anticipation that appropriate experience will occur to guide a maturation-elimination-preservation process that results in the mature sensory cortical wiring diagram. The classical example of this process, first fully elaborated by LeVay, Hubel, and Wiesel (2), is seen in layer IV of the feline and monkey visual cortex, where a set of initially overlapping geniculostriate axonal projections selectively withdraws from regions of overlap to yield the nonoverlapping ocular dominance columns that characterize the mature visual cortex. Relatively similar overproduction and withdrawal mechanisms have also been described in rodent sensory cortical development (e.g., ref. 3). During these developmental periods, postsynaptic polyribosomal aggregates, also observed in the mature cerebral cortex (4), are exhibited at elevated levels in the heads and stems of dendritic spines (5). A similar elevation is seen during synaptogenesis occurring in reaction to deafferentation (6). This elevation suggested that postsynaptic protein synthesis might play a role in the synapse formation or preservation-elimination process.
There is evidence that, subsequent to this early developmental process of experience-expectant synapse selection, “experience-dependent” synaptogenesis, in which experience seems to drive the formation of synapses, occurs (1). One classical example of this process is seen in rats reared after weaning in a complex, toy-filled environment (e.g., ref. 7) in which the number of synapses per neuron in upper layers of the visual cortex increases by 20–25% during 1 month of environmental exposure. Similar results were seen after learning in adult rats (e.g., refs. 8 and 9). We subsequently found that postsynaptic polyribosomal aggregates in the heads and stems of spines were up-regulated in the visual cortex of rats reared in complex environments (10) and in motor cortex during motor learning (J.A.Kleim, D.McNamee, E.Blankstein, and W.T.G., unpublished work). Dendritic translation of proteins in association with synaptogenesis was suggested further by the observation of postsynaptic polyribosomal aggregates in neurons that were reafferenting subsequent to destruction of axonal afferents (6). These results suggested that protein synthesis at the synapse might be an important aspect of synaptic plasticity.
|
† |
To whom reprint requests should be addressed. E-mail: greenough@uiuc.edu. |
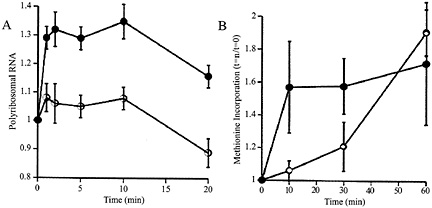
Fig. 1. (A) Relative to baseline, total amount of RNA in precipitatable polysome fraction in K+ stimulated (filled circles) and unstimulated control (open circles) synaptoneurosomes. Potassium depolarization, glutamate administration (not shown), or administration of group I metabotropic receptor agonists such as dihydroxyphenylglycine (not shown) causes a rapid shift of RNA into the polysome-associated fraction. Ordinate: ratio polyribosomal RNA (fraction of total RNA) at t=1, 2, 5, 10, or 20 min to polyribosomal fraction at t=0. (B) The RNA shift to polysomes is associated with increased protein translation, as demonstrated by rapidly increased incorporation of radiolabeled methionine into the K+ stimulated (filled circles) synaptoneurosomes. Ordinate: data expressed as ratio of value at t=10, 20 or 30 min to that at t=0. (Modified from ref. 12.)
Synaptically Regulated Protein Synthesis. To examine this phenomenon further, we developed a synaptoneurosome preparation following the method of Hollingsworth (11); this preparation consisted, as confirmed by electron microscopy, of pinched-off and resealed presynaptic terminals attached to resealed postsynaptic processes, along with other membrane-bound compartments of less identifiable origin. We found that stimulation (by K+ depolarization or glutamate administration) of such synaptoneurosomes from young rat cerebral cortex resulted in a rapid rise in the association of ribosomes with mRNAs, accompanied by a brief acceleration in protein translation, as shown in Fig. 1 (12). This effect was not blocked by antagonists to N-methyl-D-aspartate or aminomethyl phosphonic acid-kainate receptors or by extracellular calcium chelators. It was driven by specific agonists for group I mGluRs and was blocked by intracellular calcium chelators (13). Thus, this neurotransmitter-evoked protein synthesis is not based on enzymatic reactions occurring in the suspension buffer; indeed, disruption of the synaptoneurosomes, either by sonication or by flash freezing, completely abrogated the response.
The mGluR1 postsynaptic signaling response is well understood; it involves G protein-linked activation of phospholipase C, which hydrolyzes membrane phosphatidyl inositol into inositol triphosphate (which in turn liberates Ca2+ ion from stores in the endoplasmic reticulum) and diacylglycerol, which activates protein kinase C. We were able to mimic this kinase cascade by administering phorbol ester as a protein kinase C activator or alternatively by administering a membrane-permeable analog of diacylglycerol, 1-oleoyl-2-acetyl glycerol. The protein kinase C blocker calphostin reduced the strength of the response (13). The synaptoneurosome suspension contains less than 20% glial components (11), in contrast with total brain homogenates; these glia are rich in polyribosomes. Group I mGluR5 have been reported to be present on glial cells (14–16), such that a contribution to observed polyribosomal aggregation or protein synthesis by glial contaminants cannot be ruled out entirely. There are also fragments of dendrites in the preparation, and a contribution from the nonsynaptic mGluRs seen in dendritic membranes and dendritic ribosomes (15) is also possible.
We reasoned that only a subset of mRNAs would likely be involved in this response of increased translation. If we examined the polyribosomes by fractionating them on a continuous sucrose gradient and by using labeled oligonucleotides to probe the RNA along the gradient, we could identify mRNAs that were present in small polyribosomes at a higher level after stimulation than before. For this purpose, we used cDNA clones from a library of mRNA isolated from distal processes of cultured hippocampal neurons by Jim Eberwine’s group (17). Among these clones was one that showed a striking increase in polyribosomal association after mGluR1 stimulation and that showed sequence homology to both FMR-1 (the fragile X mental retardation gene) and the related molecule FXR1. An oligonucleotide probe made to the 3' region of FMR1 (nucleotides 2,023–2,070), a region that has no homology to other known fragile X-related family members, also revealed a shift of mRNA into polyribosomes after mGluR1 stimulation (18). Thus, we concluded that the FMR-1 mRNA is taken up into translational complexes in response to mGluR1 agonist application.
Fragile X syndrome is the most common form of inherited mental retardation, affecting, by one recent estimate, nearly 1 in 2,000 males and roughly half as many females (19). It is caused by the insertion of extra repeats of (CGG)n DNA into the 5' untranslated region, which in turn leads to hypermethylation of CpG residues and transcriptional silencing of the FMR-1 gene. Phenotypic traits include facial abnormalities, macroorchidism, developmental delay, mental retardation, and autistic-like behaviors (20).
To test whether the mRNA shift was accompanied by translation of the FMR protein, we took samples from fresh synaptoneurosome suspensions at short intervals after stimulation by the mGluR1 agonist dihydroxyphenylglycine and compared them with unstimulated samples by Western blot analysis with the antibody 1C3 and by comparing staining intensity standardized to lane loading by restaining the same samples with antibody to glial fibrillary acidic protein. In repeated experiments, we consistently observed an increase in FMRP within 2–5 min after stimulation (18). Six subsequent experiments have all replicated these findings; in the presence of the protein synthesis inhibitor cycloheximide, the effect is not observed. It has been objected (21) that, because mRNA for FMRP has not been observed by in situ hybridization (although it has been detected by reverse transcription-PCR; C.Bagni, personal communication, and by in situ hybridization with multiple probes; ref. 22), the amount of protein must likewise be small; this assertion is a reasonable one. However, it in no way implies that the amount of protein cannot
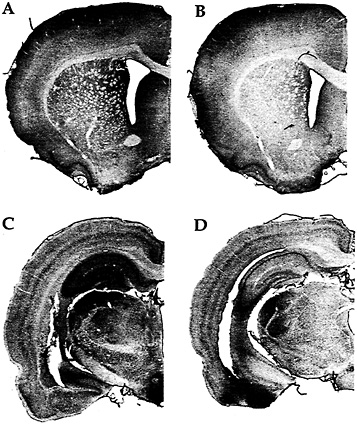
Fig. 2. Immunohistochemistry with an antibody against FMRP in a rat trained on a motor skill learning task for 7 days (A); a rat maintained inactive in its cage for 7 days except for a brief daily period of handling (B); a rat exposed to a complex, social, and toy-filled environment for 20 days (C); and a rat similar to the one described in B but housed for 20 days (D). (Modified from ref. 25.)
be increased rapidly; at 37°C a complete ß-hemoglobin chain is synthesized in about 21 s (23, 24). Indeed, an increase is more easily observed on a low background level. Direct measurement of the absolute amount of new protein must await the development of an anti-FMRP antibody functional for immunoprecipitation.
Our in vitro reports of activation-induced FMRP synthesis are paralleled by reports of activity-induced FMRP synthesis in vivo. Irwin et al. (25), for example, found that FMRP levels were elevated in animals learning new motor skills or being reared in a complex environment as illustrated in Fig. 2. Similarly, in an especially precise in vivo model, Todd and Mack (26) asked whether expression of FMRP might be altered by unilateral whisker stimulation, a model of experience-dependent plasticity. Immunoblots of subcellular fractions of the rat somatosensory cortex showed that the level of FMRP increased in the stimulated areas between 2 and 8 h after stimulation. In contrast, FMRP levels showed either a decrease or no change after a kainic acid-induced seizure. Thus FMRP levels seem to be modulated in vivo in response to physiologically normal levels of neuronal activity.
Is FMRP Required for Synaptic Protein Synthesis? We have investigated synaptoneurosomal protein translation in an fmr-1 knockout mouse model, in which insertion of an inverted neomycin cassette 3' of exon 5 prevents the production of full-length FMRP (27). These mice show immature dendritic spine morphology similar to that observed in human patients with fragile X (ref. 28; see below). We used these mice for a direct test of a role for FMRP in protein synthesis near synapses. We found that, unlike wild-type mice of the same background strain, synaptoneurosomes from fmr-1 knockout mice do not exhibit neurotransmitter-induced rapid formation of polyribosomes or accelerated methionine incorporation into proteins (C.C.Spangler, A.Y.K., V.Bertaina-Anglade, C.K.Base, I.J.Weiler, and W.T.G., unpublished work).
The absence of neurotransmitter-evoked protein synthesis in the synaptoneurosome preparation is paralleled by evidence for reduced protein synthesis at synapses in vivo. A.Y.K. examined visual cortex of FVB/129 knockout and FVB/129 wild-type mice at the electron microscopic level to compare the numbers of postsynaptic polyribosomal aggregates. The density of axospinous synapses in the neuropil of layer IV visual cortex was estimated on postnatal days 15 and 25, ages at which polyribosomal aggregates in spines are elevated, compared with adulthood.** A striking difference emerged: the number of axospi-
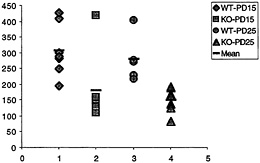
Fig. 3. Number of synapses (ordinate, y axis) with polyribosomal aggregates present in the spine for wild-type (WT) and fmr-1 knockout (KO) sighted FVB mice at 15 and 25 postnatal days (data from C.C.Spangler, A.Y.K., V.Bertaina-Anglade, C.K.Base, I.J.Weiler, and W.T.G., unpublished work). With the exception of one outlier, there is no overlap in the values for knockout and wild type, suggesting a pronounced reduction of synaptic protein synthesis in vivo in the knockout mice.
nous synapses with polyribosomal aggregates was on average twice as high in wild-type animals as in knockout, at both time points, as shown in Fig. 3. This in vivo correlate of the measurement of translation in synaptoneurosomes in vitro greatly enhances the credibility of the synaptoneurosome preparation as a good model for in vivo translational activity (C.C.Spangler, A.Y.K., V.Bertaina-Anglade, C.K.Base, I.J.Weiler, and W.T.G., unpublished work). Thus, one role of FMRP in normal brains seems to be either a permissive role with regard to or
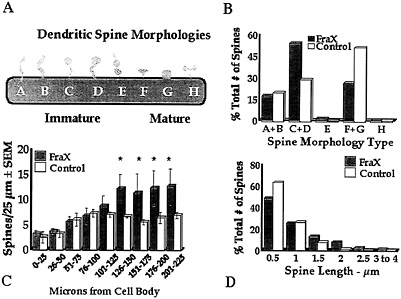
Fig. 4. Summary of measurements of apical dendritic spines of layer V pyramidal neurons in temporal cortex of human autopsy samples of male patients with fragile X syndrome (FraX) and age- and sex-matched controls. (A) Arbitrary spine shape categorization scheme. Each spine was categorized as falling into one of the eight shape categories. (B) Relative numbers of spines of each type. Overall x2 is significant (P < 0.05). Principal differences are relatively greater immature spine types C and D and fewer mature spine types F and G in affected individuals. (C) Numerical density of spines per unit length of dendrite is higher in affected individuals (*, P<0.05). (D) Affected individuals have fewer short (0.5–µm) and more long (=1.5–µm) spines. Overall x2 is significant (P<0.05). Data are from ref. 28.
regulation of a rapid localized translational response to synaptic stimulation.
Neuronal Structural Phenotype of Fragile X Syndrome. Nonquantitative observations of rapid Golgi-stained human autopsy material from a single patient with fragile X syndrome described long, thin, tortuous, dendritic spines with prominent heads and irregular dilations on apical dendrites of pyramidal cells in layers III and V of parieto-occipital cerebral cortex (29, 30). Reduced mean synaptic contact area was also reported, based on electron microscopic observations. No other major neuropathologies were noted. Two additional patients with fragile X syndrome were added to this sample (total n=3) by Hinton et al. (31); similar dendritic spine characteristics were noted, and no differences in neuronal density between patients with fragile X syndrome and controls were found by using a stereological method that would have reported higher density if neurons and their nuclei were larger in one group. The absence of detectable differences suggests relatively normal developmental neurogenesis and cell migration in patients with fragile X syndrome as well as the absence of detectable gross pathology. Subtle differences in the gross size of structures such as cerebellar vermis and hippocampus have been reported by Reiss and colleagues (32, 33) who used structural magnetic resonance imaging; Reyniers et al. (34) were not able to confirm these differences with physical measurement of autopsy specimens from a different set of patients.
We followed up this work with quantitative measurements on layer V pyramidal neurons of Golgi-Kopsch-impregnated human autopsy material from temporal and occipital cerebral cortex from three adult male patients with fragile X syndrome (for each area) and three age-matched male controls (for
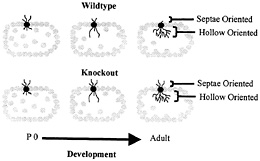
Fig. 5. Schematic depiction of dendritic development in wild-type and fragile X knockout mouse somatosensory whisker barrel cortex. In normal development in the wild type, dendrites initially extend both toward the interior hollow of the dendrite and toward the exterior septae region. As development progresses, hollow-oriented dendrites proliferate, while outwardly oriented dendrites regress. In the knockout mouse, the hollow-oriented dendrites proliferate normally, but the outwardly oriented dendrites exhibit impaired regression. P 0, postnatal day 0. Data are from Galvez et al. (R.Galvez, A.R.Gopal, and W.T.G., unpublished work).
temporal cortex; ref. 28), as illustrated in Fig. 4. Spines in the fragile X samples were significantly longer overall and exhibited a morphology consistent with that of early development: a greater number of long spines with heads and fewer short, stubby, and mushroom-shaped spines were evident in the fragile X cases. No attempt was made to eliminate noninnervated “filopodia,” which cannot be identified in Golgi preparations, but there was no statistically significant difference between groups in the relative frequency of long spines without apparent heads (types A and B in Fig. 4). In addition, the density (number per unit dendrite length) of spines was higher in the patient samples, suggesting a greater number of excitatory inputs to these neurons. The same basic pattern of results was evident on apical shafts, branches from the apical shaft, and basilar branches of the pyramidal neurons examined. We obtained similar findings from analysis of the fmr-1 knockout vs. wild-type FVB/129 hybrid mouse, except that, in the second study, which used animals screened for (and eliminated from the study) the retinal degeneration mutation characteristic of the FVB strain, there was not a significantly greater spine density in the knockout animals (ref. 35 and S.A.I., M.Idupulapati, M.E.Gilbert, J.B.Harris, A.Chakravarti, A.B.Mehta, E.J.Rogers, R.A.Crisostomo, B.P. Larsen, C.J.Alcantara, et al., unpublished work). In these studies of layer V pyramidal neurons, we found no significant differences in the size of the dendritic field or in its pattern of branching in either mouse or human samples (ref. 28 and S.A.I., M.Idupulapati, M.E.Gilbert, J.B.Harris, A.Chakravarti, A.B.Mehta, E.J.Rogers, R.A.Crisostomo, B.P. Larsen, C.J.Alcantara, et al., unpublished work).
One interpretation consistent with these findings is that the knockout mouse has failed, at least in part, to follow the normal maturational pattern of eliminating underused synapses and altering the retained synapses to a more matureappearing form of shorter, fuller spines. It is possible that FMRP or proteins dependent on FMRP for their synthesis are required, either permissively or directly involved, in the synapse stabilization and maturation process. Alternatively, it may be that the FMRP-deficient brain is in a constant state of synaptogenesis, generating new, immature-appearing spines long after elevated rates of synaptogenesis have subsided in the FMRP-containing brain.
A recent experiment may provide a partial answer to this question. Galvez et al. (R.Galvez, A.R.Gopal, and W.T.G., unpublished work) examined the “barrels” in somatosensory cortex that process information from the large facial whiskers in fmr-1 knockout vs. wild-type mice. This structure is one in which the overproduction and regression of dendrites during development is particularly evident, because dendrites of layer IV spiny stellate neurons that initially extend in the wrong direction, toward the septae outside of the barrel rather than toward the hollow at its center, are subsequently retracted, contributing to the asymmetric branching pattern exhibited by these neurons in adult mice (3). Galvez et al. (R.Galvez, A.R. Gopal, and W.T.G., unpublished work) compared the extent of both the properly directed hollow-oriented dendrites and the improperly directed dendrites that grew toward the outside of the barrel. They found the extent of properly oriented dendrites to be statistically identical in knockout and wild-type mice, whereas the knockout mice had retained a greater amount of dendrites oriented in the improper direction (Fig. 5). Thus, in this case, the fmr-1 knockout mice exhibited what seemed to be a failure to undergo the normal dendritic retraction process characteristic of these structures. This result is compatible, at a dendritic level, with what may seem to be a failure of the synapse elimination process when one compares spine density in the two types of animals. Thus, our working hypothesis remains that there is an impairment of mechanisms that promote synapse maturation and pruning in the fmr-1 knockout mouse and that FMRP plays a permissive or directive role in the neural maturation process.
This work was supported by grants from the FRAXA Research Foundation, HD37175 from the National Institute of Child Health and Human Development, MH35321 from the National Institute of Mental Health, and AG10154 from the National Institute on Aging.
1. Greenough, W.T., Black, J.E. & Wallace, C. (1987) Child Dev. 58, 539–559.
2. LeVay, S., Wiesel, T.N. & Hubel, D.H. (1980) J. Comp. Neurol 191, 1–51.
3. Greenough, W.T. & Chang, F.-L.F. (1988) Dev. Brain Res. 43, 148–152.
4. Steward, O. & Levy, W. (1982) J. Neurosci. 2, 284–291.
5. Steward, O. & Falk, P. (1986) J. Neurosci. 6, 412–423.
6. Steward, O. (1983) J. Neurosci. 3, 177–188.
7. Turner, A.M. & Greenough, W.T. (1985) Brain Res. 329, 195–203.
8. Black, J.E., Isaacs, K.R., Anderson, B.J., Alcantara, A.A. & Greenough, W.T. (1990) Proc. Natl. Acad. Sci. USA 87, 5568–5572.
9. Kleim, J.A., Lussnig, E., Schwarz, E.R., Comery, T.A. & Greenough, W.T. (1996) J. Neurosci. 16, 4529–4535.
10. Greenough, W.T., Hwang, H.-M. & German, C. (1985) Proc. Natl. Acad. Sci. USA 82, 4549–4552.
11. Hollingsworth, E.B., McNeal, E.T., Burton, J.L., Williams, R.J., Daly, J.W. & Creveling, C.R. (1985) J. Neurosci. 5, 2240–2253.
12. Weiler, I.J. & Greenough, W.T. (1991) Mol. Cell. Neurosci. 2, 305–314.
13. Weiler, I.J. & Greenough, W.T. (1993) Proc. Natl. Acad. Sci. USA 90, 7168–7171.
14. Berthele, A., Platzer, S., Laurie, D.J., Weis, S., Sommer, B., Zieglgansberger, W., Conrad, B. & Tolle, T.R. (1999) NeuroReport 10, 3861–3867.
15. Liu, X.B., Munoz, A. & Jones, E.G. (1998) J. Comp. Neurol. 395, 450–465.
16. Porter, J.T. & McCarthy, K.D. (1995) Glia 13, 101–112.
17. Miyashiro, K., Dichter, M. & Eberwine, J. (1994) Proc. Natl. Acad. Sci. USA 91, 10800–10804.
18. Weiler, I.J., Irwin, S.A., Klintsova, A.Y., Spencer, C.M., Brazelton, A.D., Miyashiro, K., Comery, T.A., Patel, B., Eberwine, J. & Greenough, W.T. (1997) Proc. Natl. Acad. Sci. USA 94, 5395–5400.
19. Brown, W.T. (1996) Am.J.Hum. Genet. 58, 903–905.
20. Hagerman, R.J. & Cronister, A., eds. (1996) Fragile X Syndrome: Diagnosis, Treatment, and Research (Johns Hopkins Univ. Press, Baltimore).
21. Steward, O. & Schuman, E.M. (2001) Ann. Rev. Neurosci. 24, 299–325.
22. Shestakova, E.A., Singer, R.H. & Condeelis, J. (2001) Proc. Natl. Acad. Sci. USA 98, 7045–7050.
23. Knopf, P.M. & Dintzis, H.M. (1965) Biochemistry 4, 1427–1434.
24. Hunt, T., Hunter, T. & Munro, A. (1969) J. Mol. Biol. 43, 123–133.
25. Irwin, S.A., Swain, R.A., Christmon, C.A., Chakravarti, A., Weiler, I.J. & Greenough, W.T. (2000) Neurobiol. Learn. Mem. 73, 87–93 and erratum (2000) 74, 80–87.
26. Todd, P.K. & Mack, K.J. (2000) Brain Res. Mol. Brain Res. 80, 17–25.
27. Consortium, D.-B.F.X. (1994) Cell 78, 23–33.
28. Irwin, S.A., Patel, B., Idupulapati, M., Harris, J.B., Cristostomo, R., Larsen, B.P., Kooy, F., Willems, P.J., Cras, P., Kozlowski, P.B., et al. (2001) Am.J. Med. Genet. 98, 161–167.
29. Rudelli, R.D., Brown, W.T., Wisniewski, K., Jenkins, E.C., Laure-Kamionowska, M., Connell, F. & Wisniewski, H.M. (1985) Acta Newopathol. 67, 289–295.
30. Wisniewski, K.E., Segan, S.M., Miezejeski, C.M., Sersen, E.A & Rudelli, R.D. (1991) Am.J. Med. Genet. 38, 476–280.
31. Hinton, V.J., Brown, W.T., Wisniewski, D. & Rudelli, R.D. (1991).Am.J.Med. Genet. 41, 239–294.
32. Reiss, A.L., Aylward, E., Freund, L.S., Joshi, P.K. & Bryan, R.N. (1991) Ann. Neurol. 29, 26–32.
33. Reiss, A.L., Lee, J. & Freund, L. (1994) Neurology 44, 1317–1324.
34. Reyniers, E., Martin, J.J., Cras, P., Van Marck, E., Handig, I., Jorens, H.Z., Oostra, B.A., Kooy, R.F. & Willems, P.J. (1999) Am.J. Med. Genet. 84, 245–249.
35. Comery, T.A., Harris, J.B., Willems, P.J., Oostra, B.A., Irwin, S.A., Weiler, I.J. & Greenough, W.T. (1997) Proc. Natl. Acad. Sci. USA 94, 5401–5404.






