
Ribonucleoprotein infrastructure regulating the flow of genetic information between the genome and the proteome
Jack D.Keene*
Department of Microbiology, Duke University Medical Center, Durham, NC 27710
Following transcription and splicing, each mRNA of a mammalian cell passes into the cytoplasm where its fate is in the hands of a complex network of ribonucleoproteins (mRNPs). The success or failure of a gene to be expressed depends on the performance of this mRNP infrastructure. The entry, gating, processing, and transit of each mRNA through an mRNP network helps determine the composition of a cell’s proteome. The machinery that regulates storage, turnover, and translational activation of mRNAs is not well understood, in part, because of the heterogeneous nature of mRNPs. Recently, subsets of cellular mRNAs clustered as members of mRNP complexes have been identified by using antibodies reactive with RNA-binding proteins, including ELAV/Hu, elF-4E, and poly(A)-binding proteins. Cytoplasmic ELAV/Hu proteins are involved in the stability and translation of early response gene (ERG) transcripts and are expressed predominately in neurons. mRNAs recovered from ELAV/Hu mRNP complexes were found to have similar sequence elements, suggesting a common structural linkage among them. This approach opens the possibility of identifying transcripts physically clustered in vivo that may have similar fates or functions. Moreover, the proteins encoded by physically organized mRNAs may participate in the same biological process or structural outcome, not unlike operons and their polycistronic mRNAs do in prokaryotic organisms. Our goal is to understand the organization and flow of genetic information on an integrative systems level by analyzing the collective properties of proteins and mRNAs associated with mRNPs in vivo.
Understanding the physical organization of gene transcripts in mammalian cells has presented significant difficulties for several reasons: (i) mRNA is generally unstable, (ii) each mRNA is relatively inabundant, and (iii) the association of each mRNA with proteins results in the formation of heterogeneous complexes with diverse biophysical properties. These qualities, together with the lack of suitable technologies to purify ribonucleoprotein (mRNP) complexes, have precluded molecular dissection of their component parts. Recently, immunological and biochemical techniques, combined with genomic methods, have allowed the elucidation of mRNAs associated with ELAV/Hu and other mRNP complexes, and the subsequent analysis of their structural and functional properties. This approach to understanding the collective properties of the mRNP infrastructure, and the organized networks of transcripts that are regulated posttranscriptionally has been termed ribonomics (1).
ELAV/Hu RNA Recognition Motif (RRM) Proteins Associate with a Distinct Subset of mRNAs. ELAV/Hu proteins were useful for developing ribonomic methods because they bind in vitro to a class of messenger RNA containing AU-rich sequences (2–6). Although many early response gene (ERG) mRNAs that contain AU-rich elements (ARE) in their 3' UTRs tend to be unstable in vivo, many other mRNAs that are not considered unstable also contain AU-rich regions. Several different proteins have been reported to bind AU-rich elements in mRNA, but the ELAV/Hu proteins are unique in that they have been shown to stabilize and/or activate translation of target mRNAs (reviewed in refs. 7 and 8). Three of the four known types of ELAV/Hu protein (HuB, HuC, and HuD) are expressed specifically in neurons or gonads and are predominately cytoplasmic, which is consistent with a role in mRNA stability and translation (reviewed in ref. 9). As shown in Fig. 1A, ELAV/Hu proteins reside in cytoplasmic granules that extend out of the cell body and along dendrites (10, 11). It is presumed that these granules represent mRNPs containing ELAV/Hu proteins bound to mRNAs. An early clue that suggested a role for ELAV/Hu proteins in translational control was the altered distribution of the protein in cortical neurons following treatment with puromycin, an antibiotic that blocks the elongation of mRNAs on polysomes (Fig. 1B; ref. 11). The presence of ELAV/Hu proteins in dendritic granules is consistent with their playing a localized role in translation. Ectopic expression of HuB in 3T3L1 preadipocytes and in hNT2 preneuronal teratocarcinoma cells resulted in translational activation of target mRNAs encoding glucose transporter-1 protein (12) and neurofilament M protein (NF-M) (13), respectively. Moreover, in the hNT2 preneuronal cells (13), and in chicken neural crest cells (14), forced expression of Hu proteins resulted in the spontaneous development of neurites.
In addition to having a profound biological effect on cell morphology, stability, and translation of specific target mRNAs, ELAV/Hu proteins appear to be multitargeted toward a broad range of AU-rich and ERG-type mRNAs. Based on in vitro selection of an AU-rich consensus sequence, Levine et al. (2) tested the binding of HuB in vitro to transcripts representing c-myc, c-fos, and GM-CSF and found high affinity binding. To define the larger mRNA-binding population, methods were subsequently developed to select mRNAs from cDNA libraries by using HuB (3). This resulted in the identification of at least one hundred putative mRNA targets for the HuB protein. In nearly every case, these mRNAs represented members of a subset of cellular growth regulatory proteins containing AREs. This result opened the intriguing possibility that dozens of ELAV/Hu targeted mRNAs containing AREs could be stabilized and/or translationally activated as a group in response to ELAV/Hu protein. mRNAs shown to be affected following overexpression of Hu proteins include glucose trans-
|
* |
To whom reprint requests should be addressed at: Department of Microbiology, Box 3020, Duke University Medical Center, Durham, NC 27710. E-mail: keene001@mc.duke.edu. |
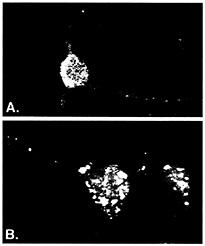
Fig. 1. ELAV/Hu RRM proteins form distinct granules in cell body and dendrites of neurons. Rabbit polyclonal serum prepared against recombinant HuB was used to visualize ELAV/Hu proteins in isolated rat embryonal cortical neurons by using confocal microscopy (reprinted from ref. 11). Prebleed serum showed no appreciable fluorescence of any neuronal samples (10, 11). The granules containing Hu proteins (A) coalesced following treatment with puromycin (B) to disrupt translation. [Reproduced with permission from ref. 11 (Copyright 1998, J. Cell Sci.).]
porter 1 (12), NF-M (13), GAP43 (15), VEGF (16), c-fos (17–19), c-myc (unpublished results), TNF-a (19), GM-CSF (19), and tau (20). With the exception of NF-M mRNA (13), the binding of these mRNA targets to ELAV/Hu proteins has only been demonstrated when using in vitro methods.
Messenger RNAs Are Generally Inabundant and Unstable. The average number of any particular mRNA species present in a mammalian cell varies over a range from less than one to as many as 1,000. This is in contrast to the U1 snRNA that is present in approximately 1 million copies per cell. In human cells, an average of about six copies of each mRNA per cell has been approximated with very few genes having at steady state as high as 50 to 100 copies per cell (21). In yeast, this number is approximately an order of magnitude lower. It is striking that so few copies of each mRNA are maintained in the steady state, and this suggests that mRNAs are continuously supplied and destroyed during normal cell metabolism. It is likely that a constant flux of mRNA through the mRNP infrastructure provides agility to the gene expression program. In profiling the expression of mRNAs by using techniques like microarray analysis or Serial Analysis of Gene Expression (SAGE), the steady-state level of each mRNA can be quantitated (22, 23). However, these procedures do not distinguish translationally active messages from inactive messages, and the relative turnover rate of each message can significantly affect protein output (23). Furthermore, the organization of mRNAs into functional complexes may influence their state of expression.
The instability of many mRNAs in comparison to ribosomal RNAs, transfer RNAs, and small nuclear RNAs, as well as their inabundance, has made analysis of their in vivo-associated protein interactions particularly difficult. As a result, most of what is currently known about mRNA-protein interactions has been derived from in vitro binding experiments. Nonetheless, the stability of endogenous mRNAs has been studied by using a variety of analytical tools (24–26). The relative stability of mRNAs involved in various biological processes can be depicted
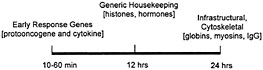
Fig. 2. The relative stability of some diverse cellular mRNAs.
on a time line along which the half lives of ERG mRNAs (such as protooncogene and cytokine transcripts) is as short as a few minutes, and housekeeping proteins like cytoskeletal components and histones have half-lives equivalent to one full cell cycle (Fig. 2). It is generally true that whereas mRNAs that encode highly abundant and stable housekeeping proteins appear to be stable themselves, mRNAs encoding many growth regulatory proteins are very unstable (25). This instability is presumably due to the powerful and possibly undesirable effects on normal cell growth and differentiation that these gene products can have. The necessity to retain tight control over growth stimulatory proteins begins at the level of transcription, but is usually maintained also at the posttranscriptional level. Short half-lives for mRNAs encoding growth factors such as c-fos or c-myc allow cells to retain tighter control at the level of transcription, and therefore, the final production of the protein can be regulated with greater precision (24–26). In keeping with this line of reasoning, the ERG (also known as the immediate early gene) products encode growth regulatory proteins, and include mRNAs with short half-lives. The ability of the ELAV/Hu proteins to bind certain AU-rich-region-containing ERG mRNAs suggested to us that a large target set of AU-richregion-containing mRNAs might be captured by using ELAV/Hu proteins to identify en masse a unique subset of the total cell mRNA population (3). More recently, a direct in vivo approach has been possible by isolating mRNP complexes and identifying the mRNA subsets by using nucleic acid hybridization (1).
The Heterogeneous Nature of mRNPs. Heterogeneous nuclear RNA (hnRNA) and the correspondingly diverse hnRNPs have been recognized for many years (reviewed in refs. 27–30). Analysis on density and velocity gradients has revealed that whole-cell mRNA, and mRNA-binding proteins, often spread across a gradient making it difficult to discern specific proteins or mRNAs that might associate with one another (Fig. 3). This heterogeneity has caused the field to rely heavily on in vitro binding methods for study of the interactions between individual proteins and the sequence elements found in mRNAs (30, 31). It has been possible to analyze the migration of individual mRNAs on sucrose velocity gradients by using Northern blotting of gradient fractions. Indeed, several studies have used gradient analysis to localize translationally engaged mRNAs in fractions containing active polysomes (12, 13, 32, 33). However, mRNPs that are not associated with the assembled translation apparatus often remain widely distributed between the free mRNA and the assembled polysomes as exemplified with ELAV/Hu proteins (Fig. 3A). It has been assumed that these widely distributed mRNPs represent complexes containing mRNAs that are competent for, but not engaged in, translation. However, as shown in Fig. 3, treatment of cell extracts with EDTA can release the ELAV/Hu proteins from the region of active translation (ß complexes) and shift them to an intermediate position (a complexes). Similar results were evident when cells were treated with puromycin to inhibit translation without disrupting polysomes (Fig. 3C). As described by Tenenbaum et al. (1), mRNP complexes that are recovered by immunoprecipitation of tagged HuB from transfected P19 cells following treatment with EDTA
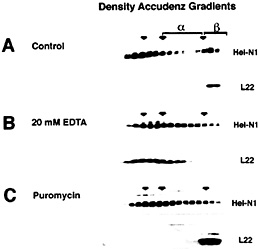
Fig. 3. Distribution of ELAV/Hu proteins following separation on Accudenz density gradients. (A) ELAV/Hu proteins that migrate in the upper (Left) and in the lower (Right) regions of the gradient represent free protein and polysome-associated protein, respectively. In the region between these populations (a-complexes) are ELAV/Hu mRNPs. Treatment with EDTA (B) or puromycin (C) to disrupt the translation apparatus results in the appearance of ELAV/Hu protein in the a-complex region. The release of ELAV/Hu protein from ß-complexes following treatment with EDTA (B) was monitored by tracking the ribosomal protein L22. Puromycin treatment (C) also resulted in the appearance of a-complexes with a concomitant loss of ß-complexes, but as expected, did not disrupt polysomes as shown by the migration of L22. [Reproduced with permission from ref. 11 (Copyright 1998, J. Cell Sci.).].
contained a unique subset of mRNAs possessing AU-rich sequences. This offers a useful convention for discerning aspects of the physical organization of mRNAs in mRNP complexes.
Combinatorial Interactions in mRNPs. The composition of mRNP complexes includes the mRNA(s) and the proteins that assemble onto the mRNAs in various combinations. Not unlike transcription, splicing, and polyadenylation, translation also involves combinatorial interactions of RNAs and proteins (34, 35). For example, in the case of translation factors, the circularization of the engaged mRNA is mediated by interactions between eIF-4E and eIF-4G. Whereas some proteins like the ELAV/Hu are bound directly to AU-rich-region-containing mRNAs, other factors are not directly bound, but participate in forming RNP complexes through protein-protein interactions with RNA-binding proteins. The availability of a variety of combinations of assembled factors is thought to allow specificity of recognition, and possibly specificity of localization of transcripts. Although it has yet to be demonstrated, thousands of different combinations of transcripts may be organized into distinct mRNP classes by using different combinations of proteins. Via this process, it would not be necessary to access thousands of different RNA binding proteins to govern the organization of thousands of different mRNAs. However, it has not yet been determined for any mRNP complex whether multiple mRNAs are clustered together into the same physical particle. Nor is it known whether the regulation of expression of clustered mRNAs is coordinated. Although the physical organization of mRNAs within the mRNP infrastructure involves both direct and indirect RNA binding, the factors that regulate each mRNA are yet to be determined. Regardless of the exact structure of ELAV/Hu mRNPs, the ERG mRNAs that successfully exit the nucleus are hypothesized
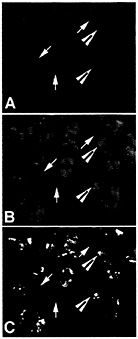
Fig. 4. ELAV/Hu protein colocalizes with poly(A)+ mRNA in distinct cytoplasmic granules. Total poly(A)+ mRNA in puromycin-treated human medulloblastoma cells was stained with an antibody to a digoxigenin-labeled oligo dT probe (A) and ELAV/Hu protein was stained (B) with the same recombinant HuB antibody used in Fig. 1 (reprinted from ref. 11). Large arrowheads show the coalesced ELAV/Hu protein granules that overlap a region stained for the total poly(A)+ mRNA population (C), whereas arrows show regions stained for poly(A)+ mRNA but without detectable ELAV/Hu protein. Recovery of ELAV/Hu complexes is expected to reveal those mRNAs within the population of total cell mRNA that are specifically associated with ELAV/Hu mRNPs. [Reproduced with permission from ref. 11 (Copyright 1998, J. Cell Sci.).]
to pass into a cytoplasmic infrastructure where their fate is determined by the organizational properties of mRNA-binding proteins and mRNP-associated factors (reviewed in ref. 7).
As shown in Fig. 4, ELAV/Hu mRNP complexes visually overlap with polyadenylated mRNA in the cytoplasm of medulloblastoma cells (11). As expected, only a fraction of poly(A)+ mRNA colocalizes with ELAV/Hu protein. For example, Fig. 4C shows the overlap of both poly(A)+ mRNA and the ELAV/Hu proteins as yellow granules following treatment with puromycin. These data are consistent with the demonstrated role of HuB in activating the translation and stability of target mRNAs (7), but also illustrate the clusters of mRNP complexes containing both ELAV/Hu proteins and polyadenylated mRNAs. These findings led to experiments designed to isolate ELAV/Hu mRNP complexes following their release from polysomes to characterize the associated mRNAs (1). It is presumed that these treatments release mRNPs similar to the a complexes shown in Fig. 3. Therefore, the limitations imposed by in vitro binding and selection (3) can be overcome by the isolation of endogenous mRNPs from cell extracts and identification of the mRNAs contained in mRNPs by directly identifying the mRNA subset on microarrays (1). Ribonomics provides a set of biochemical conventions for isolating mRNPs that can be applied systematically to determine the clustering of mRNAs that have
structural commonality, and potentially functional relationships among their gene products. Is it possible that multicellular organisms derive genetic complexity by organizing various combinations of mRNAs as mRNPs rather than using polycistronic transcripts from operons?
Posttranscriptional Regulation of Gene Expression Involving Hu mRNPs. Why is it valuable to identify mRNA subsets in messenger RNP complexes? One reason is that the expression of gene products encoded by mRNAs may need to be temporally controlled, whether sequentially or simultaneously. For example, during neuronal differentiation new proteins including neurofilaments, MAP proteins, and tau proteins are expressed sequentially following addition of retinoic acid to embryonic carcinoma cells (36). Although the transcription program is essential to neuronal differentiation, posttranscriptional regulation also has been documented to be important during growth and differentiation in neuronal and other systems (37–42). As noted above, mRNAs encoding neurofilament M (13) and tau (22) can bind in their noncoding regions to ELAV/Hu proteins that are, in turn, induced by retinoic acid before the appearance of NF-M or tau. In the case of NF-M, it has been shown that the HuB protein recruits the mRNA to active polysomes where protein production is up-regulated (13). Other examples of posttranscriptional regulation of mRNA expression by the ELAV/Hu proteins have been demonstrated with other early response gene (ERG) transcripts (7). It appears that many neuron-specific mRNAs are contained in mRNP complexes with the ELAV/Hu proteins and other RNA-binding proteins, and that their expression is activated during differentiation (1). The clustering of mRNAs as subsets that are expressed during neuronal differentiation and captured in these mRNP complexes may indicate that they are posttranscriptionally regulated in parallel. It is hypothesized that ELAV/Hu mRNPs represent a critical node in a pathway of posttranscriptional gene regulation in which decisions to stabilize, degrade, or translate multiple members of a subset of mRNAs can affect neuronal differentiation (refs. 2 and 3; reviewed in ref. 7).
Because many of the ARE-containing mRNAs shown to bind ELAV/Hu proteins encode transcription factors such as fos, myc, Id, and CREB (2–9, 43), there is potential for posttranscriptional events to feedback and alter the transcriptional program during differentiation. Likewise, secreted cytokines whose mRNAs also contain AREs have the potential to affect the growth and activation of T cells in an autocrine or paracrine manner (44). Trans-acting mRNA-binding proteins that affect the expression of cytokine mRNAs as a distinct subset have not been identified, but are thought to exist. The ubiquitously expressed ELAV/Hu protein, HuA (HuR), can bind to cytokine mRNAs in T cells (ref. 45; unpublished results), but has not been shown to have a direct regulatory role on cytokine expression. It is likely that specialized ARE-binding proteins regulate subclasses of cytokine mRNAs because different cytokines are regulated independently of one another at the posttranscriptional level (44, 45).
It is unlikely that the ARE represents the only cis-acting sequence among cellular transcripts that defines a structurally or functionally related subset of mRNAs. By using RNA-binding or RNP-associated proteins to isolate mRNPs, it should be possible to discover such relationships. Our laboratory has isolated mRNPs containing cap-binding protein, poly(A)-binding protein, and other mRNP proteins, and detected mRNAs, which appear to have relationships with one another (ref. 1; unpublished results). For example, we have used antibodies reactive with the p62 RRM/KH protein, which is a member of the insulin-like growth factor mRNA-binding protein (IMP) family (46), and detected a distinct set of mRNAs (S.Tenenbaum, C. Carson, P.Lager, E.Tan, and J.D.K., unpublished results). Among the subset were three mRNAs reported to bind members of the IMP RNA-binding protein family: c-myc, ß-actin, and insulin-like growth factor mRNAs. One implication of identifying mRNA subsets that encode functionally linked proteins is that they may be involved in the same biochemical pathway or form the same macromolecular structure. Thus, coordinated expression may be regulated at the posttranscriptional level much like operons are regulated at the transcriptional level in prokaryotic systems.
Gene Expression May Be Regulated Posttranscriptionally in Dendrites. One intriguing hypothesis regarding the organization of mRNAs in neurons is that posttranscriptional events in the cytoplasm may affect transcriptional events in the nucleus. For example, mRNAs encoding transcription factors appear to be packaged in the cytoplasm at distances far from the nucleus, and their localized expression in response to external stimuli may influence cellular mechanisms in the nucleus (39, 40, 47). As noted above, many of the mRNAs to which the ELAV/Hu proteins bind encode transcription factors, including CREB, ERG-1, fos, myc, and Id (1–9). Eberwine and colleagues (39) have suggested that “nuclear imprinting” is a phenomenon in which the production of transcription factors is regulated posttranscriptionally in dendrites. The expression of these factors is activated locally following stimulation of neurons, thus leading to secondary activation of nuclear genes when the transcription factors are transported back to the nucleus (39, 47). The advantages of such a regulatory pathway may include direct activation of specific genes (e.g., ERG) without the potential complications involved in activating multiple signal transduction cascades intended to activate multiple downstream functions. We have proposed that the ELAV/Hu proteins could be involved in multifunctional activation in neurons by regulating not only transcription factor mRNAs, but also other ERG-type mRNAs that participate in intracellular signaling, cytoskeletal assembly, and membrane activity (reviewed in refs. 7 and 9).
Parallel Analysis of mRNPs Implicated in Posttranscriptional Gene Expression: A Ribonomic Approach. It is possible to classify mRNA-binding proteins into three groups: those that are global and bind nearly all mRNAs without distinguishing unique sequences, the group-specific mRNA-binding proteins that associate with subsets of the global mRNA population, and those that are type-specific because they recognize a highly unique mRNA sequence, perhaps present in only one mRNA, with high specificity. We suggest that in some cases, the group-specific mRNA-binding proteins associate with multiple mRNAs that are structurally, and/or perhaps, functionally related. The functional relationships may concern RNA stability or instability, translational activation, transport, or the mRNA subset may encode a group of proteins involved in a common pathway or phenotypic outcome. Whereas the ARE-containing mRNAs represent an example of a group-specific subset of mRNAs that are regulated at the level of stability and translation, the iron response element (IRE)-binding protein (48) and the histone mRNA stem-loop-binding protein (49, 50) represent type-specific mRNA-binding proteins that are also involved in RNA stability and translation. The type-specific proteins recognize sequence elements that tend to encode protein products needed in large amounts within short time intervals during biological processes such as the cell cycle (50).
As suggested above, it is not likely that every mRNA transcript has its own unique binding protein because tens of thousands of cell proteins would have to be dedicated to controlling posttranscriptional gene expression. Because mRNA-protein interactions in vivo are likely to be combinatorial, it is reasonable to predict that most mRNAs can be grouped into structurally and/or functionally related subsets that associate with a limited
set of protein components. Gaining access to these putative mRNA subsets requires an ability to isolate mRNPs by using biochemical procedures. A major goal of our ribonomic approaches is to understand the dynamics of these mRNA subsets and their structural and/or functional clustering during growth and development.
Structural and Functional Linkages Among mRNAs Clustered in mRNP Complexes. mRNAs that share common sequence elements in their untranslated or coding regions have the potential to interact with the same RNA-binding proteins at those sites. With the exception of the ARE, few sequence elements common to a collection of mRNAs have been identified. Searching for homologous sequence elements by computer may reveal common features among multiple mRNAs, but a more concrete approach would be to design methods that allow RNA-binding proteins to find such elements. The method of Gao et al. (3) provided an in vitro approach to partitioning mRNA populations with related sequence elements. More recently, the ribonomic approach of Tenenbaum et al. (1) has opened the possibility of identifying mRNAs with related protein binding elements by immunoprecipitation of proteins present in mRNP complexes followed by microarray analysis. Via this approach, ARE-binding proteins of the Hu family have made it possible to identify multiple mRNAs with common Hu protein binding elements. Broader applications using many other mRNP proteins are expected to identify additional structural linkages among subsets of mRNAs.
The most important feature in common among a group of physically clustered mRNAs associated with mRNPs would be a functional relationship among their encoded proteins. If mRNAs that are isolated by purifying mRNP complexes encode proteins that function in a common pathway, it is logical to conclude there is a functional linkage. On the other hand, if functional relationships among the protein products encoded by an mRNA subset were not readily apparent from the literature, it would be necessary to investigate their individual functions. This raises the question of what properties define functional relationships among gene products. Our hypothesis is that mRNP complexes contain mRNAs that encode proteins that work together in a biological process or form a biological structure such as a ribosome or a spliceosome. In many cases, the encoded products may appear to be of diverse function, perhaps because they regulate complex biological outcomes. In some cases, these diverse gene products may be required for biological remodeling during growth and differentiation. For example, the ELAV/Hu proteins are associated with mRNAs that encode early response gene products (often transcription factors), signaling proteins that can activate downstream pathways, cytoskeletal proteins, and glucose transporters that can mobilize cellular energetics. These, and other activated functions, can provide gene products needed to construct new cell structures such as neuronal processes or dendritic spines (13–15). Therefore, if functional linkage is defined as the mobilization of a variety of gene products needed to remodel cell structure or behavior, it is expected that mRNA subsets clustered in mRNP complexes would encode proteins with seemingly diverse properties.
How Many Different RNA-Binding Proteins Exist in Model Organisms? The complexity of mRNPs and their potential roles in posttranscriptional regulation can be approximated by considering the number of RNA-binding proteins available for interactions with RNA. As one indication of their abundance, the RRM RNA-binding proteins are one of the largest families of proteins found in the genomic databases (51, 52). Moreover, based on the number of annotated RNA-binding proteins identified in more exhaustively studied model systems such as yeast, nematode, and fly, one can estimate the number of RNA-binding proteins in the human genome. For example, S. cerevisiae has over 6,700 genes with 471 annotated RNA-binding proteins in the databases. However, only 312 of the 471 annotated proteins have been designated to have a role in RNA processing, modification, splicing, or turnover (Yeast Proteome Database at www.proteome.com). These numbers suggest that an estimated range of 5–8% of the genes encode proteins involved in RNA processing. On the other hand, C. elegans and D. melanogaster have fewer annotated RNA-binding proteins in the databases than yeast, but the values range from 2–3% of the total number of known genes. Because the number of human genes is estimated to be 32,000, the estimated number of RNA-binding proteins encoded in the human genome would be as low as 640 (2% of 32,000) or as high as 2,560 (8% of 32,000). The most probable and conservative estimates would place the number of human RNA-binding proteins at ˜1,500, but their distribution across the global, group-specific, and type-specific classes is unknown. It is also not known which of these proteins might interact with small RNAs, ribosomal RNAs, or mRNAs. Although the significance of this estimated number of human RNA-binding proteins is not clear at the present time, it does suggest that ribonomic analyses have the potential to elucidate many structurally and/or functionally linked mRNA subsets.
Global mRNA-binding proteins such as poly (A)-binding protein recognize a large set of mRNAs, whereas the group-specific and type-specific mRNAs encompass subsets or even subsets of subsets of mRNAs. If the technology was available to isolate every cellular mRNP, one should be able to account quantitatively for every mRNA in the cell. The ability to define a ribonome by categorizing all mRNAs into overlapping subsets of mRNP complexes is expected to reveal a structural network for organizing genetic information (1). Furthermore, alterations of the ribonomic network may be characteristic of particular diseases. Likewise, the effects of drugs, chemicals, or toxins, as well as states of differentiation or aging should be reflected in the ribonomic analysis of a cell or tissue. For example, Tenenbaum et al. (1) demonstrated that upon treatment of ELAV/Hu-transfected P19 cells with retinoic acid to induce neuronal differentiation, new mRNAs entered into the ELAV/Hu mRNP complexes. By quantitative analysis, it was evident that these mRNAs were uniquely compartmentalized in ELAV/Hu mRNP complexes and would not all have been identified by using standard transcriptomics.
Cell Type-Specific Gene Expression Profiling. One of the potential applications of ribonomics is the recovery of mRNPs that are cell type-specific (1). The ability to recover mRNPs from whole tissue extracts that contain certain RNA-binding proteins only in a single cell type within the tissue should allow recovery of the mRNAs from that single cell type. For example, because ELAV/Hu proteins are expressed in neurons, but are not expressed in glial cells (10), one would expect to find only the neuronal mRNAs in Hu mRNP immunoprecipitates from whole brain extracts. Moreover, ELAV/Hu proteins are ectopically expressed in small cell lung tumor cells and in medulloblastoma cells (10, 11), making it possible to recover mRNAs that are present in the ELAV/Hu mRNPs of the tumor cells by using whole tumor extracts. Therefore, the potential to perform gene expression profiling of single cell types within complex tissues or tumors by using ribonomic approaches could shed light on how cell-cell communication affects the gene expression of neighboring cells. This could provide a means for understanding the crosstalk among cells within tumors, and the effects of antiangiogenesis factors on endothelial cell versus tumor cell gene expression (53).
Multiplexing RNA Processing During Growth and Differentiation. The genomics era has brought powerful tools to analyze expressed genes by using a variety of techniques, including sequencing by
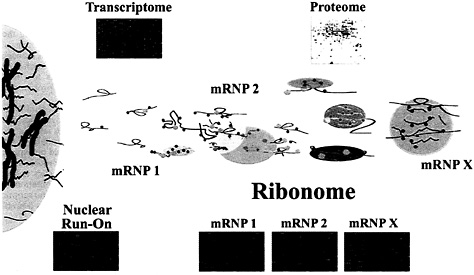
Fig. 5. Depiction of the possible organization of mRNAs in mRNP complexes in the cell cytoplasm. Messenger RNP complexes may contain single mRNAs or multiple mRNAs in association with proteins that are either directly or indirectly associated. The Upper Insets depict the total mRNA expressed in the cell (transcriptome) as a microarray and the proteome of the cell is depicted as a two-dimensional gel. The microarrays below the mRNP complexes (Ribonome) are labeled mRNP-1 through mRNP-X and depict multiple mRNAs found in mRNP complexes isolated by using antibodies reactive with mRNA-associated proteins. Microarrays representing nuclear run-on experiments (Left) can be derived by transcription using isolated nuclei (unpublished results) and analysis on Atlas arrays (CLONTECH). As opposed to transcriptomic profiles that are the result of both transcriptional and posttranscriptional contributions and represent accumulated steady-state levels of mRNA, the mRNAs detected by nuclear run-on represent only the transcriptional contribution of genes before the influence of posttranscriptional events in the ribonome.
hybridization and computational analysis of gene sequences (53, 54). However, it is clear that the protein output of cells and tissues does not always correlate precisely with the mRNA content for a variety of reasons (21, 23, 30, 33, 54, 55). In particular, posttranscriptional regulation and posttranslational modifications can significantly affect the quality and quantity of protein that a given gene will generate. To account for post-transcriptional effects, methods are needed to assess the organization of mRNAs in mRNP complexes and their corresponding functional relationships. Investigations generally examine single gene products for RNA splicing, transport, stability, or translation, but could benefit from the ability to capture mRNP complexes en masse so that the functional complexity and dynamics of posttranscriptional control can be studied. Therefore, the ability to isolate mRNP complexes from cell extracts may provide a means by which to multiplex the analysis of mRNAs as subsets based on structural and/or functional relatedness. Examples of parallel analysis of mRNA targets for RNA-binding proteins have included cDNA libraries prepared from mRNA subsets isolated by iterative in vitro selection using ELAV/Hu RRM proteins (3); and immunoprecipitation of various mRNP complexes followed by mRNA identification using microarray analysis (1). These approaches have wide applicability to other RNA-binding proteins or to other mRNP-associated proteins involved in the processing and localization of mRNA.
Mammalian cells need a robust and dynamic infrastructure to convey, channel, and rout messenger RNA transcripts with precision. Whether the information in a message is translated immediately, stored for later use, or routed to other locations, the inherent signals in the mRNAs or the corresponding RNA-binding proteins must be elucidated before the mechanisms of information transfer can be understood. Messenger RNP complexes consisting of RNA-binding proteins and/or structurally related mRNAs likely represent nodes of information transfer and accumulation. Decisions as to routing, activation, or disposal of individual transcripts involves the recognition of signals in coding or noncoding portions of each mRNA (24–31). The finding that multiple mRNAs can be identified in mRNPs containing specific proteins such as the ELAV/Hu family (1, 3) suggests that precise routing and information flow, whether individually or in clusters, represents organizational nodes of information transfer. Understanding the network of interacting RNAs and proteins that form a ribonome will require parallel multiplexing of global, group-specific, and type-specific mRNA-binding proteins because current methods of evaluating individual protein-RNA interactions are too limiting. Therefore, the dynamic interactions between RNAs and proteins involved in splicing, transport, and translation need to be evaluated in parallel and en masse so that regulatory loops and feedback mechanisms can be better understood.
How might one distinguish transcriptional from posttranscriptional regulation of multiple genes simultaneously? Fig. 5 illustrates our approach to multiplexing posttranscriptional events by categorizing mRNAs associated with mRNP complexes (1) and comparing their levels with transcriptional output. The conventional approach to distinguishing transcriptional contributions from posttranscriptional regulation of single gene transcripts often involves nuclear run-on experiments. Our laboratory has used nuclear run-on analysis en masse, employing P19 embryonic carcinoma cells, HeLa cells, and EL4 thymoma cells (C.Carson and J.D.K., unpublished results). By radiolabeling elongating transcripts in nuclear extracts, the transcriptionally active genes were identified on microarrays and compared with global mRNAs detected in the whole cell population. This approach allows a large number of candidate genes to be analyzed in parallel for
posttranscriptional effects such as mRNA stability by distinguishing those genes exclusively regulated at the level of transcription. Similar approaches for multiplexing splicing reactions and translation are imaginable, and will be necessary to fully understand the posttranscriptional network operating system.
One can imagine databases in which the functional linkages between multiple mRNAs can be accessed based on their membership in one or more mRNP complexes. For example, it should be possible to account for 100% of any given mRNA within a cell whether it is a member of a structurally or functionally related group of mRNAs, or a member of a small subset of a larger set of physically clustered mRNAs. In the future, when all of the RNA-binding proteins associated with every mRNA are known, it should be possible to describe, and ultimately simulate the organization and flow of genetic information within cells. Thus, by identifying mRNAs that are members of a physically clustered mRNP subset, the functions of proteins encoded by the mRNAs in the subset may become readily apparent through “guilt by association.” As a specific case in point, growth regulatory proteins like those encoded by the mRNAs associated with ELAV/Hu proteins are believed to have related functional properties (1). In addition to the functions of the encoded proteins, mRNAs may be clustered in vivo to optimize regulatory control of their expression, including mRNA stability, translation, and localization (1–3). Ribonomic databases may be constructed based on physical clustering of mRNAs and the functional relationships among their protein products. Such databases would allow tracking of mRNAs although their unique nodes of information management and transfer. Therefore, being able to organize each mRNP cluster into a relational database that accounts for the functional networking among its mRNAs and their protein products may offer insights into functional genomics.
A challenge for ribonomics will be to account for a full set of cellular transcripts, and to assess the dynamics of activation, repression, and product feedback that are inherent in an mRNP network. Functional perturbations by mutation, antisense expression, RNAi, or small molecules would be expected to alter the mRNP ribonomic network with a discernable outcome in the composition of the proteome. Like genomics and proteomics, ribonomics will require sophisticated computational systems to simulate the cellular dynamics of the posttranscriptional infrastructure during development. Indeed, this is a problem suited for the complexity sciences.
Many thanks to Craig Carson and Scott Tenenbaum for intellectual input and help in the preparation of figures.
1. Tenenbaum, S.A., Carson, C.C, Lager, P.J. & Keene, J.D. (2000) Proc. Natl. Acad. Sci. USA 97, 14085–14090.
2. Levine, T.D., Gao, F., King, P.H., Andrews, L.G. & Keene, J.D. (1993) Mol. Cell. Biol. 13, 3494–3504.
3. Gao, F.B., Carson, C.C., Levine, T. & Keene, J.D. (1994) Proc. Natl. Acad. Sci. USA 91, 11207–11211.
4. Liu, J., Dalmau, J., Szabo, A., Rosenfeld, M., Huber, J. & Furneaux, H. (1995) Neurology 45, 544–550.
5. Myer, V.E., Fan, X.C. & Steitz, J.A. (1997) EMBO J. 16, 2130–2139.
6. Fan, X.C., Myer, V.E. & Steitz, J.A. (1997) Genes Dev. 11, 2557–2568.
7. Keene, J.D. (1999) Proc. Natl. Acad. Sci. USA 96, 5–7.
8. Brennan, C.M. & Steitz, J.A. (2000) Cell Mol. Life Sci., in press.
9. Antic, D. & Keene, J.D. (1997) Am. J.Hum. Genet. 61, 273–278.
10. Gao, F.B. & Keene, J.D. (1996) J. Cell Sci. 109, 579–589.
11. Antic, D. & Keene, J.D. (1998) J. Cell Sci. 111, 183–19J.
12. Jain, R.G., Andrews, L.G., McGowan, K.M., Pekala, P.H. & Keene, J.D. (1997) Mol. Cell. Biol. 17, 954–962.
13. Antic, D., Lu, N. & Keene, J.D. (1999) Genes Dev. 13, 449–461.
14. Wakamatsu, Y. & Weston, J.A. (1997) Development 124, 3449–3460.
15. Chung, S., Eckrich, M., Perrone-Bizzozero, N., Kohn, D.T. & Furneaux, H. (1997) J. Biol. Chem. 272, 6593–6598.
16. Levy, N.S., Chung, S., Furneaux, H. & Levy, A.P. (1998) J. Biol. Chem. 273, 6417–6423.
17. Peng, S.S., Chen, C.Y., Xu, N. & Shyu, A.B. (1998) EMBO J. 17, 3461–3470.
18. Fan, X.C. & Steitz, J.A. (1998) EMBO J. 17, 3448–3460.
19. Ford, L.P., Watson, J., Keene, J.D. & Wilusz, J. (1999) Genes Dev. 13, 188–201.
20. Aranda-Abreu, G.E., Behar, L., Chung, S., Furneaux, H. & Ginzburg, I. (1999) J. Neurosci. 19, 6907–6917.
21. Lockhart, D.J. & Winzeler, E.A. (2000) Nature (London) 405, 827–836.
22. Velculescu, V.E., Zhang, L., Zhou, W., Vogelstein, J. & Kinzler, K.W. (1997) Cell 88, 243–251.
23. Eisen, M.B., Spellman, P.T., Brown, P.O. & Botstein, D. (1998) Proc. Natl. Acad. Sci. USA 95, 14863–14868.
24. Schiavi, S.C., Belasco, J.G. & Greenberg, M.E. (1992) Biochim. Biophys. Acta 1114, 95–106.
25. Ross, J. (1995) Microbiol. Rev. 59, 423–450.
26. Chen, C.Y. & Shyu, A.B. (1995) Trends Biochem. Sci. 20, 465–470.
27. Pinol-Roma, S. & Dreyfuss, G. (1993) Trends Cell Biol. 3, 151–155.
28. St. Johnston, D. (1995) Cell 81, 161–170.
29. Dreyfuss, G., Matunis, M.J., Pinol-Roma, S. & Burd, C.G. (1993) Annu. Rev. Biochem. 62, 289–321.
30. Richter, J.D. (1997) mRNA Formation and Function (Academic, New York).
31. Haynes, S.R. (1999) RNA-Protein Interactions Protocols (Humana, Totowa, NJ).
32. Savant-Bhonsale, S. & Cleveland, D.W. (1992) Genes Dev. 6, 1927–1939.
33. Zong, Q., Schummer, M., Hood, L. & Morris, D.R. (1999) Proc. Natl. Acad. Sci. USA 96, 10632–10636.
34. Gingras, A.-C., Raught, B. & Sonenberg, N. (1999) Annu. Rev. Biochem. 68, 913–963.
35. Gale, M., Tan, S.-L. & Katze, M.G. (2000) Micro. Mol. Biol. Rev. 64, 239–280.
36. Pleasure, S.J. & Lee, V.M. (1993) J. Neurosci. Res. 35, 585–602.
37. Steward, O., Wallace, C.S., Lyford, G.L. & Worley, P.F. (1998) Neuron 21, 741–751.
38. Steward, O. & Banker, G.A. (1992) Trends Neurosci. 15, 180–186.
39. Crino, P., Khodakhah, K., Becker, K., Ginsberg, S., Hemby, S. & Eberwine, J. (1998) Proc. Natl. Acad. Sci. USA 95, 2313–2318.
40. Mayford, M., Baranes, D., Podsypanina, K. & Kandel, E.R. (1996) Proc. Natl. Acad. Sci. USA 93, 13250–13255.
41. Schuman, E. (1999) Neuron 23, 645–648.
42. Bassell, G.J., Oleynikov, Y. & Singer, R.H. (1999) FASEB J. 13, 447–454.
43. King, P.H., Levine, T.D., Fremeau, R.T. & Keene, J.D. (1994) J. Neurosci. 14, 1943–1952.
44. Lindstren, T., June, C.H., Ledbetter, J.A., Stella, G. & Thompson, C.R. (1989) Science 244, 339–343.
45. Atasoy, U., Watson, J., Patel, D. & Keene, J.D. (1998) J. Cell Sci. 111, 3145–3156.
46. Zhuang, J.-Y., Chan, E.K.E., Peng, X.-X. & Tan, E.M. (1999) J. Exp. Med. 189, 1101–1110.
47. Albright, T.D., Jessell, T.M., Kandel, E.R. & Posner, M.I. (2000) Cell 100, S1-S55.
48. Rouault, T.A. & Klausner, R.D. (1997) Curr. Top. Cell. Regul. 35, 1–19.
49. Wang, Z.F., Ingledue, T.C., Dominski, Z., Sanchez, R. & Marzluff, W.F. (1999) Mol. Cell. Biol. 19, 835–845.
50. Whitfield, M.L., Zheng, L.X., Baldwin, A., Ohta, T., Hurt, M.M. & Marzluff, W.F. (2000) Mol. Cell. Biol. 20, 4188–4198.
51. Kenan, D.J., Query, C.C. & Keene, J.D. (1991) Trends Biochem. Sci. 16, 214–220.
52. Burd, C.G. & Dreyfuss, G. (1994) Science 265, 615–621.
53. Hanahan, D. & Weinberg, R.A. (2000) Cell 100, 57–70.
54. Lander, E.S. (1999) Nat. Genet. 21, 3–4.
55. Brown, P.O. & Botstein, D. (1999) Nat. Genet. 21, 38–41.







