
Molecular mechanisms of translation initiation in eukaryotes
Tatyana V.Pestova*†, Victoria G.Kolupaeva*, Ivan B.Lomakin*, Evgeny V.Pilipenko*‡§, Ivan N.Shatsky†, Vadim I.Agol†‡, and Christopher U.T.Hellen*¶
*Department of Microbiology and Immunology, State University of New York Health Science Center at Brooklyn, Brooklyn, NY 11203; †A.N.Belozersky Institute of Physico-chemical Biology, Moscow State University, Moscow 119899, Russia; and ‡Institute of Poliomyelitis and Viral Encephalitides, Russian Academy of Medical Sciences, Moscow Region 142782, Russia
Translation initiation is a complex process in which initiator tRNA, 40S, and 605 ribosomal subunits are assembled by eukaryotic initiation factors (eIFs) into an 80S ribosome at the initiation codon of mRNA. The cap-binding complex eIF4F and the factors eIF4A and eIF4B are required for binding of 43S complexes (comprising a 40S subunit, eIF2/GTP/Met-tRNAi and eIF3) to the 5′ end of capped mRNA but are not sufficient to promote ribosomal scanning to the initiation codon. eIF1A enhances the ability of eIF1 to dissociate aberrantly assembled complexes from mRNA, and these factors synergistically mediate 48S complex assembly at the initiation codon. Joining of 48S complexes to 60S subunits to form 80S ribosomes requires eIF5B, which has an essential ribosome-dependent GTPase activity and hydrolysis of eIF2-bound GTP induced by eIF5. Initiation on a few mRNAs is cap-independent and occurs instead by internal ribosomal entry. Encephalomyocarditis virus (EMCV) and hepatitis C virus epitomize distinct mechanisms of internal ribosomal entry site (IRES)-mediated initiation. The eIF4A and eIF4G subunits of eIF4F bind immediately upstream of the EMCV initiation codon and promote binding of 43S complexes. EMCV initiation does not involve scanning and does not require eIF1, eIF1A, and the eIF4E subunit of eIF4F. Initiation on some EMCV-like IRESs requires additional noncanonical initiation factors, which alter IRES conformation and promote binding of eIF4A/4G. Initiation on the hepatitis C virus IRES is even simpler: 43S complexes containing only eIF2 and eIF3 bind directly to the initiation codon as a result of specific interaction of the IRES and the 40S subunit.
Translation of mRNA into protein begins after assembly of initiator tRNA (Met-tRNAi), mRNA, and separated 40S and 60S ribosomal subunits into an 80S ribosome in which MettRNAi is positioned in the ribosomal P site at the initiation codon. The complex initiation process that leads to 80S ribosome formation consists of several linked stages that are mediated by eukaryotic initiation factors. These stages are:
-
Selection of initiator tRNA from the pool of elongator tRNAs by eukaryotic initiation factor (eIF)2 and binding of an eIF2/GTP/Met-tRNAi ternary complex and other eIFs to the 40S subunit to form a 43S preinitiation complex.
-
Binding of the 43S complex to mRNA, which in most instances occurs by a mechanism that involves initial recognition of the m7G cap at the mRNA 5′-terminus by the eIF4E (cap-binding) subunit of eIF4F. Ribosomes bind to a subset of cellular and viral mRNAs as a result of cap- and end-independent internal ribosomal entry.
-
Movement of the mRNA-bound ribosomal complex along the 5′ nontranslated region (5′NTR) from its initial binding site to the initiation codon to form a 48S initiation complex in which the initiation codon is base paired to the anticodon of initiator tRNA.
-
Displacement of factors from the 48S complex and joining of the 60S subunit to form an 80S ribosome, leaving Met-tRNAi in the ribosomal P site.
Research in our laboratory has addressed the molecular mechanisms of these different stages in translation initiation and the means by which they are bypassed during initiation by internal ribosomal entry. We have reconstituted each of these stages in vitro using purified translation components to identify the minimum set of eIFs that is required for each stage and to provide a framework for more detailed mechanistic analysis.
Factor Requirements for Ribosomal Attachment and Scanning of 43S Ribosomal Complexes on ß-Globin mRNA. The initiation codon of a eukaryotic mRNA is normally the first AUG triplet downstream of the 5′-terminal cap and is usually separated from it by 50–100 nt. After cap-mediated attachment to mRNA, a 43S complex is thought to scan downstream from the 5′-end until it encounters the initiation codon. We used native capped ß-globin mRNA as a model in in vitro reconstitution experiments to address three basic questions. (i) Which eIFs are required for a 43S complex to bind capped mRNA? (ii) Which eIFs are required for the bound complex to move downstream to the initiation codon? (iii) How does the scanning 43S complex recognize and reject mismatched interactions between the Met-tRNAi anticodon, and triplets in the 5′ NTR until the correct initiation codon is reached and recognized? In these experiments, the position of the leading edge of bound ribosomal complexes on mRNA was mapped by primer extension inhibition (“toeprinting”). The estimated length of the mRNA-binding cleft in 40S subunits is ≈30nt, and 48S complexes usually yield toeprints at positions +15—+17 downstream of the A of the initiation codon.
Ribosomal binding at the 5′-end of the mRNA required eIF3, the eIF2/GTP/Met-tRNAi complex, ATP, and the eIF4F cap-binding complex, and was enhanced by eIF4B (1). eIF4F is a heterotrimeric factor, and its eIF4A (ATP-dependent RNA helicase) and eIF4E subunits and the eIF4G550–1090 fragment of its 1,560-amino acid eIF4G subunit constitute the core of eIF4F
|
§ |
Present address: Department of Neurology, University of Chicago Medical Center, Chicago, IL 60637. |
|
¶ |
To whom reprint requests should be addressed at: Department of Microbiology and Immunology, State University of New York Health Science Center at Brooklyn, 450 Clarkson Avenue, Box 44, Brooklyn, NY 11203. E-mail: chellen@netmail.hscbkyn.edu. |
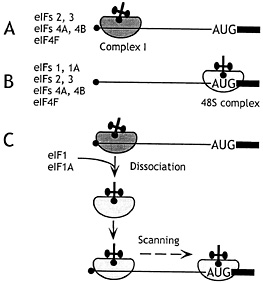
Fig. 1. The mechanism of action of eIF1 and eIF1A in promoting assembly of 48S ribosomal complexes at the authentic initiation codon of a conventional capped mRNA. The 5' terminal m7G residue is shown as a filled black circle, the 5' NTR as a black line, and the ORF downstream of the AUG initiation codon as a black rectangle. (A) In the presence of eIFs 2, 3, 4A, 4B, and 4F, an aberrant ribosomal complex (“complex I”) assembles at a cap-proximal position but is unable to scan downstream to the initiation codon. (B) In the presence of eIFs 1, 1A, 2, 3, 4A, 4B, and 4F, 48S ribosomal complexes assemble exclusively at the authentic initiation codon. (C) Addition of eIF1 and eIF1A to complex I promotes its complete conversion to correctly assembled 48S complexes after dissociation of complex I and rebinding of 43S ribosomal complexes in a scanning-competent form.
sufficient for efficient ribosomal attachment to capped mRNA (2). This fragment of eIF4G binds both eIF4E and eIF4A and probably coordinates their activities so that a cap-proximal region of mRNA is unwound and is thus rendered accessible to an incoming 43S complex so it can bind productively. The molecular interactions that enable the incoming 43S complex to bind this “prepared” template are not known but are thought to involve interaction of the eIF3 component of 43S complexes with cap-associated eIF4G. The bound ribosomal “complex I” was arrested in a cap-proximal position and did not reach the initiation codon (Fig. 1 A).
Two additional activities present in rabbit reticulocyte lysate (RRL) enabled 43S complexes to reach the initiation codon, forming “complex II” without being arrested at the initial binding site (Fig. 1 B). These small factors were purified and identified by sequencing as eIF1 (13.5 kDa) and eIF1A (19 kDa) and could be functionally replaced by corresponding recombinant polypeptides. These two factors acted synergistically; eIF1 A without eIF1 enhanced eIF4F-mediated binding of 43S complexes to mRNA but did not enable these complexes to reach the initiation codon, whereas eIF1 without eIF1A reduced the prominence of the cap-proximal complex I and promoted formation of low levels of 48S complexes. The interaction with mRNA of 48S complexes assembled in the absence of eIF1A differed subtly from complexes formed in their presence, in that only two (+16–+17) rather than three toeprints (+15–+17) were apparent. eIF1A therefore increases the competence of 43S complexes to bind mRNA and the processivity of scanning 43S/eIF1/mRNA complexes. eIF1A also stabilizes binding of the ternary complex to 40S subunits in the absence of mRNA (3, 4), presumably by an allosteric mechanism, because it is not known to interact directly with any component of the ternary complex. This stabilization by eIF1A is weak but might be indicative of a role for eIF1A in ensuring that initiator tRNA and mRNA adopt the correct relative orientation on the scanning ribosomal complex.
eIF1A comprises an oligonucleotide-binding (OB) ß-barrel fold that closely resembles prokaryotic initiation factor IF1 (and corresponds to the region of sequence homology between them) and an additional C-terminal domain (4). The experimentally determined RNA-binding surface of eIF1A is large, extending over the OB fold and the adjacent groove leading to the second domain. Mutations at multiple positions on this surface resulted in a reduced ability of eIF1A to promote assembly of 48S initiation complexes at the initiation codon. The RNA ligand for eIF1A is not known, but by analogy with IF1 (5), eIF1A might bind 18S ribosomal RNA in the ribosomal A site.
In the absence of eIF1 and eIF1A, the mRNA-binding cleft on 40S subunits appears to be open, because they can bind mRNA in an end-independent manner during initiation by internal ribosomal entry (see below). eIF1 and eIF1 A may contribute to the correct interactions of components of the 43S complex with mRNA that enable it to enter a processive mode, for example by closing this cleft directly or indirectly and possibly even by forming part of the channel on the 40S subunit through which mRNA moves during ribosomal scanning.
Experiments done by using competitor mRNAs indicated that complex I cannot be “chased” directly into complex II and is therefore not its immediate precursor. Complex I is aberrantly assembled (because it is arrested at a non-AUG triplet and is unable to scan to the initiation codon) and is intrinsically unstable. eIF1 and eIF1A together (but not individually) promote dissociation of complex I and enable the released 43S complex to rebind mRNA in a competent state to scan to the initiation codon (Fig. 1 C). eIF1 alone is able to recognize and destabilize ribosomal complexes incorrectly assembled by internal ribosomal entry (see below). Identification of this activity of mammalian eIF1 is consistent with characterization of its yeast homologue Sui1 as a monitor of translation accuracy. Mutations in Sui1 allow aberrant initiation in vivo at non-AUG codons by mismatch base pairing with Met-tRNAi (e.g., ref. 6). Determination of the solution structure of eIF1 by NMR (7) has revealed that these mutated residues form part of a surface that is almost perfectly conserved among all eIF1 homologues and that is likely directly involved in initiation codon selection by eIF1.
In summary, we have determined the set of factors required for binding of a 43S complex to a model native capped mRNA and for it to scan to the initiation codon. These experiments were done by using ß-globin mRNA, and it is possible that ribosomal scanning on longer or more highly structured 5' NTRs may require additional as-yet-unidentified factors, for example to enhance processivity or to promote unwinding of stable secondary structures. Almost all aspects of the mechanism of ribosomal scanning remain uncharacterized (8). For example, scanning is an ATP-dependent process, but it is not known whether ribosomal movement itself involves hydrolysis of ATP or whether chemical energy is required only to unwind secondary structure in the 5' NTR to permit ribosomal movement by one-dimensional diffusion from its initial 5'-terminal attachment site. The ability to reconstitute this process in vitro will enable this and other outstanding questions to be addressed.
Factor Displacement from the 48S Complex and Joining to a 60S Subunit to Form Active 80S Ribosomes. The 48S complex assembled at the initiation codon of ß-globin mRNA is bound by factors that must be displaced before the 40S subunit/mRNA/Met-
tRNAi complex can join with a 60S subunit. Substitution of GTP by guanosine 5′-[ß,γ-imido] triphosphate (GMP-PNP) (a nonhydrolyzable analogue) arrests initiation at the stage of 48S complex formation, indicating that displacement of factors and subunit joining both require hydrolysis of GTP bound to eIF2 in 48S complexes. GTP hydrolysis by eIF2 is activated by eIF5, a 49-kDa polypeptide that interacts specifically with eIF2 and eIF3 (9, 10).
Recent data suggest that eIF5 is a component of multifactor complex comprising eIF1, eIF3, eIF5, and the eIF2/GTP/Met-tRNAi ternary complex that can exist free of the ribosome and probably binds to it as a whole rather than sequentially (10). eIF5 binds strongly to eIF2 but induces its GTPase activity only when eIF2 is associated with the 40S subunit. GTP hydrolysis, which leads to dissociation of eIF2-GDP, is thought to be induced in response to base pairing between the initiation codon and the anticodon of Met-tRNAi, thereby ensuring stringent selection of the initiation codon during the scanning process (11).
Until recently, the hydrolysis of eIF2-bound GTP was considered the only requirement for the joining of a 60S subunit to the 48S complex (see ref. 12 for a review). However, we found that addition of 60S subunits and recombinant eIF5 to 48S complexes assembled on globin mRNA did not lead to formation of 80S ribosomes (13). A partially purified ribosomal salt wash fraction from mouse ascites cells was active in promoting 80S ribosome assembly and was therefore used as a source for purification of additional factors. We purified two proteins to apparent homogeneity, which together but not separately were able to mediate assembly of 48S complexes and 60S subunits into 80S ribosomes. The smaller (49 kDa) protein could be functionally replaced by recombinant eIF5. The second protein had an apparent molecular mass of 175 kDa, and its N-terminal sequence identified it as a mouse homologue of prokaryotic initiation factor IF2 (Fig. 2). A role for a eukaryotic homologue of IF2 was first revealed by studies in yeast (14). Analysis of polyribosome profiles showed that deletion of the yeast IF2 homologue led to a reduction in formation of larger polysomes and an accumulation of inactive 80S ribosomal particles, and in vitro translation assays confirmed that this deletion led to a defect in translation initiation on the majority, if not all, cellular mRNAs. This defect could be rescued by adding back purified recombinant protein (14). These results indicated that this protein is a general translation factor in yeast. Human, Drosophila, and archaeal homologues have also been identified (15, 16). In light of its function in subunit joining, we named this factor eIF5B (13). Recombinant human eIF5B587–1220 lacking amino acids 1–586 could substitute for yeast eIF5B in vivo (15) and for native mammalian eIF5B in subunit joining in our in vitro reconstitution experiments (13). It is almost certain that eIF5B is a protein that was previously implicated in subunit joining but subsequently erroneously discounted as an inactive contaminant of eIF5 (12).
Puromycin resembles the 3′-end of aminoacylated tRNA and can bind to the ribosomal A site to react with Met-tRNAi in the P site to form methionylpuromycin. This reaction mimics formation of the first peptide bond, and we therefore used it to confirm that 80S ribosomes assembled by using eIF5 and eIF5B were active. Assembly of 48S complexes on AUG triplets is much simpler than on native mRNA, because it involves neither 5′-end-dependent attachment nor scanning. 48S complex formation on AUG triplets requires only a 40S subunit and the eIF2/GTP/Met-tRNAi complex, which enabled us to investigate the influence of other factors on the requirements for subunit joining (13). A requirement for both eIF5 and eIF5B for 80S assembly was apparent only when 48S complexes were assembled by using eIF1, eIF1A, eIF2, and eIF3. These four factors are all normally associated with a 48S complex at the initiation codon. Individually, eIF5 and eIF5B were equally active in subunit
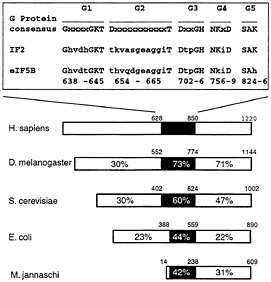
Fig. 2. Sequence and structural conservation of eukaryotic eIF5B proteins from Homo sapiens (15), Drosophila melanogaster (16), and Saccharomyces cerevisiae (14), archaeal IF2 from Methanococcus jannaschii and prokaryotic IF2 from Escherichia coli (12). The percentages of amino acid identities to human eIF5B in the N-terminal region of the protein, the GTP-binding domain, and the C-terminal region of the protein are shown. The black rectangle in the schematic representation identifies the position of the GTP-binding domains in these proteins with the indicated GTP-binding protein consensus sequence motifs G1-G5 aligned with sequence motifs G1-G5 of E. coli IF2 and human eIF5B. Numbers above the domains of eIF5B/IF2 proteins refer to the amino acid residues in each protein; numbers below the aligned sequences refer to the amino acid residues in G1-G5 motifs of human eIF5B.
joining in reactions lacking eIF1 and eIF3, but inclusion of eIF1 and eIF3 together reduced the individual activities of both eIF5 and eIF5B. The requirement for both eIF5 and eIF5B in these circumstances indicates that they have complementary functions.
Hydrolysis of GTP bound to 48S complexes is a prerequisite for subunit joining and was therefore also compared in the presence and absence of eIF1 and eIF3 (13). eIF5 and eIF5B stimulated GTP hydrolysis by eIF2 equally when 48S complexes contained only eIF1A and eIF2, but inclusion of eIF1 and eIF3 inhibited the stimulatory activity of eIF5B without affecting that of eIF5. This effect can account for the reduced ability of eIF5B to promote methionylpuromycin synthesis in the presence of eIF1 and eIF3. We conclude that, although eIF5 is active in inducing GTP hydrolysis on 48S complexes in the presence of a full set of factors (including eIF1 and eIF3), this is insufficient for subunit joining. Under these circumstances (when all factors associated with 48S complexes are present, which corresponds to the normal situation for initiation on capped mRNAs), eIF5B is also required.
The central domain of eIF5B contains sequence motifs characteristic of GTP-binding proteins (Fig. 2). By UV crosslinking, we found that [32P]GTP bound directly to eIF5B independently of ribosomal subunits, and that bound [32P]GTP exchanged readily with unlabeled GTP, GMP-PNP, or GDP. eIF5B had no detectable intrinsic GTPase activity, but its ability to hydrolyze GTP was activated by 60S subunits and considerably more by 40S and 60S subunits together. Interestingly, prokaryotic IF2 is also
a GTPase that is specifically activated by large and small ribosomal subunits together (17). This similarity between the homologous factors eIF5B and IF2 suggests that ribosomal activation of their GTPase activity may occur by a common mechanism.
Binding of GTP to eIF5B may be required for it to adopt an active conformation. To test this hypothesis, 48S complexes were assembled with GTP, separated from unincorporated GTP by gel filtration, and then incubated with eIF5, 60S subunits, different nucleotides, and either full-length native eIF5B or recombinant eIF5B587–1220. The degree of dependence of eIF5B’s activity in 80S assembly on binding GTP was determined by the integrity of the protein. eIF5B587–1220 was completely GTP-dependent, whereas native eIF5B retained low activity in the absence of GTP but was nevertheless stimulated 3-fold by GTP (T.V.P., unpublished work). This result suggests that eIF5B adopts the active conformation required for subunit joining when it binds GTP. eIF5B acts catalytically in the presence of GTP, promoting multiple rounds of subunit joining. 80S complexes were also formed by eIF5B bound to GMP-PNP, but eIF5B-GMP-PNP acted stoichiometrically rather than catalytically.
This defect in the activity of eIF5B in the presence of GMP-PNP could be because hydrolysis of GTP bound to eIF5B is required for the release of eIF5B from assembled 80S ribosomes, for the release of other factors, or for both. The proportion of Met-tRNAi in 80S ribosomes assembled in the presence of GTP (60%) that reacted with puromycin was significantly greater than in complexes assembled by using GMP-PNP (8%). Methionylpuromycin synthesis by purified 80S ribosomes assembled in the presence of GTP was completely inhibited by addition of eIF5B587–1220 with GMP-PNP but not by either eIF5B587–1220 or GMP-PNP alone. This result indicates that eIF5B-GMP-PNP can interact with preassembled 80S complexes, blocking their ability to react with puromycin (13). The specific inhibition of this reaction suggests that eIF5B binds to the ribosomal A site. When ribosomal complexes assembled by using GTP were resolved on sucrose density gradients, no eIF5B587–1220 was detected on 40S, 48S, 60S, or 80S complexes. However, a large amount of eIF5B587–1220 was bound to 80S complexes assembled in the presence of GMP-PNP. The inability of eIF5B587–1220 to hydrolyze GMP-PNP therefore locks the factor on 80S complexes and renders them inactive in methionylpuromycin synthesis. eIF1, eIF2, and eIF3 were detected in 48S complexes but not in 80S complexes assembled with GTP or GMP-PNP. GTP hydrolysis by eIF5B is therefore not required for the release of these factors during subunit joining but is needed for release of eIF5B itself. The inability of eIF5B587–1220/GMP-PNP to dissociate from 80S ribosomes explains the requirement for stoichiometric rather than catalytic amounts of this factor in assembly reactions in the presence of GMP-PNP. Neither the stage during the initiation process at which eIF1, eIF1 A, eIF2, eIF3, and eIF5 are released nor the mechanism by which release occurs during initiation on native mRNAs has yet been established.
Ribosomal subunit joining to form active 80S ribosomes that are competent to begin elongation therefore involves two successive GTP hydrolysis events: activation by eIF5 of hydrolysis of eIF2-bound GTP and ribosome-activated hydrolysis of eIF5B-bound GTP. Remarkably, eIF5B is a homologue of prokaryotic IF2, which also mediates a similar subunit-joining step that also involves ribosome-activated hydrolysis of factor-bound GTP.
Initiation of Picornavirus Translation by Internal Ribosomal Entry: The Role of Canonical Initiation Factors. Picornavirus RNA genomes are uncapped and have highly structured 5' NTRs that are barriers to scanning ribosomes. Initiation on these mRNAs is end-independent and is instead mediated by a ˜400-nt internal ribosomal entry site (IRES) in the 5' NTR (18, 19). The activity of an IRES depends on its structural integrity, and even point mutations can cause general or cell type-specific loss of function.
Picornavirus IRESs are divided into two major groups on the basis of sequence and structural similarities (20, 21). One group contains poliovirus and rhinovirus, and the other group contains encephalomyocarditis virus (EMCV), Theiler’s murine encephalomyelitis virus (TMEV), and foot-and-mouth disease virus (FMDV). The EMCV and TMEV initiation codons are located at the 3' border of the IRES, and ribosomes bind directly to them without scanning (22, 23). In poliovirus, the initiation codon is ˜160 nt from the 3' border of the IRES, and it is possible that the ribosome reaches it either by scanning or by discontinuous transfer (“shunting”) after initial attachment to the IRES (24). Picornavirus infection often leads to shutoff of cap-mediated translation initiation, for example by rhinovirus protease cleavage of eIF4G at R641/G642, such that the N-terminal domain of eIF4G that binds eIF4E and the poly (A)-binding protein is separated from the C-terminal domain that binds eIF3 and eIF4A (25). This cleavage impairs eIF4F’s function in initiation on capped mRNAs. However, as described below, this cleavage yields a fragment of eIF4F that retains functions necessary for picornavirus IRES-mediated initiation.
We reconstituted initiation in vitro on the EMCV IRES and found that it is ATP-dependent and requires only eIFs 2, 3, and either eIF4F or eIF4A and the central third of eIF4G to which eIF4A binds (26, 27). The requirement for eIF4A and the cognate domain of eIF4G is consistent with the profound inhibition of EMCV translation caused by dominant negative eIF4A mutant polypeptides (28). The inhibition caused by these mutants is thought to be because of their failure to exit the eIF4F complex and recycle efficiently, thereby trapping it in an inactive form. In this model, eIF4A therefore plays its role in initiation as part of a complex with eIF4G rather than as a singular polypeptide.
48S complex formation was enhanced 4-fold by eIF4B and less than 2-fold by the pyrimidine tract-binding protein (PTB), a noncanonical mRNA-specific initiation factor (see below). Together, these factors promoted 48S complex formation equally at AUG834 (the authentic initiation codon) and at AUG826 (which is virtually unused in vivo). Remarkably, inclusion of eIF1 in assembly reactions or even addition of eIF1 to preformed complexes led to dissociation of the ribosomal complex at AUG826 (13). This observation is consistent with the previously noted function of eIF1 in enhancing the fidelity of initiation codon selection. The principal difference between the factor requirements for initiation on ß-globin mRNA and on the EMCV IRES is that the latter has no requirement for eIF4E or the fragment of eIF4G to which it binds and is therefore not impaired by cleavage of eIF4G by viral proteases. eIF4E is a major focus of mechanisms that regulate initiation of translation in vivo (29). The EMCV IRES and other IRESs that do not require eIF4E are therefore active in circumstances that lead to inhibition of cap-mediated initiation by impairment of eIF4E function.
A significant insight to the mechanism of initiation on the EMCV IRES came from the observations that eIF4F bound to the J-K domain of the EMCV IRES a little upstream of the initiation codon (Fig. 3), and that this interaction is essential for initiation (26). The essential central eIF4G722–949 domain binds specifically to the IRES, and its binding is strongly enhanced by eIF4A (33). The interaction of eIF4G with this IRES may play a role analogous to that of eIF4E on capped mRNAs, that is, to recruit the eIF4F complex and associated factors and to promote ribosomal attachment at a defined location on an mRNA.
The Role of IRES Transacting Factors in Initiation of Picornavirus Translation. The activity of a number of picornavirus IRESs is subject to cell-type-specific restriction: for example, the atten-
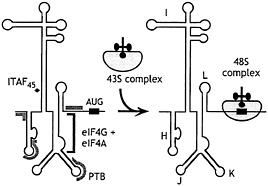
Fig. 3. Schematic representation of EMCV, FMDV, and TMEV IRES domains H-L, showing binding sites for PTB (thick gray line) and ITAF45 (black icosahedron), as determined by footprinting (30, 31), and for the eIF4G/eIF4A complex, as determined by footprinting (31, 32) and toeprinting (26, 27, 33). The interaction of eIF4G/eIF4A with the J-K domain, which is essential for recruitment of the 43S complex to the initiation codon, is enhanced by PTB and ITAF45 in an IRES-specific manner, as discussed in the text.
uation of poliovirus vaccine strains is in part because of a defect in translation in neuronal cells. Poliovirus and rhinovirus IRESs mediate initiation of translation efficiently in HeLa and Krebs 2 cells, and their restricted activity in RRL in vitro can be alleviated by deletion of the IRES or by supplementation of RRL by ribosomal salt wash fractions from HeLa or Krebs cells (e.g., refs. 34, 35). Translation mediated by poliovirus and rhinovirus IRESs thus depends on the interaction with these IRESs of noncanonical IRES transacting factors (ITAFs) that are either absent from RRL or significantly less abundant in RRL than in permissive cells. In early experiments to identify ITAFs required for picornavirus IRES function, we and others identified a 57-kDa protein that bound specifically to all picornavirus IRESs as the PTB, a cellular polypeptide that contains four RNA-recognition motif -like domains (36–39).
The PTB dependence of initiation on the wild-type EMCV IRES is small (27, 40) but was significantly enhanced by a single nucleotide insertion in the eIF4G-binding site or by alteration of the sequence downstream of the initiation codon (40). Foot-printing analysis indicated that PTB binds multiple noncontiguous sites on the EMCV IRES (ref. 30; Fig. 3). Taken together, these observations suggested a model in which binding of ITAFs such as PTB stabilizes an IRES in an optimal conformation for binding of essential factors and the 43S complex. Our analysis of initiation on the related TMEV and FMDV IRESs provided strong support for this hypothesis (31).
TMEV (GDVII strain) and FMDV (type 01K) are neurotropic and epitheliotropic picornaviruses, respectively. Their IRESs are ˜40% identical and are closely related to the EMCV IRES. Substitution of the IRES in an infectious genomic TMEV clone by that of FMDV strongly attenuated the neurovirulence of the resulting chimeric virus without significantly affecting its ability to replicate in cultured BHK-21 cells or to be translated in vitro in RRL (31). We used biochemical reconstitution of the initiation process on these mRNAs to define the molecular basis for this cell type-specific difference in the function of these IRESs.
Initiation on the FMDV and TMEV IRESs had identical requirements for canonical initiation factors to those described above for EMCV. However, whereas initiation on the EMCV IRES was only weakly stimulated by PTB, initiation on the TMEV IRES depended strongly on PTB, and initiation on the FMDV IRES required both PTB and a 45kDa ITAF (ITAF45). We purified and sequenced ITAF45 and found that it is identical to a previously identified proliferation-dependent protein that is not expressed in nonproliferating cells such as neurons (31). The absence of this factor may account for the inability of the chimeric TMEV virus to replicate in neurons.
The activities of PTB and ITAF45 in promoting 48S complex formation on the FMDV IRES were strongly synergistic. These ITAFs bound to nonoverlapping sites on the IRES (Fig. 3) and together caused localized conformational changes in it, specifically in regions adjacent to the binding site for the eIF4G/eIF4A complex. The interaction of the IRES with the eIF4G/eIF4A complex is essential for initiation and, significantly, this interaction was specifically enhanced by these two ITAFs. EMCV, FMDV, and TMEV IRESs all bind to PTB and ITAF45, so it is the requirement for them rather than their ability to interact that differs as a consequence of sequence differences between these IRESs. Similar observations have also been made for the second group of picornavirus IRESs, members of which are also closely related to each other yet also appear to have different ITAF requirements. For example, poliovirus and rhinovirus IRESs both bind to PTB, to the poly(rC)-binding protein 2 (PCBP2), and to unr (35, 41). PTB contains four RNA-recognition motiflike domains, PCBP2 has three KH domains that likely constitute its RNA-binding surface, and unr contains five cold-shock RNA-binding domains. These polypeptides therefore all have the potential to make multipoint interactions with these IRESs and to stabilize their folding in an active conformation. However, whereas initiation on the rhinovirus IRES depends on unr, strongly enhanced by PTB and less responsive to PCBP2, the poliovirus IRES depends on PTB and PCBP2 and does not respond to unr (35).
Our analysis of initiation on EMCV-like IRESs suggests a model that will likely be applicable to poliovirus-like IRESs and possibly to some other viral and cellular IRESs. Specific binding of eIF4F (or its eIF4G and eIF4A subunits) to the IRES is required to mediate internal ribosomal entry and itself depends on the eIF4F and ribosomal binding sites having the correct conformation. The role of ITAFs is to bind an IRES to enable it to attain or maintain this conformation, for binding both of eIF4G/eIF4A and possibly of other components of the 43S complex. The diversity of IRES sequences and structures leads to the requirement for a variety of ITAFs.
Internal Initiation by Factor-Independent Binding of Ribosomes to the Initiation Codon. The 5' NTRs of HCV and of the related classical swine fever virus (CSFV) and bovine viral diarrhea virus (BVDV) also promote translation by cap-independent internal ribosomal entry (e.g., ref. 42). IRESs are defined solely by functional criteria and cannot yet be predicted by the presence of characteristic RNA sequence or structural motifs. As a rule, there are no significant similarities between individual IRESs (unless they are from related viruses). The related BVDV, CSFV, and HCV IRESs are the best characterized members of an IRES group that is wholly distinct from both the EMCV-like and poliovirus-like groups of IRESs with regard to length, sequence, and structure. We investigated initiation on BVDV, CSFV, and HCV IRESs to determine whether all IRESs mediate internal initiation of translation by a single common mechanism irrespective of sequence variation and, if not, to characterize unique aspects of initiation on this group of HCV-like IRESs.
The BVDV, CSFV, and HCV 5' NTRs are 385, 372, and 342 nt long, respectively, and although they differ from each other at 35–50% of base positions, many of these nucleotide differences are covariant substitutions, indicative of conserved higher order structure. Even minor mutations in structural elements substantially reduced IRES activity, but this could in most instances be restored by compensatory second site mutations that restored
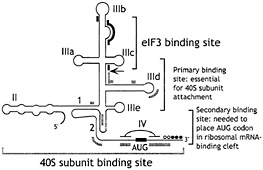
Fig. 4. Schematic secondary structure of domains II, III, and IV of HCV-like IRESs, showing sites of interaction with eIF3 (thick black lines) (47) and with 40S subunits (thick gray lines) (48, 49). The toeprints detected at the leading edge of bound eIF3 (46, 47, 49, 50) are indicated by an arrow. The toeprints at the leading edge of 40S subunits in binary IRES:40S subunit complexes are indicated by open circles and in 48S complexes formed on inclusion of eIF2/ GTP/Met-tRNAi with 40S subunits by filled circles (46, 50). Toeprints caused by ribosomal contact with the pseudoknot are not shown. Sequences flanking the initiation codon are base paired to form domain IV in HCV but not in BVDV and CSFV. BVDV and CSFV contain two hairpins (IIId1 and IIId2) at an analogous position to HCV IIId. The nomenclature of helices in the pseudoknot and of domains is as in ref. 48.
secondary structure (43–46). The most highly conserved residues are often unpaired and may thus be able to interact with components of the translation apparatus. These results and observations have led to a model for IRES function in which structural elements in the IRES act as a scaffold that orients these potential binding sites in such a way that their interaction with factors and ribosomes leads to assembly of functional ribosomal initiation complexes.
These HCV-like 5' NTRs consist of four major structural domains (I–IV) and a complex pseudoknotted structure between domains II, III, and IV (Fig. 4). HCV domain IV is base-paired, whereas equivalent residues in BVDV and CSFV are not. The boundaries of these IRESs extend from the 3' border of the basal helix of domain II to the initiation codon, and IRES activity is affected by the coding sequence downstream of the initiation codon. Minor mutations in domain II, domain III, and the pseudoknot can cause substantial reductions in IRES activity.
We determined the minimum set of factors required for assembly of 48S complexes on these IRESs by in vitro reconstitution by using purified translation components (46, 50). The most remarkable aspect of initiation on these IRESs is that they bind 40S subunits specifically and stably, in the absence of initiation factors, so that the ribosomal P site is placed in the immediate proximity of the initiation codon. Addition of eIF2/GTP/Met-tRNAi is sufficient for the bound 40S subunit to lock onto the initiation codon. The direct attachment of the 43S complex to the initiation codon is consistent with earlier reports that translation initiation on HCV and CSFV IRESs in RRL does not involve ribosomal scanning after initial attachment (44, 45). Although eIF3 is not required for assembly of this minimal 48S complex, eIF3 has been reported to be associated with free 40S subunits in the cytoplasm, and it is therefore likely that, in vivo, it is also a constituent of 48S complexes on these IRESs. eIF3 itself also binds specifically and stably to the IRES; the independent interaction of two different components of the 43S complex with the IRES may enhance the affinity and specificity of binding. The binding site for eIF3 has been mapped by toeprinting and chemical/enzymatic footprinting to the apical half of domain III (ref. 47; Fig. 4) and includes subdomain IIIb and junction IIIabc.
Notably, 48S complex formation on HCV-like IRESs has no requirement for eIF4A, 4B, 4E, or 4G, nor any requirement for ATP hydrolysis. Translation mediated by these IRESs is also not inhibited by dominant negative eIF4A mutants (46, 50). It thus differs fundamentally from cap-mediated initiation and initiation mediated by picornavirus IRESs of both the EMCV and poliovirus-like groups. The initiation factors eIF4A, 4B, 4E, and 4G do not influence initiation complex formation on HCV-like IRESs, and indeed these factors are probably unable to gain access to and unwind the region of the IRES immediately upstream and downstream of the initiation codon that enters the mRNA-binding cleft of the 40S subunit. For example, translation efficiency is strongly reduced by mutations that increase base pairing in HCV domain IV (which contains the initiation codon) and thus stabilize it (51) and by introduction of a hairpin immediately downstream of the CSFV initiation codon (52). Secondary structures of equivalent stability are readily unwound during cap-mediated initiation.
Initiation on prokaryotic mRNAs involves factor-independent binding of small (30S) ribosomal subunits as a result of base pairing between the linear Shine-Dalgarno (SD) sequence in mRNAs and complementary linear anti-SD sequences in the ribosomal 16S rRNA (52). Although there are striking analogies between this mechanism and the factor-independent binding of 40S subunits to HCV-like IRESs, it is evident that binding of 40S subunits is determined by multiple noncontiguous sequences in the IRES rather than by a single linear sequence. We do not yet know whether binding of an IRES to the 40S subunit involves RNA-RNA base pairing with 18S rRNA. The only contact identified so far is with a ribosomal protein, but this interaction likely is not a primary determinant of the IRES/40S subunit interaction (46, 48).
Toeprinting and deletion analyses indicated that a 40S subunit interacts with the IRES at multiple sites; primer extension is arrested by bound 40S subunits in the pseudoknot and downstream of the initiation codon (Fig. 4). We used enzymatic footprinting to identify the principal sites in HCV and CSFV IRESs that are protected from cleavage by bound 40S subunits (48, 49). Similar interaction sites were identified in these two IRESs, and they are located in regions of high sequence conservation in HCV-like IRESs. These sites include the apex of HCV subdomain IIId and the equivalent CSFV subdomain IIId1, the pseudoknot, and nucleotides flanking the initiation codon. The number of protected residues in the last of these sites corresponds closely to the length estimated for the mRNA-binding cleft in 40S subunits, and it is therefore likely that additional upstream contacts between the IRES and the 40S subunit involve regions of the 40S subunit outside this cleft. The ribosomal binding surface of the IRES is therefore extensive; these footprinting and mutational analyses (see below) suggest that it does not overlap the eIF3-binding site except in subdomain IIIa.
The importance of these sites of interaction with the 40S subunit for IRES function is supported by the results of mutational analysis. The apical residues GGG266–268 in HCV IIId and analogous residues (GGG268–270) in CSFV IIId1 are essential for ribosomal binding and IRES function (49, 53). The pseudoknot has long been known to be functionally important (43, 45, 46). We found that substitutions in its 5' helical segment abrogate ribosomal binding, whereas substitutions in its 3' helix do not prevent ribosomal attachment to the IRES but impair binding of sequences flanking the initiation codon to the ribosomal mRNA-binding cleft (46, 49). Consistent with this conclusion, we found that residues flanking the initiation codon are also not required for ribosomal
attachment to the IRES to form a stable binary complex, even though they constitute a major site of interaction between these IRESs and the 40S subunit. Similarly, deletion of domain II or mutations in domain IIIa also impaired binding of the initiation codon region to the ribosomal mRNA-binding cleft but did not prevent binary complex formation. These parts of the IRES therefore do not contain primary determinants of ribosomal binding (48, 49). We conclude that HCV-like IRESs contain one set of determinants that is required for initial ribosomal attachment (including subdomain IIId/IIId1, adjacent regions of domain III, and the 5' helical segment of the pseudoknot) and a second set of determinants (including domain II, the 3' helical segment of the pseudoknot, and downstream sequences) that is required for, or at least promotes, subsequent accurate placement of the initiation codon in the ribosomal P site. The IRES is not a static structure, and it is likely that it undergoes structural transitions during these two stages in ribosomal binding and subsequently during subunit joining.
The mechanism of initiation on HCV-like IRESs is therefore distinct from both cap-mediated and EMCV IRES-mediated initiation in having no requirement for ATP or for any member of the eIF4F group of factors. HCV-like IRESs bypass the requirement for these factors and for eIF1 and eIF1A by virtue of their ability to recruit 43S complexes directly to the initiation codon by binding specifically to eIF3 and to the 40S subunit. The importance of the integrity of the structure of these IRESs for this mode of translation initiation suggests that these IRESs constitute valid targets for potential chemotherapeutic agents such as antibiotics that could bind the IRES and distort the structure of binding sites for these components of the translation apparatus.
Perspectives. We have characterized the outlines of three different mechanisms of translation initiation by using biochemical reconstitution to determine the minimum set of factors required for assembly of 48S and 80S ribosomal complexes on three distinct types of eukaryotic mRNA and by using toeprinting and footprinting to map the location of translation components on these mRNAs. The findings reported here raise both general and specific questions about translation initiation.
The finding that internal ribosomal entry on the two types of IRES that we have examined occurs by very different mechanisms indicates there is no single mode of internal ribosomal entry. Indeed, the implicit possibility that there are yet other mechanisms for initiation directed by an IRES has been borne out by recent analysis of initiation on the intergenic IRES of cricket paralysis virus (CrPV), which remarkably requires neither initiator tRNA nor initiation factors (54). Like HCV and related IRESs, this CrPV IRES also binds directly to 40S subunits but in a significantly different manner, such that the P site is apparently filled by a pseudoknot and is inaccessible to the eIF2/GTP/Met-tRNAi complex. Because the number of known cellular and viral IRESs is constantly growing, we cannot rule out that additional mechanisms of internal ribosomal entry exist that are distinct from those used by EMCV, HCV, or CrPV-like IRESs. It seems probable that even those IRESs on which initiation occurs by a mechanism fundamentally similar to one of these three groups of IRESs will nevertheless require ITAFs different from those identified to date. It will be interesting to see whether the “induced active conformation” model for ITAF function described for the FMDV IRES (31) will be more generally applicable.
Just as it is unlikely that initiation on all IRESs will be described by one of the three models described above, so it would be premature to assume that initiation on all capped mRNAs occurs by the mechanism that we have described for ß-globin mRNA. More specifically, our knowledge of the scanning process is very rudimentary, and a number of open questions need to be addressed in the near future. These questions include: (i) What are the molecular interactions and conformational changes that lead to binding of a 43S complex to the capped eIF4F-bound 5' end of an mRNA? (ii) How and when are interactions between cap-bound factors and the 43S complex dissociated as this complex begins to scan from the cap-proximal region of an mRNA? (iii) Is ribosomal movement on the 5' NTR obligatorily linked to “melting” secondary structure in the 5' NTR, and, if these processes can be uncoupled, is the 43S complex intrinsically capable of movement on mRNA without concomitant ATP hydrolysis? (iv) Which factors influence the processivity of scanning? (v) How does the local sequence context of an initiation codon influence the efficiency of initiation at that codon? (vi) How does recognition of the initiation codon trigger all of the events associated with subunit joining? Answers to these questions not only will lead to a more detailed understanding of the molecular mechanism of the initiation process but also will offer insights into how structural differences between different mRNAs determine when and how efficiently they are translated.
Research done in our laboratories was supported Grants AI44108–01 and GM59660 from the National Institutes of Health (to C.U.T.H. and T.V.P.), by Grant MCB-9726958 from the National Science Foundation (to C.U.T.H.), and by grants from the Council for Tobacco Research Council (to C.U.T.H.), the Howard Hughes Medical Institute (to I.N.S. and C.U.T.H.), and the Russian Foundation of Basic Research (to V.I.A. and I.N.S.).
1. Pestova, T.V., Borukhov, S.I. & Hellen, C.U.T. (1998) Nature (London) 394, 854–859.
2. Morino, S., Imataka, H., Svitkin, Y.V., Pestova, T.V. & Sonenberg, N. (2000) Mol. Cell Biol. 20, 468–477.
3. Chaudhuri, J., Chowdhury, D. & Maitra, U. (1999) J. Biol. Chem. 274, 17975–17980.
4. Battiste, J.L., Pestova, T.V., Hellen, C.U.T. & Wagner, G. (2000) Mol. Cell 5, 109–119.
5. Dahlquist, K.D. & Puglisi, J.D. (2000) J. Mol. Biol. 299, 1–15.
6. Yoon, H. & Donahue, T.F. (1992) Mol. Cell Biol. 12, 248–260.
7. Fletcher, C.M., Pestova, T.V., Hellen, C.U.T. & Wagner, G. (1999) EMBO J. 18, 2631–2637.
8. Pestova, T.V. & Hellen, C.U.T. (1999) Trends Biochem. Sci. 24, 85–87.
9. Chakrabarti, A. & Maitra, U. (1991) J. Biol. Chem. 266, 14039–14045.
10. Asano, K., Clayton, J., Shalev, A. & Hinnebusch, A.G. (2000) Genes Dev. 14, 2534–2546.
11. Huang, H.K., Yoon, H., Hannig, E.M. & Donahue, T.F. (1997) Genes Dev. 11, 2396–2413.
12. Pestova, T.V., Hellen, C.U.T. & Dever, T.E. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Mathews, M.B. & Hershey, J.W.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 425–445.
13. Pestova, T.V., Lomakin, I.B., Lee, J.H., Choi, S.K., Dever, T.E. & Hellen, C.U.T. (2000) Nature (London) 403, 332–335.
14. Choi, S.K., Lee, J.H., Zoll, W.L., Merrick, W.C. & Dever, T.E. (1998) Science 280, 1757–1760.
15. Lee, J.H., Choi, S.K., Roll-Mecak, A., Burley, S.K. & Dever, T.E. (1999) Proc. Natl. Acad. Sci. USA 96, 1066–1070.
16. Carrera P., Johnstone, O., Nakamura, A., Casanova, J., Jackle, H. & Lasko P. (2000) Mol. Cell 5, 181–187.
17. Kolakofsky, D., Ohta, T. & Thach, R.E. (1968) Nature (London) 220, 244–247.
18. Jang, S.-K., Kräusslich, H.-G., Nicklin, M.J.H., Duke, G.M., Palmenberg, A.C. & Wimmer, E. (1988) J. Virol. 62, 2636–2643.
19. Pelletier, J. & Sonenberg, N. (1988) Nature (London) 334, 320–325.
20. Pilipenko, E.V., Blinov, V.M., Chernov, B.K., Dmitrieva, T.M. & Agol, V.I. (1989) Nucleic Acids Res. 17, 5701–5711.
21. Pilipenko, E.V., Blinov, V.M., Romanova, L.I., Sinyakov, A.N., Maslova, S.V. & Agol, V.I. (1989) Virology 168, 201–209.
22. Kaminski, A., Howell, M.T. & Jackson, R.J. (1990) EMBO J. 9, 3753–3759.
23. Pilipenko, E.V., Gmyl, A.P., Maslova, S.V., Belov, G.A., Sinyakov, A.N., Huang, M., Brown, T.D.K. & Agol, V.I. (1994) J. Mol. Biol. 241, 398–414.
24. Hellen, C.U.T., Pestova, T.V. & Wimmer, E. (1994) J. Virol 68, 6312–6322.
25. Lamphear, B.J., Kirchenweger, R., Skern, T. & Rhoads, R.E. (1995) J. Biol. Chem. 270, 21975–21982.
26. Pestova, T.V., Hellen, C.U.T. & Shatsky, I.N. (1996) Mol. Cell. Biol. 16, 6859–6869.
27. Pestova, T.V., Shatsky, I.N. & Hellen, C.U.T. (1996) Mol. Cell. Biol. 16, 6870–6878.
28. Pause, A., Methot, N., Svitkin, Y., Merrick, W.C. & Sonenberg, N. (1994) EMBO J. 13, 1205–1215.
29. Gingras, A.C., Raught, B. & Sonenberg, N. (1999) Annu. Rev. Biochem. 68, 913–963.
30. Kolupaeva, V.G., Hellen, C.U.T. & Shatsky, I.N. (1996) RNA 2, 1199–1212.
31. Pilipenko, E.V., Pestova, T.V., Kolupaeva, V.G., Khitrina, E.V., Poperechnaya, A.N., Agol, V.I. & Hellen, C.U.T. (2000) Genes Dev. 14, 2028–2045.
32. Kolupaeva, V.G., Pestova, T.V, Hellen, C.U.T. & Shatsky, I.N. (1998) J. Biol. Chem. 273, 18599–18604.
33. Lomakin, I.B., Hellen, C.U.T. & Pestova, T.V. (2000) Mol. Cell. Biol. 20, 6019–6029.
34. Svitkin, Y.V., Pestova, T.V., Maslova, S.V. & Agol, V.I. (1988) Virology 166, 394–404.
35. Hunt, S.L., Hsuan, J.J., Totty, N. & Jackson, R.J. (1999) Genes Dev. 13, 437–448.
36. Borovjagin, A.V., Evstafieva, A.G., Ugarova, T.Y. & Shatsky, I.N. (1990) FEBS Lett. 261, 237–240.
37. Jang, S.K. & Wimmer, E. (1990) Genes Dev. 4, 1560–1572.
38. Hellen, C.U.T., Witherell, G.W., Schmid, M., Shin, S.H., Pestova, T.V., Gil, A. & Wimmer, E. (1993) Proc. Natl. Acad. Sci. USA 90, 7642–7646.
39. Borman, A., Howell, M.T., Patton, J. & Jackson, R.J. (1993) J. Gen. Virol. 74, 1775–1788.
40. Kaminski, A. & Jackson, R.J. (1998) RNA 4, 626–638.
41. Walter, B.L., Nguyen, J.H., Ehrenfeld, E. & Semler, B.L. (1999) RNA 5, 1570–1585.
42. Tsukiyama-Kohara, K., lizuka, N., Kohara, M. & Nomoto, A. (1992) J.Virol. 66, 1476–1483.
43. Wang, C., Le, S.Y., Ali, N. & Siddiqui, A. (1995) RNA 1, 526–537.
44. Reynolds, J.E., Kaminski, A., Carroll, A.R., Clarke, B.E., Rowlands, D.J. & Jackson, R.J. (1996) RNA 2, 867–878.
45. Rijnbrand, R., van der Straaten, T., van Rijn, P.A., Spaan, W.J.M. & Bredenbeek, P.J. (1997) J.Virol. 71, 451–457.
46. Pestova, T.V., Shatsky, I.N., Fletcher, S.P., Jackson, R.J. & Hellen, C.U.T. (1998) Genes Dev. 12, 67–83.
47. Sizova, D.V., Kolupaeva, V.G., Pestova, T.V., Shatsky, I.N. & Hellen, C.U.T. (1998) J.Virol 72, 4775–4782.
48. Kolupaeva, V.G., Pestova, T.V. & Hellen, C.U. T. (2000) J. Virol 74, 6242–6250.
49. Kolupaeva, V.G., Pestova, T.V. & Hellen, C.U.T. (2000) RNA 6, 1791–1807.
50. Pestova, T.V. & Hellen, C.U.T. (1999) Virology 258, 249–256.
51. Honda, M., Brown, E.A. & Lemon, S.M. (1996) RNA 2, 955–968.
52. Jackson, R.J. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Mathews, M.B. & Hershey, J.W.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 127–183.
53. Jubin, R., Vantuno, N.E., Kieft, J.S., Murray, M.G., Doudna, J.A., Lau, J.Y. & Baroudy, B.M. (2000) J. Virol 74, 10430–10437.
54. Wilson, J.E., Pestova, T.V., Hellen, C.U.T. & Sarnow, P. (2000) Cell 102, 511–520.








