
The target of rapamycin (TOR) proteins
Brian Raught*, Anne-Claude Gingras*, and Nahum Sonenberg†
Department of Biochemistry and McGill Cancer Centre, McGill University, 3655 Promenade Sir-William-Osler, Montréal, QC H3G 1Y6 Canada
Rapamycin potently inhibits downstream signaling from the target of rapamycin (TOR) proteins. These evolutionary conserved protein kinases coordinate the balance between protein synthesis and protein degradation in response to nutrient quality and quantity. The TOR proteins regulate (i) the initiation and elongation phases of translation, (ii) ribosome biosynthesis, (iii) amino acid import, (iv) the transcription of numerous enzymes involved in multiple metabolic pathways, and (v) autophagy. Intriguingly, recent studies have also suggested that TOR signaling plays a critical role in brain development, learning, and memory formation.
Rapamycin Inhibits Long-Term Facilitation
Synaptic plasticity, the capacity of neurons to modulate the strength of synaptic connections, is believed to be critical for learning and memory formation. Long-term synaptic plasticity (necessary for the formation of long-term memory) requires alterations in gene expression and the establishment of new synaptic connections (1–3). These findings presented an interesting dilemma: That is, how can changes in gene expression in the cell body alter the strength of individual synaptic connections? Recent data suggest that stimulated synapses are “tagged” to capture mRNAs produced in the soma and exported throughout the cell (4). Synaptic tagging thus results in localization of mRNAs only to those synapses marked by previous activity. This model also presupposes that long-term plasticity depends on local translation of the localized mRNAs. Indeed, ribosomes, tRNAs, translation initiation factors, and translation elongation factors are found in dendrites (5, 6), and protein synthesis has been demonstrated to occur in isolated synaptic bodies (7, 8). Functional studies have demonstrated that protein synthesis is required for potentiation of synaptic transmission elicited by neurotrophic factors in hippocampal slices (9), and for the establishment of long-term facilitation in Aplysia neurons (10). Kandel and coworkers implicated a specific intracellular signaling pathway in this process by demonstrating that serotonin-stimulated synaptic protein synthesis can be blocked with rapamycin, an inhibitor of the target of rapamycin (TOR) proteins (11).
The aim of this review is to outline a current model regarding the intracellular signaling pathway inhibited by rapamycin, to detail known downstream targets of this signaling module, and to discuss putative links between TOR signaling and localized protein synthesis in neurons.
Rapamycin and TOR
Rapamycin is a lipophilic macrolide, isolated from a strain of Streptomyces hygroscopicus indigenous to Easter Island (known as Rapa Nui to the inhabitants; ref. 12). The intracellular rapamycin receptor in all eukaryotes is a small, ubiquitous protein termed FKBP12 (FK506-binding protein, molecular mass of 12 kDa; refs. 13, 14, 15). A rapamycin-FKBP12 “gain-of-function” complex interacts specifically with the evolutionarily conserved TOR proteins, to potently inhibit signaling to downstream targets. Two Saccharomyces cerevisiae TOR genes code for two large molecules (>280 kDa) sharing 67% identity at the amino acid level (16–19). Two Tor orthologs have also been isolated from Schizosaccharomyces pombe (20). Metazoans appear to possess only one TOR protein. A single Drosophila melanogaster ortholog, dTOR, is present in the completed fly genome, and shares 38% identity with the S. cerevisiae Tor2 protein (21, 22). A single mammalian TOR protein has been cloned from several species, and alternatively termed mTOR, FRAP (FKBP12 and rapamycin associated protein), RAFT (rapamycin and FKBP12 target), SEP (sirolimus effector protein), or RAPT (rapamycin target; refs. 23–27). Here, we refer to the mammalian protein as mTOR. mTOR is 289 kDa and shares ˜45% identity with the S. cerevisiae Tor1 and -2 proteins, and 56% identity with dTOR (21–23, 26, 27). The human, rat, and mouse mTOR proteins share >95% identity at the amino acid level (reviewed in ref. 28).
TOR Signaling
The TOR proteins have been assigned to a protein family termed the phosphatidylinositol kinase-related kinases (or PIKKs), a large group of signaling molecules that also includes the ataxiatelangiectasia mutated (ATM) protein, ATR/FRP (ataxia-telangiectasia- and rad3-related/FRAP related protein), and DNA-dependent protein kinase (DNA-PK; e.g., ref. 29). Despite significant homology to lipid kinases, the TOR proteins (as well as the other PIKKs) function as Ser/Thr protein kinases (reviewed in refs. 30 and 31).
How Does Rapamycin Inhibit TOR Signaling? The rapamycin-FKBP12 gain-of-function complex inhibits downstream signaling from the TOR proteins in vivo. However, whether this complex directly inhibits the kinase activity of the TOR proteins is an unresolved issue. Rapamycin was reported to inhibit a moderate stimulation of mTOR kinase activity (measured in vitro, using an mTOR immunoprecipitate) in response to insulin treatment (32), and rapamycin-FKBP12 can inhibit mTOR autokinase activity in vitro. However, it appears that a much higher concentration of rapamycin than is required in vivo is necessary to elicit this effect (ref. 33; and references therein). Furthermore, only very modest differences, or no change at all, in the kinase activity of TOR immunoprecipitates have been reported after mitogenic stimulation, amino acid withdrawal, or rapamycin treatment (refs. 22 and 33; and references therein). Rapamycin treatment of cells in culture does not inhibit autophosphorylation at S2481, as determined with a phosphospecific antibody directed against this site (33). Finally, in S. cerevisiae, a mutation in the kinase domain of the Tor2 protein is lethal, yet rapamycin treatment of yeast leads only to G1 arrest. If rapamycin were to inhibit Tor2p kinase activity, mutation of the Tor2p kinase
|
* |
B.R. and A.-C.G. contributed equally to this report. |
|
† |
To whom reprint requests should be addressed. E-mail: nsonen@med.mcgill.ca. |
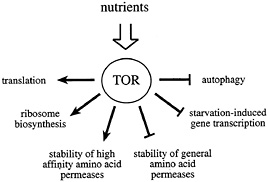
Fig. 1. The Tor proteins regulate the balance between protein synthesis and protein degradation. TOR signaling is active in the presence of sufficient nutrients to fuel protein synthesis. The TOR signal allows for the translation of mRNAs coding for components of the translation machinery, ribosome biosynthesis, and the stabilization of high affinity amino acid permeases. At the same time, TOR signaling destabilizes general amino acid permeases, inhibits autophagy, and represses the transcription of a subset of genes required for amino acid biosynthesis.
region and rapamycin treatment should elicit identical phenotypes. Thus, whereas it is clear that rapamycin functions through an inhibition of downstream signaling from the TOR proteins, this repression may involve mechanisms other than a direct suppression of TOR kinase activity.
What Signals to TOR? The TOR proteins do not appear to function as components of a conventional linear signaling pathway. Rather, several lines of evidence suggest that the TOR proteins function in a nutrient-sensing checkpoint control capacity. As discussed further below, both TOR and phosphoinositide 3-kinase (PI3K) signaling are required for the activation (or inactivation) of several downstream effector proteins. However, whether TOR activity is regulated by PI3K, or whether the two signaling pathways function independently, is unknown. Over-expression of a membrane-targeted Akt/PKB protein (a downstream effector of PI3K) in mammalian cells leads only to a modest increase (or no change) in mTOR kinase activity (as assayed in vitro), and moderately increases mTOR autophosphorylation in vivo, as assessed with the S2481 phosphospecific antibody (32–34).
A putative Akt consensus phosphorylation site, S2448, was observed to be phosphorylated on mTOR in vivo, as determined with a phosphospecific antibody. Addition of insulin or IL-3 engenders an increase in S2448 phosphorylation in a PI3K- and Akt-dependent manner (34, 35). However, an mTOR mutant protein possessing an alanine substitution at this site retains the ability to activate S6K1 (a downstream effector of mTOR, see below) after growth factor stimulation (34). Thus, the role of this phosphorylation event in the regulation of mTOR activity is not clear.
Inactivation of the TOR proteins, or rapamycin treatment, mimics nutrient deprivation in yeast, Drosophila, and mammalian cells (21, 36–38). Thus, a current working model for TOR signaling proposes that these kinases relay a permissive signal to downstream targets only in the presence of sufficient nutrients to fuel protein synthesis (Fig. 1). In some cases, the TOR proteins appear to function in a coregulatory capacity with other conventional, linear signaling pathways (such as the PI3K pathway; see below). In this way, a passive nutrient sufficiency signal may be combined with stimulatory signaling from a second pathway to coordinate cellular processes that require the uptake of nutrients. The absence of either signal is predicted to prohibit activation of downstream targets.
A Model for TOR Signaling. How does TOR signal to downstream effectors? TOR signaling is thought to be effected through a combination of direct phosphorylation of downstream targets, and repression of phosphatase activity (Fig. 2). Genetic screening in S. cerevisiae has identified the PP2A-like phosphatase Sit4p, two PP2A regulatory subunits (CDC55 and TPD3), and a phosphatase-associated protein (Tap42p), as components of a rapamycin-sensitive signaling pathway (38, 39). Tap42p interacts directly with the catalytic subunits of PP2A and Sit4p. S. cerevisiae expressing a temperature-sensitive Tap42 mutant protein exhibit a dramatic defect in translation initiation at the nonpermissive temperature (39). Thus, Tap42p is thought to repress PP2A (or Sit4p) activity (also see refs. 40 and 41).
Phosphorylation of Tap42p regulates its interaction with phosphatases. Whereas phosphorylated Tap42p competes with the phosphatase adapter (A) subunit for binding to the catalytic subunit, dephosphorylated Tap42p does not efficiently compete for binding (42). Tap42p phosphorylation is modulated by Tor signaling. The Tap42p-PP2A association in vivo is disrupted by nutrient deprivation or rapamycin treatment (39, 42). Further, a yeast Tor2p immunoprecipitate can phosphorylate Tap42p in vitro (42), and Tap42p phosphorylation is rendered rapamycin resistant in yeast strains expressing a rapamycin-resistant Tor1 protein (42).
Tap42 orthologs are found in Arabidopsis (43), Drosophila, (GenBank accession number AAF53289), and mammalian cells (44, 45). The B cell receptor binding protein a4 (a.k.a Ig binding protein 1, IGBP1) is the mammalian ortholog of Tap42p (44, 45). The ability of this protein to interact with PP2A-like phosphatases is conserved in mammals, as a4 binds directly to the catalytic subunits of PP2A (46, 47), PP4, and PP6 (48, 49). Like Tap42p, a4 is also a phosphoprotein, and the a4-PP2A interaction was reported to be abrogated by rapamycin treatment (although this finding remains somewhat controversial; refs. 46 and 47). These observations suggest that Tap42p/a4 phosphorylation, and PP2A binding, are regulated by TOR signaling, and that an inhibition in TOR signaling leads to Tap42p/a4 dephosphorylation, dissociation of the Tap42p/a4-phosphatase complex, and phosphatase derepression.
Interestingly, mTOR was reported to undergo nucleocytoplasmic shuttling (50). Abrogation of shuttling (by treatment with leptomycin B, a specific inhibitor of the nuclear export receptor Crml, or by transfection of mTOR tagged with exogenous nuclear export or import signals) was demonstrated to inhibit signaling to S6K1 and 4E-BP1 (50). Why mTOR shuttling may be important for 4E-BP1 and S6K1 activity is unknown (50).
TOR Signaling Modulates the Phosphorylation State of Proteins Involved in Translational Control
Tor and Translation in S. cerevisiae. Inhibition of Tor activity in yeast potently represses translation initiation, concomitant with polysome disaggregation and cell cycle arrest in G1 (36). The mechanism for this translational repression is not understood, but could be due, at least in some strains, to the degradation of the initiation factor eIF4G (51, 52). A putative regulator of yeast eIF4E function, termed Eap1p (eIF4E-associated protein 1), may also be involved in this process, as disruption of the EAP1 gene results in partial rapamycin resistance (53). The G1 arrest in response to Tor inactivation was suggested to be due to the inhibition of translation of an mRNA coding for a cyclin involved in G1 to S progression, CLN3, because the cell cycle block can be overcome by forced expression of CLN3 (54–56).
TOR and Translation in Mammalian Cells. TOR activity also regulates translation in mammalian cells (reviewed in refs. 57, 58, and
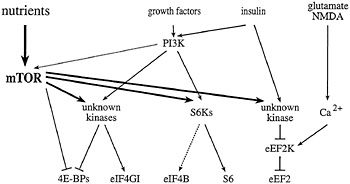
Fig. 2. Signaling to eukaryotic translation initiation and elongation factors. mTOR signaling, in combination with the PI3K pathway, activates the translation of rapamycin-sensitive mRNAs. In the presence of sufficient nutrients to fuel protein synthesis, mTOR and PI3K signaling activate the S6Ks, and one or more unknown kinases, to effect phosphorylation of the ribosomal S6 protein, eIF4B, eIF4GI, and the4E-BPs. In response to agents that raise intracellular Ca2+ (such as glutamate or NMDA), a specific Ca2+/CaM-dependent kinase effects the phosphorylation of eEF2 to inhibit elongation. mTOR signaling has been reported to inhibit eEF2 phosphorylation (possibly via inhibition of the eEF2 kinase), and thus, to increase elongation rates. Phosphatases have been implicated in the dephosphorylation of several translation effectors, but are not depicted in this figure.
59). However, whereas rapamycin treatment of S. cerevisiae leads to a precipitous disaggregation of polysomes, rapamycin treatment of mammalian cells specifically inhibits only the translation of certain classes of mRNAs. As detailed below, mTOR is thought to modulate translation of these mRNAs via the regulation of the phosphorylation state of several different translation effector proteins (Fig. 2).
The S6Ks. The ribosomal S6 kinases (S6K1 and S6K2) regulate the translation of a group of mRNAs possessing a 5' terminal oligopyrimidine tract (5’TOP), a stretch of 4–14 pyrimidines found at the extreme 5' terminus of ribosomal protein mRNAs, and mRNAs coding for other components of the translation machinery (reviewed in ref. 60). When nutrient levels are low, the translation of 5TOP-containing mRNAs is repressed. Even in the presence of sufficient nutrients, translation of 5TOP-containing mRNAs is inhibited by rapamycin treatment (reviewed in ref. 59). 5'TOP-containing mRNAs are present in mammalian and Drosophila cells (61), and comprise a significant amount of the total mRNA. The mechanism for 5'TOP regulation is not understood; however, two S6K substrates that could play a role in the modulation of 5 TOP translation are the ribosomal S6 protein and the translation initiation factor eIF4B (see below; reviewed in refs. 59 and 62).
S6K activity is inhibited by both PI3K inhibitors and rapamycin, indicating that both PI3K and mTOR signaling are required for S6K activation (63). A key finding in the understanding of this signaling module is that the PI3K and mTOR inputs to S6K1 can be separated. Deletion of an N-terminal S6K1 fragment confers rapamycin resistance to the S6K1 protein, yet this truncation mutant remains sensitive to treatment with PI3K inhibitors (64, 65). These data thus argue against a linear pathway to S6K1 comprised of PI3K and mTOR, but instead suggest that two separate inputs are required for S6K activation.
4E-BPs. Protein synthesis is regulated in many instances at the initiation phase (Fig. 3), the stage during which a ribosome is recruited to the 5' end of an mRNA, and positioned at a start codon (reviewed in ref. 66). The eukaryotic ribosome does not have the ability to locate and bind to the 5' end of an mRNA; it must rely on a number of translation initiation factors to guide it there. The mRNA 5' end is distinguished by the presence of a “cap” (the structure m7GpppN, in which m is a methyl group and N any nucleotide), which is specifically recognized by the eukaryotic translation initiation factor 4E (eIF4E). eIF4E, via an interaction with one of two large scaffolding proteins, termed eIF4GI and eIF4GII, directs the translation machinery to the 5' end of the mRNA (reviewed in refs. 57, 58, and 62). The interaction between eIF4E and eIF4G is regulated in mammalian and Drosophila cells by a family of translation repressor peptides, the eIF4E-binding proteins (4E-BPs; refs. 67 and 68–71). The 4E-BPs compete with the eIF4G proteins for an overlapping binding site on eIF4E, such that binding of a 4E-BP or an eIF4G protein to eIF4E is mutually exclusive (72–74).
Binding of the mammalian and Drosophila 4E-BPs to eIF4E is regulated by phosphorylation (67, 68, 70). Whereas hypophosphorylated 4E-BPs bind with high affinity to eIF4E, 4E-BP hyperphosphorylation abrogates this interaction. As is the case with S6K1, the PI3K and TOR signaling pathways modulate 4E-BP phosphorylation. Immunoprecipitates of mTOR phosphorylate two “priming” sites in the mammalian 4E-BP1 protein in vitro (75–77). This phosphorylation event is thought to be required for subsequent PI3K-dependent phosphorylation of other S/T residues, resulting in release from eIF4E (refs. 77, 79, and 80; A.-C.G., B.R., S.P. Gygi, A.Niedzwiecka, M.Miron, S. K.Burley, R.D.Polakiewicz, A.Wyslouch-Cieszynska, and R. Aebersold, unpublished observations). Using a panel of pharmacological inhibitors, the D. melanogaster 4E-BP ortholog was also demonstrated to lie downstream of dTOR and dPI3K (70).
eIF4GI. Two eIF4G homologs have been identified in mammalian cells (81, 82). Both eIF4GI and eIF4GII are phosphoproteins (83). Whereas the intracellular signaling pathways that modulate the phosphorylation of eIF4GII have not been elucidated, phosphorylation of the eIF4GI isoform is regulated by mTOR and PI3K signaling. Three phosphorylation sites (S1108, S1148, and S1 192) were demarcated in a C-terminal eIF4GI “hinge” region. Phosphorylation of the hinge residues is elevated by serum or insulin treatment, and is inhibited by rapamycin or PI3K inhibitors (83). However, neither mTOR nor S6Ks can directly phosphorylate the hinge residues in vitro. Interestingly, eIF4GI proteins truncated at their N termini are constitutively phosphorylated on the hinge residues, even in the presence of PI3K or mTOR inhibitors (83). Thus, rapamycin-insensitive kinases appear to phosphorylate these residues, but an amino-terminal domain could regulate the accessibility of the hinge phosphorylation sites to these kinases in a rapamycin-sensitive manner. The function of these phosphorylation events is unclear.
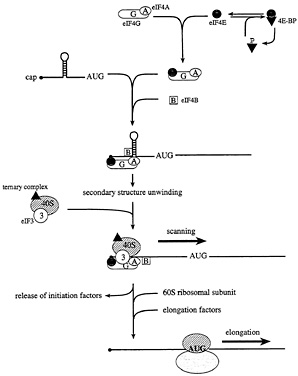
Fig. 3. The initiation and elongation phases of translation in eukaryotes. In starved or stressed cells, the cap binding protein eIF4E is sequestered by hypophosphorylated 4E-BPs. In growing or stimulated cells, the 4E-BPs are hyperphosphorylated to release eIF4E, such that it can interact with the scaffolding protein, eIF4G. In conjunction with the RNA helicase eIF4A and the cofactor eIF4B, 5' secondary structure is melted, and a small ribosomal subunit is recruited to a single-stranded, cap-proximal region of an mRNA via an interaction between eIF4G and the ribosome-associated factor eIF3. The small ribosomal subunit, along with a ternary complex composed of eIF2, GTP, and Met-tRNAi, then scans the mRNA in a 5' to 3' direction until an AUG start codon in the proper sequence context is encountered. At this point, initiation factors are released, and the large ribosomal subunit is recruited. The elongation factors catalyze aminoacyl-tRNA binding to ribosomes, and the translocation of the mRNA from the ribosomal A site to the P site.
The hinge region residues do not overlap with binding sites for any known eIF4GI interacting protein, and no differences in the interaction of eIF4GI with several known binding partners were observed for eIF4GI isolated from serum-starved vs. serumstimulated cells. It was thus suggested that phosphorylation could effect changes in eIF4GI structure, to increase eIF4GI activity toward rapamycin-sensitive mRNAs (83).
eIF4B. eIF4B is a ubiquitous protein that dramatically stimulates the activity of eIF4A, an RNA helicase thought to unwind mRNA 5' secondary structure (84). Mammalian eIF4B is a phosphoprotein (85), and treatment of cells with serum, insulin, or phorbol esters results in eIF4B hyperphosphorylation (62, 86). eIF4B can be phosphorylated in vitro with several different kinases, including S6K1 (refs. 87 and 88; F.Peiretti and J.W.B. Hershey, personal communication). Two-dimensional tryptic phosphopeptide mapping has revealed that eIF4B possesses at least one serum-stimulated phosphorylation site that is sensitive to rapamycin and inhibitors of PI3K (B.R., F.Peiretti, A.-C.G., and J.W.B.Hershey, unpublished observations). Thus, PI3K and mTOR also appear to signal to eIF4B. Unlike the 4E-BPs and eIF4GI, however, eIF4B appears to be a direct target of the S6Ks.
eEF2. Another level at which translation may be modulated in eukaryotes is the elongation phase (Fig. 3). The eukaryotic elongation factors (eEFs) 1 and 2 regulate this process (reviewed in ref. 89). eEF1 promotes aminoacyl-tRNA binding to ribosomes, whereas eEF2 promotes the translocation of the mRNA from the ribosomal A site to the P site (90). Phosphorylation of eEF2 by a specific Ca2+/CaM-dependent kinase inhibits eEF2 activity (reviewed in ref. 89). Amino acid withdrawal from cultured mammalian cells results in a marked increase in eEF2 phosphorylation, accompanied by a decrease in elongation rates (e.g., ref. 91). Many agents that raise intracellular Ca2+ concentrations also bring about eEF2 phosphorylation, including histamine treatment of epithelial cells (92, 93), or glutamate or N-methyl-D-aspartate (NMDA) treatment of neurons (see below). Conversely, insulin stimulation leads to eEF2 dephosphorylation, resulting in a decrease in ribosomal transit time, and an increase in elongation rates (94–96). Rapamycin treatment inhibits the insulin-stimulated dephosphorylation of eEF2 (91, 96). Thus, eEF2 phosphorylation also appears to be modulated by mTOR signaling. How mTOR signaling regulates eEF2 activity is unknown; mTOR has been proposed to regulate the activity of the eEF2 kinase and/or to modulate the dephosphorylation of eEF2 via regulation of PP2A activity (89, 97). As discussed further below, eEF2 activity has been implicated in the control of protein synthesis in neurons.
In sum, mTOR signaling regulates the phosphorylation state of many proteins involved in translation control, including the S6 kinases, the translation initiation factors eIF4B and eIF4GI, the translation elongation factor eEF2, and a family of translation inhibitory proteins, the 4E-BPs. Many other translation factors are also known to be phosphoproteins (62), but the pathways modulating the phosphorylation state of these factors have not been studied. Additional proteins involved in translation control may thus also be downstream of mTOR.
TOR Regulates the Abundance of the Translation Machinery
In addition to its effect on the phosphorylation state of proteins involved in translational control, TOR signaling regulates the abundance of the components of the translation machinery (Fig.1), at both the transcriptional and translational levels. The
number of ribosomes in a given cell can vary dramatically, according to growth conditions (reviewed in ref. 98). Actively growing cells require numerous ribosomes (e.g., logarithmically dividing yeast cells produce 2000 ribosomes/minute), and ribosome synthesis represents a major energy expenditure for the cell (98). In S. cerevisiae, ribosome biosynthesis requires the transcription of over 100 different genes, involving all three RNA polymerases (98). In response to nutrient availability, TOR signaling in S. cerevisiae regulates the transcription of rRNA by Pol I and Pol III (52, 99), and the transcription of ribosomal protein mRNAs by Pol II (41, 52, 100, 101). TOR signaling has also been implicated in the processing of the ribosomal 35S precursor rRNA (52). When nutrients are limiting, ribosome production is curtailed (or a cell may even begin to degrade ribosomes, in a scavenging process termed autophagy; see below). The abundance of several yeast translation factors was also demonstrated to be regulated by the TOR pathway (52). Transcriptional modulation in S. cerevisiae is responsible for a decrease in the mRNA levels of initiation and elongation factors after rapamycin treatment, although the extent of this transcriptional inhibition is less than that observed for the ribosomal proteins (52).
Through the S6Ks, mTOR signaling regulates the translation of ribosomal protein mRNAs in mammalian cells (102, 103). In Drosophila and mammalian cells, translation of the elongation factor mRNAs, and mRNAs coding for other proteins involved in translation, such as the poly(A) binding protein, is also regulated by the presence of the 5′TOP element (reviewed in ref. 60). Thus, the TOR pathway simultaneously regulates the abundance and activity of the translation machinery in both unicellular and multicellular organisms.
TOR as a Master Switch for Catabolism vs. Anabolism
In yeast, TOR signaling has been demonstrated to coordinate the activity of various metabolic pathways in response to nutrient quality (Fig. 1). In particular, TOR signaling modulates the transcription of genes involved in amino acid biosynthesis, and regulates the activity of amino acid permeases. In both yeast and mammalian cells, TOR signaling regulates autophagy.
Nutrient-Sensitive Transcriptional Regulation. Switching yeast cells to a poor carbon or nitrogen source induces a state of quiescence (G0). Whereas the transcription of many genes is inhibited after a switch from a rich to a poor nitrogen or carbon source (or after rapamycin treatment), global mRNA profiling has revealed that the transcription of mRNAs coding for proteins involved in nutrient utilization, respiration, and protein degradation is actually augmented (41, 100, 101, 104). Tor signaling modulates gene expression via cytoplasmic sequestration of several nutrient-responsive transcription factors. For example, the GATA transcription factor Gln3p is retained in the cytoplasm through an interaction with the Ure2 protein, whereas the zinc-fingercontaining transcription factors Msn2p and Msn4p are sequestered in the cytoplasm via an interaction with the 14–3-3 protein Bmh2p (reviewed in ref. 38). Starvation abrogates Tor signaling and results in a loss of cytoplasmic retention of Gln3p, Msn2p, and Msn4p, followed by nuclear translocation and transcription of various target genes (38, 41). Tor signals to several specific effectors (Tap42, Mks1p, Ure2p, Gln3p, and Gat1p) to elicit changes in the expression levels of enzymes involved in several different metabolic pathways (104, 105). How TOR signaling may affect the transcription rates of metabolic enzymes in multicellular organisms has not yet been elucidated.
Amino Acid Permeases. Permeases are necessary for nutrient uptake, and may be divided into two functional classes. One class is regulated in response to the available nitrogen source (e.g., the general amino acid permease Gap1p), and members of this class transport amino acids to be used as a nitrogen source. The second class mainly consists of high affinity permeases, which specifically transport one or a small group of related amino acids to be used as building blocks for protein synthesis. In starved yeast cells, or in cells treated with rapamycin, ubiquitination and degradation of the high affinity tryptophan permease Tat2p is induced, leading to a decrease in tryptophan import (40, 106). This phenomenon is not unique to Tat2p, as a histidine permease (Hip1p) is also degraded upon nutrient deprivation or rapamycin treatment (106). In contrast, rapamycin treatment increases the abundance of the general permease Gap1p, indicating that TOR signaling inversely regulates the two classes of permeases (106). TOR regulation of permeases is mediated through the serine/ threonine kinase Npr1p, whose phosphorylation is regulated by the Tor proteins and Tap42p, in a manner similar to the regulation of S6Ks and 4E-BPs in mammalian cells (40).
Autophagy. When nutrient levels are low, eukaryotic cells degrade cytoplasmic proteins and organelles to scavenge amino acids, in a process termed autophagy (107–109). Autophagy involves the sequestration of a portion of cytoplasm by a double (or multi) layered membrane structure termed the autophagosome or autophagic vacuole. This structure fuses with lysosomal or endosomal membranes, resulting in the degradation of cytoplasmic components. The TOR proteins regulate autophagy. Rapamycin addition to yeast cultures or to mammalian cells in culture induces autophagy, even in a nutrient-rich medium (110, 111). Shifting a temperature-sensitive TOR2 yeast mutant to the nonpermissive temperature also induces autophagy (110). In mammalian cells, autophagy is inhibited by amino acids and insulin. Activation of S6K is associated with inhibition of autophagy in rat hepatocytes, and the inhibition of autophagy by amino acids could be partially prevented by rapamycin treatment (111, 112).
In sum, the TOR proteins appear to act as master regulators of the balance between protein synthesis and degradation. In the presence of sufficient nutrients to fuel protein synthesis, TOR provides a permissive signal to translation, ribosome biosynthesis, and high affinity amino acid permeases, while repressing autophagy and the general amino acid permeases. In the absence of TOR signaling, the translation of mRNAs coding for components of the translation machinery is specifically inhibited, ribosome biosynthesis is blocked, and autophagy is activated.
How Might TOR Signaling Be Involved in Learning and Memory?
The observation that rapamycin can inhibit long-term facilitation in Aplysia neurons has implicated TOR signaling in the control of neuronal protein synthesis (11). How might a kinase involved in the regulation of protein metabolism also be involved in learning and memory? In fact, several putative links have been established between TOR and neuronal function.
Several types of neurotransmitters were described to affect the activity of the rapamycin-sensitive pathway leading to S6K1 and 4E-BP1 phosphorylation. Serotonin (5-HT) addition to Aplysia neurons or Chinese hamster ovary (CHO) cells expressing the 5-HT1B receptor increases phosphorylation of S6K1 in a rapamycin-dependent manner (113, 114). Dopamine addition to CHO cells also activates S6K1 in a rapamycin-dependent manner (115). Finally, both S6K1 and 4E-BP1 phosphorylation is induced by stimulation of the µ-opioid receptors (which mediate the analgesic and addictive properties of morphine) by the agonist [D-Ala2, N-MePhe4,Gly5-ol] enkephalin (DAMGO; ref. 116).
mTOR interacts with gephyrin, a tubulin-binding protein involved in neuronal γ-aminobutyric acid type A (GABAA) and glycine receptor clustering (117–120). Gephyrin binding was reported to be required for signaling to S6K1 and 4E-BP1, and, consistent with a role in localized protein synthesis, a fraction-
ation experiment demonstrated that mTOR and gephyrin were enriched in the synaptosomal fraction, but not the postsynaptic density fraction (117).
Another possible connection between mTOR signaling and localized translation is via the modulation of eEF2 phosphorylation. Several studies have noted an increase in eEF2 phosphorylation in response to various neurotransmitters. For example, glutamate or NMDA treatment of cortical neurons in culture leads to a rapid and pronounced increase in eEF2 phosphorylation, and a decrease in translation rates in cell bodies and proximal (but not distal) cell processes (121). Activation of the NMDA receptor also leads to eEF2 phosphorylation, in tadpole tecta (122). It is tempting to speculate that mTOR could inhibit eEF2 phosphorylation in active synapses to locally derepress translation. It has also been suggested that eEF2 phosphorylation could actually enhance the translation of specific mRNAs localized to dendrites by driving these mRNAs from untranslated ribonucleotide particles or small polysomes into larger polysomes (122–125).
Another possible link between TOR and neuronal function is the regulation of autophagy. In addition to nutrient scavenging during starvation, autophagy has been demonstrated to play an important role in developmental processes that involve cellular remodeling, such as insect metamorphosis (126) or luteal regression (127). Whereas neuronal death certainly involves apoptosis (128), several reports have suggested that an alternative form of cell death may occur in some nerve cells. For example, nerve growth factor (NGF)-deprivation of sympathetic neurons was reported to induce a rapid, 30-fold increase in autophagic particles, before any signs of DNA fragmentation (a hallmark of apoptosis) were observed. Treatment of these cells with the anti-autophagic drug 3-methyladenine delayed cell death (129). In another study, autophagic vacuoles were observed in PC12 cells 3 h after serum starvation, whereas chromatin condensation did not occur until 6 h poststarvation (130). Finally, the removal of specific spinal cord neurons in Xenopus tadpoles (a normal developmental process during metamorphosis) was also suggested to occur through autophagy-directed cell death (131). Intriguingly, elevated levels of autophagy have been reported to be associated with neurodegenerative disorders such as Parkinson’s disease (132).
TOR Activity Is Required for Murine Forebrain Development
A recently described mouse mutant suggests that mTOR plays a critical role in embryonic brain development (133; K.Hentges and A.Peterson, personal communication). The murine flat top mutation was isolated in a chemical mutagenesis screen designed to identify genes involved in embryonic telencephalic development (133). Flat top defects include a failure of the embryo to up-regulate proliferation in the telencephalic primordium, and a failure to establish dorsal and ventral domains of gene expression in the developing telencephalon. Homozygous mutant embryos fail to rotate around the body axis, and die in utero (78). The flat top mutation was mapped to a single nucleotide change in an mTOR intron, which leads to aberrant splicing. The protein products derived from these abnormally spliced mRNAs appear to be inactive (or much less active), because of the presence of a 3-aa insertion or 3-aa deletion at the intron-exon junction. Transgenic rescue experiments confirmed that mTOR is the affected gene in this animal, and a rapamycin injection regimen during pregnancy yields embryos with an identical phenotype (K. Hentges and A.Peterson, personal communication). Whether the brain defect is the result of a failure to inhibit autophagy, or is elicited through some other function of mTOR is unknown. S6K1 activity was demonstrated to be significantly lower (17% of wild-type levels) in flat-top embryos, but effects on other translation factors have not yet been determined. The flat top mouse should provide a very valuable tool for the study of TOR function in mammalian cells.
Summary and Future Prospects
The TORs are evolutionarily conserved protein kinases that regulate the balance between protein synthesis and degradation in unicellular and multicellular organisms. This complex balance is maintained via the regulation of translation initiation and elongation factor activity, the modulation of ribosome biosynthesis at both the transcriptional and translational levels, the control of amino acid permease activity, the coordination of the transcription of many enzymes involved in various metabolic pathways, and the control of autophagy. An interesting and unexpected finding was that mTOR also appears to play a critical role in embryonic brain development, learning, and memory formation.
There is still much to be learned. For instance, how the TOR proteins sense the quality or quantity of nutrients is unknown. The mammalian GCN2 kinase, which senses intracellular amino acid levels by binding to deacylated tRNAs, does not appear to play a role in this process, because amino acid withdrawal leads to S6K1 and 4E-BP1 dephosphorylation even in GCN2 null cells (C.Jousse and D.Ron, personal communication). Further, whereas the role of the TOR proteins in the control of metabolic enzymes and amino acid permeases in S. cerevisiae is now well documented, similar studies have not been conducted for the mammalian and Drosophila systems. The recent description of the Drosophila TOR homolog (dTOR; refs. 21 and 22) and the isolation of the murine flat top mTOR mutant (133) should provide invaluable tools for further dissection of the TOR signaling module in multicellular organisms.
We thank A.Peterson, K.Hentges, C.Jousse, D.Ron, F.Peiretti, and J.W.B.Hershey for sharing unpublished data, and W.S.Sossin, F. Poulin, P.F.Cho-Park, and M.Miron for critical reading of the manuscript. Work in the authors’ laboratory is supported by grants from the Canadian Institutes of Health Research, the National Cancer Institute of Canada, the Howard Hughes Medical Institute (HHMI), and the Human Frontier Science Program. B.R. is supported by a Medical Research Council (MRC) of Canada postdoctoral fellowship. A.-C.G. is supported by an MRC of Canada doctoral fellowship. N.S. is an MRC of Canada Distinguished Scientist and an HHMI International Scholar.
1. Goelet, P., Castellucci, V.F., Schacher, S. & Kandel, E.R. (1986) Nature (London) 322, 419–422.
2. Bailey, C.H., Bartsch, D. & Kandel, E.R. (1996) Proc. Natl. Acad. Sci. USA 93, 13445–13452.
3. Sossin, W. S. (1996) Trends Neurosci. 19, 215–218.
4. Frey, U. & Morris, R.G. (1998) Trends Neurosci. 21, 181–188.
5. Steward, O. & Levy, W.B. (1982) J. Neurosci. 2, 284–291.
6. Tiedge, H. & Brosius, J. (1996) J. Neurosci. 16, 7171–7181.
7. Torre, E.R. & Steward, O. (1992) J. Neurosci. 12, 762–772.
8. Weiler, I.J. & Greenough, W.T. (1993) Proc. Natl. Acad. Sci. USA 90, 7168–7171.
9. Kang, H. & Schuman, E.M. (1996) Science 273, 1402–1406.
10. Martin, K. C., Casadio, A., Zhu, H., E, Y., Rose, J.C, Chen, M., Bailey, C.H. & Kandel, E.R. (1997) Cell 91, 927–938.
11. Casadio, A., Martin, K.C., Giustetto, M., Zhu, H., Chen, M., Bartsch, D., Bailey, C.H. & Kandel, E.R. (1999) Cell 99, 221–237.
12. Vezina, C., Kudelski, A. & Sehgal, S.N. (1975) J. Antibiot. (Tokyo) 28, 721–726.
13. Harding, M.W., Galat, A., Uehling, D.E. & Schreiber, S.L. (1989) Nature (London) 341, 758–760.
14. Siekierka, J.J., Hung, S.H., Poe, M., Lin, C.S. & Sigal, N.H. (1989) Nature (London) 341, 755–757.
15. Siekierka, J.J., Wiederrecht, G., Greulich, H., Boulton, D., Hung, S.H., Cryan, J., Hodges, P.J. & Sigal, N.H. (1990) J. Biol. Chem. 265, 21011–21015.
16. Cafferkey, R., Young, P.R., McLaughlin, M.M., Bergsma, D.J., Koltin, Y., Sathe, G.M., Faucette, L., Eng, W.K., Johnson, R.K. & Livi, G.P. (1993) Mol. Cell. Biol. 13, 6012–6023.
17. Heitman, J., Movva, N.R. & Hall, M.N. (1991) Science 253, 905–909.
18. Helliwell, S.B., Wagner, P., Kunz, J., Deuter-Reinhard, M., Henriquez, R. & Hall, M.N. (1994) Mol. Biol. Cell 5, 105–118.
19. Kunz, J., Henriquez, R., Schneider, U., Deuter-Reinhard, M., Movva, N.R. & Hall, M.N. (1993) Cell 73, 585–596.
20. Weisman, R. & Choder, M. (2001) J. Biol. Chem. 276, 7027–7032.
21. Zhang, H., Stallock, J.P., Ng, J.C, Reinhard, C. & Neufeld, T.P. (2000) Genes Dev. 14, 2712–2724.
22. Oldham, S., Montagne, J., Radimerski, T., Thomas, G. & Hafen, E. (2000) Genes Dev. 14, 2689–2694.
23. Brown, E. J., Albers, M.W., Shin, T.B., Ichikawa, K., Keith, C.T., Lane, W.S. & Schreiber, S.L. (1994) Nature (London) 369, 756–758.
24. Chen, Y., Chen, H., Rhoad, A.E., Warner, L., Caggiano, T.J., Failli, A., Zhang, H., Hsiao, C.L., Nakanishi, K. & Molnar-Kimber, K.L. (1994) Biochem. Biophys. Res. Commun. 203, 1–7.
25. Chiu, M.I., Katz, H. & Berlin, V. (1994) Proc. Natl. Acad. Sci. USA 91, 12574–12578.
26. Sabatini, D.M., Erdjument-Bromage, H., Lui, M., Tempst, P. & Snyder, S.H. (1994) Cell 78, 35–43.
27. Sabers, C.J., Martin, M.M., Brunn, G.J., Williams, J.M., Dumont, F.J., Wiederrecht, G. & Abraham, R.T. (1995) J. Biol. Chem. 270, 815–822.
28. Abraham, R.T. & Wiederrecht, G.J. (1996) Annu. Rev. Immunol. 14, 483–510.
29. Keith, C.T. & Schreiber, S.L. (1995) Science 270, 50–51.
30. Hunter, T. (1995) Cell 83, 1–4.
31. Hoekstra, M.F. (1997) Curr. Opin. Genet. Dev. 7, 170–175.
32. Scott, P.H., Brunn, G.J., Kohn, A.D., Roth, R.A. & Lawrence, J.C., Jr. (1998) Proc. Natl. Acad. Sci. USA 95, 7772–7777.
33. Peterson, R.T., Beal, P.A., Comb, M.J. & Schreiber, S.L. (2000) J. Biol. Chem. 275, 7416–7423.
34. Sekulic, A., Hudson, C.C., Homme, J.L., Yin, P., Otterness, D.M., Karnitz, L.M. & Abraham, R.T. (2000) Cancer Res. 60, 3504–3513.
35. Navé, B.T., Ouwens, M., Withers, D.J., Alessi, D.R. & Shepherd, P.R. (1999) Biochem. J. 344, 427–431.
36. Barbet, N.C., Schneider, U., Helliwell, S.B., Stansfield, I., Tuite, M.F. & Hall, M.N. (1996) Mol. Biol. Cell 7, 25–42.
37. Dennis, P.B., Fumagalli, S. & Thomas, G. (1999) Curr. Opin. Genet. Dev. 9, 49–54.
38. Schmelzle, T. & Hall, M.N. (2000) Cell 103, 253–262.
39. Di Como, C.J. & Arndt, K.T. (1996) Genes Dev. 10, 1904–1916.
40. Schmidt, A., Beck, T., Koller, A., Kunz, J. & Hall, M.N. (1998) EMBO J. 17, 6924–6931.
41. Beck, T. & Hall, M.N. (1999) Nature (London) 402, 689–692.
42. Jiang, Y. & Broach, J.R. (1999) EMBO J. 18, 2782–2792.
43. Harris, D.M., Myrick, T.L. & Rundle, S.J. (1999) Plant Physiol. 121, 609–617.
44. Inui, S., Kuwahara, K., Mizutani, J., Maeda, K., Kawai, T., Nakayasu, H. & Sakaguchi, N. (1995) J. Immunol. 154, 2714–2723.
45. Onda, M., Inui, S., Maeda, K., Suzuki, M., Takahashi, E. & Sakaguchi, N. (1997) Genomics 46, 373–378.
46. Inui, S., Sanjo, H., Maeda, K., Yamamoto, H., Miyamoto, E. & Sakaguchi, N. (1998) Blood 92, 539–546.
47. Murata, K., Wu, J. & Brautigan, D.L. (1997) Proc. Natl. Acad. Sci. USA 94, 10624–10629.
48. Chen, J., Peterson, R.T. & Schreiber, S.L. (1998) Biochem. Biophys. Res. Commun. 247, 827–832.
49. Nanahoshi, M., Tsujishita, Y., Tokunaga, C., Inui, S., Sakaguchi, N., Hara, K. & Yonezawa, K. (1999) FEBS Lett. 446, 108–112.
50. Kim, J.E. & Chen, J. (2000) Proc. Natl. Acad. Sci. USA 97, 14340–14345. (First Published December 12, 2000; 10.1073/pnas.011511898)
51. Berset, C., Trachsel, H. & Altmann, M. (1998) Proc. Natl. Acad. Sci. USA 95, 4264–4269.
52. Powers, T. & Walter, P. (1999) Mol. Biol. Cell 10, 987–1000.
53. Cosentino, G.P., Schmelzle, T., Haghighat, A., Helliwell, S.B., Hall, M.N. & Sonenberg, N. (2000) Mol. Cell. Biol. 20, 4604–4613.
54. Polymenis, M. & Schmidt, E.V. (1997) Genes Dev. 11, 2522–2531.
55. Gallego, C., Gari, E., Colomina, N., Herrero, E. & Aldea, M. (1997) EMBO J. 16, 7196–7206.
56. Danaie, P., Altmann, M., Hall, M.N., Trachsel, H. & Helliwell, S.B. (1999) Biochem. J. 340, 135–141.
57. Gingras, A.-C., Raught, B. & Sonenberg, N. (1999) Annu. Rev. Biochem. 68, 913–963.
58. Raught, B., Gingras, A.-C. & Sonenberg, N. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 245–294.
59. Fumagalli, S. & Thomas, G. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 695–718.
60. Meyuhas, O. & Hornstein, E. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 671–694.
61. Meyuhas, O., Avni, D. & Shama, S. (1996) in Translational Control, eds. Hershey, J.W.B., Mathews, M. & Sonenberg, N. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 363–388.
62. Hershey, J.W.B. & Merrick, W.C. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 33–88.
63. Thomas, G. & Hall, M.N. (1997) Curr. Opin. Cell Biol. 9, 782–787.
64. Mahalingam, M. & Templeton, D. (1996) Mol. Cell. Biol. 16, 405–413.
65. Dennis, P.B., Pullen, N., Kozma, S.C. & Thomas, G. (1996) Mol. Cell. Biol. 16, 6242–6251.
66. Mathews, M.B., Sonenberg, N. & Hershey, J.W.B. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), Vol. 1, pp. 1–31.
67. Pause, A., Belsham, G.J., Gingras, A.-C., Donzé, O., Lin, T.A., Lawrence, J.C., Jr., & Sonenberg, N. (1994) Nature (London) 371, 762–767.
68. Lin, T.A., Kong, X., Haystead, T.A., Pause, A., Belsham, G., Sonenberg, N. & Lawrence, J. C., Jr. (1994) Science 266, 653–656.
69. Bernal, A. & Kimbrell, D.A. (2000) Proc. Natl. Acad. Sci. USA 97, 6019–6024. (First Published May 16, 2000; 10.1073/pnas.100391597)
70. Miron, M., Verdœ, J., Lachance, P.E.D., Birnbaum, M.J., Lasko, P.F. & Sonenberg, N. (2001) Nat. Cell Biol., in press.
71. Poulin, F., Gingras, A.-C., Olsen, H., Chevalier, S. & Sonenberg, N. (1998) J. Biol. Chem. 273, 14002–14007.
72. Mader, S., Lee, H., Pause, A. & Sonenberg, N. (1995) Mol. Cell. Biol. 15, 4990–4997.
73. Haghighat, A., Mader, S., Pause, A. & Sonenberg, N. (1995) EMBO J. 14, 5701–5709.
74. Marcotrigiano, J., Gingras, A.-C., Sonenberg, N. & Burley, S.K. (1999) Mol. Cell 3, 707–716.
75. Brunn, G.J., Hudson, C.C., Sekulic, A., Williams, J.M., Hosoi, H., Houghton, P.J., Lawrence, J.C, Jr., & Abraham, R.T. (1997) Science 277, 99–101.
76. Burnett, P.E., Barrow, R.K., Cohen, N.A., Snyder, S.H. & Sabatini, D.M. (1998) Proc. Natl. Acad. Sci. USA 95, 1432–1437.
77. Gingras, A.-C., Gygi, S.P., Raught, B., Polakiewicz, R.D., Abraham, R.T., Hoekstra, M.F., Aebersold, R. & Sonenberg, N. (1999) Genes Dev. 13, 1422–1437.
78. Hentges, K., Thompson, K. & Peterson, A. (1999) Development 126, 1601– 1609.
79. Heesom, K.J. & Denton, R.M. (1999) FEBS Lett. 457, 489–493.
80. Mothe-Satney, I., Yang, D., Fadden, P., Haystead, T.A. & Lawrence, J.C., Jr. (2000) Mol. Cell. Biol. 20, 3558–3567.
81. Yan, R., Rychlik, W., Etchison, D. & Rhoads, R.E. (1992) J. Biol. Chem. 267, 23226–23231.
82. Gradi, A., Imataka, H., Svitkin, Y.V., Rom, E., Raught, B., Morino, S. & Sonenberg, N. (1998) Mol. Cell. Biol. 18, 334–342.
83. Raught, B., Gingras, A.-C., Gygi, S.P., Imataka, H., Morino, S., Gradi, A., Aebersold, R. & Sonenberg, N. (2000) EMBO J. 19, 434–444.
84. Rozen, F., Edery, I., Meerovitch, K., Dever, T.E., Merrick, W.C. & Sonenberg, N. (1990) Mol. Cell. Biol. 10, 1134–1144.
85. Benne, R., Edman, J., Traut, R.R. & Hershey, J.W.B. (1978) Proc. Natl. Acad. Sci. USA 75, 108–112.
86. Duncan, R. & Hershey, J.W. (1985) J. Biol. Chem. 260, 5493–5497.
87. Morley, S.J. & Traugh, J.A. (1990) J. Biol. Chem. 265, 10611–10616.
88. Morley, S.J. & Traugh, J.A. (1989) J. Biol. Chem. 264, 2401–2404.
89. Proud, C.G. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 719–739.
90. Merrick, W.C. & Nyborg, J. (2000) in Translational Control of Gene Expression, eds. Sonenberg, N., Hershey, J.W.B. & Mathews, M.B. (Cold Spring Harbor Lab. Press, Plainview, NY), pp. 89–125.
91. Wang, X., Campbell, L.E., Miller, C.M. & Proud, C.G. (1998) Biochem. J. 334, 261–267.
92. Mackie, K.P., Nairn, A.C., Hampel, G., Lam, G. & Jaffe, E.A. (1989) J. Biol. Chem. 264, 1748–1753.
93. Marino, M.W., Dunbar, J.D., Wu, L.W., Ngaiza, J.R, Han, H.M., Guo, D., Matsushita, M., Nairn, A.C., Zhang, Y., Kolesnick, R., et al. (1996) J. Biol. Chem. 271, 28624–28629.
94. Nielsen, P.J. & McConkey, E.H. (1980) J. Cell. Physiol. 104, 269–281.
95. Chang, Y.W. & Traugh, J.A. (1997) J. Biol. Chem. 272, 28252–28257.
96. Redpath, N.T., Foulstone, E.J. & Proud, C.G. (1996) EMBO J. 15, 2291–2297.
97. Wang, L., Wang, X. & Proud, C.G. (2000) Am. J. Physiol. Heart Circ. Physiol. 278, H1056-H1068.
98. Warner, J.R. (1999) Trends Biochem. Sci. 24, 437–440.
99. Zaragoza, D., Ghavidel, A., Heitman, J. & Schultz, M.C. (1998) Mol. Cell. Biol. 18, 4463–4470.
100. Hardwick, J. S., Kuruvilla, F.G., Tong, J.K., Shamji, A.F. & Schreiber, S.L. (1999) Proc. Natl. Acad. Sci. USA 96, 14866–14870.
101. Cardenas, M.E., Cutler, N.S., Lorenz, M.C., Di Como, C.J. & Heitman, J. (1999) Genes Dev. 13, 3271–3279.
102. Leicht, M., Simm, A., Bertsch, G. & Hoppe, J. (1996) Cell Growth Differ. 7, 1199–1209.
103. Mahajan, P.B. (1994) Int. J. Immunopharmacol. 16, 711–721.
104. Shamji, A.F., Kuruvilla, F.G. & Schreiber, S.L. (2000) Curr. Biol. 10, 1574–1581.
105. Chan, T.-F., Carvalho, J., Riles, L. & Zheng, X.F.S. (2000) Proc. Natl. Acad. Sci. USA 97, 13227–13232. (First Published November 14, 2000; 10.1073/ pnas.240444197)
106. Beck, T., Schmidt, A. & Hall, M.N. (1999) J. Cell Biol. 146, 1227–1238.
107. Klionsky, D.J. & Emr, S.D. (2000) Science 290, 1717–1721.
108. Kim, J. & Klionsky, D.J. (2000) Annu. Rev. Biochem. 69, 303–342.
109. Klionsky, D.J. & Ohsumi, Y. (1999) Annu. Rev. Cell Dev. Biol. 15, 1–32.
110. Noda, T. & Ohsumi, Y. (1998) J. Biol. Chem. 273, 3963–3966.
111. Blommaart, E.F., Luiken, J.J., Blommaart, P.J., van Woerkom, G.M. & Meijer, A.J. (1995) J. Biol. Chem. 270, 2320–2326.
112. Shigemitsu, K., Tsujishita, Y., Kara, K., Nanahoshi, M., Avruch, J. & Yonezawa, K. (1999) J. Biol. Chem. 274, 1058–1065.
113. Khan, A., Pepio, A. & Sossin, W. (2001) J. Neurosci. 21, 382–391.
114. Pullarkat, S.R., Mysels, D.J., Tan, M. & Cowen, D.S. (1998) J. Neurochem. 71, 1059–1067.
115. Welsh, G.I., Hall, D.A., Warnes, A., Strange, P.G. & Proud, C.G. (1998) J. Neurochem. 70, 2139–2146.
116. Polakiewicz, R.D., Schieferl, S.M., Gingras, A.C., Sonenberg, N. & Comb, M.J. (1998) J. Biol. Chem. 273, 23534–23541.
117. Sabatini, D.M., Barrow, R.K., Blackshaw, S., Burnett, P.E., Lai, M.M., Field, M.E., Bahr, B.A., Kirsch, J., Betz, H. & Snyder, S.H. (1999) Science 284, 1161–1164.
118. Scheper, G.C., Voorma, H.O. & Thomas, A.A. (1992) J. Biol. Chem. 267, 7269–7274.
119. Essrich, C., Lorez, M., Benson, J.A., Fritschy, J.M. & Luscher, B. (1998) Nat. Neurosci. 1, 563–571.
120. Kirsch, J., Wolters, I., Triller, A. & Betz, H. (1993) Nature (London) 366, 745–748.
121. Marin, P., Nastiuk, K.L., Daniel, N., Girault, J.A., Czernik, A.J., Glowinski, J., Nairn, A.C. & Premont, J. (1997) J. Neurosci. 17, 3445–3454.
122. Scheetz, A.J., Nairn, A.C. & Constantine-Paton, M. (1997) Proc. Natl. Acad. Sci. USA 94, 14770–14775.
123. Scheetz, A.J., Nairn, A.C. & Constantine-Paton, M. (2000) Nat. Neurosci. 3, 211–216.
124. Walden, W.E. & Thach, R.E. (1986) Biochemistry 25, 2033–2041.
125. Ray, A., Walden, W.E, Brendler, T., Zenger, V.E. & Thach, R.E. (1985) Biochemistry 24, 7525–7532.
126. Schwartz, L.M., Smith, S.W., Jones, M.E. & Osborne, B.A. (1993) Proc. Natl. Acad. Sci. USA 90, 980–984.
127. Paavola, L.G. (1978) J. Cell Biol. 79, 59–73.
128. Dudek, H., Datta, S.R., Franke, T.F., Birnbaum, M.J., Yao, R., Cooper, G.M., Segal, R.A., Kaplan, D.R. & Greenberg, M.E. (1997) Science 275, 661–665.
129. Xue, L., Fletcher, G.C. & Tolkovsky, A.M. (1999) Mol. Cell. Neurosci. 14, 180–198.
130. Isahara, K., Ohsawa, Y., Kanamori, S., Shibata, M., Waguri, S., Sato, N., Gotow, T., Watanabe, T., Momoi, T., Urase, K., Kominami, E. & Uchiyama, Y. (1999) Neuroscience 91, 233–249.
131. Lamborghini, J.E. (1987) J. Comp. Neurol. 264, 47–55.
132. Anglade, P., Vyas, S., Javoy-Agid, F., Herrero, M.T., Michel, P.P., Marquez, J., Mouatt-Prigent, A., Ruberg, M., Hirsch, E.C. & Agid, Y. (1997) Histol. Histopathol. 12, 25–31.








