
Kinesin molecular motors: Transport pathways, receptors, and human disease
Lawrence S.B.Goldstein*
Howard Hughes Medical Institute, Department of Cellular and Molecular Medicine, University of California at San Diego School of Medicine, 9500 Gilman Drive, La Jolla, CA 92093–0683
Kinesin molecular motor proteins are responsible for many of the major microtubule-dependent transport pathways in neuronal and non-neuronal cells. Elucidating the transport pathways mediated by kinesins, the identity of the cargoes moved, and the nature of the proteins that link kinesin motors to cargoes are areas of intense investigation. Kinesin-II recently was found to be required for transport in motile and nonmotile cilia and flagella where it is essential for proper left-right determination in mammalian development, sensory function in ciliated neurons, and opsin transport and viability in photoreceptors. Thus, these pathways and proteins may be prominent contributors to several human diseases including ciliary dyskinesias, situs inversus, and retinitis pigmentosa. Kinesin-I is needed to move many different types of cargoes in neuronal axons. Two candidates for receptor proteins that attach kinesin-I to vesicular cargoes were recently found. One candidate, Sunday driver, is proposed to both link kinesin-I to an unknown vesicular cargo and to bind and organize the mitogen-activated protein kinase components of a c-Jun N-terminal kinase signaling module. A second candidate, amyloid precursor protein, is proposed to link kinesin-I to a different, also unknown, class of axonal vesicles. The finding of a possible functional interaction between kinesin-I and amyloid precursor protein may implicate kinesin-I based transport in the development of Alzheimer’s disease.
The large size and extreme polarity of neurons presents these cells with an unusual and substantial transport challenge. Materials synthesized in the cell body must be transported down long axons to presynaptic sites of utilization. These distances can reach 1 m or more in the case of humans and larger animals, and axonal volumes can exceed the volume of the cell body by 1,000-fold or more. In addition, axons and dendrites can be highly branched, and in some cases have very small diameters, which can limit transport rate and volume. The polarity of neurons presents analogous problems. Structural and signaling components destined for the axon must somehow be sorted from components needed in dendrites; the transport system appears to play a critical role in these processes (1). The combination of the substantial pressure of distance and volume, coupled to the enormous branching and narrow caliber of many neuronal processes, suggests that the intracellular transport system could be the “Achilles heel” of these large, complex cells—easily disturbed by environmental insult, mutation, or other trauma to cause neurodegenerative disease. This possibility has been suggested repeatedly over the past decades, but without a great deal of supporting evidence (e.g., refs. 2 and 3). This article revisits these themes and discusses data that suggest a possible interplay of kinesin molecular motor-based neuronal transport pathways and human disease.
Lessons from Green Algae: Possible Links of Intraflagellar Transport to Human Disease
A non-neuronal transport system that has the potential to teach us a great deal about neuronal transport recently was discovered in the green alga, Chlamydomonas reinhardii (reviewed in ref. 4). These small, free-living, unicellular organisms have long flagella that are used to swim. Flagellar assembly appears to occur at the site most distant from the cell body, and there is strong evidence that a kinesin-based transport pathway is responsible for moving key membrane and flagellar components from sites of synthesis in the cell body to sites of assembly. This system uses an evolutionarily conserved kinesin called kinesin-II to power the movement of proteinaceous “rafts.” These rafts are closely apposed to the flagellar plasma membrane as they move along the outer surface of flagellar microtubules. Kinesin-II is composed of two related motor polypeptides, KIF3A and KIF3B in mammals, coupled to a nonmotor kinase-associated protein subunit (reviewed in ref. 5). Several raft complex proteins also have been identified and found to be highly conserved from algae to mammals (6, 7). Mutations in the gene encoding the KIF3A or KIF3B subunits in mice cause an embryonic lethal phenotype (8–10). Strikingly, the cilia normally present on cells of the embryonic node fail to form in these mutants, confirming the broad evolutionary requirement for a kinesin-II based transport pathway for flagellar assembly. In addition to missing nodal cilia, embryos lacking KIF3A and KIF3B exhibit defective left-right body axis determination, providing strong experimental support for the long-standing hypothesis that cilia are crucial to left-right body axis determination in mammals. Similarly, mouse mutants lacking a homologue of a raft complex protein also fail to form embryonic nodal cilia and have defective left-right body axis determination (7, 11). It is noteworthy that a complex of heterogeneous human diseases called Kartagener’s triad or primary ciliary dyskinesia have been known for some time and appear to result from defects in bronchial cilia and sperm flagella. Thus, these diseases generally present with male infertility (sperm motility defects), bronchial abnormalities (bronchial ciliary defects), and situs inversus (defects in left-right body axis determination, causes previously unknown). These syndromes were previously suggested, with little supporting evidence, to alter embryonic cilia in human embryos (12). An intriguing possibility is that the components of the flagellar transport pathways may identify susceptibility loci for this class of human diseases.
In addition to typical, usually motile, cilia, eukaryotes have an array of cells that bear modified nonmotile cilia, often to serve sensory functions. Among these are so-called primary cilia whose functions are unknown (reviewed in ref. 13). Recently, a mouse homologue of a raft complex protein surfaced as a gene that when mutant causes polycystic kidney disease and leads to
|
* |
E-mail: lgoldstein@ucsd.edu. |
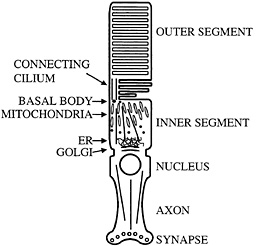
Fig. 1. Schematic diagram of mammalian photoreceptor. Microtubule organization and location of major cellular organelles are shown. In the inner segment, microtubules have their minus ends located near the basal bodies; connecting cilium microtubules have their minus ends at the basal body as well. ER, endoplasmic reticulum.
shorter primary cilia in the kidney (7, 11). It was suggested that these cilia function in the kidney to sense ionic concentrations, disturbance of which leads to disease. Similar mutants lacking kinesin-II motor or raft complex homologues in Caenorhabditis elegans disturb the structure and function of nonmotile chemosensory cilia (6).
Perhaps the most distinctive use of nonmotile cilia is presented by the vertebrate photoreceptor. This neuronal cell has an axon, but in place of a typical dendritic arbor it has a cellular compartment called the inner segment in which most biosynthesis takes place. Components such as opsin that are needed to sense light then are transported to sites of utilization in the disks of the outer segment (Fig. 1). Transport appears to occur through a narrow isthmus or connecting cilium, which is structurally a typical nonmotile cilium. A substantial amount of material must be moved through the connecting cilium because the photoreceptor turns over ca. 10% of its mass daily. Thus, it is perhaps not surprising that kinesin-II has been reported by a number of groups to be localized in the connecting cilium of the photoreceptor (14–16). These observations suggested that the transport system found in more typical cilia and flagella might be harnessed to move opsin, and perhaps other photoreceptor components, from the inner segment to the outer segment through the connecting cilium. Recently, specific removal of kinesin-II from photoreceptors using the lox-cre system was found to cause a substantial accumulation of opsin and arrestin in the inner segment accompanied by apoptosis. It was suggested that this phenotype was caused by a defect in transport of opsin and arrestin from the inner segment to the outer segment (17). Similar phenotypes have been seen in a particular class of opsin mutants that cause retinitis pigmentosa in humans. These mutants have been suggested to interfere with opsin transport and cause opsin accumulation in the inner segment and apoptosis (18–20). The region of opsin to which these mutants map also appears to interact with the dynein molecular motor (21), further suggesting a role for transport dysfunction in the development of degenerative retinal diseases such as retinitis pigmentosa. As with primary ciliary dyskinesia, it is tempting to speculate that
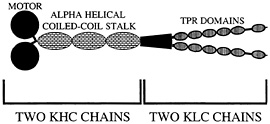
Fig. 2. Organization of kinesin-I. Two heavy chain components (KHC) and two light chain components (KLC) form the native heterotetramer. Proposed TPR domains are thought to mediate cargo binding via protein-protein interactions.
the collection of genes encoding the components required for transport from the inner segment to the outer segment, and in particular for opsin transport, may represent susceptibility loci for retinitis pigmentosa and other diseases where photoreceptor degeneration is a central feature. Indeed, it is intriguing that myosin VIIA, which when mutant can cause retinitis pigmentosa in humans (but curiously not mice), has been suggested to play a minor role in opsin transport and to be localized in the connecting cilium of the photoreceptor in addition to the retinal pigment epithelium (22, 23).
Finally, in thinking about neuronal transport pathways, it is striking that kinesin-II has been found in many typical neurons that lack cilia (24–27). In Drosophila, mutants lacking a kinesin-II subunit exhibit defects in axonal transport of choline acetyltransferase, a possibly cytosolic enzyme (28). In mammals, antibody inhibition, two-hybrid and biochemical experiments suggest a direct functional linkage between kinesin-II and non-erythroid spectrin (fodrin) in neurons (29). Perhaps nonciliated neurons also use a raft-based kinesin-II transport system to move cytoplasmic proteins in association with membrane-associated rafts or vesicles. An intriguing possibility is that kinesin-II and associated raft complexes might play an important role in the movement of cytosolic proteins by the slow axonal transport system. Further experimental work is needed to test this idea.
Lessons from Fruit Flies: Anterograde Axonal Transport and Mitogen-Activated Protein Kinase Signaling
Conventional kinesin, kinesin-I, was first discovered in a squid fast axoplasmic transport system, prompting early suggestions that kinesin-I would be an important motor protein to power fast anterograde axonal transport. This suggestion has been amply supported by a large number of antibody, antisense, and genetic experiments that support a general role of kinesin-I in axonal transport, but have not clearly linked this motor protein to a particular type of vesicular cargo (reviewed in ref. 30). It is thus not surprising that a “receptor” that mediates the attachment of kinesin-I to vesicular cargoes and other organelles has been elusive. In addition, whether it is the kinesin heavy chain (KHC) or the kinesin light chain (KLC) subunit of kinesin-I (Fig. 2) that binds to cargo has been unclear. Although a protein called kinectin has been suggested to play a role in linking kinesin-I to vesicles in non-neuronal cells (31, 32), its apparent absence in mammalian axons, Drosophila, and Caenorhabditis (33–35) has motivated additional searches for kinesin-I cargo receptors. Two serious candidates recently have emerged. One called Sunday driver was found in a genetic screen for axonal transport mutants in Drosophila (36). The other called amyloid precursor protein (APP) was identified initially in biochemical experiments (37).
The genetic screen that identified syd was based on work in Drosophila that revealed a constellation of phenotypes common to mutants defective in components of the anterograde or
retrograde axonal transport systems (3, 38, 39). This phenotype includes a relatively late larval lethality coupled to asymmetric paralysis of the animal. This paralysis manifests as either an upward tail “flip” during larval crawling or frank posterior paralysis of the motile larva. The underlying cellular phenotype is an accumulation of vesicles and organelles in apparent “traffic jams” or “clogs” in long narrow caliber axons. The first such example was presented by mutants lacking KHC, followed by mutants lacking KLC, dynein, and dynactin components, all components of the motor proteins themselves. The first nonmotor protein subunit found to cause this phenotype when missing is encoded by the Sunday driver gene, syd, which was found in a screen for mutants with the axonal transport phenotypic constellation (36).
The syd gene was found to encode an evolutionarily highly conserved protein predicted to be a type II transmembrane protein. Similar proteins are found in Caenorhabditis and mammals, which have two related genes encoding syd homologues. Because antibodies specific for syd are thus far of poor quality, localization of syd could not be accomplished, but transaction experiments with green fluorescent protein-tagged mouse syd in cultured mammalian COS cells revealed that syd could target to tubulovesicular organelles and small vesicles. These structures costain with antibodies recognizing both a marker of the secretory pathway and KLC, but not with probes for mitochondria or the endoplasmic reticulum-Golgi intermediate compartment. Two-hybrid coimmunoprecipitation and direct binding analyses demonstrated that the syd protein can bind directly to the KLC subunit of kinesin-I. Strikingly the interaction appeared to be with the predicted TPR repeat domains of KLC, which have previously been implicated in kinesin-I attachment to vesicular cargoes in axons (40). The combination of the axonal transport defective phenotype of syd mutants, the tubulovesicular localization of the protein in transfected cells, and direct binding of syd to KLC lead to the proposal that syd has a function as a kinesin-I receptor for at least one class of vesicles transported in the axon. In a surprising development, it turns out that mammalian syd is identical to a previously discovered gene called JIP3 or JSAP1, which was found to encode a protein having a protein kinase scaffold function that can bind and organize the mitogenactivated protein kinase components of a c-Jun N-terminal kinase signaling module (41, 42). Although not previously recognized as a membrane-associated protein the data suggest some role of syd (JIP3/JSAP1) in signaling networks in addition to kinesin-I attachment. Although a simple possibility is that a signaling protein has a dual function as a kinesin-I motor receptor, it is also possible that syd serves to integrate mitogenactivated protein kinase signaling with the regulation of some kinesin-I transport pathways.
Lessons from Humans: From Alzheimer’s Disease to Kinesin-I Receptors
APP was identified because of its possible role in the initiation or progression of Alzheimer’s disease (reviewed in refs. 43 and 44). APP is a type I transmembrane protein whose normal cellular function is poorly understood. Null mutants in both Drosophila and mice are viable and have relatively minor neuronal phenotypes (45, 46). However, proteolytic fragments of the APP are an abundant component of the plaques found throughout the brains of people afflicted with Alzheimer’s disease. Missense mutants in the gene encoding APP cause some forms of familial Alzheimer’s disease whereas mutants in presenilin genes cause others. Both types of mutants appear to increase the number of plaques and the abundance of toxic proteolytic fragments of APP. The presenilin genes may encode one of the key proteases, thus accounting for their role in disease. There also have been suggestions that axonal transport dysfunction or aberrant trafficking of APP might be an important element in causing disease.
The possibility that APP might have a kinesin-I receptor function was suggested initially by coimmunoprecipitation studies (37). Subsequent analyses of velocity gradient sedimentation, microtubule-binding, and direct binding analyses with expressed proteins confirmed this interaction and revealed that like syd, APP binds directly and tightly to the tetratrico peptide repeat region of KLC (37). In addition, although previous antisense experiments revealed that kinesin-I was needed for APP transport in neurons, whether this was direct or indirect, or a reflection of axonal versus preaxonal events was unclear (47–49). Analysis of mice lacking the neuron-enriched form of KLC, KLC1, revealed that APP transport in sciatic nerve axons strongly depended on KLC1, showing dramatic reduction in its absence. Thus, based on the tight binding of APP to KLC, and the strong dependence of APP axonal transport on KLC1, it was proposed that APP has a function as a kinesin-I receptor for a class of vesicular cargoes in the axon, perhaps distinct from vesicles whose transport is mediated by syd.
Kinesin Receptors and Kinesin Regulation
Taken together, these data on potential kinesin receptors suggest that proteins that have other roles in the cell may function to attach kinesin-I to cellular vesicles. This suggestion fits nicely with recent findings that other previously recognized proteins with nontransport functions may have dual roles as receptors and adaptors for motor proteins (reviewed in ref. 50). In fact, an intriguing possibility is that there are many cellular proteins that interact directly with the transport machinery to mediate movement. This view is an interesting alternative to the possibility that there are only a few proteins that interact directly with motor proteins, and that many proteins depend on these few “motor receptor” proteins for their transport. Further work is needed to evaluate these ideas.
What then are the relative roles of KLC and the KHC tail in binding cargo and regulating motor activity? Formulation of a compelling model is complicated by the apparent contradictions in the experimental literature to date (reviewed in ref. 37). In brief, the tail domain of KHC has been reported both to repress the KHC motor activity and to bind membranes and perhaps cargoes in the absence of KLC. Some fungi also appear to have KHC but not KLC. Yet, mutants that lack KLC in flies and mice have significant phenotypes, and antibodies that bind KLC can block membrane binding of kinesin-I. KLC also has been suggested to have a function as either an activator or represser of KHC motor activity (51, 52). Although it is possible that one or more of these observations is incorrect, a model that accounts for most of the data has been proposed (37). In this model, both KHC and KLC are suggested to repress the KHC motor activity and both KHC and KLC have membrane binding activity. Binding to both is suggested to derepress the motor and initiate transport. Thus, in the absence of KLC in organisms that ordinarily have it, KHC cannot initiate transport of classes of cargo that require KLC for attachment and derepression. Organisms that ordinarily lack KLC naturally may rely solely on the KHC tail for membrane binding and repression.
A Speculative Proposal for the Relationship of Axonal Transport to the Initiation of Alzheimer’s Disease
At present, most workers accept the hypothesis that inappropriate proteolytic processing of APP to generate aggregates of Aß is an important early event in the pathogenesis of Alzheimer’s disease. Sorely lacking, however, is an understanding of whether inappropriate processing of APP is the initiating event in disease, and if so, why it occurs. Additional important holes in our understanding of disease include knowledge of where in the neuron this inappropriate processing takes place, i.e., in the
axonal, dendritic, synaptic, or cell body compartment, and why Aß is neurotoxic.
Several groups have suggested that axonal transport defects may occur later in the pathogenesis of the disease (e.g., ref. 53), but have left unanswered the question of whether it might be an initiating event as well. Could the transport function of APP be related to the initiation or progression of Alzheimer’s disease? Several observations suggest that the answer to this question could be yes. First, as just discussed, normal transport of the APP protein in mammalian axons appears to depend on a direct interaction with the KLC subunit of the kinesin-I molecular motor protein (37). Thus, the disease causing protein may have a kinesin-I receptor function and thus be in close apposition to the transport machinery. Second, overexpression of the Drosophila homologue of APP called APPL in Drosophila (54) causes axonal clogs analogous to those found in syd and many other mutants with defective axonal transport. Perhaps overexpression of a protein such as APP with a motor receptor function either titrates out needed kinesin motor function in the axon or unbalances traffic in these narrow caliber axons leading to transport dysfunction, clogging, and other abnormalities. Third, humans bearing trisomy 21 suffer from premature onset of the symptoms characteristic of Alzheimer’s disease, perhaps because of overproduction of Aß (55). Although other genes are clearly present in excess in trisomy 21, it is striking that the gene encoding APP is located on chromosome 21 and thus is certainly one of the genes overexpressed in these people. Experiments in mouse models that overexpress APP have given equivocal results, but in some cases similar phenotypes have been reported (reviewed in ref. 56). Fourth, one of the early phenotypes of mouse models of Alzheimer’s disease, and perhaps human Alzheimer’s disease, is “dystrophic neurites,” whose morphology includes organelle and vesicle accumulations in axons (e.g., ref. 57). This phenotype is strikingly reminiscent of the axonal clogging observed after disturbance of axonal transport in Drosophila. Fifth, while it is unclear where in neurons Aß production is prominent, it is striking that even though APP is widely expressed, Alzheimer’s disease is primarily a neuronal disease. One feature that sets neurons apart from other cells is long axonal and dendritic processes. That a critical function of these processes is movement and transport of vesicular cargoes may be important to the development of disease.
How could alterations in axonal transport of APP lead to the generation of excess Aß and Alzheimer’s disease? Perhaps enhanced proteolysis of APP caused by axonal damage, presenilin or APP mutations, or elevated APP levels, cause impairments of APP transport efficiency in axons and increase the time spent by APP and proteases in a common axonal vesicular transport compartment. Time-dependent or damage-induced generation and accumulation of Aß by proteolysis of APP in this compartment might lead to aggregates of Aß that impair or block axonal transport and further stimulate Aß production in an autocatalytic spiral. Such a process could lead to neuronal dysfunction and progressive, age-related neurodegeneration and disease. In fact, although not measured directly by any researcher, it is possible that the populations of neurons affected first in Alzheimer’s disease could be those that combine the narrowest caliber with a higher than usual transport burden. Such features might predispose these axons to aggregation, or reduction in velocity, of their axonal transport cargoes, analogous to what has been observed in genetic models of axonal transport disturbance in Drosophila. Ultimately, neurotrophic signaling in neurons could be blocked by formation of axonal clogs, leading to apoptotic neuronal cell death. This proposal also may explain why some people appear to be more susceptible to Alzheimer’s disease than others. Perhaps the degree of axonal branching and caliber and perhaps allelic state at crucial molecular motor subunit genes will be found to be important once explored. If correct, this view also can account for the observation that it generally takes decades for Alzheimer’s disease to develop. Slight decrements in transport rate or efficiency could lead to slightly enhanced proteolysis rates that will in turn eventually lead to Alzheimer’s disease. Clearly, further work to test these ideas is needed.
Concluding Remarks
We may be at the beginning of an era in which neuronal transport is recognized as a major cellular target for the development of neurodegenerative disease. Although ciliary dyskinesias, retinitis pigmentosa, and Alzheimer’s disease are the major examples discussed above, there are also suggestions that amyotrophic lateral sclerosis may be caused or complicated by transport defects in motor neurons (58, 59). Similarly, it may be more than a coincidence that huntingtin, tau, and ApoE4, all of which are implicated in causation or susceptibility to neurodegenerative disease, all have been suggested to modulate transport when experimentally manipulated or to interact directly with the transport machinery (60–65). Finally, it is possible that for those neurodegenerative diseases in which formation of aggregates is an important feature, inhibition of axonal transport by the aggregates may be an important element in disease progression. In this regard, a recent report that axonal blockages and possible impairment of axonal transport may be present in CreutzfeldtJacob disease is intriguing (66).
I am an Investigator of the Howard Hughes Medical Institute. Some of the work discussed in this article was supported by National Institutes of Health Grant GM35252.
1. Burack, M.A., Silverman, M.A. & Banker, G. (2000) Neuron 26, 465–472.
2. Griffin, J.W. & Watson, D.F. (1988) Ann. Neurol. 23, 3–13.
3. Hurd, D.D. & Saxton, W.M. (1996) Genetics 144, 1075–1085.
4. Rosenbaum, J.L., Cole, D.G. & Diener, D.R. (1999) J. Cell Biol. 144, 385–388.
5. Marszalek, J.R. & Goldstein, L.S. (2000) Biochim. Biophys. Acta 1496, 142–150.
6. Cole, D.G., Diener, D.R., Himelblau, A.L., Beech, P.L., Fuster, J.C. & Rosenbaum, J.L. (1998) J. Cell Biol. 141, 993–1008.
7. Pazour, G.J., Dickert, B.L., Vucica, Y., Seeley, E.S., Rosenbaum, J.L., Witman, G.B. & Cole, D.G. (2000) J. Cell Biol. 151, 709–718.
8. Nonaka, S., Tanaka, Y., Okada, Y., Takeda, S., Harada, A., Kanai, Y., Kido, M. & Hirokawa, N. (1998) Cell 95, 829–837.
9. Marszalek, J.R., Ruiz-Lozano, P., Roberts, E., Chien, K.R. & Goldstein, L.S. (1999) Proc. Natl. Acad. Sci. USA 96, 5043–5048.
10. Takeda, S., Yonekawa, Y., Tanaka, Y., Okada, Y., Nonaka, S. & Hirokawa, N. (1999) J.Cell Biol. 145, 825–836.
11. Murcia, N.S., Richards, W.G., Yoder, B.K., Mucenski, M.L., Dunlap, J.R. & Woychik, R.P. (2000) Development (Cambridge, U.K.) 127, 2347–2355.
12. Afzelius, B.A. (1985) CRC Crit. Rev. Biochem. 19, 63–87.
13. Wheatley, D.N., Wang, A.M. & Strugnell, G.E. (1996) Cell Biol. Int. 20, 73–81.
14. Beech, P.L., Pagh-Roehl, K., Noda, Y., Hirokawa, N., Burnside, B. & Rosenbaum, J.L. (1996) J. Cell Sci. 109, 889–897.
15. Muresan, V., Bendala-Tufanisco, E., Hollander, B.A. & Besharse, J.C. (1997) Exp. Eye Res. 64, 895–903.
16. Muresan, V., Lyass, A. & Schnapp, B.J. (1999) J. Neurosci. 19, 1027–1037.
17. Marszalek, J.R., Liu, X., Roberts, E.A., Chui, D., Marth, J.D., Williams, D.S. & Goldstein, L.S. (2000) Cell 102, 175–187.
18. Sung, C.H., Makino, C., Baylor, D. & Nathans, J. (1994) J. Neurosci. 14, 5818–5833.
19. Portera-Cailliau, C., Sung, C.H., Nathans, J. & Adler, R. (1994) Proc. Natl. Acad. Sci. USA 91, 974–978.
20. Sung, C.H., Davenport, C.M. & Nathans, J. (1993) J. Biol. Chem. 268, 26645–26649.
21. Tai, A.W., Chuang, J.Z., Bode, C., Wolfrum, U. & Sung, C.H. (1999) Cell 97, 877–887.
22. Liu, X., Vansant, G., Udovichenko, I.P., Wolfrum, U. & Williams, D.S. (1997) Cell Motil. Cytoskeleton 37, 240–252.
23. Liu, X., Udovichenko, I.P., Brown, S.D., Steel, K.P. & Williams, D.S. (1999) J. Neurosci. 19, 6267–6274.
24. Kondo, S., Sato-Yoshitake, R., Noda, Y., Aizawa, H., Nakata, T., Matsuura, Y. & Hirokawa, N. (1994) J. Cell Biol. 125, 1095–1107.
25. Muresan, V., Abramson, T., Lyass, A., Winter, D., Porro, E., Hong, F., Chamberlin, N.L. & Schnapp, B.J. (1998) Mol. Biol. Cell 9, 637–652.
26. Yamazaki, H., Nakata, T., Okada, Y. & Hirokawa, N. (1995) J. Cell Biol. 130, 1387–1399.
27. Yang, Z. & Goldstein, L.S. (1998) Mol. Biol. Cell 9, 249–261.
28. Ray, K., Perez, S.E., Yang, Z., Xu, J., Ritchings, B.W., Steller, H. & Goldstein, L.S. (1999) J. Cell Biol. 147, 507–518.
29. Takeda, S., Yamazaki, H., Seog, D.H., Kanai, Y., Terada, S. & Hirokawa, N. (2000) J. Cell Biol. 148, 1255–1265.
30. Goldstein, L.S. & Philp, A.V. (1999) Annu. Rev. Cell Dev. Biol. 15, 141–183.
31. Yu, H., Nicchitta, C.V., Kumar, J., Becker, M., Toyoshima, I. & Sheetz, M.P. (1995) Mol. Biol. Cell 6, 171–183.
32. Toyoshima, I., Yu, H., Steuer, E.R. & Sheetz, M.P. (1992) J. Cell Biol. 118, 1121–1131.
33. Toyoshima, I. & Sheetz, M.P. (1996) Neurosci. Lett. 211, 171–174.
34. Goldstein, L.S. & Gunawardena, S. (2000) J.Cell Biol. 150, F63-F68.
35. Rubin, G.M., Yandell, M.D., Wortman, J.R., Gabor Miklos, G. L, Nelson, C.R., Hariharan, I.K., Fortini, M.E., Li, P.W., Apweiler, R., Fleischmann, W., et al. (2000) Science 287, 2204–2215.
36. Bowman, A.B., Kamal, A., Ritchings, B.W., Philp, A.V., McGrail, M., Gindhart, J.G. & Goldstein, L.S. (2000) Cell 103, 583–594.
37. Kamal, A., Stokin, G.B., Yang, Z., Xia, C. & Goldstein, L.S. (2000) Neuron 28, 449–459.
38. Bowman, A.B., Patel-King, R.S., Benashski, S.E., McCaffery, J.M., Goldstein, L.S. & King, S.M. (1999) J. Cell Biol. 146, 165–180.
39. Martin, M., Iyadurai, S.J., Gassman, A., Gindhart, J.G., Jr., Hays, T.S. & Saxton, W.M. (1999) Mol. Biol. Cell 10, 3717–3728.
40. Stenoien, D.L. & Brady, S.T. (1997) Mol. Biol. Cell 8, 675–689.
41. Kelkar, N., Gupta, S., Dickens, M. & Davis, R.J. (2000) Mol. Cell. Biol. 20, 1030–1043.
42. Ito, M., Yoshioka, K., Akechi, M., Yamashita, S., Takamatsu, N., Sugiyama, K., Hibi, M., Nakabeppu, Y., Shiba, T. & Yamamoto, K.I. (1999) Mol. Cell. Biol. 19, 7539–7548.
43. De Strooper, B. & Annaert, W. (2000) J. Cell Sci. 113, 1857–1870.
44. Price, D.L. & Sisodia, S.S. (1998) Annu. Rev. Neurosci. 21, 479–505.
45. Zheng, H., Jiang, M., Trumbauer, M.E., Hopkins, R., Sirinathsinghji, D.J., Stevens, K.A., Conner, M.W., Slunt, H.H., Sisodia, S.S., Chen, H.Y. & Van der Ploeg, L.H. (1996) Ann. N.Y. Acad. Sci. 777, 421–426.
46. Luo, L., Tully, T. & White, K. (1992) Neuron 9, 595–605.
47. Feiguin, F., Ferreira, A., Kosik, K.S. & Caceres, A. (1994) J. Cell Biol. 127, 1021–1039.
48. Ferreira, A., Caceres, A. & Kosik, K.S. (1993) J. Neurosci. 13, 3112–3123.
49. Ferreira, A., Niclas, J., Vale, R.D, Banker, G. & Kosik, K.S. (1992) J. Cell Biol. 117, 595–606.
50. Klopfenstein, D.R., Vale, R.D. & Rogers, S.L. (2000) Cell 103, 537–540.
51. Rahman, A., Kamal, A., Roberts, E.A. & Goldstein, L.S. (1999) J. Cell Biol. 146, 1277–1288.
52. Verhey, K.J., Lizotte, D.L., Abramson, T., Barenboim, L., Schnapp, B.J. & Rapoport, T.A. (1998) J. Cell Biol. 143, 1053–1066.
53. Kasa, P., Papp, H., Kovacs, I., Forgon, M., Penke, B. & Yamaguchi, H. (2000) Neurosci. Lett. 278, 117–119.
54. Torroja, L., Chu, H., Kotovsky, I. & White, K. (1999) Curr. Biol. 9, 489–492.
55. Takashima, S. (1997) Curr. Opin. Neurol. 10, 148–152.
56. Price, D.L., Tanzi, R.E., Borchelt, D.R. & Sisodia, S.S. (1998) Annu. Rev. Genet. 32, 461–493.
57. Masliah, E., Sisk, A., Mallory, M., Mucke, L., Schenk, D. & Games, D. (1996) J. Neurosci. 16, 5795–5811.
58. Williamson, T.L. & Cleveland, D.W. (1999) Nat. Neurosci. 2, 50–56.
59. Xu, Z., Cork, L.C., Griffin, J.W. & Cleveland, D.W. (1993) Cell 73, 23–33.
60. Li, S.H., Gutekunst, C.A., Hersch, S.M. & Li, X.J. (1998) J. Neurosci. 18, 1261–1269.
61. Block-Galarza, J., Chase, K.O., Sapp, E., Vaughn, K.T., Vallee, R.B., DiFiglia, M. & Aronin, N. (1997) NeuroReport 8, 2247–2251.
62. Engelender, S., Sharp, A.H., Colomer, V., Tokito, M.K., Lanahan, A., Worley, P., Holzbaur, E.L. & Ross, C.A. (1997) Hum. Mol. Genet. 6, 2205–2212.
63. Ebneth, A., Godemann, R., Stamer, K., Illenberger, S., Trinczek, B. & Mandelkow, E. (1998) J. Cell Biol. 143, 777–794.
64. Tesseur, I., Van Dorpe, J., Bruynseels, K., Bronfman, F., Sciot, R., Van Lommel, A. & Van Leuven, F. (2000) Am.J.Pathol. 157, 1495–1510.
65. DiFiglia, M., Sapp, E., Chase, K.O., Davies, S.W., Bates, G.P., Vonsattel, J.P. & Aronin, N. (1997) Science 277, 1990–1993.
66. Liberski, P.P. & Budka, H. (1999) Acta Neuropathol. 97, 329–334.





