
Tracking the estrogen receptor in neurons: Implications for estrogen-induced synapse formation
Bruce McEwen*†, Keith Akama*, Stephen Alves*, Wayne G. Brake*, Karen Bulloch*, Susan Lee*, Chenjian Li*, Genevieve Yuen*, and Teresa A.Milner‡
*Laboratory of Neuroendocrinology, The Rockefeller University, New York, NY 10021; and ‡Department of Neurology and Neuroscience, Weill Medical College of Cornell University, New York, NY, 10021
Estrogens (E) and progestins regulate synaptogenesis in the CA1 region of the dorsal hippocampus during the estrous cycle of the female rat, and the functional consequences include changes in neurotransmission and memory. Synapse formation has been demonstrated by using the Golgi technique, dye filling of cells, electron microscopy, and radioimmunocytochemistry. N-methyl-D-aspartate (NMDA) receptor activation is required, and inhibitory interneurons play a pivotal role as they express nuclear estrogen receptor alpha (ERα) and show E-induced decreases of GABAergic activity. Although global decreases in inhibitory tone may be important, a more local role for E in CA1 neurons seems likely. The rat hippocampus expresses both ERα and ERß mRNA. At the light microscopic level, autoradiography shows cell nuclear [3H]estrogen and [125I]estrogen uptake according to a distribution that primarily reflects the localization of ERα-immunoreactive interneurons in the hippocampus. However, recent ultrastructural studies have revealed extranuclear ERα immunoreactivity (IR) within select dendritic spines on hippocampal principal cells, axon terminals, and glial processes, localizations that would not be detectable by using standard light microscopic methods. Based on recent studies showing that both types of ER are expressed in a form that activates second messenger systems, these findings support a testable model in which local, non-genomic regulation by estrogen participates along with genomic actions of estrogens in the regulation of synapse formation.
The brain is widely responsive to gonadal hormones. Not only is the hypothalamus regulated by these hormones in relation to reproductive behavior and neuroendocrine physiology, but also structures like the hippocampus and midbrain serotonin system undergo sexual differentiation during perinatal development and are hormone responsive in maturity (1, 2). One of the processes regulated by ovarian hormones is the cyclic formation and breakdown of excitatory synapses on dendritic spines in the hippocampus (3). This finding was surprising because, until recently, the hippocampus was known as a brain region in which cell nuclear estrogen receptors (ER) are present in scattered inhibitory interneurons but not in principal neurons where spine formation occurs (4). Yet the effects of ovarian hormones on synaptic turnover were as impressive in the hippocampus as those in the ventromedial hypothalamus (5–7), a classic estrogen (E) target area of the brain for female sexual behavior (8). Moreover, effects of estrogens on hippocampal-dependent cognitive function are now recognized in rodents (9) and humans (10).
Recent electron microscopic studies have revealed that ERs are expressed in hippocampus in non-nuclear locations within principal cells (11). This fact, along with the discovery that ER can couple to second messenger systems (12–14), has raised the possibility that ER may be involved in local signaling within neurons as well as regulating expression of genes via nuclear receptors in interneurons. Among the possible targets of local signaling is the translation of RNAs found in dendrites of hippocampal and other neurons. This review paper presents the state of current knowledge about the location of ER and progesterone receptors (PR) in hippocampus and the regulation of synapse formation by estradiol and removal by progesterone in CA1 pyramidal neurons. We start with a discussion of the functional significance of hippocampal synaptogenesis and then review what is known about the mechanism of synapse formation and the location of ER and their cellular mechanisms of action. The discovery of non-nuclear ER in dendritic spines, presynaptic nerve endings, and spine-associated glial cell processes has led us to propose a testable model for understanding the role of nuclear and non-nuclear ER in synapse formation.
Functional Significance of Actions of Estradiol in the Hippocampus
The functional significance of estrogen actions in the hippocampal CA1 region is evident from electrophysiological studies indicating that E treatment of ovariectomized rats produces a delayed facilitation of synaptic transmission in CA1 neurons that is N-methyl-D-aspartate (NMDA) mediated (15) and leads to an enhancement of voltage-gated Ca2+ currents (15, 16). This approach was significantly advanced by Woolley et al. (17), who used biocytin injection after recording from CA1 pyramidal neurons to visualize E induction of dendritic spines (17). Spine density correlated negatively with input resistance, and input/ output curves showed an increased slope under conditions where NMDA receptor-mediated currents predominated, whereas there was no increased slope where α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor currents predominated (17).
Other studies have shown that long-term potentiation sensitivity peaks on the afternoon of proestrus in intact female rats at exactly the time when excitatory synapse density has reached its peak (18). Proestrus is also the time of the estrous cycle when seizure thresholds in dorsal hippocampus are the lowest (19). Although activation of NMDA receptors in hippocampus is enhanced via AMPA receptors in some cases but not in others
|
† |
To whom reprint requests should be addressed. E-mail: mcewen@rockvax.rockefeller.edu. |
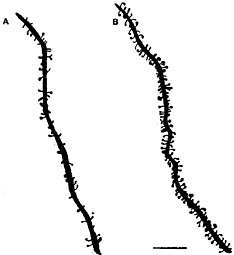
Fig. 1. Camera lucida drawings of apical dendrites of CA1 pyramidal neurons from ovariectomized rats either untreated (A) or treated (B) with estradiol and progesterone to induce spines. Scale bar = 10 µm. [Reproduced with permission from ref. 29 (Copyright 1990, Society for Neuroscience)].
(20), the involvement of AMPA receptors in response to ovarian steroid manipulations is not known. It remains to be determined whether the E-induced synapses are so-called “silent” synapses with only NMDA receptors (21) or ones that contain AMPA receptors as well. In contrast to the efficacy of NMDA receptor inhibition for synapse formation (see below), blockade of AMPA receptors with the antagonist NBQX during E treatment failed to block synaptogenesis (22).
Besides increasing NMDA currents, reducing seizure thresh-olds, and enhancing long-term potentiation in hippocampus, E treatment exerts effects on hippocampal-dependent learning and memory. Three types of effects have been reported. First, in the natural estrous cycle of the rat, a recent study has used a delayed matching-to-place task in female rats to show a close parallel between the temporal conditions by which E improves memory and the conditions for E to induce new excitatory synaptic connections in the hippocampus (9). Second, E treatment of ovariectomized rats has been reported to improve acquisition on a radial maze task as well as in a reinforced T-maze alternation task (23, 24). Third, sustained E treatment is reported to improve performance in a working memory task (25), as well as in the radial arm maze (24, 26). The effects of E replacement in rats are reminiscent of the effects of E treatment in women whose E levels have been suppressed by a gonadotrophin-releasing hormone agonist used to shrink the size of fibroids before surgery (10, 27).
Excitatory Synapse Formation in the Hippocampus
E treatment increases dendritic spine density on CA1 pyramidal neurons (Figs. 1 and 2). As observed by electron microscopy, E also induces new synapses on spines and not on dendritic shafts of CA1 neurons (28). There were no E effects on dendritic length or branching (3, 28, 29). Progesterone (P) treatment acutely enhances spine formation (Fig. 1). But, over a 12- to 24-h period, P caused the down-regulation of E-induced synapses (29, 30), as will be discussed further below.
Estrogens do not act alone, and, in fact, ongoing excitatory neurotransmission is required for synapse induction, as shown by the finding that antagonists of NMDA receptors block
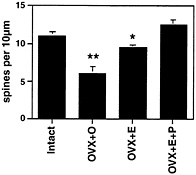
Fig. 2. Number of dendritic spines per 10 µm obtained from the apical portion of the CA1 pyramidal cell dendritic tree. Values are the mean ± SEM for estrogen and estrogen plus 5-h progesterone treatment. E induces increased spine density, an effect that is enhanced by 5-h progesterone. **, Different from other groups, P< 0.01; *, different from E+P group, P< 0.05. [Reproduced with permission from ref. 29 (Copyright 1990, Society for Neuroscience)].
E-induced synaptogenesis on dendritic spines in ovariectomized female rats (ref. 22 and Fig. 3). Because E treatment increases the density of NMDA receptors in the CA1 region of the hippocampus (17, 31), the activation of NMDA receptors by glutamate may lead the way in causing new excitatory synapses to develop.
Spines are occupied by asymmetric, excitatory synapses and are sites of Ca2+ ion accumulation and contain NMDA receptors (32). NMDA receptors are expressed in large amounts in CA1 pyramidal neurons and can be imaged by conventional immunocytochemistry as well as by confocal imaging, in which individual dendrites and spines can be studied for colocalization with other markers (33–35). Confocal microscopic imaging showed that E treatment up-regulates immunoreactivity for the largest NMDA receptor subunit, NR1, on dendrites and cell bodies of CA1 pyramidal neurons, whereas NR1 mRNA levels did not change after E treatment that induces new synapses (35), suggesting the possibility that NR1 expression is regulated posttranscriptionally by E (Fig. 4).
Nuclear Estrogen Receptors in the Hippocampus
Adult CA1 pyramidal cells appear to lack detectable nuclear ER as shown by tritium autoradiography (36) and light microscopic immunocytochemistry (4, 37), whereas they show low levels of ERa and -ß mRNA by in situ hybridization (38, 39). Autoradiographic mapping studies of [3H]estradiol uptake in hippocampus showed a sparse distribution of interneurons in the CA1 region, as well as other regions of Ammon’s horn that contain nuclear E binding sites (36). This observation was confirmed by immunocytochemistry for ERa in the guinea pig hippocampus (37) and subsequently in the rat hippocampus (ref. 4 and Fig. 5). These findings and those from cell culture studies described below led to a hypothesis regarding the role of interneurons as trans-synaptic regulators of synapse formation. Two other mechanisms will then be considered: (i) that E acts via a novel non-genomic mechanism; or (ii) that there are low levels of genomic ERs that are undetectable by conventional immunocytochemistry.
Cell Culture Model of Synapse Formation
Recent studies revealed that E induces spines on dendrites of dissociated hippocampal neurons in culture by a process that is blocked by an NMDA receptor antagonist and not by an AMPA/ kainate receptor blocker (40). In a subsequent study, E treatment was found to increase the phosphorylation of cAMP response
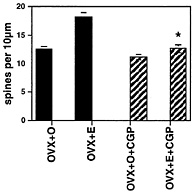
Fig. 3. Number of dendritic spines per 10 µm obtained from the apical portion of the CA1 pyramidal cell dendritic tree. Values are the mean ± SEM for treatment of ovariectomized rats with either vehicle or E in the presence or absence of the competitive NMDA receptor blocker, CGP 43 487. NMDA blockade prevents E induction of spines. *, Different from E alone, P < 0.01. [Reproduced with permission from ref. 22 (Copyright 1994, Society for Neuroscience)].
element-binding protein (CREB), and a specific antisense to CREB prevented both the formation of dendritic spines and the elevation in phosphoCREB immunoreactivity (IR; ref. 41).
In agreement with the in vivo data (4), ERα was located in the cultures on glutamic acid decarboxylase (GAD)-immunoreactive cells that constituted around 20% of total neurons; E treatment caused decreases in GAD content and the number of neurons expressing GAD. Mimicking this decrease with an inhibitor of γ-aminobutyric acid (GABA) synthesis, mercaptopropionic acid, caused an up-regulation of dendritic spine density, paralleling the effects of E (42).
An additional factor in the formation of dendritic spines in the cell culture model is the neurotrophin, brain-derived neurotrophic factor (BDNF; ref. 43). Besides down-regulating GABA in inhibitory interneurons, E treatment also reduced BDNF by 60% within 24 h (43). This neurotrophin appears to be a negative regulator of dendritic spines; exogenous BDNF blocks E induction of dendritic spines whereas BDNF depletion mimicks E in inducing spine density (43). Interestingly, neurotrophins such as BDNF and neurotrophin-3 (NT-3) also increase the function of inhibitory and excitatory synapses in hippocampal cell cultures; moreover, BDNF causes an increase in axonal branching and length of GABAergic interneurons (44).
Non-Nuclear Estrogen Receptors
Besides exerting delayed and prolonged effects via nuclear receptors, estrogens can have rapid effects on hippocampal and other neurons, sometimes involving coupling to second messenger systems, such as the phosphorylation of CREB (12, 13, 45). Our recent findings have compelled us to consider such a mechanism in relation to hippocampal synapse formation. What is becoming evident is that, besides the indirect, transsynaptic mechanism described above, local signaling by E also may be involved. A seminal study using transfection of ERα and ERß into Chinese hamster ovarian cells (46) revealed that both ERs are expressed in a form that couples to second messenger systems that are stimulated by E and blocked at least partially by non-steroidal estrogen antagonists. Previous studies had indicated that non-nuclear ERs can be seen at the light microscopic level in cultured cells (47) and also at the electron microscopic level in hypothalamus (48).
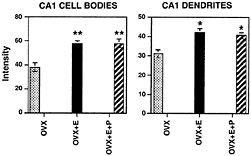
Fig. 4. Bar graphs depicting NMDA subunit R1 immunofluorescence intensity measurements in the somata (Left) and dendrites (Right) of the CA field of the hippocampus. For somatic intensity measurements (Left), there is a significant increase when comparing E and E+P with OVX control. **, P< 0.0001. In dendritic fields (Right), E and E+P treatments were increased compared with OVX control; *, P<0.05. [Reproduced with permission from ref. 35 (Copyright 1996, Society for Neuroscience)].
We used electron microscopy to examine ERα localization in rat hippocampal formation (11), with four antibodies to different parts of the ERα structure (2 polyclonal; 2 monoclonal). The specificity of these antibodies was determined by preabsorption with the full-length ER protein, which abolished labeling in all sites examined, both nuclear and non-nuclear. We confirmed at the EM level the cell nuclear labeling seen by light microscopy in some select GABA interneurons. We also found that some pyramidal and granule neuron perikarya have small amounts of ERα IR in the nuclear membrane, although not in the nucleus itself. This finding may help explain a recent report that [125I]estradiol labels the cell nuclei of hippocampal principal cells weakly in dorsal and more abundantly in ventral hippocampus (39).
We also identified extranuclear ERα-immunolabeling within axons and axon terminals associated with unlabeled dendritic spines, within dendritic spines and spine apparati of principal cells, as well as some select glial processes adjacent to spines (11). The most abundant labeling was seen in the CA1 stratum radiatum, where the E-mediated spine induction is most clearly evident. Around 50% of the ERα-IR profiles in stratum radiatum of CA1 were in unmyelinated axons and axon terminals containing small synaptic vesicles (Fig. 6 A), supporting findings that E can rapidly influence neurotransmitter release (49–51) or reuptake (52, 53); ERα-IR was found in synaptic terminals that formed both asymmetric and symmetric synapses on dendritic shafts and spines, suggesting that both excitatory and inhibitory transmitter systems express ERα (54).
Around 25% of the ERα IR was found in dendritic spines of principal cells. Within spines, ERα was often associated with spine apparati and/or postsynaptic densities, suggesting that E might act locally to regulate calcium availability, phosphorylation, and/or protein synthesis (ref. 55 and Fig. 6 B). The remaining 25% of ERα IR was found in astrocytic profiles, often located near the spines of principal cells (Fig. 6 C). Whereas these findings corroborate existing evidence for an indirect GABAergic mediation of E actions (56, 57), the close association between the ERα-IR and dendritic spines suggests a possible local, non-genomic role for this ER in regulation of dendritic spine density via second messenger systems.
Initial in vivo and in vitro studies in hippocampus involving the phosphorylation of the transcription regulator, CREB, have indicated that E has rapid effects that are evident within as little as 15′ to increase phosphorylated CREB immunoreactivity in cell nuclei of hippocampal pyramidal neurons (S.L., S.A., and

Fig. 5. By light microscopy, ERa immunoreactivity (IR) is found in scattered interneurons in the hippocampal formation. (A) Schematic diagram of regions examined by light and electron microscopy. (B) In CA1, a few interneurons with cell nuclear ERa are found primarily in stratum radiatum (sr) and occasionally in the pyramidal cell layer (pcl). (C) Scattered interneurons located within the infragranular regions of the hilus (hil) contain ERa IR associated with their cell nuclei. DG, dentate gyrus; gcl, granule cell layer; CA1, CA3 regions of Ammon’s horn; ml, molecular layer; so, stratum oriens; slm, stratum lacunosum moleculare. Scale bars=40 µm. [Reproduced with permission from ref. 11 (Copyright 2001, Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc)].
B.M., unpublished observations). One pathway by which CREB phosphorylation occurs involves the phosphoinositol-3 (PI3) kinase/Akt system (58). Cell culture studies indicate that E rapidly stimulates phosphorylation of Akt (58) in a pathway leading to CREB phosphorylation (59). PhosphoAkt-IR is evident in cell nuclei as well as dendrites of CA1 pyramidal neurons (S.L., T.A.M., K.A., and B.M., unpublished observations). Akt is known to affect phosphorylation events in the cytoplasm (60) as well as expression of phosphorylated CREB in cell nuclei (59). Studies are underway to try to connect these events together in the early actions of E on hippocampal neurons that precede the induction of synapse formation. We next consider some of the cellular and molecular events associated with the formation of synapses in which E actions may be involved.
Cellular and Molecular Events Associated with Synapse Formation
The E-induced increase in dendritic spines on CA1 neurons parallels an increase in synapse density on spines without any decrease in shaft synapses (28), implying that new spine synapses are formed. Whether this event occurs by a division process or by de novo growth of new spines, new protein components are likely to be formed. We, therefore, discuss the current status of mechanisms of synaptogenesis and the role of protein synthesis. Synapse formation on dendritic spines is a collaborative process involving in-growth of a presynaptic element on a site where a postsynaptic spine is either present or ready to form (32, 61, 62). Because the direct observations of synapse formation are done on neurons in cell culture, one must extrapolate to the situation in the adult hippocampus where new synapses are formed under the control of estrogens. In cultured cells studied by time-lapse photography, filopodia extend from dendrites reaching out to establish contact with nearby axons (63, 64), implying an active role for the dendrite in forming synaptic contacts. When synaptic contacts form, excitatory and inhibitory neurotransmitter receptors move to form clusters opposite to synaptic terminals (65) but only after the initial events of contact and differentiation have taken place (61, 66). Division of dendritic spines has been a postulated mechanism for spine formation, and actin filaments may assist in the division process (32, 67). Vacant spines are not seen in vivo, and spine-like processes in cells in culture are much longer than normal spines when they are unoccupied by synapses (32, 63, 68). Dendritic spine synapses are overwhelmingly of the Gray type 1, or asymmetric type, and therefore excitatory (32). The following discussion concerns the time course, sequence of steps, and key gene products and events in synapse formation.
Sequence and Time Course of Steps in Synapse Formation. In cell culture, individual synapses are reported to form within 1–2 h (61, 62). The cadherin/catenin and CNR (cadherin-like neuronal receptors) systems are postulated to play a role in the recognition between presynaptic growth cones and dendritic filopodia (66). After the initial contact is established, recruit-
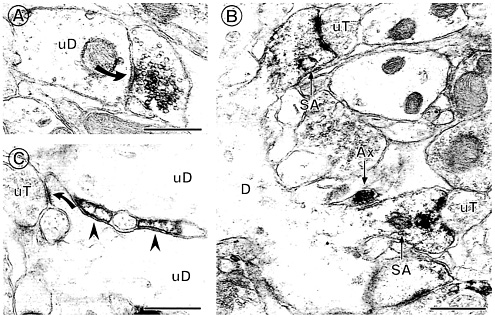
Fig. 6. ERa IR is found in several types of extranuclear sites within the hippocampal formation. (A) A terminal with ERa IR forms a symmetric synapse (solid curved arrow) with an unlabeled dendrite (uD). (B) ERa IR is found in two dendritic spines identifiable by the presence of spine apparati (SA), which arise from the same dendrite (D). Both labeled spines are contacted by unlabeled terminals (uT). An ERa-labeled axon (Ax) is found nearby. (C) ERa-labeled astrocytic profiles (arrowheads) are found in between two unlabeled dendrites (uD) near a region where an unlabeled terminal (uT) contacts a dendritic spine (solid curved arrow). Bars=0.5 µM. [Reproduced with permission from ref. 11 (Copyright 2001, Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc)].
ment of pre- and postsynaptic proteins leads to the formation of a synapse at the site of initial contact (66). The immediate early genes, Narp (69, 70), Arc (71), and synaptotagmin IV (72) are activated by synaptic firing and are candidates for the recruitment and localization of protein components of the synapse. The neuroligin/neurexin system is believed to play an important role in the recruitment and localization of pre- and postsynaptic components of the forming synapse (66). Neuroligin-1 and -2 can induce presynaptic differentiation in contacting axons, suggesting that the postsynaptic cell has a strong influence on presynaptic differentiation (73). The effects of E treatment on these gene products remain to be determined.
Presynaptic Markers of Synapses. There are a number of presynaptic molecular markers of synapse formation. Growth-associated protein-43 (GAP-43) is a marker of the growth cone and has been shown to increase in the hypothalamus after E treatment (74); however, no studies of this type have been done on the hippocampus. Synaptosomal-associated protein-25 (SNAP-25) is a marker of presynaptic terminals (75), as are syntaxin (76), synaptotagmins (77, 78), synaptoporin (79, 80), synaptophysin (81), and the synapsins (82–84). Although mRNAs for these proteins are most likely found in neuron cell bodies, growth cones of hippocampal neurons in culture have been reported to have mRNAs for proteins such as GAP-43 and Arc, and perhaps other presynaptic proteins; these mRNAs can be translated in the growth cone (85). Initial results using radioimmunocytochemistry indicate that E treatment increases expression of synaptophysin and syntaxin in the CA1 region by exactly the same magnitude as synapse induction determined by electron microscopy and Golgi staining (86).
Components of the Spine Apparatus and Postsynaptic Density. Gene products characterizing dendritic spines include microtubule associated protein-2 (MAP-2), actin, and spinophilin (87–89). Spinophilin, a protein that helps to bundle actin filaments in the dendritic spine, regulates many of the properties of spines (89). Initial results using radioimmunocytochemistry indicate that E treatment increases expression of spinophilin in the CA1 region by exactly the same magnitude as synapse induction determined by electron microscopy and Golgi staining (86). The calcium-calmodulin kinase II (CaMKII) is a major protein of the postsynaptic density (90–94) that plays an important role in long-term potentiation and synaptic differentiation. Recent evidence indicates that CaMKII plays a key role in the formation of synapses and localization of receptors in synapses (90, 93, 95). Glutamatergic synapses contain other key proteins in the postsynaptic density besides CaMKII; these include post-synaptic density-95 (PSD-95), densin-180, and citron, a rac/rho effector protein (90). Rac and Rho are GTPases that regulate spine structure and dendritic branching (96). PSD-95 plays a key role in the anchoring of the NMDA receptor within the synapse (90). The NMDA R1 (NR1) receptor subunit is one of those proteins that may be translated from mRNA located in the dendrites (97).
Dendritic mRNAs Transport and Protein Synthesis. Protein synthesis is an essential component of de novo synapse formation, and neurons have at least three strategies for activity-dependent regulation of protein synthesis and targeting of those proteins to pre- and postsynaptic sites (85, 98–100): (i) translation of mRNA
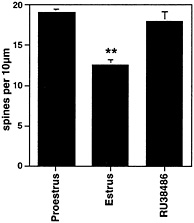
Fig. 7. Number of dendritic spines per 10 µm obtained from the apical portion of the CA1 pyramidal cell dendritic tree. Values are the mean ± SEM for different stages of the estrous cycle. Normally, spine density decreases after the progesterone surge at the time of ovulation; hence, the decrease in the 24 h between the day of proestrus and the day of estrus. The progesterone receptor antagonist, RU38486, given on proestrus, prevented the decline of spine density. **, Different from other groups, P<0.01. [Reproduced with permission from ref. 30 (Copyright 1993, Wiley-Liss, Inc., a subsidiary of John Wiley & Sons, Inc.)].
in the cell soma and trafficking of proteins to “tagged” synapses; (ii) transport of mRNA into the dendrites or growth cones and local translation into protein on polyribosomal clusters such as are found at the base of spines (101); and (iii) local regulation of the translation of transported mRNAs. Dendrites contain transported mRNAs for gene products such as microtubule associated protein-2, CaMKII, NMDA R1 subunit, Arc, GAP-43, and BC1 (102). A key feature of the regulation of translation is that the dendritic mRNAs are deficient in poly (A), and, therefore, the regulation of polyadenylation by cytoplasmic polyadenylation element-binding protein (CPEB) is able to rapidly activate translation (99). A prime example of this process is the effect of visual experience in causing CPEB-dependent cytoplasmic polyadenylation of the alpha-CaMKII mRNA, which is known to reside in dendrites, followed by the rapid activation of the translation of this mRNA (103). We have made an initial attempt to see whether E treatment increases mRNA polyadenylation in whole hippocampus, and the results were negative (G.Y., K.A., and B.M., unpublished observations). It is conceivable, that, in contrast to visual experience, the E effects are much more discrete and not evident in the whole hippocampus.
Involvement of Glial Cells. Glial cells respond to gonadal hormones and may play a role in synapse formation in response to estradiol and down-regulation in response to progesterone. Astrocytic volume in the CA1 region fluctuates in an opposite manner to synapse density, being lowest on proestrus when synapse density is highest (104). On the other hand, in the hilus of the dentate gyrus, the surface area/volume occupied by cell staining for an astrocyte marker, glial fibrillary acidic protein (GFAP) are increased on the afternoon and evening of proestrus, more or less in parallel with the increased synapse density (105). Because astrocytes produce apolipoprotein E (106), they are likely to play a role in the formation of membranes via their regulation of cholesterol and fatty acid availability: e.g., ApoE mRNA levels increase rapidly in response to entorhinal cortex lesions that cause denervation and collateral sprouting within the hippocam
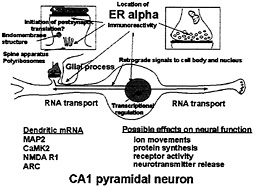
Fig. 8. Schematic depiction of ER localization in CA1 pyramidal neurons that respond to E with synapse formation. ERa is found in dendrites, presynaptic terminals, glia, and the nuclear envelope of some principal cells, as well as in cell nuclei of inhibitory interneurons (not shown). Glia may be involved in synapse formation and/or removal. Dendrites are sites of protein synthesis on polyribosomes and at endomembrane structures using RNAs transported from the cell body (see text). Non-nuclear ER may be involved in other E effects linked to second messenger activation on processes such as neurotransmitter release and phosphorylaton of neurotransmitter receptors and ion channels. Second messenger activation by E in nerve terminals, dendrites, and glial cell processes may result in retrograde second messenger signals, such as phoshoCREB and P-Akt, that return to signal the genome.
pus (107). E treatment has been reported to increase ApoE expression both in vivo and in vitro (108, 109).
Role of Progesterone in Synapse Down-Regulation
At the end of the estrous cycle, the down-regulation of E-induced synapses in the hippocampus is triggered by P.However, as noted above, P administration initially potentiated E-induced spine formation, within 5 h, but then triggered the decrease of spines on CA1 neurons within 8–12 h (Fig. 2). In the absence of P, the disappearance of dendritic spines was much slower and occurred over many days when E was withdrawn (30). Moreover, the natural down-regulation of dendritic spines that occurs between the proestrus peak of spine density and the trough 24 h later on the day of estrus was blocked by the P antagonist, RU38486 (ref. 30 and Fig. 7). This finding is consistent with the involvement of intracellular progestin receptors (PR) and is compatible with the finding, noted above, of estrogen-inducible PR in the CA1 region of hippocampus (110). Curiously, however, nuclear PR is not evident by light microscopic immunocytochemistry in ERa-expressing interneurons or in principal cells in the rat hippocampus, except possibly after prolonged E treatment or damage. Nevertheless, data from in situ hybridization revealed the presence of low levels of PR mRNA in both the CA1 and CA3 regions of Ammon’s horn (111). Initial electron microscopic immunocytochemistry has revealed the presence of non-nuclear PR in glial processes and dendritic spines, although this result needs to be confirmed with several antibodies to different parts of the PR molecule (T.A.M., S.A., and B.M., unpublished observations). In the mouse brain, however, there is evidence for E-induced nuclear PR in interneurons in the hippocampus that also express ERa (112). The mouse hippocampus, however, differs from the rat hippocampus and shows a different response to E treatment that may be better described as synapse maturation as opposed to de novo synapse formation (C.L., W.G.B., and B.M., unpublished observations).
Microglial and astroglial cells must also be considered for a role in the down-regulation of synapses in response to P.Synaptic stripping is a phenomenon seen after noninflammatory neuronal
injury in which microglia attach to the dendrites and displace and then remove synaptic boutons (113, 114). In the injured hamster facial nucleus, the testosterone attenuated the amount of synaptic stripping while increasing regeneration of facial nucleus neurons (115).
Conclusions: A Model of Estrogen Action
Our current knowledge of ovarian hormone actions on hippocampal synapse formation and breakdown has led us to a testable, working model (Fig. 8) in which possible sites of E action are delineated in relation to the location of nuclear and non-nuclear ER. The present discussion pertains to ERa, but further studies of ERß may reveal that it is also present in non-nuclear sites within the hippocampus and may participate in some of the processes outlined in Fig. 8. ER in the dendritic spine may be associated with the activation of mRNA translation from polyribosomes (100, 101) or endomembrane structures found in spines (116). In addition, other second messenger signaling effects might include the phosphorylation of neurotransmitter receptors or ion channels. ER in certain presynaptic terminals might modulate neurotransmitter release(49–51) or reuptake (52). In addition, ER-mediated activation of second messenger systems in dendritic spines and presynaptic endings might lead to retrograde signal transduction back to the cell nucleus, perhaps via Akt or CREB, providing another pathway through which E could regulate gene expression. In addition, as noted above, ER in glial cells might modulate both the formation of constituents of the plasma membrane or the induction of progestin receptors, activation of which may be involved in synapse down-regulation. We consider that these postulated actions of E operate synergistically with the actions of E via nuclear receptors in interneurons, discussed above, that modulate the inhibitory tone on the CA1 pyramidal neurons where synapse formation occurs.
Research support is acknowledged from the National Institutes of Health/National Institute of Neurological Disorders and Stroke (NS 070880) and from the National Institute on Aging (PO1AG16765) to B.M.
1. Chadwick, D.J. & Goode, J.A. (2000) Neuronal and Cognitive Effects of Oestrogens (Wiley, London).
2. McEwen, B.S. & Alves, S.H. (1999) Endocr. Rev. 20, 279–307.
3. Weiland, N.G., Orikasa, C., Hayashi, S. & McEwen, B.S. (1997) J. Comp. Neurol. 388, 603–612.
4. Woolley, C., Gould, E., Frankfurt, M. & McEwen, B.S. (1990) J. Neurosci. 10, 4035–4039.
5. Carrer, H. & Aoki, A. (1982) Brain Res. 240, 221–233.
6. Frankfurt, M., Gould, E., Wolley, C. & McEwen, B.S. (1990) Neuroendocrinology 51, 530–535.
7. Calizo, L.H. & Flanagan-Cato, L.M. (2000) J. Neurosci. 20, 1589–1596.
8. Pfaff, D.W. (1980) Estrogens and Brain Function (Springer, New York).
9. Sandstrom, N.J. & Williams, C.L. (2000) Behav. Neurosci., 115, 384–393.
10. Sherwin, B.B. & Tulandi, T. (1996) J. Clin. Endocrinol. Metab. 81, 2545–2549.
11. Milner, T.A., McEwen, B.S., Hayashi, S., Li, C.J., Reagen, L. & Alves, S.E. (2001) J. Comp. Neurol. 429, 355–371.
12. Levin, E.R. (1999) Trends Endocrinol. Metab. 10, 374–377.
13. Kelly, M.J. & Wagner, E.J. (1999) Trends Endocrinol. Metab. 10, 369–374.
14. Simoncini, T., Hafezi-Moghadam, A., Brazil, D.P., Ley, K., Chin, W.W. & Liao, J.K. (2000) Nature (London) 407, 538–541.
15. Wong, M. & Moss, R.L. (1992) J. Neurosci. 12, 3217–3225.
16. Wong, M. & Moss, R.L. (1991) Brain Res. 543, 148–152.
17. Woolley, C.S., Weiland, N.G., McEwen, B.S. & Schwartzkroin, P.A. (1997) J. Neurosci. 17, 1848–1859.
18. Warren, S.G., Humphreys, A.G., Juraska, J.M. & Greenough, W.T. (1995) Brain Res. 703, 26–30.
19. Terasawa, E. & Timiras, P. (1968) Endocrinology 83, 207–216.
20. Takumi, Y., Ramirez-Leon, V., Laake, P., Rinvik, E. & Ottersen, O.P. (1999) Nat. Neurosci. 2, 618–624.
21. Malgaroli, A. (1999) Nat. Neurosci. 2, 3–5.
22. Woolley, C. & McEwen, B.S. (1994) J. Neurosci. 14, 7680–7687.
23. Fader, A.J., Hendricson, A.W. & Dohanich, G.P. (1998) Neurobiol. Learn. Mem. 69, 225–240.
24. Daniel, J.M., Roberts, S.L. & Dohanich, G.P. (1999) Physiol. Behav. 66, 11–20.
25. O’Neal, M.F., Means, L.W., Poole, M.C. & Hamm, R.J. (1996) Psychoneuroendocrinology 21, 51–65.
26. Luine, V.N., Richards, S.T., Wu, V.Y. & Beck, K.D. (1998) Horm. Behav. 34, 149–162.
27. Sherwin, B.B. (1994) Ann. N.Y.Acad. Sci. 743, 213–231.
28. Woolley, C. & McEwen, B.S. (1992) J. Neurosci. 12, 2549–2554.
29. Gould, E., Woolley, C., Frankfurt, M. & McEwen, B.S. (1990) J. Neurosci. 10, 1286–1291.
30. Woolley, C. & McEwen, B.S. (1993) J. Comp. Neurol. 336, 293–306.
31. Weiland, N.G. (1992) Endocrinology 131, 662–668.
32. Horner, C.H. (1993) Prog. Neurobiol. 41, 281–321.
33. Siegel, S.J., Brose, N., Janssen, W.G., Gasic, P., Jahn, R., Heinemann, S. & Morrison, J.H. (1994) Proc. Natl. Acad. Sci USA 91, 564–568.
34. Gazzaley, A.H., Siegel, S.J., Kordower, J.H., Mufson, E.J. & Morrison, J.H. (1996) Proc. Natl. Acad. Sci. USA 93, 3121–3125.
35. Gazzaley, A.H., Weiland, N.G., McEwen, B.S. & Morrison, J.H. (1996) J. Neurosci. 16, 6830–6838.
36. Loy, R., Gerlach, J. & McEwen, B.S. (1988) Dev. Brain Res. 39, 245–251.
37. DonCarlos, L.L., Monroy, E. & Morrell, J.I. (1991) J. Comp. Neurol. 305, 591–612.
38. Simerly, R.B., Chang, C., Muramastsu, M. & Swanson, L.W. (1990) J. Comp. Neurol. 29, 76–95.
39. Shughrue, P.J. & Merchenthaler, I. (2000) Neuroscience 99, 605–612.
40. Murphy, D.D. & Segal, M. (1996) J. Neurosci. 16, 4059–4068.
41. Murphy, D.D. & Segal, M. (1997) Proc. Natl. Acad. Sci. USA 94, 1482–1487.
42. Willeit, M., Praschak-Rieder, N., Neumeister, A., Pirker, W., Asenbaum, S., Vitouch, O., Tauscher, J., Hilger, E., Stastny, J., Brucke, T. & Kasper, S. (2000) Biol. Psychiatry 47, 482–489.
43. Murphy, D.D., Cole, N.B. & Segal, M. (1998) Proc. Natl. Acad. Sci. USA 95, 11412–11417.
44. Vicario-Abejon, C., Collin, C., McKay, R.D.G. & Segal, M. (1998) J. Neurosci 18, 7256–7271.
45. Toran-Allerand, C.D., Singh, M. & Setalo, G., Jr. (1999) Front. Neuroendocrinol. 20, 97–121.
46. Razandi, M., Pedram, A., Greene, G.L. & Levin, E.R. (1999) Mol. Endocrinol. 13, 307–319.
47. Clarke, C.H., Norfleet, A.M., Clarke, M.S.F., Watson, C.S., Cunningham, K.A. & Thomas, M.L. (2000) Neuroendocrinology 71, 34–42.
48. Blaustein, J.D., Lehman, M.N., Turcotte, J.C. & Greene, G. (1992) Endocrinology 131, 281–290.
49. Nixon, R.L., Janowsky, D.S. & Davis, J.M. (1979) Res. Commun. Chem. Pathol. Pharmacol. 7, 233–236.
50. Packard, M.G., Kohlmaier, J.R. & Alexander, G.M. (1996) Behav. Neurosci. 110, 626–632.
51. Becker, J.B. (1990) Synapse 5, 157–164.
52. O’Malley, C.A., Hautamaki, M., Kelley, M. & Meyer, E.M. (1987) Brain Res. 403, 389–392.
53. Singh, M., Meyer, E.M., Millard, W.J. & Simpkins, J.W. (1994) Brain Res. 644, 305–312.
54. Peters, A., Palay, S.L. & Webster, H.D. (1991) The Fine Structure of the Nervous System (Oxford Univ. Press, New York), 3rd Ed.
55. Steward, O. & Falk, P.M. (1991) J. Comp. Neurol. 314, 545–557.
56. Murphy, D.D., Cole, N.B., Greenberger, V. & Segal, M. (1998) J. Neurosci. 18, 2550–2559.
57. Rudick, C.N. & Woolley, C.S. (2000) Hippocampus 10, 274–283.
58. Datta, S.R., Brunet, A. & Greenberg, M.E. (1999) Genes Dev. 13, 2905–2927.
59. Du, K.I. & Montminy, M. (1998) J. Biol. Chem. 273, 32377–32379.
60. Datta, S.R., Dudek, H., Tao, X., Masters, S., Fu, H., Gotoh, Y. & Greer, M.E. (1997) Cell 91, 231–241.
61. Rao, A. & Levi, S. (2000) Neuron 27, 3–5.
62. Friedman, H.V., Bresler, T., Garner, C.C. & Ziv, N.E. (2000) Neuron 27, 57–69.
63. Ziv, N.E. & Smith, S.J. (1996) Neuron 17, 91–102.
64. Smith, S.J. (1999) Science 283, 1860–1861.
65. Craig, A.M., Blackstone, C.D., Huganir, R.L. & Banker, G. (1994) Proc. Natl. Acad. Sci. USA 91, 12373–12377.
66. Brose, N. (1999) Naturwissenschaften 86, 516–524.
67. Carlin, R.K. & Siekevitz, P. (1983) Proc. Natl. Acad. Sci USA 80, 3517–3521.
68. Segal, M., Korkotian, E. & Murphy, D.D. (2000) Trends Neurosci. 23, 53–57.
69. Fong, D.K. & Craig, A.M. (1999) Neuron 23, 195–197.
70. O’Brien, R.J., Xu, D., Petralia, R.S., Steward, O., Huganir, R.L. & Worley, P. (1999) Neuron 23, 309–323.
71. Guzowski, J.F., McNaughton, B.L., Barnes, C.A. & Worley, P.F. (1999) Nat. Neurosci. 2, 1120–1124.
72. Vician, L., Lim, I.K., Ferguson, G., Tocco, G., Baudry, M. & Herschman, H.R. (1995) Neurobiology 92, 2164–2168.
73. Scheiffele, P., Fan, J., Choih, J., Fetter, R. & Serafini, T. (2000) Cell 101, 657–669.
74. Lustig, R.H., Hua, P., Wilson, M.C. & Frederoff, H.J. (1993) Mol. Brain Res. 20, 101–110.
75. Oyler, G.A., Higgins, G.A., Hart, R.A., Battenberg, E., Billingsley, M., Bloom, F.E. & Wilson, M.C. (1989) J. Cell Biol. 109, 3039–3052.
76. Bennet, M.K., Calakos, N. & Scheller, R.H. (1992) Science 257, 255–259.
77. Lou, X.-J. & Bixby, J.L. (1995) Mol. Cell. Neurosci. 6, 252–262.
78. Berton, F., Cornet, V., Iborra, C., Garrido, J., Dargent, B., Fukuda, M., Seagar, M. & Marqueze, B. (2000) Eur. J. Neurosci. 12, 1294–1302.
79. Knaus, P., Marqueze-Pouey, B., Scherer, H. & Betz, H. (1990) Neuron 5, 453–462.
80. Marqueze-Pouey, B., Wisden, W., Malosio Luisa, M. & Betz, H. (1991) J. Neurosci. 11, 3388–3397.
81. Wiedenmann, B. & Franke, W.W. (1985) Cell 41, 1017–1028.
82. Greengard, P., Valtorta, F., Czernik, A.J. & Benfanati, F. (1993) Science 259, 780–785.
83. Han, H.-Q. & Greengard, P. (1994) Cell Biol. 91, 8557–8561.
84. Ferreira, A., Kao, H.-T., Feng, J., Rapoport, M. & Greengard, P. (2000) J. Neurosci. 20, 3736–3744.
85. Crino, P.B. & Eberwine, J. (1996) Neuron 17, 1173–1187.
86. Brake, W., Alves, S., Dunlop, J., Lee, S., Bulloch, K., Allen, P., Greengard, P. & McEwen, B. (2001) Endocrinology 142, 1284–1289.
87. Matus, A., Ackermann, M., Pehling, G., Byers, H.R. & Fujiwara, K. (1982) Proc. Natl. Acad. Sci. USA 79, 7590–7594.
88. Caceres, A., Payne, M.R., Binder, L.I. & Steward, O. (1983) Proc. Natl. Acad. Sci. USA 80, 1738–1742.
89. Feng, J., Yan, Z., Ferreira, A., Tomizawa, K., Liauw, J.A., Zhuo, M., Allen, P.B., Ouimet, C.C. & Greengard, P. (2000) Proc. Natl. Acad. Sci. USA 97, 9287–9292.
90. Kennedy, M.B. (1998) Brain Res. Rev. 26, 243–257.
91. Benson, D.L., Gall, C.M. & Isackson, P.J. (1992) Neuroscience 46, 851–857.
92. Shen, K. & Meyer, T. (1999) Science 284, 162–166.
93. Rongo, C. & Kaplan, J.M. (1999) Nature (London) 402, 195–199.
94. Ouyang, Y., Kantor, D., Harris, K.M., Schuman, E.M. & Kennedy, M.B. (1997) J. Neurosci. 17, 5416–5427.
95. Hayashi, Y., Shi, S.-H., Esteban, J.A., Piccini, A., Poncer, J.-C. & Malinow, R. (2000) Science 287, 2262–2267.
96. Nakayama, A.Y., Harms, M.B. & Luo, L. (2000) J. Neurosci. 20, 5329–5338.
97. Gazzaley, A.H., Benson, D.L., Huntley, G.W. & Morrison, J.H. (1997) J. Neurosci. 17, 2006–2017.
98. Steward, O. (1997) Neuron 18, 9–12.
99. Wells, D.G., Richter, J.D. & Fallen, J.R. (2000) Curr. Opin. Neurobiol. 10, 132–137.
100. Tiedge, H., Bloom, F.E. & Richter, D. (1999) Science 283, 186–187.
101. Kiebler, M.A. & DesGroseillers, L. (2000) Neuron 25, 19–28.
102. Gao, F.-B. (1998) BioEssays 20, 70–78.
103. Wu, L., Wells, D., Tay, J., Mendis, D., Abbott, M.-A., Barnitt, A., Quinlan, E., Heynen, A., Fallen, J.R. & Richter, J.D. (1998) Neuron 21, 1129–1139.
104. Klintsova, A., Levy, W.B. & Desmond, N.L. (1995) Brain Res. 690, 269–274.
105. Luquin, S., Naftolin, F. & Garcia-Segura, L.M. (1992) J. Neurobiol. 24, 913–924.
106. Poirier, J. (1994) Trends Neurosci. 17, 525–530.
107. Poirier, J., Hess, M., May, P.C. & Finch, C.E. (1991) Mol. Brain Res. 11, 97–106.
108. Stone, D.J., Rozovsky, I., Morgan, T.E., Anderson, C.P., Hajian, H. & Finch, C.E. (1997) Exp. Neurol. 143, 313–318.
109. Stone, D.J., Rozovsky, I., Morgan, T.E., Anderson, C.P. & Finch, C.E. (1998) J. Neurosci. 18, 3180–3185.
110. Parsons, B., Rainbow, T.C., MacLusky, N. & McEwen, B.S. (1982) J. Neurosci. 2, 1446–1452.
111. Hagihara, K., Hirata, S., Osada, T., Hirai, M. & Kato, J. (1992) Mol. Brain Res. 14, 239–249.
112. Alves, S.E., McEwen, B.S., Hayashi, S., Korach, K.S., Pfaff, D.W. & Ogawa, S. (2000) J. Comp. Neurol. 427, 185–195.
113. Blinzinger, K. & Kreutzberg, G. (1968) Z.Zellforsch. Mikrosk. Anat. 85, 145–157.
114. Pastor, A.M., Moreno-Lopez, B., De La Cruz, R.R. & Delgado-Garcia, J.M. (1997) Neuroscience 81, 457–478.
115. Jones, K.J., Durica, T.E. & Jacob, S.K. (1997) J. Neurocytol. 26, 257–266.
116. Pierce, J.P., van Leyen, K. & McCarthy, J.B. (2000) Nat. Neurosci. 3, 311–313.








