H
Atmospheric Deposition and Air-Sea Exchange of Petroleum Hydrocarbons to the Ocean
In this analysis, petroleum hydrocarbons are defined as consisting of n-alkanes between C10 and C33 chain length and polycyclic aromatic hydrocarbons (PAH). Total hydrocarbon concentrations and loadings are calculated as the sum of these 24 alkanes and 21 PAH. Unburned petroleum contains a myriad of compounds beyond these alkanes and PAH and there are other, nonpetrogenic sources of both of these classes of hydrocarbons. This analysis focuses on these petroleum components because they make up a large fraction of petroleum and because information is available in the literature on their concentrations in marine atmospheres and surface waters and on their physical properties. Though the use of C10 through C33 alkanes and unsubstituted PAH as total hydrocarbons undoubtedly underestimates the total mass of petroleum loadings, this bias is likely less than two-fold. The second caveat, that nonpetroleum sources of n-alkanes and PAH are important, is more problematic. Many species produce and release these hydrocarbons into surface waters and the atmosphere, and n-alkane patterns have been used extensively to trace biogenic aerosols over regional and global scales. Many species of plankton produce n-alkanes, contributing to the observed levels of these compounds in marine waters. Combustion of biomass and fossil fuels are a substantial source of PAH to the global atmosphere, especially in highly populated areas. While many methods have been proposed to estimate the relative contributions of petrogenic, pyrogenic, and biogenic hydrocarbons to the atmosphere, surface waters, and sediments, the necessary data required to apportion the n-alkane levels by source in this analysis were not available. The objective of this analysis is to quantify air-sea exchange of n-alkanes and PAH, regardless of their source.
Temporal Scale of Estimates
This analysis began with the data used by Duce and Gagosian (1982) in a National Research Council report. Published literature and known on-going studies were then used to update the estimates of hydrocarbon concentration in the marine atmosphere and in surface waters. Wherever possible, the most recent reliable data were used. The paucity of data often required that all reliable data, regardless of age, be used. Because there are no consistent monitoring programs for hydrocarbons in surface waters or the atmosphere, it is not possible to determine temporal trends in these data. In general, hydrocarbon levels, and therefore loadings, represent conditions in the early- to mid-1990s.
Spatial Scale
This analysis was conducted on two spatial scales. The earlier method used by Duce and Gagosian (1982) in the 1985 NRC report, in which the world’s ocean was divided into impacted (Case A) and remote (Case B) zones, was used to estimate hydrocarbon and polycyclic aromatic hydrocarbon (PAH) air-sea exchange worldwide. In addition, the North American coastline was divided into 17 zones, each of which was further divided into zones 0-3 miles and 3-200 miles from shore (For discussion of zones used in this study, see Figure 1-7 and Table B-1) As part of this analysis, each of these zones was described as urban-influenced or rural and assigned consensus values for gas, aerosol particle, and dissolved hydrocarbon and PAH concentrations based on our review of the literature. Deposition was assumed to be uniform within each North American zone and that the concentrations represent annual averages. Assessing seasonality, which certainly influences both the concentrations and depositional processes, was not considered in this analysis.
METHODOLOGY
The general approach used here was similar to that employed by Duce and Gagosian (1982).
Sources of Data
Ambient gas phase, aerosol-bound and dissolved concentrations of each hydrocarbon in the atmosphere and surface waters of each North American model segment and in the global background were estimated from the current literature. Due to the scarcity of data for the atmospheric petroleum hydrocarbons in the atmosphere bordering North America, the selection of representative distributions of PAH and n-alkanes was developed from the current available literature. For this assessment, petroleum hydrocarbons were defined as n-alkanes with carbon lengths ranging from C10 to C33. To develop an accurate assessment of the contaminant burden to the coastal waters via atmospheric deposition, the various coastal structure and representative contaminant loadings had to be determined. Five zones were assembled based on the degree of urbanization along the zone’s shoreline: (1) urban coastline 0-3 miles from shore (U0-3), (2) urban coastline 3-200 miles from shore (U3-200), (3) rural coastline 0-3 miles from shore (R0-3), (4) rural coastline 3-200 miles from shore (R3-200), and (5) background (BG) contaminant levels that would represent the open ocean. In most cases, adjoining 0-3 and 3-200 mile zones had the same designation (rural or urban) except along the west coast of North America, where are 3-200 zones were ‘rural’ to reflect the predominant westerly air flows off the Pacific Ocean.
Literature on atmospheric hydrocarbons in North America is sparse. This compilation includes those endeavors that have measured concentrations in various selected areas on the United States (Hoff and Chan, 1987; Fraser, 1997; Doskey and Andren, 1986; Foreman and Bidleman, 1990; Ligocki and Pankow, 1989; Mazurek et al., 1991; Simoneit, and Mazurek, 1984). Even fewer atmospheric n-alkane data were available for the North American coast that reported vapor phase alkanes per homologue (Fraser, 1997, 1998; Hoff, 1987). Sampling methods were somewhat consistent throughout the literature. All of the authors use a high-volume air sampler to pull ambient air at specified flow rates through a sample train that contains a glass or quartz fiber filter to retain atmospheric particles. When the vapor phase is collected, a polyurethane plug or PUF is placed beyond the filter in the sampling train. Doskey and Andren (1986) deviated from this format by using XAD-8 resin to collect the vapor phase n-alkanes. Lighter hydrocarbons can also be collected using an evacuated stainless steel canister filled with ambient air and directly injected into a gas chromatograph via sorbent thermal desorption (Fraser, 1997). Sample preparation usually involved the extraction of the filter, PUF or XAD with nonpolar organic solvent by soxhlation or sonication. Extracts may be fractionated over an alumina silicic-acid column (Foreman and Bidleman, 1990; Cotham and Bidleman, 1995) or applied to thin-layer chromatography (Daisey et al., 1981) to separate alkanes. The various forms of instrumentation used in the quantification of hydrocarbons ranged from high performance liquid chromatography coupled with a fluorescence detector (Daisey, 1981; Foreman and Bidleman, 1990) and confirmed by GC/MS (Foreman and Bidleman, 1990) or secondary spectroscopic techniques (Daisey et al., 1981). While Doskey and Andren, (1986) quantified their extracts using gas chromatography equipped with a flame ionization detector, the remainder of the most recent work relies on the resolving power of gas chromatography and the instant confirmation of mass selective detector technology (Hoff and Chan, 1987, Baker and Eisenreich, 1990; Cotham and Bidleman, 1995; Fraser et al., 1997, 1998; Offenberg, 1998; Bamford et al., 1999a; Giglitotti et al., 2000).
The data set acquired from Hoff (1987) collected along the Niagara River between Lake Erie and Ontario, contained values for C16, C22, C24, and C28 n-alkanes in the vapor and particulate phase. Fraser et al., (1997) performed a thorough analysis of the distribution of various hydrocarbons in California during a photochemical smog episode. Aerosol and vapor phase data were taken from San Nicolas Island, Long Beach, central Los Angeles, Azusa, and Claremont. Gaseous and particulate samples were taken using a standard high volume air sampler fitted with a quartz fiber filter (aerosols) and polyurethane plug (vapor). Concurrent gas samples were also taken with 6-L stainless steel canisters. Reported concentrations for the filters, PUF, and canister samples ranged from C18 to C36, from C14 to C28, and from C2 to C13 alkane homologues, respectively. There seems to be some discrepancy between the canister and PUF concentration values from C13 to C14. This may be due to breakthrough of the lighter alkanes in the PUF portion of the high-volume sampler, which consisted of a series of five PUF plugs in series. Separate analysis of the individual PUF plug series showed that the last PUF contained no more than 15 percent of the total alkanes collected on the previous four, which does not explain the factor of 10 difference between the C13 (canister) measurement and the C14 (PUF) measurement. Despite these discrepancies, this data set constitutes the most expansive coverage of the alkane distribution along the urbanized North American coast. The mean of the four coastal California cities, Long Beach, central Los Angeles, Azusa, and Claremont, has been selected to represent the highly impacted coastline of North America in terms of alkane vapor and particulate concentrations (U0-3). San Nicolas Island, which lies approximately 100 km west of the California coast, will constitute the typical atmospheric concentrations offshore (3-200 miles) of a highly urbanized coastline (U3-200).
A second, urbanized coastline alkane distribution was also selected. For the purpose of this analysis Los Angeles, California, is considered to represent an extreme example of an urbanized coastline. Thus, a second alkane distribution, representative of a less urbanized area, was needed. As no well-documented coastal setting was available, this analysis uses data obtained from an interior urban center. Denver,
Colorado was thus selected as being reflective of a more moderately impacted urban area. Foreman and Bidleman (1990) characterized the Denver atmosphere for alkanes. The authors note the major sources of airborne contaminants in the Denver area may be due to coal-driven electric power generation and wood burning, unlike Los Angeles, where the mean concentrations found by Fraser (1997) in southern California incorporate several alkane sources, from dense automobile traffic from major freeways to active shipping ports. For the purposes of this study the southern California levels will be retained as a highly impacted urban coastline with the note that nonrural, urban coastlines may more accurately be reflected in the Denver alkane distribution.
The rural sectors of the coastline are expected to have less of a petrogenic alkane signal. Doskey and Andren (1986) conducted a study in northern Wisconsin approximately 200 km from urbanization (Green Bay, Wisconsin) and 4.5 mils from any major roadway. Similar methods of sampling were performed, as mention above, using a high-volume sampler fitted with a filter and gaseous sorbent. The PUF was replaced with XAD-2 resin for the collection of vapor alkane constituents. Winter total alkanes were taken from this study to minimize the biogenic contribution via plant waxes from spring pollen that may falsely elevate hydrocarbon concentrations in a rural environment. Rural offshore (R3-200 and BG) particulate phase alkane levels were obtained from an expansive aerosol characterization endeavor performed along the western U.S. coast by Simoneit (1982). Aerosol samples were collected from Crater Lake, Oregon. These particulate values were chosen due to the low value obtained and the remote nature of the sampling site. Totals were also only reported for this particular sampling site. The distribution was back calculated using the alkane distribution from Fraser et al. (1997).
The rural areas consisted of the northern Chesapeake Bay (Offenberg, 1998; Leister and Baker, 1994; Dickhut and Gustafson, 1995) Isle Royal, Lake Superior (McVeety and Hites, 1988); and Sandy Hook, New Jersey (Gigliotti et al., 2000). Isle Royal, the main island in Lake Superior, represents a remote signal; the winds are predominantly from the west, and the nearest urban center is approximately 300 km from the sampling site. Sandy Hook, New Jersey, lies on a peninsula approximately 10 km south of New York and 30 km southeast of Newark, lending to direct urban influence and from elevated populations to the west, south, and southwest (Gigliotti et al., 2000). The rural coastal compartment (R0-3) can be best characterized by the sampling performed on the eastern shore of the northern Chesapeake Bay (Offenberg, 1998). No observable urban influence (via Baltimore) was observed from air parcels from Baltimore, the nearest urban center. Therefore this station has minimal urban influence, representative of a rural coastline. The offshore rural or background values (BG) were selected from Ellsmere Island and Alert, Canada, and Tangish in the Yukon Territories (Hallsall et al., 1997; Fellin et al., 1996); Barrow, Alaska (Daisey et al., 1981); Narwahl Island (Daisey et al., 1981); and Isle Royal, Lake Superior (McVeety and Hites, 1988).
VOC Emissions
Crude oils contain a variety of volatile organic compounds (VOC) that evaporate quickly into the atmosphere. Significant quantities of VOC can be released during cargo loading and unloading, during transport, and during crude oil washing operations on board crude oil carriers. Methane makes up approximately 80 percent of these VOC emissions. Methane released into the atmosphere will not deposit, and while it may be a “greenhouse gas” concern, does not appreciably impact the volume of oil entering the sea. Of the remaining VOC, only a small fraction is likely deposited to the sea, as detailed later in this chapter.
Precise measurement of VOC loss from tankers is difficult. The best measure currently available is derived from the fact that cargo insurance companies will typically exclude coverage for loss of 0.5 percent of a crude oil cargo as normal variation between loading and unloading ports. This is the upper range of potential uncovered loss, and it is generally assumed that average loss is probably about half that amount or 0.25 percent most of which can be attributed either to cargo tank gauging variations or, more likely, to VOC emissions.
Approximately 3.3 billion tonnes of cargo oil was moved on tank vessels in 1999. Thus, VOC emissions during crude oil shipment can be estimated as follows:
billion tonnes • 0.0025 tonnes lost/tonne shipped = 8,250,000 tonnes
The VOC emissions (heavier than butane) are therefore:
8,250,000 tonnes • 0.2 non-methane tonnes/tonne = 1,650,000 tonnes
This is a worldwide estimate from shipping. The best estimate of VOC emissions (heavier than butane) from platforms in coastal North American waters is 60,000 tonnes, and the worldwide estimate is 649,000 tonnes (see Section B).
Deposition Calculations
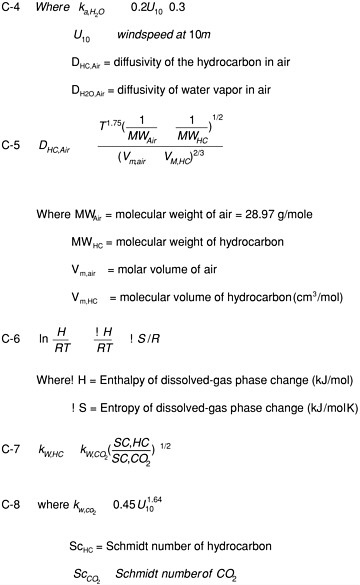
Deposition models were used to estimate depositional fluxes (mass deposited per unit area per year) from these concentrations, and these fluxes were integrated over the area of each model segment to calculate the annual loading. Equations used in these calculations are shown in Figure H-1 and have been used extensively to estimate exchange of semivolatile organic chemicals between the atmosphere and surface waters (Baker and Eisenreich, 1990; Iwata et al., 1993; Achman et al., 1993; Hornbuckle et al., 1994; 1995; Nelson et al., 1998; Bamford et al., 1999a,b; Zhang et al.,
1999; Harman-Fetcho et al., 2000; Bamford et al., 2000). Wet deposition results from the scavenging of gases and particles, which were modeled from the temperature-corrected Henry’s law constant and the aerosol scavenging ratios, respectively (Equations A-1 through A-3). Henry’s Law constants for the hydrocarbons are identical to those used by Duce and Gagosian (1982). Each Henry’s law constant was corrected to 11º C using the enthalpies of phase change reported by Bamford et al., (1999a, 2000), as detailed in Equation C-6. Global annual precipitation was assumed to be 100 cm/year. While spatial and temporal variability in temperature and precipitation rate will alter atmospheric deposition rates, any bias resulting from using uniform global temperature (11º C) and precipitation rates here is likely within the error of these estimates.
Dry aerosol deposition fluxes were calculated as the product of the estimated aerosol-bound hydrocarbon and the dry deposition velocity (Equation B-1). Estimates of deposition velocity range from <0.01 cm/sec to >1 cm/sec and depend on particle size, relative humidity, and surface turbulence. Most studies of organic chemical dry aerosol deposition suggest a deposition velocity in the range of 0.1 cm/sec is conservative. For this analysis, a deposition velocity of 0.1 cm/ sec was used, corresponding to a 0.5-μm particle depositing under average wind conditions. Annual dry deposition velocity was assumed to be spatially invariant.
Gross gas absorption deposition fluxes were calculated by dividing the estimated gas phase hydrocarbon concentrations by their respective temperature-corrected dimensionless Henry’s law constants and multiplying the result by the air-sea exchange mass transfer coefficient (Equations C1-C12). The mass transfer coefficient for each compound was estimated using the two-film model, using relationships between wind speed and tracer exchange rates to parameterize surface turbulence and the compound-specific diffusivities and Henry’s law constants (see Nelson et al., 1997, and Bamford et al., 1999 for details of this calculation). Gross gas deposition fluxes are only one-half of the net bidirectional diffusive exchange of gases across the air-water interface. The corresponding gross volatilization fluxes for each compound were calculated as the product of the estimated dissolved phase hydrocarbon concentration and the air-sea mass transfer coefficient described above (Equation D-1). Since the analysis is calculating loads from the atmosphere to the ocean, volatilization fluxes are negative by convention.
Deposition of Fugitive Emissions From Tankers and Production Platforms
For fugitive emissions from tankers (loss of volatile product during transport) and emissions of volatile species from platforms, see Section B), a simple box model calculation is used to estimate the fraction of the emission that may be deposited into the surface waters. While the exact composition of volatile organic carbon (VOC) lost by volatilization is poorly known, it is very likely that it is dominated by one to three carbon hydrocarbons that are significantly more volatile than the C10 to C33n-alkanes considered in this report. To assess the potential loading of hydrocarbons from fugitive emissions of VOC, this analysis considers the following conservative calculation. We assume (1) that 80% of the VOC released from tankers is methane and other light hydrocarbons that do not deposit to the sea surface; (2) that the remaining VOC mix (20% of total VOC emitted from tankers) has a Henry’s law constant equal to that of decane (which certainly underestimates its volatility and, therefore, overestimates deposition); (3) that the released VOC do not react in the atmosphere or in surface waters (which ignores the substantial degradation due to hydroxyl radical attack in the troposphere); and (4) that the released VOC rapidly partition between the atmosphere, which is well mixed to 1000 m altitude, and the surface ocean, which is well mixed to 100 m depth. Ignoring the substantial atmospheric reactions and using the physical properties of decane result in a very conservative calculation, likely overestimating hydrocarbon loadings to the oceans from these sources. Under this simple scenario, equilibrium calculations show that less than 0.2 percent of the released VOC are deposited to surface waters, even under these very conservative conditions. Based on the VOC emissions of 60,000 metric tonnes (VOC heavier than butane) to North American coastal waters from production platforms (Section B), on the order of 120 metric tonnes (60,000 tonnes x 0.002 tonnes deposited/tonne released) of VOC may enter the coastal ocean as a result of VOC release from platforms. This compares to the 2,100 metric tonnes of hydrocarbons calculated to enter the North American coastal ocean (0-200 miles) from the atmosphere (see below). VOC emissions (heavier than butane) from tankers during loading, transit, and offloading worldwide (1,650,000 tonnes) may result in a VOC loading of 3,300 metric tonnes globally.
Calculation of Total Net Loads
Annual total net loads of individual hydrocarbons were calculated as the sum of wet and dry aerosol deposition and gas absorption minus gross volatilization, integrated over the area of each zone. Total hydrocarbon loads were calculated as the sum of C10 to C33n-alkanes. Finally, total loads for North American waters and the world’s oceans were calculated by summing the loads to the component areas.
ESTIMATES OF ATMOSPHERIC DEPOSITION LOADINGS OF PETROLEUM HYDROCARBONS TO THE OCEAN
Based on the data and methodologies detailed above, wet, dry aerosol, gas absorption, and volatilization fluxes for each of the hydrocarbons were determined (contact NRC staff to
obtain detailed information describing how specific determinations were made, including initial data and intermediate calculated values for individual compounds of interest). Globally, wet deposition and dry aerosol deposition supply 43,000 and 13,600 metric tonnes of C10 to C33n-alkanes to the ocean, respectively. This estimate of 56,600 metric tonnes is within the range of 40,000 to 400,000 metric tonnes reported earlier by Duce and Gagosian, (1982). Most importantly, this loading is overwhelmed by the volatilization of 3,000,000 metric tonnes of these compounds. Clearly these results indicate that the world’s oceans are a net source of hydrocarbons to the atmosphere, where they are transported globally and degraded. In North American coastal waters, 16,000 and 5,030 metric tonnes of C10 to C33n-alkanes are delivered from the atmosphere to the water’s surface, and 1.76 million metric tonnes volatilize (See Table H-1). It is apparent that terrestrial hydrocarbon loadings and near shore sources support dissolved hydrocarbon loadings in coastal waters that far exceed that in equilibrium with the atmosphere. Hydrocarbon degassing from coastal waters is a major geochemical process.
TABLE H-1 Summary of Alkanes and PAH Included in Net Annual Atmospheric Deposition Calculations
|
Alkanes |
PAH |
|
C-10 |
Naphthalene |
|
C-11 |
Acenapthylene |
|
C-12 |
Fluorene |
|
C-13 |
Phenanthrene |
|
C-14 |
Anthracene |
|
C-15 |
Fluoranthene |
|
C-16 |
Pyrene |
|
C-17 |
Benzo[a]fluorene |
|
C-18 |
Benzo[b]fluorene |
|
C-19 |
Benz[a]anthracene |
|
C-20 |
Chrysene/Triphenylene |
|
C-21 |
Benzo[k]fluoranthene |
|
C-22 |
Benzo[b]fluoranthene |
|
C-23 |
Benzo[e]pyrene |
|
C-24 |
Benzo[a]pyrene |
|
C-25 |
Perylene |
|
C-26 |
Indeno[1,2,3-cd]pyrene |
|
C-27 |
Indeno[1,2,3-cd]fluoranthene |
|
C-28 |
Dibenz[ah]anthracene |
|
C-29 |
Benzo[ghi]perylene |
|
C-30 |
Coronene |
|
C-31 |
|
|
C-32 |
|
|
C-33 |
|
TABLE H-2 Calculated Net Annual Atmospheric Deposition Loadings to Each Geographic Zone, Tonnes/Year
|
Zone |
Area (m2) |
Zone Type |
Alkanes |
PAH |
Total By Zone |
|
Global Case A |
8.0E+13 |
Case A |
−2,177,515 |
−14,253 |
−2,191,768 |
|
Global Case B |
2.8E+14 |
Case B |
−865,923 |
−4,989 |
−870,912 |
|
A 0-3 miles |
1.7E+11 |
R3 |
−41,149 |
38 |
−41,111 |
|
A 3-200 miles |
1.9E+12 |
R200 |
−33,709 |
42 |
−33,667 |
|
B 0-3 miles |
1.1E+11 |
R3 |
−27,273 |
25 |
−27,248 |
|
B 3-200 miles |
1.9E+12 |
R200 |
−34,028 |
42 |
−33,985 |
|
C 0-3 miles |
7.6E+10 |
R3 |
−18,795 |
17 |
−18,778 |
|
C 3-200 miles |
1.3E+12 |
R200 |
−24,137 |
30 |
−24,107 |
|
D 0-3 miles |
2.4E+10 |
U3 |
−195,398 |
1 |
−195,398 |
|
D 3-200 miles |
3.8E+11 |
U200 |
−97,813 |
45 |
−97,768 |
|
E 0-3 miles |
1.7E+10 |
U3 |
−138,365 |
1 |
−138,364 |
|
E 3-200 miles |
7.7E+11 |
U200 |
−197,633 |
90 |
−197,543 |
|
F 0-3 miles |
8.7E+09 |
U3 |
−70,175 |
0 |
−70,175 |
|
F 3-200 miles |
3.6E+11 |
U200 |
−92,366 |
42 |
−92,324 |
|
G 0-3 miles |
1.4E+10 |
U3 |
−110,435 |
1 |
−110,435 |
|
G 3-200 miles |
2.6E+11 |
U200 |
−67,579 |
31 |
−67,548 |
|
H 0-3 miles |
1.4E+10 |
U3 |
−113,761 |
1 |
−113,760 |
|
H 3-200 miles |
7.8E+11 |
U200 |
−200,888 |
92 |
−200,796 |
|
I 0-3 miles |
4.0E+09 |
R3 |
−1,003 |
1 |
−1,002 |
|
I 3-200 miles |
6.1E+11 |
R200 |
−11,010 |
14 |
−10,996 |
|
J 0-3 miles |
3.5E+10 |
R3 |
−8,606 |
8 |
−8,599 |
|
J 3-200 miles |
1.2E+12 |
R200 |
−21,460 |
27 |
−21,433 |
|
K 0-3 miles |
2.1E+09 |
U3 |
−16,764 |
0 |
−16,764 |
|
K 3-200 miles |
6.3E+10 |
R200 |
−1,155 |
1 |
−1,154 |
|
L 0-3 miles |
6.5E+09 |
U3 |
−52,498 |
0 |
−52,497 |
|
L 3-200 miles |
3.3E+11 |
R200 |
−6,060 |
105 |
−5,955 |
|
M 0-3 miles |
1.0E+10 |
R3 |
−2,501 |
2 |
−2,499 |
|
M 3-200 miles |
2.8E+11 |
R200 |
−5,053 |
6 |
−5,046 |
|
N 0-3 miles |
2.3E+10 |
R3 |
−5,672 |
5 |
−5,667 |
|
N 3-200 miles |
3.1E+11 |
R200 |
−5,711 |
7 |
−5,704 |
|
O 0-3 miles |
1.2E+10 |
R3 |
−70 |
3 |
−67 |
|
O 3-200 miles |
3.2E+12 |
R200 |
−58,156 |
72 |
−58,084 |
|
P 0-3 miles |
7.3E+10 |
R3 |
−18,123 |
17 |
−18,106 |
|
P 3-200 miles |
1.8E+12 |
R200 |
−31,926 |
40 |
−31,886 |
|
Q 0-3 miles |
4.8E+10 |
R3 |
−11,836 |
11 |
−11,825 |
|
Q 3-200 miles |
1.9E+12 |
R200 |
−34,621 |
43 |
−34,578 |
|
North America Total |
|
|
−4,799,167 |
−18,383 |
−4,817,550 |