
1
Introduction
Since the beginning of modern history, we have sought cures for disease and injury and searched for ways to improve the well-being of societies through the understanding of cultures and civilizations. Scientific progress has been central to these efforts, leading to vast improvements in the way we live. Often, this progress has occurred by studying humans and the human condition. Thus, those who participate as the subjects of research studies should share in the accolades usually accorded great scientists. In some studies, research participants1 assume great risks, even though the prospect for personal benefit is slim or nonexistent. By volunteering to participate, they provide researchers with a capability that they would otherwise lack. In return, research participants deserve to be fully informed, treated with respect, listened to, and protected from foreseeable harms. At the very least, they deserve respect and the highest level of consideration for their safety and well-being. Concern for their rights and welfare should permeate every aspect of the research process, from protocol design to dissemination of results.
A series of events in the late 1990s involving mishaps and errors—some tragic—in the conduct of human research focused renewed attention on the ethical requirement to protect the rights and welfare of those who volunteer
to participate in research. The rapid growth in the size and breadth of the research enterprise in the United States makes it imperative to determine how improvements can be made in the system of protections to ensure that, given the volume and sometimes complex nature of research, institutions and investigators fulfill their ethical responsibilities to research participants.
CONTEXT FOR THIS REPORT
In October 2000, the Department of Health and Human Services (DHHS) asked the Institute of Medicine (IOM) to conduct a two-phase study to address concerns about protecting the rights and interests of research participants. The first-phase report, Preserving Public Trust: Accreditation and Human Research Participant Protection Programs, was released in April 2001 (IOM, 2001a). In that report, the Committee on Assessing the System for Protecting Human Research Participants (“the committee”) developed terminology to describe a set of activities and functions critical to protecting research participants. The term “Human Research Participant Protection Program” (HRPPP), although perhaps unwieldy, reflects the committee’s vision of a system of components, functions, and accountability that should exist, at a minimum, when human research is conducted.
In its first report, the committee addressed the potential for accreditation of HRPPPs to enhance the function of the current protection system. The committee also outlined the basic elements of an HRPPP, envisioning a system with appropriate functions within which roles and accountability would be articulated. The committee suggested that HRPPPs are the appropriate units for accreditation, that human research participant protection should be integral to every aspect of the research effort, and that it can most effectively be provided through an HRPPP. However, it will be critical to evaluate the effects of accreditation to determine whether it actually improves protections.
In this second-phase report, the committee broadens its focus, refining the concept of an HRPPP and examining the overall system of protections within which accreditation is merely one factor. The two primary questions addressed in this phase are, “What should be the functioning units of the protection system?” and “How can performance be assessed to ensure the public safety and effectively maintain public trust?”
It should be noted that the current system is a moving target, and the committee acknowledges that a number of individuals and groups are working within this framework to improve protections for human research participants. It could be said that many institutions already have an HRPPP in place, some more fully developed than others and perhaps applying different names and functions while seeking to achieve the same goals.
In this report the committee was specifically charged with the following tasks:
-
Review the ethical foundations for protecting human participants in research.
-
Assess and describe the current system for protecting human participants and make recommendations for potential enhancements and improvements to
-
ensure informed consent,
-
monitor ongoing research,
-
accommodate private Institutional Review Board (IRBs), multicenter research, and nonmedical research,
-
ensure continuous improvement in the system, and
-
educate researchers, participants, and others involved in research with human participants.
-
-
Assess the potential impact of recommended changes on resource needs and determine how to address them.
-
Consider the effects of accreditation on improving human participant protection activities.
-
Determine the need and develop potential mechanisms for continual independent review of the national system.
BASIC TENETS OF ETHICAL HUMAN RESEARCH
It is widely recognized that research involving humans must follow general ethical principles. The National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (1974-1978) (National Commission) was charged by the U.S. Congress to investigate the ethics of research and to study how research was conducted and reviewed in U.S. institutions; it was also charged with determining the basic ethical principles that should govern research with humans. In response, the National Commission developed a schema of basic ethical principles and related it to the subject areas of research ethics to which the principles apply. The principle of respect for persons states that informed consent should be received from subjects before their involvement in research. The purpose of consent provisions is the protection of autonomy and personal dignity, including the dignity of incompetent persons who are incapable of acting autonomously. The principle of beneficence requires that an appropriate risk-benefit assessment be conducted in order to protect subjects from harm, and the principle of justice requires that there be an appropriate selection of subjects so that certain populations of participants are not over- or underused (National Commission, 1979). These three principles have come to form the ethical foundation upon which participant protection mecha-
nisms are built. As described within this report, the core functions necessary to provide adequate protection are comprehensive protocol review, ethically sound investigator-participant interactions—including an appropriate informed consent process—ongoing safety monitoring, and quality improvement and compliance.
The principles discussed above are necessary conditions of justified research involving humans, but they do not speak directly to the justification of research as a collective social enterprise. The ultimate justification for placing persons at (some level of) risk as research participants is the creation of new and beneficial knowledge. This is the only tenet that allows research investigators or institutions to balance the assumption of risk by the research participant against the individual and social benefits reasonably expected from the research itself. Inherent in this tenet is the responsible use of resources (time, money, people). In addition, although it is not possible to completely eliminate the possibility of harm to participants when conducting research, every effort should be made to minimize risks to the extent possible.
The requirement of a reasonable expectation that the research will benefit society may be more direct when the research is federally funded. However, this requirement still applies to privately funded research, because it also involves the assumption of risk by the participant.
The moral imperative that answering the research question should be of justifiable value to society is articulated in every professional code of research ethics, including the Nuremberg Code and the Declaration of Helsinki, and in federal regulations governing human research (Nuremberg Code, 1949; World Medical Association, 2000; 45 CFR 46; 21 CFR 50 and 56) (see Box 1.1).
THE RANGE OF HUMAN RESEARCH
The term “human research” often evokes the image of experimental studies in biomedicine, such as clinical trials. Although these studies may well represent those for which the most frequent ethical concerns arise, this report considers other types of studies as well.
Biomedical Research
Biomedical research studies can be classified in two ways—experimental or observational. In experimental studies, the investigator manipulates the participants in some way, either to learn more about biological mechanisms or to observe a clinical effect. Examples of studies of biological mechanisms might be the administration of a substance to examine its bioavailability or the administration of a substance to allow the study of the
|
Box 1.1 Thomas Percival’s Code of Medical Ethics (1803) “Whenever cases occur, attended with circumstances not heretofore observed, or in which the ordinary modes of practice have been attempted without success, it is for the public good, and in especial degree advantageous to the poor (who, being the most numerous class of this society, are the greatest beneficiaries of the healing art) that new remedies and new methods of chirurgical treatment should be devised but, in the accomplishment of the salutary purpose, the gentlemen of the faculty should be scrupulously and conscientiously governed by sound reason, just analogy, or well-authenticated facts. And no such trials should be instituted without a previous consultation of the physicians or surgeons according to the nature of the case” (Annas and Grodin, 1992, p. 124; CIOMS, 1982, p.1435). William Beaumont’s Code of Ethics (1833) “There must be recognition of an area where experimentation in man is needed… Some experimental studies in man are justifiable when the information cannot otherwise be obtained; The investigator must be conscientious and responsible…for a well-considered, methodological approach is required so that as much information as possible will be obtained whenever a human subject is used. No random studies are to be made” (Annas and Grodin, 1992, p.125; CIOMS, 1982, p.66). The Nuremberg Code (1949) “The experiment should be such as to yield fruitful results for the good of society, unprocurable by other methods or means of study, and not random and unnecessary in nature; The experiment should be so designed and based on the results of animal experimentation and a knowledge of the natural history of the disease or other problem that the anticipated results will justify the performance of the experiment… The degree of risk to be taken should never exceed that determined by the humanitarian importance of the problem to be solved by the experiment… The experiment should be conducted only by scientifically qualified persons. The highest degree of skill and care should be required through all stages of the experiment of those who conduct or engage in the experiment” (Nuremberg Code, 1949, Principles 2,3,6,8). The Declaration of Helsinki (1964, revised 2000) “Medical research involving human subjects must conform to generally accepted scientific principles, be based on a thorough knowledge of the scientific literature, other relevant sources of information… Medical research involving human subjects should be conducted only by scientifically qualified persons and under the supervision of a clinically competent medical person. The responsibility for the human subject must always rest with a medically qualified person and never rest on the subject of the research, even though the subject has given his or her consent… |
|
Medical research involving human subjects should only be conducted if the objective outweighs the inherent risks and burdens to the subject…” (World Medical Association, 2000, Principles 11,15,18). Council for International Organizations of Medical Sciences Guidelines (1993) The objectives of the research are directed to a justifiable advancement in biomedical knowledge that is consonant with prevailing community interests and priorities; The interventions are justifiable in terms of these objectives: the required information cannot be obtained from animal models; and the study has been designed with a view to obtaining this information from as few subjects as possible who will be exposed to a minimum of risk and inconvenience; The responsible investigator is appropriately qualified and experienced, and commands facilities to ensure that all aspects of the work will be undertaken with due discretion and precaution to protect the safety of the subjects (CIOMS, 1993). Code of Federal Regulations “Risks to subjects are reasonable in relation to the anticipated benefits, if any, to subjects, and the importance of the knowledge that may reasonably be expected to result” [45 CFR 46.111(a)(2); 21 CFR 56.111(a)(2)]. |
biochemical events associated with the symptoms provoked. These types of experimental studies are basic science involving humans that may involve either minor or serious risks.
Another type of experimental study is the clinical trial, which typically involves the administration of an intervention for diagnosis, treatment, or prevention. The intervention could be a drug or biologic; a device; a behavioral intervention, such as counseling or education; a procedure, such as surgery, laser treatment, or a diagnostic test; or a specific service, such as home or hospice care. A clinical trial can be designed and supported for commercial reasons, such as approval of a new drug, or in response to interest by an individual investigator or research group.
Drug companies are required to submit data from clinical trials in order to receive new drug approval. New drug trials have four generally recognized sequential phases. Phase 1 trials test dosage and safety and typically involve a small number of people who are either healthy, paid volunteers or patients with the condition for which the treatment is being developed— cancer patients in the final stages of their illness, for example. Phase 1 studies can involve serious risks and, perhaps most importantly, are not designed to benefit the participant volunteer’s health. Phase 2 trials con-
tinue testing of the dosage and safety of the new drug and also look for evidence of the intervention’s efficacy. Phase 2 studies involve more patients than Phase 1 studies, may involve a comparison group, and may be randomized. Phase 3 trials are randomized and are designed to test the drug’s efficacy. Phase 4 trials occur after approval and examine the long-term benefits and risks of the new drug.
In observational studies, the investigator does not perform any intervention on the study participant, but instead observes or studies the experiences of the participant. Observational studies can include focus groups; surveys or cross-sectional studies; studies involving analysis of large patient datasets, such as those collected by Medicare; studies that follow a cohort of individuals, examining or surveying them at regular intervals (e.g., the Framingham Heart Study); studies in which medical records are reviewed (e.g., to learn the outcome for women treated for osteoporosis); or studies in which cases and unaffected controls are compared to examine a possible association with a past exposure or characteristic (e.g., comparison of young women with and without vaginal cancer who had in utero exposure to diethylstilbestrol). The primary potential harms in observational studies are those related to confidentiality of medical records and information collected as part of the study, the effect of the interactions related to the study itself (e.g., the interview process), and the time and resources (e.g., extra doctor visits or telephone calls) that could be required of participants.
Clinical trials constitute only a subset of research, but because of the heightened concerns that surround them, they are an important subset and are the focus of much of the discussion in this report. Clinical trials comprise a sizeable fraction of the studies that entail medical risks to participants and are a large and growing segment of medical research. Also, on the basis of the growth of organizations dedicated to managing clinical trials and other evidence, it appears that the number of privately financed clinical trials has grown dramatically over the past decade (Rettig, 2000). Furthermore, many trials are multicenter trials involving participants drawn from academic medical centers, private physicians’ practices, community hospitals, clinics, and other institutions and are therefore not subject to a single formal oversight structure.
Genetic research is one form of biological or medical research, and like other types of medical information, it can reveal sensitive information about an individual, his or her family, or even entire groups of people. Moreover, because DNA can be stored, samples studied at later dates can provide new information about individuals or groups in unanticipated ways. Some believe that the major distinguishing characteristics of genetic research and the uncovering of genetic information are its predictive capabilities and its implications for family members and future generations (IOM, 1994a). Thus, any harms that might occur from participating in research could
include emotional, psychological, social, and even economic harms (if such information resulted in discrimination in employment or insurance). Others argue that genetic information is not inherently different from other types of medical information and that caution should be exercised to protect research participants from psychosocial harm in all forms of research (Murray, 1997).
Non-biomedical Research
The social sciences, which include sociology, psychology, and anthropology, also employ both experimental and observational methods. Observational studies and survey work tend to predominate in these disciplines. Traditional research methods include qualitative as well as quantitative approaches, with qualitative methods more prevalent than they are in biomedical research. In terms of subject matter, the boundaries between biomedicine (especially public health) and the social sciences are not always clear. For example, issues such as violence, the health effects of poverty or racism, depression, and child development are encompassed by both fields.
Finally, some research in the humanities may also involve human participants. For example, research in the fields of history, English, and other disciplines might involve interviews with individuals and groups about their past or current experiences.
The United States requires review of federally funded research in disciplines both inside and outside of medicine, but many other countries review only medical research. Although the principles of informed consent and the importance of oversight apply to all research, the principles may be applied in different ways when the risks are social rather than medical and when the goals of research may not be the prevention, detection, or treatment of disease. Therefore, the risks and benefits of such projects might be analyzed differently from those of clinical trials, and such projects might require the application of different kinds of expertise and sensitivities to different categories of research participants. (See Appendix B for a more in-depth discussion of issues related to the social and behavioral sciences.)
RECENT EVENTS
Much of the recent debate and analysis about the protection of research participants has focused on the federal and local institutions and agencies charged with this task, including the federal regulatory agencies, academic and industrial laboratories, IRBs, and funding organizations. To a great extent, examinations have focused on IRBs—the bodies responsible for reviewing the ethical acceptability of proposed human research. In June 1998, the Office of the Inspector General (OIG) of DHHS issued a report,
Institutional Review Boards: A Time for Reform (1998a). Its foremost finding was that “the effectiveness of IRBs is in jeopardy,” and it found that IRBs are facing overwhelming demands (1998a, p. ii). OIG concluded that the system originally devised as a volunteer effort to oversee a much smaller research effort in the 1970s was having difficulty contending with its growing and broadening workload with scant resources.
But the focus of national attention has not been exclusively on IRBs; the institutions in which research is conducted have also been in the spotlight. In May 1999, the federal office charged with overseeing federally funded research, the Office for Protection from Research Risks (OPRR), halted human research studies at Duke University Medical Center. This was shocking to the research community and the public, for if a highly respected institution such as Duke could be noncompliant, then problems were likely to be more widespread than previously imagined. From October 1998 through December 2001, OPRR and its successor, the DHHS Office for Human Research Protections (OHRP), restricted or suspended a number of multiple project assurances and cited 113 research organizations for non-compliance (OHRP, 2001); the Food and Drug Administration (FDA) has also suspended clinical research at other organizations. In addition, issues concerning conflicts of interest at the investigator and institutional levels have forced professional groups and academic institutions to revisit or create policies to ensure that research participants are not placed in harm’s way because the financial interests of those funding or conducting the research conflict with the need to assure participant protection.
Attention was already focused on the protection of human research participants in 1999 when 18-year-old Jesse Gelsinger died in a Phase 1 gene transfer study at the University of Pennsylvania. He was a relatively healthy (i.e., medically stable) young adult with a genetic condition—ornithine transcarbamylase deficiency—who had suffered intermittent health crises throughout his life but was responding relatively well to medications when he entered the gene transfer trial (Gelsinger, 2000; Lehrman, 2000a,b). The details of the case are complex and to some extent contested. Although Gelsinger was aware that he was participating in a gene transfer study, FDA found that the consent form had been altered from the original approved document and that data relevant to safety had not been reported. Questions were raised about whether some participants in the trial, including Gelsinger, fit the revised inclusion criteria and whether the IRB and relevant federal agencies were notified of adverse events that had occurred in studies with animals and in previous participants (Weiss and Nelson, 1999).
The Gelsinger case was heavily reported in the national media and drew the attention of clinical investigators and research administrators throughout the world. It also became the focus of a Senate hearing and commanded direct attention from the Secretary of DHHS, who subsequently
requested the IOM study presented in this report (Shalala, 2000; U.S. Congress, Senate, Subcommittee on Public Health, Committee Health, Education, Labor, and Pensions, 2000). Gelsinger’s death brought a sharp escalation in attention to problems with the system of research participant protections because it resulted more from the experimental intervention and failures in the system of protections than from his underlying condition. The failure to protect this young man in many ways was paradigmatic of failures in the system of protections itself—lack of accountability, conflicts of interest of the investigators and the institutions, insufficient monitoring upon trial commencement, a questionable scientific review procedure, and inadequate resources for comprehensive and stringent review and oversight.
At the institutional level, OPRR/OHRP sanctions have been imposed when systematic deficiencies and concerns regarding systemic protections for human research participants have been found. The deficiencies could be in such areas as IRB membership, education of IRB members and investigators, institutional commitment, initial and continuing review of protocols by IRBs, review of protocols involving vulnerable persons, or procedures for obtaining voluntary informed consent. Although the federal government has been finding fault at the institutional level, some have turned attention to the federal system itself, suggesting that it is in need of consolidation, harmonization, clarity, and a change in organizational culture.
In 2001, the National Bioethics Advisory Commission issued a comprehensive report on ethical and policy issues in human research. The report recommended that federal oversight be centralized and that various components of the oversight system be revised to clarify regulatory responsibilities and to provide more guidance to assist institutions in formulating and implementing policies (2001b).
Calls for such reform are not new. Since the 1974 formation of the National Commission and the activities of the President’s Commission for the Study of Ethical Problems in Medicine and Biomedical and Behavioral Research (1980-1983), an evolving system of protections has consistently, albeit not always successfully, tried to enhance protections for human research participants.
In response to recent concerns, many groups have taken steps to improve protections for research participants. For example, Public Responsibility in Medicine and Research, the Applied Research Ethics National Association, the Association of American Medical Colleges, and the Association of American Universities have issued policy statements, instituted workshops and training, or encouraged their member organizations to strengthen their protection procedures.
Federal agencies also have moved to strengthen and streamline the oversight system. The DHHS elevated its oversight office from NIH to the
Office of the Secretary and formed the National Human Research Protections Advisory Committee to make policy recommendations regarding human participant protection issues to OHRP and DHHS. FDA has centralized and elevated its coordination of participant protection activities into a new office, the Office for Good Clinical Practice. In addition, several federal initiatives provide education and training of investigators or support for institution-based programs.
A System Under Pressure
The federal policies for protecting research participants, as codified in 45 CFR 46 (DHHS regulations) and 21 CFR 50 and 56 (FDA regulations) provide a regulatory framework through which to implement the ethical principles incumbent to human research. Based on this ethical and regulatory guidance, national and international policies have evolved to create a system of protections requiring the involvement of investigators, research sponsors, research institutions, health care providers, federal agencies, and patient and consumer groups. However, the system’s sheer size and complexity and the changing nature of research and relationships within the research enterprise have challenged its ability to fully protect research participants. There is no single cause of failure in the system. Rather, it results from a confluence of factors—a combination of stresses, weaknesses, vagaries, and lack of accountability—that has pushed the system to the point where change should occur or the public trust in the research enterprise will be further eroded.
Investigators and research institutions complain that there is a lack of national guidance on the administrative and ethical requirements of providing adequate protections and that the current federal posture is reactive and punitive rather than proactive and positive. Institutions complain about an overemphasis on documentation, which can lead to unproductive use of time that would be better spent seeking substantive protections. IRBs complain that the regulatory language is not easily understandable and is subject to wide interpretation—sometimes by federal regulators and research sponsors in ways that differ from local views. What sometimes appears to be a senseless bureaucracy has led to cynicism on the part of investigators, which could detract from a genuine commitment to ensuring protections. IRBs themselves are overburdened and at times focus on avoiding risk in the face of rising regulatory pressures. IRB members, who must also fulfill other professional duties and who are often ill rewarded for their IRB service, are reviewing growing numbers of increasingly complex studies that may be conducted at multiple sites and reviewed by multiple IRBs. In addition, IRBs are asked to address conflicts of interest, the science underlying protocols, and a number of other issues in addition to fulfilling their
primary obligations of ensuring appropriate informed consent and properly weighing the risks and benefits of the research.
Most importantly, research participants too often report that they do not understand the nature or risks of research, that they find the informed consent process confusing, and that they are frequently divorced from the decision-making processes involved in the conduct of research. Numerous articles have demonstrated that the current informed consent process is not achieving its purpose (Amdur, 2000; Bohaychuck et al., 1998; Ganter, 2002; Kass and Sugarman, 1996; Moreno et al., 1998). In our litigious society, informed consent documents have become increasingly complex and legalistic and too often are used inappropriately to protect the institution rather than the participant (Annas, 1991). As a practical matter, those participating in research are in the best position to appreciate their own wants and needs, and the principle of autonomy suggests that their wishes should be respected (Faden and Beauchamp, 1986). Although participants might not always be in a position to judge the scientific validity of a protocol, their perspectives can improve the study design, the review of protocols, and the oversight of ongoing research.
In addition, new notions of justice are emerging within the research environment. In some cases, participants now want access to research and are actively seeking protocols that are relevant to their disease or condition (Gifford et al., 2002; Kahn, et al., 1998; Levine, 1986; Mastroianni and Kahn, 2001). Although this development enhances the autonomy of research participants, it should be monitored, as it is often difficult to distinguish research from treatment when routine health care is nonexistent, inadequate, or inaccessible (NBAC, 2001b). Individuals, particularly those who are ill, should not be forced to pursue participation in research as the only means to secure treatment for their condition.
THEMES AND ORGANIZATION OF THIS REPORT
As Figure 1.1 demonstrates, the well-being and interests of participants should be considered at all phases of the research process, from conception of the research question (i.e., will answering this question serve a purpose worthy of exposing human participants to even minimal risks or unnecessary inconvenience?) to dissemination of study results (i.e., ensuring that participants’ personal information is protected and that the study information is accurately reported in order to contribute to the advancement of knowledge). The informed consent process is an ongoing expression of participant protection and should begin from the time a participant first becomes involved in the research and continue throughout his or her participation. Likewise, safety monitoring activities are essential to ensure that participants are protected throughout the entire research process. This re-
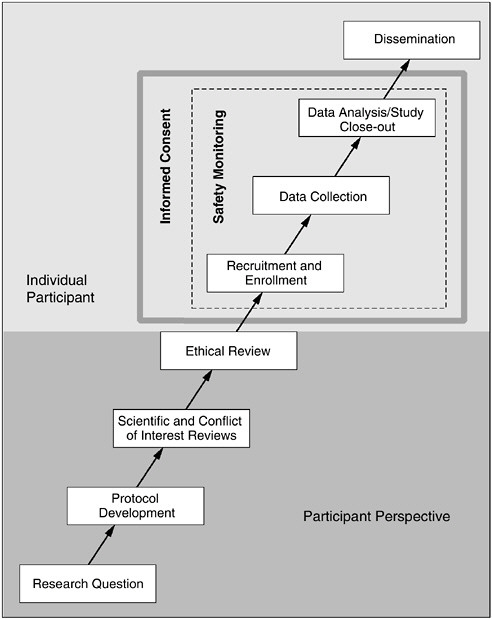
FIGURE 1.1 The Phases of Human Research
Research studies involving human participants are conceived, developed, and implemented through a serial progression of discrete phases. Consideration of the rights, interests, and safety of participants is fundamental to the ethical conduct of each of these phases. Therefore, prior to the recruitment and enrollment of participants, consideration and integration of the perspectives of potential participants is central to ensuring comprehensive protection. With the entrance of an individual participant into a study, informed consent and safety monitoring provide the most direct protection mechanisms to safeguard volunteers and facilitate responsible research.
port is organized to follow a research protocol from initial review through implementation and safety monitoring to completion. In doing so, the committee targeted the HRPPP elements and functions that should be in place at each step in the process to provide and enhance participant protections. In addition, because the greatest concerns exist for studies that pose the highest risk, much of the orientation of this report is focused on creating a system of protections commensurate with the risks involved. Minimum protection requirements exist for all studies with human participants, regardless of the level of risk. However, as risks increase, so should those requirements.
Chapter 2 presents the HRPPP and its functions as a set of organizational policies and practices that ensure adequate protection of participants in any type of research. The diligent application of HRPPP policies and practices will ensure that individual participants in any research project are protected against undue risk, that they provide informed consent to the research, and that their safety, rights, privileges, and privacy are protected. The precise composition of an HRPPP within a given organizational setting depends on the applicable circumstances and context.
Chapter 3 describes the need for independent scientific, ethical, and financial conflict of interest reviews to ensure that the proposed research is meritorious, that it does not expose participants to unnecessary risk, and that the interests of the investigator, institution, or IRB are not in conflict with those of the participants. Chapter 4 focuses on the qualifications of the investigator in designing and conducting a study, the roles of the research participant, and the primary focus of their interactions—the informed consent process. Chapter 5 addresses the need for ongoing oversight and monitoring at the federal, institutional, and local levels to increase safety during the conduct of studies.
Chapter 6 focuses on organizational responsibilities to ensure the optimum performance of HRPPPs, discussing accreditation, quality assurance and improvement, and the need for role clarification within research organizations. Also included in Chapter 6 are recommendations regarding compensation for research-related injury, a topic that has been discussed for decades but never adequately addressed. Chapter 7 addresses broader issues affecting HRPPPs that should be addressed at the national level and mechanisms to provide continuing assessment of the national protection system.
This report is not intended to be comprehensive. Many important issues in human research have received extensive analysis and review by other groups or remain unresolved and deserve further discussion. Although these issues periodically emerged during committee discussions, they were beyond the scope of the committee’s mandate or were too important to be treated in a cursory fashion. For example, although the ethical obligations of researchers in conducting studies in international settings have been
explored in depth by others, the continuing issues involved merit explicit and ongoing discussion (NBAC, 2001a; World Medical Association, 2000). In addition, the complexities of ethically involving minors or individuals with impaired decisional capacity in research continue to be reviewed by several other groups, and ethical issues in research utilizing large databases and human biological materials will continue to evolve as the volume of such research expands (NBAC, 1999).
Furthermore, a recent report by IOM demonstrated that race is important in understanding how people of color are treated by the health care system (IOM, 2002). And, given the lack of health insurance coverage for many persons in this country, research studies increasingly are seen as a way to receive access to otherwise unavailable health care services—a pattern that has been most evident in HIV/AIDS studies, but that can be seen in many other types of investigations. Although the committee recognizes the critical importance of these issues, as well as the issue of social class, to matters of participant choice in enrolling or refusing to participate in a particular study or protocol, they cannot be sufficiently considered within the scope of this report.
Finally, ongoing debates about ethical issues in social science research, as distinct from biomedical research, call for focused attention. In the process of its work, the committee heard concern that draft accreditation standards would require the elaboration of formal policies and documentation that would be irrelevant for IRBs that primarily review social science, behavioral research, anthropology, sociology, oral history, epidemiology, and population studies (Levine, 2001b; Overbey, 2001; Shopes, 2001). However, pleas to exempt nonbiomedical research from oversight were not heard; rather, the committee received suggestions to reduce paperwork, to develop criteria sensitive to social and behavioral research, and to expand the categories of research exempt from review when the risks of nonmedical research are inherently minimal and informed consent can be “presumed” (e.g., by returning a survey form or answering interview questions) (Erickson, 2001; Rubin, 2001; Rudder, 2001).
The American Association of University Professors has addressed this topic (AAUP, 2001), and at The National Academies, the Committee on National Statistics, in collaboration with the Board on Behavioral, Cognitive, and Sensory Sciences and Education (CNSTAT/BBCSS), is conducting a study of research oversight for the social and behavioral sciences that is intended to complement and inform the work of this committee. The initial conclusions and recommendations of that group can be found in Appendix B and are referenced as they apply throughout this report. The CNSTAT/ BBCSS Panel on IRBs, Surveys, and Social Science Research is expected to issue its final report in early 2003.
SUMMARY
Conducting research with human participants is a privilege granted by willing volunteers. Such research is central to the translation of scientific knowledge into societal goods and should be encouraged. However, in doing so, it should be realized that we have a solemn responsibility to protect research participants and to ensure that this protection is integral to every aspect of the research process.
Our current system of protection, however, is not functioning as intended, a message that this committee as well as a long line of analysts and observers have delivered. The committee believes that the system should be adapted to overcome its current constraints in order to ensure that the oversight of comprehensive participant safety occurs in a manner that does not curb the quality of research. Previous groups have made recommendations to the federal government, to IRBs, and to investigators. This committee also offers recommendations to those entities, but in addition it seeks to emphasize a systems approach (i.e., the HRPPP) for protecting research participants.
The envisioned HRPPP should strive to prevent research harms through systematic and interlocking protection functions within which discrete roles and accountability are clearly articulated; the program should be buttressed by an infrastructure that is adequately funded and embraced by its leadership; and the level of protection it provides should be commensurate with the anticipated risks of the research. To achieve these goals, each research entity should develop processes and procedures that are clear, efficient, and effective. Finally, clear and open communication among those who are involved in protecting research participants should be established and maintained.
















