
2
Prion Diseases and Their Challenges
ORIGINS AND DEVELOPMENT OF PRION SCIENCE
The identification of a previously unknown malady in the Fore Tribe of Papua New Guinea drew international attention to the group of brainwasting diseases called transmissible spongiform encephalopathies (TSEs). Physicians Vincent Zigas and Daniel Carleton Gajdusek in 1957 described an epidemic among the Fore people characterized by loss of balance, dementia, and death (Gajdusek and Zigas, 1957). The tribe called the illness kuru, meaning "to tremble" or "to shiver." Studies of the brains of deceased patients revealed widespread neurodegeneration marked by vacuoles in the cytoplasms of nerve cells (Klatzo et al., 1959). The vacuoles gave the victims' brains a sponge-like appearance at the microscopic level, hence the term "spongiform encephalopathy."
Ethnological and epidemiological studies indicated that kuru was transmitted during an endocannibalistic1 funeral ritual (Alpers, 1968; Gajdusek, 1977; Glasse, 1967). Women would remove the brain of a deceased relative, eat it along with other tissues, and smear it over their bodies and those of young children of both sexes (Gajdusek, 1977; Glasse, 1967). Women who fell victim to kuru outnumbered men who fell victim to the disease by more than 14 to 1 (Gajdusek and Zigas, 1957). After a 1957 ban on cannibalism in Papua New Guinea, the number of kuru cases gradually declined over decades, reaching single
figures in recent years (Huillard d'Aignaux et al., 2002; Klitzman et al., 1984).
Veterinary neuropathologist William Hadlow was the first to recognize similarities between kuru and scrapie, a TSE of sheep and goats that had been known since the 1700s (Hadlow, 1959). He pointed out in a 1959 letter to The Lancet that the brains of people with both conditions had a unique form of widespread neuronal degeneration. "Large single or multilocular 'soap-bubble' vacuoles in the cytoplasm of nerve-cells have long been regarded as a characteristic finding in scrapie," he wrote; "this extremely unusual change, apparently seldom seen in human neuropathological material, also occurs in kuru, and first aroused my curiosity about the possible similarity of the two diseases" (p. 290).
Scrapie and kuru both were endemic to specific populations in which the usual incidence was low, he added. Clinical symptoms could appear months after a victim was separated from the source community, and both diseases were found in previously healthy communities after the introduction of an individual from a known source community. Hadlow also noted that data suggested a genetic predisposition toward both diseases, which did not appear to be infectious in the traditional sense. Victims exhibited increasingly severe ataxia, tremors, and behavioral changes, yet no consistent abnormalities appeared in their blood or cerebrospinal fluid. Both diseases began insidiously, he wrote, "and usually end fatally…; only rarely have remissions and recoveries been observed" (p. 290).
On the basis of these observations, Hadlow suggested that experimental transmission of kuru into nonhuman primates might prove fruitful, since veterinary scientists were successfully investigating scrapie by inoculating healthy sheep and goats with brain tissue from animals with scrapie. After extensive work, Gajdusek and colleagues did transmit a "kuru-like syndrome" with an incubation period of 18 to 21 months to chimpanzees by inoculating them with brain suspensions from kuru patients (Gajdusek et al., 1966), indicating that a noninflammatory neurodegenerative disease could be transmissible.
Similarities in the neuropathology of kuru and a rare, fatal condition called Creutzfeldt-Jakob disease (CJD) led investigators to attempt experimental transmission of CJD to nonhuman primates. They successfully transmitted the disease to a chimpanzee, which first displayed clinical signs after a 13-month incubation period, providing more evidence that spongiform encephalopathies are transmissible (Gibbs et al., 1968).
These studies and observations generated a groundswell of interest in discovering the nature of the infectious agent or agents that caused scrapie and kuru. Many hypotheses on the nature of the agent surfaced between 1962 and 1981, ranging from a small DNA virus to a replicating polysaccharide to naked nucleic acid similar to plant viroids (Prusiner, 1982). None of these explanations gained widespread acceptance, however, and the cause of scrapie remained an enigma.
In 1982, neurologist Stanley Prusiner asserted that the infectious agent in scrapie was either a protein or a small nucleic acid surrounded by a tightly packed protein (Prusiner, 1982, 1999). He called this infectious agent a "prion," which stands for "small, proteinaceous infectious particles that are resistant to inactivation by most procedures that modify nucleic acids" (Prusiner, 1982, p. 141).
At the time, replication of microorganisms and viruses was thought to require nucleic acids. Several investigators had proposed that the infectious agent of scrapie might not require nucleic acids and could be a replicating protein (Alper et al., 1967; Griffith 1967; Lewin, 1972; Pattison and Jones, 1967). Until Prusiner's entry into the field, however, no other investigator had provided compelling data to support his hypothesis.
Applying advanced biochemical techniques, Prusiner generated a purified infectious scrapie preparation that yielded a peptide fragment. By determining the nucleic acid sequence that encoded the peptide, he located the gene where the sequence for the peptide was embedded, allowing him to decipher the full-length protein PrP (the prion protein) (Brown and Bradley, 1998). He then demonstrated that the scrapie agent resisted six different procedures known to attack nucleic acids and was susceptible to six methods of protein inactivation (Prusiner, 1982). (It was later established that prions and PrPSc [a protease-resistant protein associated with prion disease] were resistant to limited digestion by one of those methods, proteinase K digestion.) Like some investigators whose theoretical work preceded him, Prusiner correctly suggested that a prion might act as "an inducer or template for its own synthesis" (Prusiner, 1982, p. 139).
Two decades of research have borne out the prion hypothesis, leading most TSE experts to accept this theory. Prusiner won the Nobel Prize in Physiology or Medicine in 1997 for his groundbreaking work. But because Koch's postulates2 have not been demonstrated for prions,
some scientists believe that prions alone do not explain all aspects of the etiology of TSEs (Chesebro, 1998, 1999; Rohwer, 1991).
An overview of all known TSEs appears in Table 2-1.
THE NATURE OF PRIONS AND PRION DISEASES
The protein that plays a critical role in prion disease is called PrP and is encoded by the gene PRNP on chromosome 20 in humans. Like all proteins, PrP has a characteristic conformation, but under certain conditions it folds into an abnormal shape that is associated with fatal neurodegeneration after a long incubation period. In this report, the terms "prion" and "PrPSc" refer to the protease-resistant protein associated with prion disease.3
The normal, cellular prion protein, PrPC, has a glycolipid anchor and resides on the membranes of many avian and mammalian cells. Its physiologic function is poorly understood, although it is possible that its function or loss of function could contribute to some aspects of the disease state in TSEs.
Unlike PrPC, the aberrantly folded PrPSc is aggregated and insoluble, resists complete digestion by proteinase K, and contains a higher proportion of flat peptide regions called β-sheets (Prusiner, 2001). To date it appears that the immune system does not recognize and destroy PrPSc, presumably because it has the same primary structure4 as the normal isoform, despite its distinct conformation. This lack of an immune response makes it difficult to develop diagnostic tools based on antibodies, since there is no confirmed5 antibody that binds exclusively to PrPSc.
TABLE 2-1 Classification of TSEs
|
Type of TSE |
Affected Mammals |
Modes of Natural Transmission |
Date First Recognized |
|
In humans |
|||
|
Sporadic Creutzfeldt-Jakob disease (sCJD) |
|
Unknown |
1920 |
|
Sporadic fatal insomnia (sFI) |
Unknown |
1999 |
|
|
Familial Creutzfeldt-Jakob disease |
Genetic |
1924a |
|
|
Fatal familial insomnia (FFI) |
Genetic |
1986b |
|
|
Gerstmann-Straussler-Scheinker disease |
Genetic |
1936c |
|
|
Kuru |
Exposure to contaminated human tissues during endocannibalistic rituals |
1957 |
|
|
latrogenic Creutzfeldt-Jakob disease |
CJD-infected surgical equipment or tissue transplants |
1974d |
|
|
Variant Creutzfeldt-Jakob disease (vCJD) |
Eating BSE-infected tissue; other modese |
1996 |
|
|
Type of TSE |
Affected Mammals |
Modes of Natural Transmission |
Date First Recognized |
|
In animals |
|||
|
Scrapie |
Sheep, goats |
Contact with infected placenta; possibly oral exposure to environmental contamination |
18th century |
|
Transmissible mink encephalopathy |
Mink |
Eating infected tissue |
1947 |
|
Chronic wasting disease |
Deer, elk |
Unknown; likely oral |
1967 |
|
Bovine spongiform encephalopathy (BSE) |
Cattle |
Eating TSE-infected tissue |
1986 |
|
Nyala, gemsbok, Arabian oryx, eland, kudu, scimtarhorned oryx, puma, cheetah, ocelot, tigerf |
Eating BSE-infected tissue |
Mid-1980s |
|
|
Domestic catg |
Eating BSE-infected tissue |
1990 |
|
|
a Gambetti et al. (1999). b Lugaresi et al. (1986). c Kretzschmar et al. (1991). d Duffy et al. (1974). e It is unknown whether the disease is transmissible by transfusion or transplantation. f The TSE affecting these animals was called "exotic ungulate encephalopathy" until it was discovered to be BSE. These animals living in zoos became infected by eating BSE-contaminated feed. g The TSE affecting domestic cats was called "feline spongiform encephalopathy" until it was discovered to be BSE. The animals became infected by eating BSE-contaminated feed. SOURCES: Godon and Honstead (1998), Johnson and Gibbs (1998), Haywood (1997), Prusiner (1995), and E. Williams, University of Wyoming, December 2002. |
|||
In experiments in which animals received PrPSc by peritoneal inoculation or through the gastrointestinal tract, the proteins migrated to the lymphoreticular system and propagated there (Brown et al., 1999; Weissmann et al., 2001). By a poorly understood mechanism, prions convert PrPC into the abnormally folded conformation containing β-sheets. In a demonstration of the pivotal role of normal prion proteins in the progression of TSEs, mice in which PrPC expression was knocked out or ablated remained healthy after infection with pathogenic prions (Bueler et al., 1993). It is widely hypothesized that one or more so-called chaperone molecules also play an indispensable role in the conversion of PrPC to PrPSc (Chernoff et al., 1995; Telling et al., 1995).
PrPSc migrates to the brain along peripheral nerves (Beekes et al., 1998; Kimberlin et al., 1983; Oldstone et al., 2002; Race et al., 2000). Prions multiply in the lymphoreticular system, but this activity is not essential for neuroinvasion. The misfolded proteins aggregate into rodshaped fibrils, and prions at the ends of the rods continue converting normal PrP into PrPSc (Caughey, 2002). By an unknown mechanism, the aggregated prions appear to destroy nerve cells and create microscopic vacuoles in the brain. Clinically, this destruction manifests itself differently in different species, but inevitably, it seems to lead to death. Both the incubation period and the length of time between the onset of clinical symptoms and death vary widely depending on the host species and the strain of PrPSc.
Investigators initially differentiated among prion strains through clinical observations of goats that displayed either drowsy or hyperactive behaviors (Pattison and Millson, 1961). Later work with mice revealed that genetic factors play a role in determining strain differences. Dickinson and Fraser identified two different alleles called sinc (strain incubation) genes in inbred mice that consistently resulted in a long or short incubation period prior to the onset of disease (1968). The investigators later published additional findings that strains could be differentiated by the distribution of microscopic lesions (vacuoles) in the brain (Fraser and Dickinson 1973). Studies using the agent of transmissible mink encephalopathy (TME) in hamsters showed differences in clinical presentation and glycoform patterns by prion strain (Bessen and Marsh, 1992).
More recently, the use of selected inbred and transgenic mice to characterize prion strains has led to important insights, including the idea that a similar strain causes both bovine spongiform encephalopathy (BSE) and variant Creutzfeldt-Jakob disease (vCJD) (Bruce et al., 1997; Scott et al., 1999). There are about 20
phenotypically distinct, mouse-passaged strains of scrapie alone (Haywood, 1997).
Despite these advances, much about prion strains remains a mystery. It is unclear why strain variants exist in the first place. Researchers must be able to differentiate one strain from another, determine the scope of strain diversity, determine the special characteristics of a strain's conformation that make the strain unique, determine a host's susceptibility to various strains, and determine how incubation periods and patterns of disease expression vary by strain and host.
THE EPIDEMIC OF BOVINE SPONGIFORM ENCEPHALOPATHY AND THE EMERGENCE OF vCJD
Prion diseases remained obscure outside the circles of infectious disease specialists and neurologists until the mad cow epidemic struck in the United Kingdom. The illness was first recognized in 1985 when a handful of cattle from disparate locations in the United Kingdom began dying of a strange illness marked by insidious onset, rapidly progressive dementia, and death (Wells et al., 1987; Wilesmith et al., 1988). Neuropathological examinations of the sick cattle's brains revealed abnormal, microscopic vacuoles and fibrils, much like the spongiform characteristics of scrapie and kuru. A team of veterinary scientists at the United Kingdom's Ministry of Agriculture reported in 1987 that the new disease strongly resembled the so-called unconventional viral-agent encephalopathies previously observed in sheep and the Fore people, so they named it "bovine spongiform encephalopathy" (BSE) (Wells et al., 1987). The outbreak appeared mainly in dairy cows rather than beef cattle (Anderson et al., 1996) because dairy cows much more commonly consumed meat and bonemeal that was the attributed source for BSE transmission.
BSE quickly ballooned into an epidemic that peaked at more than 37,000 annual cases in the United Kingdom in 1992 (Figure 2-1) (Department for Environment, Food and Rural Affairs, United Kingdom, 2002a). It has been estimated that 840,000 to 1.25 million infected animals entered the human food chain from 1974 through 1995 (Anderson et al., 1996; Wilesmith et al., 1992).
Conscious of the fact that the transmission of BSE to humans was a possibility, in 1990 the United Kingdom increased epidemiological surveillance of a rare, human spongiform encephalopathy called CJD. It was thought that changes in the pattern of CJD could signify a link to
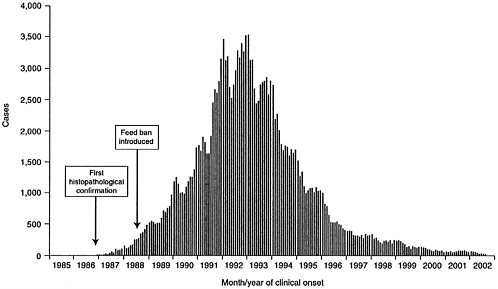
FIGURE 2-1 Confirmed cases of BSE by month and year of clinical onset. [Reprinted with permission from the Department for Environment, Food and Rural Affairs (DEFRA), United Kingdom. Copyright 2002 by DEFRA. Online: http//www.defra.gov.uk/animalh/bse/bse-statistics/graphs/epidem.pdf (2002b).]
BSE. The neuropathological profiles and age distribution of 10 of the 207 patients with CJD examined between 1990 and 1997 differed markedly from those that are typical for CJD (Will et al., 1996). This discovery led to the conclusion that a new variant of CJD had arisen in the United Kingdom.
Glycosylation studies revealed that the new variant, called vCJD, involved the same prion strain involved in BSE (Collinge et al., 1996), and transmission studies with inbred mice confirmed this (Bruce et al., 1997; Collinge et al., 1996).
There is evidence of genetic susceptibility to vCJD. A 1997 study found that a specific genotype of codon 129 in PRNP correlated with vCJD in all 26 patients tested (Zeidler et al., 1997). This codon normally codes for methionine on one allele and valine on the other, or the alleles may be homozygous, but the PRNP genes from all patients tested were consistently homozygous for methionine (Collinge et al., 1996). However, it is possible that individuals with the other genotypes are susceptible but that the incubation periods in these individuals may be longer. It is well known that substantial variation in the incubation periods of strains of a mouse-adapted scrapie agent results from their passage through mice with different PrP genotypes.
The fear of vCJD devastated the United Kingdom's beef industry. Hundreds of thousands of cattle have died of BSE or have been culled. Many other countries worldwide unknowingly imported BSE-infected cattle and contaminated meat and bonemeal from the United Kingdom and have also suffered outbreaks of BSE, public panic, financial losses, and political repercussions.
The Risk of BSE in the United States
Two recent reports suggest that the federal government should strengthen its policies designed to avert BSE and vCJD, although the first study concluded that the risks of a BSE outbreak in the United States are minimal (GAO, 2002; Harvard Center for Risk Analysis, Harvard School of Public Health, Center for Computational Epidemiology, College of Veterinary Medicine, Tuskegee University, 2001).
The 2002 report by the General Accounting Office (GAO) identified a number of holes in Food and Drug Administration (FDA) and U.S. Department of Agriculture (USDA) policies that could allow BSE to slip into the United States without prompt detection, spread to other cattle, contaminate meat in the food supply, and be consumed. The report asserts that the United States lacks sufficient capacity to
inspect all cattle imports and that mandated animal testing excludes some high-risk animals: those that die on farms. FDA does not know the full extent of industry compliance because its inspection data are "severely flawed," according to GAO (p. 3). Moreover, GAO asserts that FDA has been slow or has neglected to enforce the animal feed ban even in firms found to be noncompliant. Finally, cattle brains and other central nervous system tissue may be sold as human food without being labeled as such.
In a report commissioned by USDA, researchers at the Harvard School of Public Health and Tuskegee University determined that USDA and FDA policies prevent BSE from taking root in the United States. These have included a ban on the import of live ruminants and ruminant meat and bonemeal from the United Kingdom since 1989 and from all of Europe since 1997, as well as a 1997 ban to prevent the recycling of potentially infectious cattle tissues by prohibiting the use of cattle tissue as animal feed. There is a 20 percent chance that 173 of the 334 cattle imported from the United Kingdom before 1989 exposed U.S. cattle to BSE, the Harvard study reported, and those cases were probably so few in number that they fell under the radar of U.S. disease surveillance. (The committee notes that there is no evidence that BSE is a contagious disease and that the greatest risk would have occurred if the carcasses of any of these animals had been used to manufacture meat and bonemeal.) However, even if U.S. cattle had been exposed to the BSE agent, present policies would keep the disease from becoming established in U.S. cattle herds, the study concluded, making the risk of vCJD in the U.S. human population extremely low.
THE SPREAD OF CHRONIC WASTING DISEASE IN THE UNITED STATES
Although the BSE epidemic that struck Europe has spared the United States so far, another TSE called chronic wasting disease (CWD) is affecting free-ranging and captive deer and elk in several mid western and western states. It has also occurred in Canada.
The unusual, insidious, and fatal illness began appearing in a captive herd of mule deer in the late 1960s at a research facility in Fort Collins, Colorado (Williams and Young, 1980). The disease affected young adult deer that had been captive for approximately 2.5 to 4 years. Sick animals became listless, depressed, and anorexic and died of emaciation, secondary complications, or euthanasia within 2 weeks to 8 months after the onset of clinical symptoms. The nature of these signs led biologists to name the illness "chronic wasting disease."
The most striking and consistent pathological features of CWD were nerve cell degeneration and widespread, microscopic vacuoles in the neurons of the brain and spinal cord, trademarks of the spongiform encephalopathies previously described in sheep, goats, and humans. This led scientists Elizabeth Williams and Stuart Young to conclude in 1978 that CWD was a new spongiform encephalopathy. Captive Rocky Mountain elk living in the same Colorado and Wyoming facilities as the affected deer were diagnosed with the disease a few years later (Williams and Young, 1982).
Unlike BSE, CWD can spread efficiently from an infected animal to an uninfected animal of the same species as well as related species either directly after exposure or indirectly from the pasture occupied by an infected animal (Gross and Miller, 2001; Williams and Miller, 2002). It is clear that the disease can be transmitted among mule deer, white-tailed deer, and elk (Williams and Miller, 2002). The lack of understanding about how CWD spreads and whether it can cause disease in humans and cattle has lent urgency to research on CWD and the development of tools to detect prions in humans, animals, and the environment.
Major media outlets reported on the growing number of CWD-infected deer and elk in North America during 2002 (Blakeslee, 2002; Regalado, 2002). Although the disease appears to be spreading, the larger numbers and wider geographical distribution of CWD cases also may reflect more active surveillance during the past several years. This surveillance has resulted in the identification of foci of CWD in the wild and on game farms that have existed for a decade or two-the news is their discovery (Miller et al., 2000). Most of the current CWD epidemics in free-ranging and farmed cervids appear to be independent of each other, although they may have a common origin dating back several decades (Williams and Miller, 2002). It is unclear how the disease arose in the first place.
The spread of CWD among free-ranging cervids will likely follow the animals' predictable, natural movements. By contrast, some researchers speculate that CWD in farmed animals has spread more widely and unpredictably due to market forces (Williams and Miller, 2002).
Unique Challenges in Conducting Prion Research
Much about prion diseases remains unclear: how prions replicate, why they target neurons, and how prions kill neurons are a few examples. Prion research has progressed slowly because of a number of
challenges unique to the field. First and foremost, prions are an entirely new type of infectious molecule, precluding the use of many tools designed for studying infectious diseases. Since PrPSc and PrPC have identical amino acid sequences, the existence of a prion-specific antibody has not been confirmed to date and infected individuals do not exhibit a prion-specific immune response. In addition, prions replicate sluggishly in existing cell culture systems and incubate for several months to several years in animal models, limiting the pace of research.
TSE investigators face not only scientific challenges but logistical ones as well. Their work often must take place in laboratories designed for research with biohazardous materials that are expensive to construct, and the United States has few such facilities for prion research. In addition, standardized reagents are hard to come by: there is only one U.S.-based repository for vCJD tissue and no repository for other reagents or transgenic animals. Until recently, the federal government's limited interest in prion diseases meant that it was relatively difficult to win research grants, and this apparent lack of financial stability has discouraged young scientists from entering the field. Hence, the community of TSE researchers in the United States is small.
Sensitive, specific TSE diagnostics would help protect people and animals from fatal prion infections in the absence of prophylactics and treatments. Given that there are few tools to inactivate prions, the ability to test blood and other tissues for prions would help prevent the inadvertent transmission of vCJD by blood transfusion or organ transplantation, provided that there is actually any infectious agent to be detected in blood or the organs used for transplantation. Despite many attempts in Europe and the United States, no one has developed a reliable antemortem diagnostic test for TSEs. The next chapter describes the technologies that offer the greatest promise for reaching this important goal.
REFERENCES
Alper T, Cramp WA, Haig DA, Clarke MC. 1967. Does the agent of scrapie replicate without nucleic acid? Nature 214(90):764-766.
Alpers MP. 1968. Kuru: implications of its transmissibility for the interpretation of its changing epidemiological pattern. Bailey OT and Smith DE, editors. The Central Nervous System: Some Experimental Models of Neurological Diseases. Baltimore: Williams and Wilkins. Pp. 234-251.
Anderson RM, Donnelly CA, Ferguson NM, Woolhouse ME, Watt CJ, Udy HJ, MaWhinney S, Dunstan SP, Southwood TR, Wilesmith JW, Ryan JB, Hoinville LJ,
Hillerton JE, Austin AR, Wells GA. 1996. Transmission dynamics and epidemiology of BSE in British cattle. Nature 382(6594):779-788.
Beekes M, McBride PA, Baldauf E. 1998. Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. Journal of General Virology 79 (Pt 3):601- 607.
Bessen RA, Marsh RF. 1992. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. Journal of General Virology 73 (Pt 2):329-334.
Blakeslee S. 2002, September 3. Brain disease rises in deer, scaring hunters. The New York Times. Science, p. 1.
Brock TD, Madigan MT, Martinko JM, Parker J. 1994. Biology of Microorganisms: Seventh Edition. Englewood Cliffs, N.J.: Prentice Hall.
Brown KL, Stewart K, Ritchie DL, Mabbott NA, Williams A, Eraser H, Morrison WI, Bruce ME. 1999. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nature Medicine 5(11):1308-1312.
Brown P, Bradley R. 1998. 1755 and all that: a historical primer of transmissible spongiform encephalopathy. BMJ 317(7174):1688-1692.
Bruce ME, Will RG, Ironside JW, McConnell I, Drummond D, Suttie A, McCardle L, Chree A, Hope J, Birkett C, Cousens S, Eraser H, Bostock CJ. 1997. Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature 389(6650):498-501.
Bueler H, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. 1993. Mice devoid of PrP are resistant to scrapie. Cell 73(7):1339-1347.
Caughey B. 2002. The PrP conversion process. Presentation to the IOM Committee on Transmissible Spongiform Encephalopathies: Assessment of Relevant Science, Meeting II. The National Academies, Washington, D.C.
Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. 1995. Role of the chaperone protein Hspl04 in propagation of the yeast prion-like factor [psi+]. Science 268(5212):880-884.
Chesebro B. 1998. BSE and prions: uncertainties about the agent. Science 279(5347):42- 43.
Chesebro B. 1999. Prion protein and the transmissible spongiform encephalopathy diseases. Neuron 24(3):503-506.
Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. 1996. Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD. Nature 383(6602):685-690.
Department for Environment, Food and Rural Affairs, United Kingdom. 2002a. Confirmed cases of BSE in the United Kingdom by date of restriction (R) and by date of confirmation (C). [Online] Available: http://www.defra.gov.uk/animalh/bse/bsestatistics/bse/res-con.pdf [accessed October 2002].
Department for Environment, Food and Rural Affairs, United Kingdom. 2002b. Confirmed cases of BSE plotted by month and year of clinical onset. [Online] Available: http://www.defra.gov.uk/animalh/bse/bse-statistics/graphs/epidem.pdf [accessed October 2002].
Dickinson AG, Meikle VM, Fraser H. 1968. Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. Journal of Comparative Pathology 78(3):293-299.
Duffy P, Wolf J, Collins G, DeVoe AG, Streeten B, Cowen D. 1974. Possible person-to-person transmission of Creutzfeldt-Jakob disease. (Letter.) New England Journal of Medicine 290(12):692-693.
Fraser H, Dickinson AG. 1973. Scrapie in mice. Agent-strain differences in the distribution and intensity of grey matter vacuolation. Journal of Comparative Pathology 83(1):29-40.
Gajdusek DC. 1977. Unconventional viruses and the origin and disappearance of kuru. Science 197(4307):943-960.
Gajdusek DC, Zigas V. 1957. Degenerative disease of the central nervous system in New Guinea: the endemic occurence of "kuru" in the native population. New England Journal of Medicine 257(20):974-978.
Gajdusek DC, Gibbs CJ, Alpers M. 1966. Experimental transmission of a kuru-like syndrome to chimpanzees. Nature 209(25):794-796.
Gambetti P, Petersen RB, Parchi P, Chen SG, Capellari S, Goldfarb L, Gabizon R, Montagna P, Lugaresi E, Piccardo P, Bernardino G. 1999. Inherited prion diseases. Prusiner S, Editor. Prion Biology and Diseases. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. Pp. 509-583.
GAO (General Accounting Office). 2002. Mad Cow Disease: Improvements in the Animal Feed Ban and Other Regulatory Areas Would Strengthen U.S. Prevention Efforts. Report GAO-02-183. Washington, D.C.: General Accounting Office.
Gibbs CJ Jr., Gajdusek DC, Asher DM, Alpers MP, Beck E, Daniel PM, Matthews WB. 1968. Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161(839):388-389.
Glasse R. 1967. Cannibalism in the kuru region of New Guinea. Transactions of the New York Academy of Sciences 29:748-754.
Godon KA, Honstead J. 1998. Transmissible spongiform encephalopathies in food animals. Human food safety and animal feed safety concerns for veterinarians. Veterinary Clinics of North America. Food Animal Practice 14(1):49-70.
Griffith, J.S.1967. Self-replication and scrapie. Nature 215(105):1043-1044.
Gross J.E., Miller M.W.2001. Chronic wasting disease in mule deer: disease dynamics and control. Journal of Wildlife Management 65(2):205-215.
Hadlow W. 1959. Scrapie and kuru. Lancet ii:289-290.
Harvard Center for Risk Analysis, Harvard School of Public Health, Center for Computational Epidemiology, College of Veterinary Medicine, Tuskegee University. 2001. Executive summary. Evaluation of the Potential for BSE in the United States. Washington, D.C.: U.S. Department of Agriculture.
Haywood AM. 1997. Transmissible spongiform encephalopathies. New England Journal of Medicine 337(25):1821-1828.
Huillard d'Aignaux JN, Cousens SN, Maccario J, Costagliola D, Alpers MP, Smith PG, Alperovitch A. 2002. The incubation period of kuru. Epidemiology 13(4):402-408.
Johnson RT, Gibbs CJ Jr. 1998. Creutzfeldt-Jakob disease and related transmissible spongiform encephalopathies. New England Journal of Medicine 339(27):1994- 2004.
Kimberlin RH, Hall SM, Walker CA. 1983. Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. Journal of the Neurological Sciences 61(3):315-325.
Klatzo I, Gajdusek DC, Zigas V. 1959. Pathology of kuru. Laboratory Investigation 8:799-847.
Klitzman RL, Alpers MP, Gadjusek DC. 1984. The natural incubation period of kuru and the episodes of transmission in three clusters of patients. Neuroepidemiology 3:3- 20.
Korth C, Stierli B, Streit P, Moser M, Schaller O, Fischer R, Schulz-Schaeffer W, Kretzschmar H, Raeber A, Braun U, Ehrensperger F, Hornemann S, Glockshuber R,
Riek R, Billeter M, Wuthrich K, Oesch B. 1997. Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 390(6655):74-77.
Kretzschmar HA, Honold G, Seitelberger F, Feucht M, Wessely P, Mehraein P, Budka H. 1991. Prion protein mutation in family first reported by Gerstmann, Straussler, and Scheinker. Lancet 337(8750):1160.
Lewin PK. 1972. Scrapie: an infective peptide? [Letter] Lancet 1(7753):748-749.
Lugaresi E, Montagna P, Baruzzi A, Cortelli P, Tinuper P, Zucconi M, Gambetti PL, Medori R. 1986. [Familial insomnia with a malignant course: a new thalamic disease]. Revue Neurologique 142(10):791-792.
Miller MW, Williams ES, McCarty CW, Spraker TR, Kreeger TJ, Larsen CT, Thorne ET. 2000. Epizootiology of chronic wasting disease in free-ranging cervids in Colorado and Wyoming. Journal of Wildlife Diseases 36(4):676-690.
Oldstone MB, Race R, Thomas D, Lewicki H, Homann D, Smelt S, Holz A, Koni P, Lo D, Chesebro B, Flavell R. 2002. Lymphotoxin-alpha- and lymphotoxin-beta-deficient mice differ in susceptibility to scrapie: evidence against dendritic cell involvement in neuroinvasion. Journal of Virology 76(9):4357-4363.
Pattison IH, Jones KM. 1967. The possible nature of the transmissible agent of scrapie. Veterinary Record 80(1):2-9.
Pattison IH, Millson GC. 1961. Scrapie produced experimentally in goats with special reference to the clinical syndrome. Journal of Comparative Pathology 71:101-108.
Prusiner SB. 1982. Novel proteinaceous infectious particles cause scrapie. Science 216(4542):136-144.
Prusiner SB. 1995. The prion diseases. Scientific American 272(1):48-51, 54-57.
Prusiner SB, ed. 1999. Prion Biology and Diseases. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory Press.
Prusiner SB. 2001. Shattuck Lecture-neurodegenerative diseases and prions. New England Journal of Medicine 344(20):1516-1526.
Race R, Oldstone M, Chesebro B. 2000. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. Journal of Virology 74(2):828-833.
Regalado A. 2002, May 24. Medical mystery: growing plague of "mad deer" baffles scientists-epidemic threatens hunting in several states; the risk to humans still unclear-parallels to fatal cow ailment. The Wall Street Journal. A. p. 1.
Rohwer RG. 1991. The scrapie agent: "a virus by any other name." Current Topics in Microbiology and Immunology 172:195-232.
Scott MR, Will R, Ironside J, Nguyen HO, Tremblay P, DeArmond SJ, Prusiner SB. 1999. Compelling transgenetic evidence for transmission of bovine spongiform encephalopathy prions to humans. Proceedings of the National Academy of Sciences of the United States of America 96(26):15137-15142.
Telling GC, Scott M, Mastrianni J, Gabizon R, Torchia M, Cohen FE, DeArmond SJ, Prusiner SB. 1995. Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83(1):79-90.
Weissmann C, Raeber AJ, Montrasio F, Hegyi I, Frigg R, Klein MA, Aguzzi A. 2001. Prions and the lymphoreticular system. Philosophical Transactions of the Royal Society of London. Series B: Biological Sciences 356(1406):177-184.
Wells GA, Scott AC, Johnson CT, Gunning RF, Hancock RD, Jeffrey M, Dawson M, Bradley R. 1987. A novel progressive spongiform encephalopathy in cattle. Veterinary Record 121(18):419-420.
Wilesmith JW, Wells GA, Cranwell MP, Ryan JB. 1988. Bovine spongiform encephalopathy: epidemiological studies. Veterinary Record 123(25):638-644.
Wilesmith JW, Ryan JB, Hueston WD, Hoinville LJ. 1992. Bovine spongiform encephalopathy: epidemiological features 1985 to 1990. Veterinary Record 130(5):90-94.
Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A, Poser S, Pocchiari M, Hofman A, Smith PG. 1996. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347(9006):921-925.
Williams ES, Miller MW. 2002. Chronic wasting disease in deer and elk in North America. Revue Scientifique et Technique 21(2):305-316.
Williams ES, Young S. 1980. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. Journal of Wildlife Diseases 16(1):89-98.
Williams ES, Young S. 1982. Spongiform encephalopathy of Rocky Mountain elk. Journal of Wildlife Diseases 18(4):465-471.
Zeidler M, Stewart G, Cousens SN, Estibeiro K, Will RG. 1997. Codon 129 genotype and new variant CJD. Lancet 350(9078):668.


















