
3
INSECTICIDE TOXICOLOGY
Insecticides offer many benefits such as improved health of humans and animals, increased agricultural productivity, and reduced worldwide hunger. The use and misuse of insecticides, however, have been associated with health risks, environmental contamination, and poisoning (Ecobichon and Joy, 1994; Ecobichon, 2001; Ware, 1989). The toxicity of insecticides has been studied extensively in humans and animals (Ecobichon et al., 1990). Insecticides were used in the Gulf War to control insects that could serve as vectors for disease.
This chapter discusses the toxicity of several insecticides and classes of insecticides believed to have been used in the Gulf War including: organophosphorous compounds, carbamates, pyrethrins and pyrethroids, lindane, and N,N-diethyl-3-methylbenzamide (DEET). Although organophosphorous compounds and carbamates have similar mechanisms of toxicity and many insecticides have neurotoxic effects, it is difficult to summarize the general toxicity of insecticides. Therefore, the chemistry, toxicokinetics, genetic polymorphisms and susceptibilities, mechanism of action, human health effects of acute exposure, experimental data (including animal toxicity data and mutagenicity data), and available information on interactions with other agents are presented separately for each chemical class. Epidemiologic studies of the effects of chronic exposure to insecticides are discussed in the chapters on specific health effects (Chapters 5–9).
Although this chapter focuses on the active ingredients of insecticides, it is important to remember that the toxicity of an insecticide can be altered by its formulation. Agents contributing to formulation of an insecticide are often listed as “inert ingredients” (for example, petroleum products, xylenes, oils, and surfactants), but they can alter the toxicokinetics of an insecticide, potentially increase the absorption of active ingredients, and be toxic themselves (Petrelli et al., 1993; Ware, 1989).
ORGANOPHOSPHOROUS COMPOUNDS
Exposure to organophosphorous compounds can occur under a variety of conditions. Organophosphorous compounds are used as contact insecticides and are applied to crops, gardens, and domestic animals. Some organophosphorous compounds are used as systemic insecticides, in that they are taken up by the roots of plants and disseminated into stems and leaves. Others are used as ophthalmic agents, industrial chemicals (plasticizers and
lubricants), and chemical-warfare agents. Organophosphorous compounds used as contact insecticides during the Gulf War include malathion, diazinon, chlorpyrifos, dichlorvos, and azamethiphos (Abou-Donia, 1995; Chambers and Levi, 1992; Ecobichon, 2001; Kamrin, 1997; Ware, 1989).
Chemistry
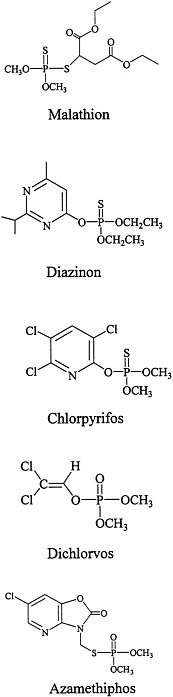
The structures of the organophosphorous compounds used as contact insecticides in the Gulf War are shown in Figure 3.1. Organophosphorous compounds that are used as insecticides contain a pentavalent phosphorus atom connected by esteratic, amide, or sulfur linkages to the organic portions of the molecule.
As esters or amides, organophosphorous compounds are chemically unstable and easily inactivated by hydrolysis. Organophosphorous compounds are lipophilic, some are oily liquids, and others are liquids that can be volatilized (Chambers and Levi, 1992; Ware, 1989).
Toxicokinetics
Toxicokinetics plays an important role in the toxicity of organophosphorous compounds. The oil-water partition coefficient, formulation, and route of exposure can affect the extent of and time needed for absorption. Dermal exposure can increase the time needed for absorption and ensuing toxicity. Almost 100% of the dermally administered dose of some of the highly lipophilic organophosphorous compounds can be absorbed. The potential for percutaneous absorption can be increased if formulations include petroleum products, oils, solvents, or surfactants or if occlusive dressings are placed on the skin (Ware, 1989).
Many organophosphorous compounds are supplied commercially in inactive forms (as protoxicants that need to be activated usually by liver mixed-function oxidases). For most organophosphorous compounds, that requires the change of the phosphorus-sulfur bond to a phosphorus-oxygen bond (for example, malathion needs to be oxidized to malaoxon, chlorpyrifos to chlorpyrifos-oxon, and diazinon to diazoxon). That bioactivation is catalyzed primarily by the P450 system. Dichlorvos and azamethiphos, however, are active without the need for biotransformation.
Detoxification of organophosphorous compounds involves hydrolysis, which can occur spontaneously in an aqueous environment. Hydrolysis can also be catalyzed by aryl and aliphatic hydrolases. Glutathione transferases and cytochrome P450s contribute to the detoxification of some organophosphorous compounds. Metabolic activation and inactivation of organophosphorous compounds occurs primarily in the liver, although other tissues also contribute. Extensive metabolism occurs via multiple pathways, and little, if any, unmetabolized organophosphorous compound is excreted.
Differences in biotransformation of organophosphorous compounds are important contributors to differences in potency and in susceptibility among species and individuals. In addition, the effects of organophosphorous compounds on biotransformation enzymes are important in interactions between the compounds and other chemicals (Ballantyne and Marrs, 1992; Chambers and Levi, 1992; Ecobichon, 2001; Ecobichon and Joy, 1994; Gallo and Lawryk, 1991).
Genetic Polymorphisms and Susceptibility
Organisms can differ in the amounts and activities of their B esterases, and the differences affect susceptibility to organophosphorous compounds. Although acetylcholinesterase shows relatively little variation in structure and activity among individuals of a species, other esterases that interact with organophosphorous insecticides differ widely among individuals in a population. For example, several variants of pseudocholinesterase have been noted in human serum, with distinguishing differences related to capability for interaction with particular molecules (for example, succinylcholine and fluoride). A single clinical case report noted that atypical pseudocholinesterase was found in a soldier who suffered adverse effects when exposed to an acetylcholinesterase inhibitor during the Gulf War (Loewenstein-Lichtenstein et al., 1995). Differences in pseudocholinesterase activities did not, however, differentiate between symptomatic and asymptomatic Gulf War veterans when more subjects were studied (Kurt, 1998).
Differences in the biotransformation of organophosphorous compounds play a role in susceptibility to them in organisms of different ages and species. The young are generally more susceptible to acetylcholinesterase inhibition because they are less likely to convert organophosphorous compounds into nontoxic metabolites. Apart from age, different species have different capabilities for organophosphorous biotransformation; for example, avians can be 10 times as susceptible as mammals.
Genetic polymorphisms of A esterases (arylesterases) might play a role in susceptibility of humans and animals to organophosphorous compounds. Although other A esterases exist, the most studied are the paraoxonases, which metabolize, in addition to paraoxon, chlorpyrifos-oxon and diazoxon, the active metabolites of chlorpyrifos and diazinon, respectively, two organophosphorous insecticides used in the Gulf War. At least three gene products exist for paraoxonase. One of the gene products, paraoxonase-1 (PON1), has at least two isozymes (Q, formerly referred to as A; and R, formerly referred to as B). Those isozymes differ in their ability to metabolize organophosphorous insecticides. Population studies have demonstrated a trimodal distribution of paraoxonase activity, reflecting QQ, RR, and QR individuals. Reported individual differences in Q activities suggest that such differences contribute to the varied responses to environmental organophosphorous compounds in people and animals (Brophy et al., 2001; Cowan et al., 2001; Hernandez et al., 1999; La Du et al., 1999). In a small sample of Gulf War veterans, individuals with the neurologic symptom complexes were more likely to have the R allele (heterozygous QR or homozygous R) than to be homozygous Q for the allele (Haley et al., 1999). Animal studies also demonstrate the role of PON1 in organophosphorous metabolism and the varying activity of the differential isozymes. PON1 knockout mice (mice without PON1) were found to be very sensitive to the toxicity of organophosphorous compounds, and following introduction of the enzyme to the knockout animals, the sensitivity to specific organophosphorous compounds varied with the isoform given to the animal. Animals given the Q isozyme were less sensitive to diazoxon while animals given the R isozyme were less sensitive to chlorpyrifos-oxon and paraoxon. It is important to note, however, that although mouse and human Q isoforms are similar, the catalytic efficiencies of their R isozymes differ (Furlong et al., 2000; Li et al., 2000).
Mechanism of Action
Organophosphorous insecticides kill insects by affecting their nervous system. Specifically, they inhibit acetylcholinesterase, the enzyme that is responsible for the breakdown of acetylcholine and therefore the termination of its activity. Acetylcholine is a neurotransmitter that acts at two major receptor subtypes, nicotinic and muscarinic. Binding of acetylcholine to receptors at neuromuscular synapses leads to the activation of muscles. Failure to break down acetylcholine results in sustained activity and consequent overstimulation of cholinergically-mediated synapses, particularly nicotinic neuromuscular synapses, muscarinic parasympathetic synapses, and cholinergic synapses of the central nervous system (CNS). That mechanism of toxicity is the same in mammals, birds, and fish, so the acute effects are similar in humans and animals (Ecobichon, 2001).
Organophosphates inhibit acetylcholinesterase when the oxygen with the coordinate bond on the organophosphate molecule (see Figure 3.1) binds to the esteratic site of the acetylcholinesterase enzyme. That binding is initially reversible, but within a matter of minutes, part of the organophosphate molecule may be cleaved from the phosphorus group, and the remainder of the molecule will become essentially irreversibly attached at the esteratic site of the enzyme. The production of the essentially irreversible bond is called aging (Abou-Donia, 1995; Ballantyne and Marrs, 1992; Chambers and Levi, 1992; Ecobichon, 2001; Ecobichon and Joy, 1994; Gallo and Lawryk, 1991; Marrs, 1996). Once aging has occurred, enzyme activity can be recovered only with the synthesis of new enzyme.
Acute Human Exposures
Immediate Effects
Clinical signs of toxicity associated with organophosphate-induced inhibition of acetylcholinesterase depend on dosage. Toxicity in humans and animals includes the signs associated with overstimulation of muscarinic receptors of the autonomic nervous system by acetylcholine (SLUD—salivation and sweating, lacrimation, urination, and defecation—as well as emesis and bradycardia). Acetylcholinesterase inhibition can also cause overstimulation (which can be followed by depression) of nicotinic receptors at neuromuscular junctions and autonomic ganglia and result in ataxia and fasciculations that, at higher dosages, can be followed by flaccid paralysis. Electromyographic changes can be observed after acute poisoning because nicotinic sites in muscles are affected; the changes include decreases in amplitude and increases in peak latencies in nerve conduction (Baker and Wilkinson, 1990; Gallo and Lawryk, 1991; Kaloianova and El Batawi, 1991). Stimulation of autonomic ganglia can also cause hypertension. As is the case at neuromuscular junctions, excess acetylcholine in the CNS causes stimulation that can be followed by depression. Overstimulation can be manifested as nervousness, delirium, hallucinations, and psychoses. Obvious signs do not generally appear until nervous system acetylcholinesterase inhibition approaches 70%.
Not all exposed people show all signs, and signs can vary with the organophosphorous compound, dose, route of exposure, and species. Signs often appear within minutes or hours, but they might not appear for several days. Duration can vary from minutes to weeks and can be followed by full recovery from obvious manifestations of
cholinergic poisoning. If death occurs, it is due to respiratory failure, usually as a result of a combination of the autonomic effects mediated by the muscarinic and nicotinic acetylcholine receptors and the effects of acetylcholine at CNS receptors. Those effects include excessive fluid in the respiratory tract, paralysis of the respiratory muscles, and depression of the respiratory centers of the CNS.
Of the organophosphorous insecticides shipped from the United States to the Gulf War, oral lethal doses (LD50 values, doses that kill 50% of the animals tested) are highest for malathion (about 1 g/kg), intermediate for diazinon and chlorpyrifos (about 150–250 mg/kg), and lowest for dichlorvos (about 50 mg/kg) (Abou-Donia, 1995; Ballantyne and Marrs, 1992; Brown and Brix, 1998; Cecchine et al., 2000; Chambers and Levi, 1992; Ecobichon, 2001; Ecobichon and Joy, 1994; Gallo and Lawryk, 1991; Kaloianova and El Batawi, 1991; Lotti, 2001; Marrs, 1996; Ware, 1989).
Diagnosis of organophosphorous-induced acute toxicity is based on exposure history, clinical manifestations of acetylcholinesterase inhibition, and laboratory findings. Erythrocyte acetylcholinesterase activity is used as an indicator of enzyme status in the nervous system. Metabolites of organophosphorous compounds to which humans and animals are exposed can also be detected in urine. Toxicity is unlikely to be overt unless blood acetylcholinesterase is substantially decreased (for example, by at least 50%; 70% inhibition is more likely to be correlated with clinical signs). Response to administration of atropine, an anticholinergic agent, has also been used as a diagnostic tool: poisoned organisms will not respond to atropine at doses that a nonpoisoned organism will respond to but require doses about 10 times higher before the expected pupil dilation, increased heart rate, and decreased secretions are noted (Ballantyne and Marrs, 1992; Ecobichon and Joy, 1994; Gallo and Lawryk, 1991; Marrs, 1996).
Treatment for organophosphorous-caused acetylcholinesterase inhibition includes administration of atropine to antagonize acetylcholine stimulation of muscarinic receptors and administration of an oxime (such as pralidoxime) to regenerate acetylcholinesterase that is inhibited but not yet irreversibly bound. In the Gulf War and elsewhere, a carbamate compound (pyridostigmine bromide) has been used prophylactically when exposure to organophosphorous nerve gases was expected, because it inhibits but does not age the enzyme and so provides time for clearance of the organophosphate before sites on acetylcholinesterase are available to bind it irreversibly. Other treatments for acute acetylcholinesterase inhibition are not specific and consist of decreasing absorption, enhancing excretion, and addressing symptoms. Time is needed for recovery of acetylcholinesterase activity after aging because recovery requires synthesis of new enzyme. Weeks of supportive treatment might be needed if acetylcholinesterase remains sufficiently inhibited to cause signs of cholinergic poisoning (Ballantyne and Marrs, 1992; Ecobichon, 2001; Ecobichon and Joy, 1994; Feldman, 1999; Gallo and Lawryk, 1991; Lotti, 2001).
Delayed Effects
Tolerance. Tolerance can occur after repeated exposure to cholinesterase-inhibiting organophosphorous insecticides. In general, tolerance can develop due to prolonged stimulation of cholinergic receptors by acetylcholine. Those receptors no longer respond as effectively to the neurotransmitter. Tolerance is more likely to occur at muscarinic than at nicotinic receptors and can develop when erythrocyte acetylcholinesterase is low but cholinergic poisoning is not overt (Bushnell et al., 1993).
Tolerance can also occur when there is an increase in the proteins other than acetylcholinesterase to which organophosphorous compounds can bind. The alternative binding sites protect animals from organophosphate-induced acetylcholinesterase inhibition. Such sites include other esterases, notably pseudocholinesterase and carboxylesterases that are found in serum, liver, and other tissues. Although their precise physiologic role is unknown, those esterases can be involved in the metabolism of drugs and other compounds that contain ester and amide groups. Quantities of the alternative esterases, especially the carboxylesterases, depend on age, tissue, species, and exposure to agents that induce or inhibit enzymes. Agents that induce or inhibit enzymes include a number of drugs and other foreign compounds. When the esterases are induced, animals and humans are likely to be less susceptible to some of the organophosphorous compounds used as insecticides (Ballantyne and Marrs, 1992; Ecobichon, 2001; Gallo and Lawryk, 1991).
Intermediate syndrome. Clinical manifestations of acute acetylcholinesterase inhibition in humans or animals are not generally long-lasting or delayed, but there are exceptions. An “intermediate syndrome” has been described after severe poisoning: muscle weakness that occurs about 16 to 120 hours after exposure and 7 to 75 hours after the onset of acute poisoning symptoms (He et al., 1998; Shailesh et al., 1994). Overstimulation of nicotinic receptors followed by depression at neuromuscular junctions and muscle necrosis might be contributing factors. The muscle weakness can become severe and result in respiratory insufficiency. If respiration can be sustained, recovery occurs, although it can take weeks. Intermediate syndrome has been reported in humans after exposure to malathion and diazinon (Gallo and Lawryk, 1991).
Organophosphorous-induced delayed neuropathy. Another type of toxicity caused by a few organophosphorous compounds is a progressive, irreversible delayed neuropathy termed organophosphate-induced delayed neuropathy (OPIDN). OPIDN can occur in many species, including humans. Clinical manifestations of OPIDN include progressive, irreversible ataxia that develops weeks to months after exposure. Lesions are found in peripheral nerves and the spinal cord (Ehrich and Jortner, 2001).
OPIDN occurs only if organophosphorous compounds sufficiently, and essentially irreversibly, inhibit neuropathic target esterase (NTE) within hours of exposure. Inhibition of NTE is not related to inhibition of acetylcholinesterase, and organophosphorous compounds used as contact insecticides generally do not inhibit NTE. Organophosphorous compounds are tested for their potential to cause OPIDN before they are registered for use as insecticides, so most commercially available insecticides do not inhibit NTE. Commercially available insecticides that do inhibit NTE, such as chlorpyrifos and dichlorvos, do so only at doses that are sufficient to cause lethal cholinergic poisoning. OPIDN can occur only after rescue from acute chlorpyrifos or dichlorvos poisoning; even then it might not occur. At least six cases of OPIDN have been documented after ingestion of near-lethal doses of chlorpyrifos or dichlorvos (Aiuto et al., 1993; Lotti et al., 1986; Martinez-Chuecos et al., 1992; Vasilescu and Florescu, 1980); all but one occurred after unsuccessful suicide attempts. The absence of documented cases of OPIDN after exposure to diazinon or malathion is consistent with their lack of NTE inhibition in animal models. In fact, the chemical structures of malathion, diazinon, and azamethiphos make them exceedingly unlikely to inhibit NTE at all, and OPIDN has not been produced in experimental animals exposed to either malathion or diazinon (Ballantyne and Marrs, 1992; Cecchine et al., 2000; Chambers and Levi, 1992; Ecobichon, 2001; Ecobichon and Joy,
1994; Ehrich and Jortner, 2001; Gallo and Lawryk, 1991; Johnson and Glynn, 2001; Kamrin, 1997; Lotti, 2001; Richardson, 1995).
Other delayed effects. Some studies have reported other persistent symptoms after poisoning with organophosphorous compounds or symptoms that appear 5–10 years after a poisoning episode, including neurologic and visual deficits, behavioral alterations, and impairment of cognition. Those effects, however, might be confounded by other factors or be the result of inappropriate study designs (Abou-Donia, 1995; Baker and Wilkinson, 1990; Chambers and Levi, 1992; Ecobichon and Joy, 1994; Eyer, 1995; Gallo and Lawryk, 1991; Jamal, 1997; Kaloianova and El Batawi, 1991; Lotti, 2001). Although some latent effects have been noted in laboratory rats, the symptoms reported in people have been difficult to verify in animal studies partly because of difficulties in replication of exposures and extrapolation of end points from humans to animals (Ballantyne and Marrs, 1992; Bushnell et al., 1993; Ecobichon and Joy, 1994; Gallo and Lawryk, 1991; Marrs, 1996; Mattsson et al., 1996; Maurissen et al., 2000).
Experimental Data
Neurotoxic Effects
As noted above, organophosphorous insecticides increase levels of the neurotransmitter acetylcholine in both the central and peripheral nervous systems. Excess acetylcholine at neuromuscular junctions causes excessive neuromuscular stimulation (such as tremors), which can be followed by neuromuscular block. Excess acetylcholine at synapses of the autonomic nervous system affects quantity of secretions, heart rate, blood pressure, gastrointestinal function, urination, and pupil size. Excess acetylcholine at synapses of the CNS can alter behavior and cognition. Studies in animals generally require substantial inhibition of acetylcholinesterase (for example, greater than 40% inhibition of erythrocyte acetylcholinesterase) before those effects are seen. Even when doses of organophosphorous compounds are sufficient to cause notable evidence of cholinergic poisoning in animals or people, it is unusual for signs and symptoms to continue after recovery of acetylcholinesterase activity (Ballantyne and Marrs, 1992; Cecchine et al., 2000; Chambers and Levi, 1992; Ecobichon, 2001; Ecobichon and Joy, 1994; Eyer, 1995; Gallo and Lawryk, 1991; Marrs, 1996; Mattsson et al., 1996).
There has also been discussion around the potential effects of organophosphorous compounds on learning and memory following fetal and childhood exposures. In animals, however, some experiments have not demonstrated an increased sensitivity during the developing periods. Mattsson and colleagues (2000) treated rats with chlorpyrifos (0.3, 1.0, and 5.0 mg/kg/day) from gestational day 6 to postnatal day 10 and measured chlorpyrifos concentrations and cholinesterase inhibition in the fetuses and the dams. The nursing pups had a lower concentration than the dams and cholinesterase activity in all tissues of the high-dose pups rapidly returned to near control levels. Another study in rats with the same dosing regimen examined learning and memory, but no effects were seen in the absence of maternal toxicity (Maurissen et al., 2000).
Animal studies have been conducted to assess the persistence of neurotoxic effects of organophosphorous compounds. The results of a study of chlorpyrifos (at 1, 3, and 10 mg/kg per day, 5 days/week for 4 weeks followed by 4 weeks of recovery) do not indicate effects on short-term memory in adult rats, but do indicate a decrease in motor activity (Maurissen
et al., 2000). Other reports noted locomotor reduction shortly after cessation of exposure and partial recovery of acetylcholinesterase inhibition in preweanling rats (Carr et al., 2001), long-term effects on cognitive end points in neonatally-exposed rats (Levin et al., 2001), and impairment of learning in preweanling rats and in rats immediately after weaning without regional brain acetylcholinesterase inhibition (Jett et al., 2001). Additional studies with relatively low, but cholinesterase-inhibiting, doses of other organophosphorous compounds have revealed behavioral and learning dysfunction in rats and in monkeys, especially after chronic administration (Eriksson and Talts, 2000; Prendergast et al., 1997, 1998).
In addition to their action as esterase inhibitors, some organophosphorous compounds have been reported to directly stimulate cholinergic receptors, including receptors of the heart and the nervous system, although concentrations might be higher than those needed for acetylcholinesterase inhibition (Chambers and Levi, 1992; Pope and Liu, 2001; Richardson, 1995).
Carcinogenicity
Organophosphorous insecticides, including those used in the Gulf War, are generally not considered carcinogenic. Long-term rodent studies of dichlorvos and malathion, however, have yielded mixed results (see ATSDR, 1997, 2001a; IARC, 1983, 1991; Kamrin, 1997; Van Maele-Fabry et al., 2000 for reviews).
Early studies in rats and mice did not show an increased incidence of tumors attributable to dichlorvos treatment (Blair et al., 1976; Horn et al., 1987, 1988). The National Toxicology Program (NTP) investigated the carcinogenicity of dichlorvos in feed in Osborne-Mendel rats (at 150 and 326 ppm) and B6C3F1 mice (at 318 and 635 ppm) (NTP, 1977); no evidence of increased tumor incidence attributable to dichlorvos was seen. More recently, NTP examined the carcinogenicity of dichlorvos given by gavage in F344/N rats (at 4 and 8 mg/kg per day, 5 days/week for 103 weeks in males and females) and B6C3F1 mice (males, at 10 and 20 mg/kg per day, 5 days/week for 103 weeks; females, at 20 and 40 mg/kg per day, 5 days/week for 103 weeks) (Chan et al., 1991; NTP, 1989). Some increased incidences of neoplastic effects were seen: in rats, adenomas of the exocrine pancreas (males and females), mononuclear cell leukemia (males), mammary gland fibroadenomas (females), combined fibroadenomas or adenomas (females), and multiple fibroadenomas (females); and in mice, squamous cell papillomas of the forestomach (males and females). In addition, two female mice developed forestomach squamous cell carcinomas. NTP concluded that there was “some evidence of carcinogenic activity of dichlorvos” in male F344/N rats and male B6C3F1 mice, “equivocal evidence of carcinogenic activity of dichlorvos” in female F344/N rats, and “clear evidence of carcinogenic activity of dichlorvos” in female B6C3F1 mice (Chan et al., 1991; NTP, 1989).
Evaluation of available data by the International Agency for Research on Cancer (IARC) resulted in the conclusion that there was inadequate evidence of the carcinogenicity of dichlorvos in humans but sufficient evidence in experimental animals. Therefore, dichlorvos was classified as a possible human carcinogen (Ballantyne and Marrs, 1992; IARC, 1991; Kamrin, 1997). A recent review of studies on the carcinogenicity of dichlorvos examining the length of the studies, confounding factors, and potential for bias has led the Health Council of Belgium to conclude that there is only sparse evidence that dichlorvos is carcinogenic in experimental animals and that it is not classifiable as to carcinogenicity in humans (Van Maele-Fabry et al., 2000).
Animal studies of the carcinogenicity of malathion also have produced mixed results (see summaries in ATSDR, 2001a). In feeding studies conducted by the National Cancer Institute (NCI), no evidence of carcinogenicity was seen in Osborne-Mendel rats (at about 359 and 622 mg/kg per day for 80 weeks) (NCI, 1978), B6C3F1 mice (at about 1490 and 2980 mg/kg per day for 80 weeks) (NCI, 1978), and Fischer 344 rats (at about 166 and 322 mg/kg per day for 103 weeks) (NCI, 1979a). In a 2-year unpublished study in Fischer 344 rats with a wider dose range (2–868 mg/kg per day), however, some evidence of hepatocarcinogenicity (a statistical trend) was seen in female rats (Daly, 1996). Slauter (1994) treated B6C3F1 mice with 17.4–3448 mg/kg per day for 80 weeks. They saw an increase in the incidence of hepatocellular tumors with a positive dose trend, in both male and female mice at the two highest doses.
NCI also investigated the effects of malaoxon, the active metabolite of malathion, in Fischer 344 rats (at about 41 and 82 mg/kg per day for 103 weeks) and B6C3F1 mice (at about 91 and 182 mg/kg per day for 103 weeks) (NCI, 1979b). There was an increase in C-cell adenomas and carcinoma of the thyroid in female rats, but historical-control data led NCI to conclude that there was no evidence of carcinogenicity attributable to malaoxon (NCI, 1979b). NTP has since re-evaluated the histopathologic findings in the NCI studies (NCI 1978, 1979a,b) and concurred with most of the NCI conclusions, but concluded that for C-cell neoplasms of the thyroid gland after malaoxon treatment there is “equivocal evidence of carcinogenicity” in male and female Fischer 344 rats (Huff et al., 1985).
A 1983 IARC evaluation of malathion concluded that available data provided no evidence that malathion was carcinogenic in animals, and that it was unlikely to present a carcinogenic risk to humans (IARC, 1983). An EPA review of more recent information provided when malathion was evaluated for reregistration, however, resulted in the conclusion that there was “suggestive evidence of carcinogenicity but not sufficient to assess human carcinogenic potential”; this was based on the appearance of liver tumors in rodents that were given doses of malathion considered excessive (US EPA, 2000).
Mutagenicity and Genotoxicity
Kamrin (1997) summarized results of mutagenicity tests of chlorpyrifos, diazinon, malathion, and dichlorvos. Mutagenicity tests of diazinon yielded inconclusive results, but malathion produced detectable mutagenesis in three types of cultured human cells. Dichlorvos binds to DNA and has been demonstrated to be mutagenic in vitro but not in vivo. Mutagenicity and genotoxicity tests yielded no evidence that chlorpyrifos has such activity (Gollapudi et al., 1995).
Reproductive and Developmental Effects
Organophosphorous insecticides have not historically been considered to be female reproductive or developmental toxicants at dosages lower than would cause acute maternal toxicity in mammals, although teratogenesis has been reported in fish and birds and endocrine changes in women (Baker and Wilkinson, 1990; Ballantyne and Marrs, 1992; Breslin et al., 1996; Kamrin, 1997).
Embryotoxicity, as indicated by decreases in body weight and skeletal size and a lag in development, has been reported in mice after administration of malathion at about 15–50% of the oral LD50 values, but no indication of maternal toxicity was provided (Asmatullah et al., 1993).
Decreased pup weight and increased pup mortality were reported after administration of chlorpyrifos to rats. That occurred when pregnant female rats were exposed to chlorpyrifos at doses that caused maternal toxicity. Acetylcholinesterase inhibition was indicated by excessive salivation and tremors (Breslin et al., 1996). Decreased pup weight and increased pup mortality have also been reported in rats exposed to acetylcholinesterase-inhibiting doses of chlorpyrifos between birth and weaning (Carr et al., 2001). Biochemical changes other than acetylcholinesterase inhibition have been reported in neonatal rats exposed to chlorpyrifos, including changes in protein synthesis, DNA synthesis, intracellular signaling, and cholinergic receptors (Dam et al., 1998; Song et al., 1997; Tang et al., 1999; Whitney et al., 1995). Changes in righting reflex, cliff avoidance, locomotor activity, and spatial learning have been reported in neonatal, weanling, and juvenile rats exposed to chlorpyrifos at doses expected to inhibit acetylcholinesterase activity; some detriments occurred without notable enzyme inhibition or continued after substantial recovery of esterase activity (Carr et al., 2001; Chanda and Pope, 1996; Jett et al., 2001). The significance of behavioral changes in young rats with regard to possible toxicity in adult animals or in other species is unknown.
Immunotoxic Effects
The modulation of the immune system by malathion and its impurities depends on the dose, specific agent, cellular target, and duration of exposure; both stimulatory and suppressive effects have been reported in exposed animals. Dichlorvos has been reported to have suppressive effects on the generation of macrophages on chronic exposure and the ability to suppress cellular and humoral immune responses at cholinergic doses (Rodgers, 2001).
Other Health Effects
Dermatitis and hypersensitivities, including bronchospasm, have been reported after exposure to organophosphorous insecticides. The contributions of contaminants and vehicles to those responses have not been differentiated from effects of the active ingredients alone. Transient effects of malathion and dichlorvos on the immune system, including hypersensitivity and dermatitis, have been reported (Baker and Wilkinson, 1990; Chambers and Levi, 1992; Gallo and Lawryk, 1991; Kaloianova and El Batawi, 1991). Chlorpyrifos has been reported to increase lymphocyte numbers (Richardson, 1995).
Effects on respiratory, cardiac, and gastrointestinal systems in humans and animals are related to the ability of the insecticides to inhibit acetylcholinesterase and increase acetylcholine-mediated neural transmission (Ballantyne and Marrs, 1992). Some organophosphorous compounds—but not the insecticides used in the Gulf War—have been reported to have endocrine effects, including dysregulation of hypothalamic releasing factors when acetylcholinesterase was substantially inhibited (Smallridge et al., 1991), decreased spermatogenesis (Somkuti et al., 1991), increased estrogen metabolism (Berger and Sultatos, 1997), and antagonism at androgen receptors (Tamura et al., 2001).
Interactions with Other Agents
Organophosphorous insecticides can inhibit esterases other than acetylcholinesterase, including pseudocholinesterase and carboxylesterases in both humans and animals.
Inhibition of those esterases can decrease the biotransformation of ester and amide drugs used in human and veterinary medicine—for example, local anesthetics, the neuromuscular blocker succinylcholine, such carbamates as neostigmine and pyridostigmine, and organophosphorous compounds used as ophthalmic agents and antiparasitic drugs. That inhibition would increase the duration of action of the drugs; sufficient inhibition of those esterases has the potential to result in drug toxicity (Ballantyne and Marrs, 1992; Chambers and Levi, 1992; Ecobichon, 2001; Ecobichon and Joy, 1994; Gallo and Lawryk, 1991; IOM, 2000).
Interactions are seen between organophosphorous insecticides and carbamates, another class of insecticides that also act by inhibiting acetylcholinesterases. The outcome of such interactions depends on dosage and sequence of exposure. Pretreatment with relatively low doses of carbamates will protect humans and animals from the irreversible acetylcholinesterase-inhibiting effects that can follow later exposure to organophosphorous compounds. That was the basis of the prophylactic use of the carbamate pyridostigmine bromide against organophosphorous nerve agents in the Gulf War (IOM, 2000). But simultaneous exposure to organophosphorous compounds and carbamates can result in exaggeration of acetylcholinesterase inhibition, which can also occur when exposure to carbamates follows exposure to organophosphorous compounds. Exposure to carbamates can also exaggerate the delayed neuropathy caused by some organophosphorous compounds (Ehrich and Jortner, 2001; Gallo and Lawryk, 1991).
It has long been known that the toxicity of cholinesterase inhibitors can increase with sequential exposure to cholinesterase inhibitors or other compounds detoxified by the same metabolic pathways. The enzymes that detoxify organophosphorous-insecticides are inhibited following the initial exposure, and would not be available for detoxification of a second organophosphorous insecticide (or other substrate) applied hours or days later because enzyme activity recovers slowly (Gallo and Lawryk, 1991). Interference with metabolism was thought to contribute to interactions seen in hens exposed to compounds used in the Gulf War, including chlorpyrifos. An increase in neurotoxicity (effects on locomotion and some neuropathologic changes) was seen when hens were treated with chlorpyrifos in combination with pyridostigmine, DEET, or both compared to hens treated with chlorpyrifos only (Abou-Donia et al., 1996). Biochemical alterations included a modest exaggeration in the decline of brain esterase activity when combinations of toxicants were used. The toxicity observed was not OPIDN but might have been the result of the combination of chemicals that have intrinsic neurotoxic potential. It should be noted, however, that the doses used were higher than those expected with human exposures and that hens are particularly sensitive to the neurotoxic effects of organophosphorous insecticides.
CARBAMATES
Chemistry
The use of carbamate insecticides began in the 1950s. About 50 carbamate compounds are in use today as insecticides or pharmaceuticals. Three carbamates were sent to the Gulf War: carbaryl, propoxur, and methomyl. Carbamates are N-substituted esters of carbamic acid. The structure of carbaryl, the prototype carbamate insecticide, is shown in
Figure 3.2. The insecticidal carbamates were synthesized as analogues of physostigmine, the first carbamate, a toxic anticholinesterase alkaloid extracted from the calabar bean, the seed of the plant Physostigma venenosum.
Toxicokinetics
Carbamates are absorbed dermally and from the gastrointestinal tract. They are also readily absorbed after inhalation at temperatures at which vapors are formed. The rate and extent of absorption depend on the vehicle; oil vehicles virtually double oral toxicity (Goncharova, 1968). LD50 values are far lower with parenteral than with oral administration (Rybakova, 1966; Yakim, 1967). Once absorbed, the carbamates are distributed rapidly to tissues. The rate of elimination differs by route of exposure, but labeled compound or metabolite can be detected in blood, urine, and feces.
Biotransformation of carbaryl has been reported to be similar in humans, rats, guinea pigs, monkeys, and sheep, with the major difference being the extent to which carbaryl is hydrolyzed to yield 1-naphthol. It appears to be the primary metabolite (Baron, 1991). Other water-soluble metabolites, including unidentified conjugates that could be hydrolyzed by acid to thioethers, have been identified in the urine and bile of rats that received intravenous or intraperitoneal ring- or carbonyl-labeled carbaryl. In a study of rats, those metabolites accounted for up to 32% of the dose secreted in bile. Some evidence also suggests that carbaryl is oxidized to CO2 in rats, guinea pigs, and humans. Small quantities of some intermediate metabolites of carbaryl are excreted in cows’ milk.
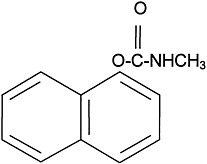
FIGURE 3.2 Structure of carbaryl.
Carbamate metabolism and excretion is relatively rapid. Mammals given naphthyl-labeled carbaryl, for example, excreted 68–74% of the label in urine and 2–11% in feces within 24 h of administration. Rats dosed with N-methyl[14C]carbaryl eliminated 12–24% of the label in exhaled air and 53–54% in urine within 48 h. In another experiment, rats given carbonyl-labeled carbaryl eliminated 34–45% of the label in urine and 8% in feces within 24 h of administration; 30% was exhaled as 14CO2 (see Baron, 1991 for review). Intratracheal instillation of radioactive carbaryl as an aerosol produced peak activity in blood within 2–5 min in rats; 90% of the radioactivity was recovered in urine and 2–5% in feces within 3 days. In a gavage study in mice, 69% of a dose of carbaryl was absorbed within 60 min, with a half-life (t1/2) for absorption of 17 min. Peak blood concentrations occurred within 35–40 min after administration. By 60 min after administration, 16.9% of the dose appeared in urine and 8.6% in exhaled CO2. In rats that received [14C]carbaryl by gavage, the percentage of the administered dose recovered per gram of tissue ranged from less than 0.1% to 0.4% after 11 h; the highest concentrations were in liver, kidneys, and fat. Following an acute oral exposure of rats to carbaryl, it could be detected in the liver, brain, and heart 48 h after exposure.
Genetic Polymorphisms and Susceptibility
Genetic polymorphisms and susceptibility have not been systematically explored with respect to carbamates. Two reports in humans might be pertinent. A pilot case-control study looked at the association between environmental exposure and acute leukemia in infants who were either positive or negative for a translocation involving chromosome band llq231 (that is, MLL+ve and MLL−ve infant acute leukemia). The data from that study indicate that fetal exposure to carbamate-based insecticides in some settings increased the odds ratio for infant acute leukemia but only in the presence of MLL+ve rearrangements (Alexander et al., 2001). Those data suggest that MLL+ve people might be more susceptible to carbamates. However, other mosquitocides might have been present, and the authors note limitations of the study, including the small number of cases and the potential nonrepresentativeness of controls. In addition, Loewenstein-Lichenstein and colleagues (1995) reported a soldier homozygous for “atypical” butyrylcholinesterase who experienced severe symptoms after pyridostigmine prophylaxis during the Gulf War.
In a rodent study, females exhibited greater sensitivity to carbaryl than males (Gaines, 1969).
Mechanism of Action
As with the organophosphorous insecticides, carbamate insecticides act by inhibiting acetylcholinesterase, the enzyme responsible for the breakdown and therefore the termination of the activity of acetylcholine. Failure to break down acetylcholine results in sustained effects of this neurotransmitter and consequent overstimulation of cholinergically mediated synapses, particularly nicotinic neuromuscular synapses, muscarinic parasympathetic synapses, and cholinergic synapses of the CNS (Ecobichon, 2001).
In contrast with the phosphorylation that occurs with organophosphorous insecticides, carbamylation of cholinesterase is reversible. Regeneration of the enzyme activity occurs within a few hours: the carbamate is cleaved and loses its ability to inhibit anticholinesterase.
Acute Human Exposures
As would be anticipated from its mechanism of action, the symptoms of carbamate-insecticide poisoning are similar to those of organophosphorous-insecticide poisoning. The clinical symptoms of acute exposure to carbaryl are derived from its effects on acetylcholine synapses, including actions at the synapses of the CNS and neuromuscular junctions, sensory nerve endings, ganglionic synapses of the parasympathetic and sympathetic nervous system, postganglionic sympathetic nerve terminals innervating sweat glands and blood vessels, sympathetic nerve terminals in the adrenal medulla, and postganglionic parasympathetic nerve terminals. Specifically, the effects stem from the accumulation of acetylcholine at those synapses. Carbamate toxicity typically involves the nervous and respiratory systems. Effects of peripheral muscarinic stimulation include increases in bronchial secretions and bronchoconstriction; excessive sweating, salivation, and lacrimation; pinpoint pupils; bradycardia; and vomiting and diarrhea. Effects of peripheral
nicotinic stimulation include fine-muscle fasciculations and tachycardia. Various CNS manifestations also occur, including headache, respiratory depression, dizziness, anxiety, and mental confusion. Effects can progress to convulsions and coma (Ecobichon, 2001).
The combination of signs and symptoms that are exhibited depends on the specific chemical and the dose, duration, and route of exposure. Mild exposures typically produce only muscarinic and nicotinic signs, but severe exposures evoke CNS signs and pulmonary edema. Adverse effects can occur after dermal, inhalation, or ingestion exposure (Ecobichon, 2001).
The effects of carbamates are typically of shorter duration and milder than those of the organophosphorous compounds. Carbamates are not associated with OPIDN, but death has resulted from intentional administration of high doses of carbaryl (Ecobichon, 2001).
Experimental Data
Neurotoxic Effects
Because of its anticholinesterase activity, the transient effects of carbamates on the nervous system resemble those of cholinergic stimulation. In addition, acute exposures appear to change a variety of neurotransmitter systems in the CNS. Thresholds of such effects have not been determined.
Dietary exposure of swine to carbaryl resulted in a carbamate-induced syndrome that included neurotoxicity (Smalley, 1970; Smalley et al., 1969). Administration at 125–300 mg carbaryl/kg per day administered via the diet for 72–83 days resulted in myasthenia, incoordination, ataxia, tremor, muscle contractions, paraplegia, and prostration. At the higher dosages, those effects had a more rapid onset. The authors observed some recovery after exposure, but the effects recurred with micropathologic findings in the nervous system (Smalley, 1970; Smalley et al., 1969).
Carbamate exposure has behavioral consequences. Few systematic studies have addressed the behavioral effects, however, and they generally have been carried out with end points not considered especially sensitive. For example, acute exposure via various routes appears to decrease locomotor activity as measured in several devices. Decreases in wheel-running activity of rats were noted after acute exposure to carbaryl at only 0.56–2.24 mg/kg administered intraperitoneally. This was less than 4% of the LD50, and the effect was reversed by atropine (Singh, 1973). Acute exposure results in persistent behavioral effects, including reductions in motor activity noted 72 h after oral doses of 20, 75, and 150 mg/kg (Moser, 1995). Those doses represent 9, 33, and 65% of the reported LD50 for oral exposure of rats. Even at the lowest dose studied, autonomic function, motor activity, neuromuscular function, sensorimotor function, and reactivity are altered. An acute dose of only 10 mg/kg increased tolerance to electric shock and thereby attenuated the suppression of operant responding that also resulted in electric shock (Sideroff and Santolucito, 1972). Somewhat longer exposures to carbaryl may have the opposite effect on locomotor activity, reportedly increasing open-field activity and lowering the rate of habituation relative to controls.
An apparently unanswered question with respect to the carbamate insecticides is their effect on cognitive functions, including learning and memory. In a study of acute exposure to carbaryl in monkeys, dose-related decreases in accuracy (increases in errors) were observed in the repeated-learning paradigm after intramuscular injections of 1–10 mg/kg, whereas oral doses up to 50 mg/kg were without effects. Carbaryl was reported to be
without effects on a working-memory paradigm in rats (Heise and Hudson, 1985a,b). In the only protracted-exposure study of carbaryl, Dési and colleagues (1974) reported a progressive increase in maze-running time and an increase in the number of associated errors in rats over the course of a 50-day exposure at 10–20 mg/kg in the diet. Additional studies with long-term, low-dose exposures and measures of effects on complex cognitive function are warranted and should include comparisons of effects after exposure by different routes.
Carcinogenicity
Several studies have examined the question of carbaryl carcinogenicity. In general, they have not provided any evidence of a relationship. The studies have been carried out in mice, rats, and dogs and have included exposures as long as 2 years (in the case of rats) (Gallo and Lawryk, 1991).
Mutagenicity and Genotoxicity
Substantial evidence has led to the assertion that methyl and dimethyl carbamates are not mutagenic as determined with a variety of assays for gene mutation, primary DNA damage, and chromosomal effects (WHO, 1986). Gene-mutation assays yielding negative results have included assessments of forward and reverse mutations in Salmonella typhimurium, Escherichia coli, Bacillus subtilis, yeast, in vitro rodent cells, host-mediated assays, and Drosophila sex-linked recessive lethal assays. DNA damage has been evaluated in bacteria, yeast, and human and rodent cells in vitro. Absence of chromosomal effects has been reported in Drosophila, in vitro rodent cells, in vivo rodent somatic cells, and dominant-lethal-mutation assays in rodents. Some reports of weak mutagenicity have been described at toxic doses, but the studies often had serious limitations that prevented interpretation of results. In in vivo cytogenetic studies in rats exposed to carbaryl, C-mitosis and additional mitotic abnormalities were noted. In 1980, the Environmental Protection Agency concluded that the totality of the available evidence did not support the assertion that carbaryl exposure is a risk factor for genotoxicity in somatic or germinal tissue of humans (Cranmer, 1986). A study of sister-chromatid exchanges (SCEs) and chromosomal aberrations in a pesticide-exposed population reported an increased frequency of SCEs in the symptomatic group, but the increase did not correlate with symptoms, and the group was exposed to a mixture of insecticides that included organophosphorous compounds, carbamates, and organochlorines (Dulout et al., 1985).
Reproductive and Developmental Effects
The reproductive and developmental toxicity of carbaryl has been examined in rats, gerbils, dogs, and primates following a variety of protocols, including multigeneration studies. Various effects have been seen. A 1-year oral exposure of rats at 3 mg/week was associated with decreases in spermatogonia and spermatozoa (Kitagawa et al., 1977). In several studies, decreases in viability, litter size, and survival have been noted. For example, in a three-generation reproductive study in rats, even the lowest dose (100 mg/kg per day) decreased weaning weights (Collins et al., 1971). In a three-generation study of gerbils, a dose of 150 mg/kg per day was found to be the no-observed-effect level; higher doses were associated with decreases in fertility, litter size, viability, and survival (Collins et al., 1971). In dogs, doses of 6–25 mg/kg per day (Smalley et al., 1968) and 2–12.5 mg/kg per day
(Imming et al., 1969) resulted in teratogenesis. Notably, that teratogenesis was seen at doses similar to those at which fetotoxicity was not seen. Doses of 0.2–20 mg/kg per day on gestational days 20–38 resulted in no consistent evidence of fetotoxicity (Coulston et al., 1974).
More recently, effects on estrogenic and progesterone systems and the ability of carbamate insecticides to act as general endocrine modulators have been described. Klotz and colleagues (1997) reported, on the basis of in vitro studies, that the carbamates alone weakly activated estrogen- and progesterone-responsive reporter genes in breast and endometrial cancer cells. In whole-cell competition binding assays, the carbamates showed little capacity to displace radiolabeled estrogen or progesterone from its receptors. The effect of two carbamates, benomyl and carbendazim (at 500 and 1000 mg/kg for 5 days), on growth of decidua (the endometrium of the pregnant uterus) in pseudopregnant rats was assessed in an in vivo study. Both produced reductions in uterine decidual weight and uterine protein content, but serum estradiol and progesterone and the binding capacities of cytosolic estrogen and progesterone receptors were unchanged. The authors interpreted the findings as suggesting that antigrowth and antimitotic activities of the compounds on the decidua were direct and did not involve steroidal or receptor mechanisms.
Several reports have described effects on sperm due to carbaryl. When carbaryl at 50 and 100 mg/kg was fed to rats 5 days/week for 60 or 90 days, dose- and age-dependent declines in sperm count and motility and increased abnormal sperm structure were observed. The effects were more pronounced in young animals than in adults (Pant et al., 1995, 1996). Luca and Balan (1987) reported genotoxic effects in a sperm-abnormality assay in rats fed carbaryl for 3, 6, 9, 12, 15, and 18 months at 12.5, 25, and 250 mg/kg per day. Effects were generally dose-related, but they were not consistently observed for all exposure durations; maximal effects occurred at 6 and 15 months.
Other Health Effects
Chronic studies of carbaryl’s effects on the immune system have revealed several effects. Doses of 2–8 mg/kg per day for 4 weeks resulted in immunosuppression in rabbits (Street and Sharma, 1975). A 9-month oral exposure of rats at 2 mg/kg per day resulted in changes in serum complement-fixing activity, lysozyme activity, and immune functions of the reticuloendothelial system, neutrophils, skin, and mucosa. That dose is less than 1% of the oral LD50 for rats. It is not clear from those reports to what extent the effects are accompanied by overt toxicity, loss of body weight, or other toxic outcomes. Furthermore, the studies did not determine the extent to which such effects are progressive or reversible.
Interactions with Other Agents
The aryl hydrocarbon receptor (AhR) is a transcription factor that activates gene expression, including expression of cytochrome P450 enzymes (such as CYP1A1), in a ligand-dependent manner. The environmental chemical dioxin is one of the most potent AhR ligands ever found. Data from an in vitro study indicate that carbaryl can induce CYP1A1 in human HepG2 and HaCaT cell lines (Ledirac et al., 1997), but that the effect occurs via AhR-independent mechanisms. More recent work (Denison et al., 1998), however, indicates that carbaryl indeed is a weak AhR ligand and inducer of AhR-dependent and dioxin-response-element-dependent gene expression in cell lines from other species. Whether that
effect occurs in humans has yet to be evaluated, but the data suggest interactions between carbaryl and compounds that act via the AhR and between carbaryl and compounds that are metabolized by AhR-induced enzymes.
Carbaryl interacts with cimetidine (Tagamet®), a drug that blocks histamine-induced acid production in the stomach and is used to treat indigestion, acid reflux, heartburn, ulcers, and Zollinger-Ellison syndrome (Ward et al., 1988). May and colleagues (1992) investigated acetylcholinesterase activity in human red cells isolated 1 h after oral administration of carbaryl (at 1 mg/kg) to four healthy people who were taking or not taking cimetidine (at 300 mg every 8 h for 3 days). The results indicate additive effects of high concentrations of cimetidine and carbaryl on the inhibition of red-cell acetylcholinesterase, but no enhanced inhibition was seen at a therapeutically relevant concentration of cimetidine (10 µg/mL). Cotreatment with cimetidine doubled the peak plasma carbaryl concentrations and reduced clearance; carbaryl half-life was unchanged. Maximal inhibition of red-cell acetylcholinesterase activity was statistically significantly reduced. The concentration of carbaryl required to produce 20% inhibition was increased to about 0.5 µg/mL from 0.02 µg/mL. Those findings suggest that carbaryl is metabolized to bioactive metabolites by enzymes that cimetidine inhibits (May et al., 1992).
Mice pretreated with the liver microsomal-enzyme inducer phenobarbital were less susceptible to carbaryl toxicity; those treated with the inhibitor SKF525A demonstrated increased susceptibility (Neskovic et al., 1978). Other studies reported increased toxicity of carbaryl after pretreatment with reserpine or chlordiazepoxide and decreased toxicity after pretreatment with chlorpromazine or meprobamate (Weiss and Orzel, 1967). Tremor induced by carbaryl was substantially reduced by pretreatment with L-dopa and exacerbated by haloperidol; this suggests central catecholaminergic-dopaminergic mechanisms associated with tremor (Ray and Poddar, 1985). Pretreatment of rats with methylmercury hydroxide or chlordane accelerated the urinary excretion of carbaryl (Lucier et al., 1972).
Carpenter and colleagues (1961) reported that the effects of carbaryl were not altered by coadministration of other organophosphorous or other noncarbamate insecticides. However, Lechner and Abdel-Rahman (1986) reported that coadministration of malathion and carbaryl altered pharmacokinetic properties of both insecticides and delayed the elimination of 14C-labeled carbaryl from the plasma of rats. Coadministration of acute equitoxic oral doses of other compounds—such as diphenyl, o-diphenyl, piperonyl butoxide, and thiabendazole—potentiated the effects of carbaryl in mice (Isshiki et al., 1983). Abu-Qare and Abou-Donia (2001) reported that combined exposures to the organophosphate chemical-warfare agent sarin (intramuscular) and the carbamate pyridostigmine bromide (oral) increased concentrations of 3-nitrotyrosine and 8-hydroxy-2-deoxyguanosine, biomarkers of oxidative stress. They also reported decreases in plasma butyrylcholinesterase activity and brain neurotoxic target esterase in hens after combined exposures to pyridostigmine (gavage), DEET (subcutaneous), and chlorpyrifos (subcutaneous) (Abou-Donia et al., 1996) and suggested the possibility that carbamates interact with other neurotoxic pesticides. In another study, however, dermal exposures to DEET did not influence absorption or dermal disposition of carbaryl (Baynes et al., 1997).
PYRETHRINS AND PYRETHROIDS
Pyrethrins are insecticidal compounds that occur naturally in pyrethrum or chrysanthemum flowers (Chrysanthemum cinerariifolium and C. coccineum). Those flowers, which are grown mainly in Kenya, contain six toxins: pyrethrin I and II, jasmolin I and II, and cinerin I and II. Dried pyrethrum flowers and pyrethrum were used extensively as insecticides before World War II. They are potent insecticides with low mammalian toxicity, and they are relatively unstable under ultraviolet radiation and therefore degrade in the environment quickly (Matsumura, 1985; O’Brien, 1967). Use of pyrethrins declined after World War II with the advent of synthetic insecticides, such as DDT. In the 1960s, however, as concern about the harmful effects of some of those synthetic insecticides and their persistence in the environment grew, interest in the pyrethrins was renewed. A large number of pyrethrin derivatives, called pyrethroids, have since been synthesized and tested for insecticidal potency, mammalian toxicity, and biodegradability (Elliott, 1977). At least 2 dozen pyrethroids are in use today, and they make up one of the most popular classes of substances for control of agricultural and household insect pests. The rest of this section deals mainly with pyrethroids.
Two pyrethroids, permethrin and d-phenothrin, were used in the Gulf War. They were sprayed on military clothing to repel and kill flies and mosquitoes (Schreck and Kline, 1989; Schreck et al., 1986; Sholdt et al., 1989). Permethrin has also been used topically for treatment of head lice and scabies in humans (Asakawa et al., 1996; Facts and Comparisons, 2001; Fuortes, 1999; Llewellyn et al., 1996).
Chemistry
Pyrethrins and pyrethroids are esters of alcohols and acids (Elliott, 1977; Matsumura, 1985; O’Brien, 1967). Pyrethrins have excellent insecticidal properties, including a higher potency for insects than for mammals, but because they are relatively unstable in ultraviolet radiation, frequent application is necessary and can become expensive. Therefore, many pyrethroids with increased photostability have been synthesized (Elliott, 1977). Those pyrethroids may be divided into two large categories: type I pyrethroids, which do not contain a cyano moiety at the α position; and type II pyrethroids, which do contain an α-cyano group. Permethrin and phenothrin, the pyrethroid insecticides in this class used in the Gulf War, are type I pyrethroids. Other type I pyrethroids are resmethrin, allethrin, and cismethrin. Examples of type II pyrethroids are cypermethrin, fenvalerate, deltamethrin, and cyphenothrin (Ecobichon, 2001). The structures of pyrethrin I, permethrin, and d-phenothrin are shown in Figure 3.3.
Pyrethroids have two or three chiral carbon atoms, and the commercial pyrethroids are mixtures of four or eight isomers. Insecticidal activity varies greatly among isomers of a pyrethroid. For example, the most potent allethrin isomer against houseflies is d-allethronyl d-trans-chrysanthemate, and the least potent allethrin isomer is l-allethronyl l-trans-chrysanthemate; they differ in potency by a factor of 150.
Toxicokinetics
Pyrethroids are hydrophobic compounds that are absorbed and distributed after ingestion by mammals and inhalation exposure (ATSDR, 2001b). Dermal absorption is
much slower than oral or inhalation exposure, and less than 2% of the applied dose is absorbed. Once absorbed, pyrethroids are distributed to most tissues, especially to those with a high lipid concentration. Pyrethroids are metabolized mainly via ester hydrolysis and oxidation at several loci of the structure. For example, permethrin is detoxified to at least 80 metabolites, including products hydroxylated at the cis and trans methyl groups (Casida et al., 1983). Metabolic enzymes include esterases and P450s. Most of the metabolites are inactive. Half-life varies among the different pyrethroid compounds, but range from 6.4 to 16.5 h in humans, with the elimination mostly complete within 5 d (ATSDR, 2001b). The metabolites are generally excreted as alcohols, phenols, carboxylic acids, and their glycine, sulfate, glucuronide and glucoside conjugates (ATSDR, 2001b).
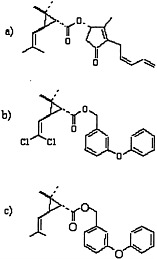
FIGURE 3.3 Structures of a) pyrethrin I, b) permethrin, and c) d-phenothrin.
Genetic Polymorphisms and Susceptibility
No information specific to pyrethroids is available on genetic polymorphisms. Young rats have been shown to be more sensitive than adults to an acute lethal dose of type II pyrethroids, but no differences in age-related sensitivity were seen in response to lower doses. No age differences in sensitivity to type I pyrethroids were seen (Sheets, 2000). Other investigators, however, have demonstrated that fetal or young rats and mice are sensitive to the neurochemical and neurobehavioral effects of pyrethroids (Aziz et al., 2001; Eriksson and Fredriksson, 1991; Husain et al., 1994; Lazarini et al., 2001).
Mechanism of Action
Type I and type II pyrethroids exert their toxicity by affecting the voltage-gated sodium channels of neurons (Narahashi, 1996, 2001; Soderlund et al., 2002; Vijverberg and van den Bercken, 1990). Slowing of the kinetics of activation and inactivation of the gates of the sodium channel by the pyrethroids results in a prolonged opening of individual sodium channels from the normal few milliseconds to several hundred milliseconds by type I pyrethroids and as long as several seconds by type II pyrethroids.
Type I and type II pyrethroids produce the T syndrome and the CS syndrome, respectively (see “Experimental Data” for descriptions of symptoms). In both syndromes, sodium currents of whole cells are greatly prolonged, and this leads to an increase in the depolarizing after-potential or a membrane depolarization. In the T syndrome, type I pyrethroids increase and prolong the depolarizing after-potential and generate repetitive after-discharges. In the CS syndrome, type II pyrethroids generally cause membrane depolarization that evokes repetitive discharges. There is also evidence that type II pyrethroids block the chloride-ion channel of the γ-aminobutyric acid-receptor channel complex (Ecobichon, 2001). To increase the depolarizing after-potential to the threshold for inducing repetitive discharges, only about 1% of the sodium-channel population of rat brain
neurons needs to be modified (Song and Narahashi, 1996); that is why pyrethroids are so potent.
Acute exposure to the type II pyrethroid deltamethrin induces apoptosis in the rat brain that might be associated with the neurodegeneration seen after exposure (Wu and Liu, 2000). Persistent increases in p53 and Bax expression and transient increases in Bcl-2 expression that were seen immunohistochemically after deltamethrin treatment of rats could contribute to the apoptotic cell death (Wu et al., 2000).
Pyrethroids are much more potent in insects than in mammals; the difference in LD50s is a factor of about 4500 (Elliott, 1976). The major factors underlying the selective toxicity of pyrethroids in insects are sodium-channel sensitivity and temperature dependence (Song and Narahashi, 1996). Insect sodium channels are 100–1000 times more sensitive to pyrethroids than are mammalian sodium channels (Narahashi, 2001; Warmke et al., 1997). Furthermore, the activity of pyrethroids is temperature-dependent; they have a greater effect on sodium channels at lower temperatures than at higher temperatures. For example, potency increases by a factor of 5 if temperature is lowered by 10°C (Song and Narahashi, 1996). Because insects have a lower body temperature (about 27°C) than mammals (about 37°C), temperature dependence contributes to the selective toxicity in insects. A difference (by a factor of about 3) in the rate of pyrethroid detoxification between mammals and insects also contributes to the selective toxicity.
Acute Human Exposures
Ray and Forshaw (2000) review pyrethroid poisonings. Accidental spilling of pyrethroids on the head, face, and eyes caused pain, lacrimation, photophobia, congestion, and edema of the conjunctiva and eyelids (He et al., 1988, 1989). Acute ingestion of pyrethroids was reported to cause epigastric pain, nausea, vomiting, headache, dizziness, anorexia, fatigue, tightness in the chest, blurred vision, paresthesia, palpitations, coarse muscular fasciculations, and disturbances of consciousness (He et al., 1988, 1989).
Cutaneous paresthesia is a general effect seen after exposure to all pyrethroids and pyrethrins. It is reversible and not accompanied by electrophysiologic, clinical, and persistent abnormalities. Type II pyrethroids tend to be more potent toxicants than type I pyrethroids (Aldridge, 1990; Flannigan and Tucker, 1985; He et al., 1988, 1989; Knox et al., 1984; Kolmodin-Hedman et al., 1982; LeQuesne et al., 1981; Litchfield, 1985; Tucker and Flannigan, 1983; Vijverberg and van den Bercken, 1990). There have been reports of contact dermatitis (Bainova, 1987; Tomova, 1982), but the dermal toxicity is not considered serious (Bradbury and Coats, 1989; Miyamoto, 1976).
Acute inhalation of type II pyrethroids has been reported to have irritating effects on the mucous membranes of respiratory passages (Vijverberg and van den Bercken, 1990). Asthma-like attacks and anaphylactic reactions with peripheral vascular collapse were reported after exposure to pyrethrins (ATSDR, 2001b).
Altenkirch and colleagues (1996) reported on the neurological examinations of 23 reported cases (out of 64) of pyrethroid poisoning. Exposures were to permethrin, deltamethrin, cyphenothrin, tetramethrin, and mixtures of various pesticides. In nine cases, severe somatic or psychiatric disorders were present that had no plausible relationship to the chemical exposure. Cullen (1987) reported that eight people developed multiple chemical sensitivity syndrome after exposure to pyrethroids. In six cases, a causal link between acute
symptoms and pyrethroid exposure seemed likely or could not be ruled out. No evidence of irreversible peripheral or central nervous system lesions was found in any case.
Experimental Data
Neurotoxic Effects
Type I and type II pyrethroids cause different acute symptoms in rats, the animal species in which most toxicity studies of pyrethroids have been conducted (Aldridge, 1990; Vijverberg and van den Bercken, 1990). Type I pyrethroids, such as those used in the Gulf War, do not have the α-cyano moiety and cause what is called the T syndrome. The T syndrome is characterized in rats by aggressive sparring, hypersensitivity to external stimuli, whole-body tremor, and prostration. Type II pyrethroids, which contain an α-cyano group, cause the CS syndrome. The CS syndrome is characterized by burrowing behavior, profuse salivation without lacrimation, coarse tremors, clonic seizures, and sinuous writhing (choreoathetosis). Type II pyrethroids are also known to increase cardiac contractility (Forshaw and Bradbury, 1983). Some pyrethroids, such as fenproponate, cause mixed or intermediate T and CS motor syndromes (Lawrence and Casida, 1982; Wright et al., 1988). Some other pyrethroids, such as fenproparthrin, produce an intermediate TS syndrome characterized by tremors and salivation.
Acute near-lethal and lethal doses of type II pyrethroids cause axonal swelling and demyelination in the sciatic nerve of the rat (FAO/WHO, 1980; Parker et al., 1985). Near-lethal doses of both type I and II pyrethroids have also been reported to cause sparse axonopathy of rat sciatic and posterior nerves (Aldridge, 1980, 1990; Vijverberg and van den Bercken, 1990).
Chronic neurotoxicity has been reported after pyrethroid exposure in some animal experiments, but the toxicity was usually either low or absent. Little or no chronic neurobehavioral or neurohistologic toxicity has been reported after administration of pyrethrins and pyrethroids, including permethrin (Aldridge, 1990; Bainova et al., 1986; Extoxnet, 1994; Thomson, 1985; Vijverberg and van den Bercken, 1990). Axonal swelling and myelin degeneration have been reported only after repeated or high doses of permethrin, cypermethrin, or fenvalerate (Extoxnet, 1994; FAO/WHO, 1980; Rose and Dewar, 1983). Increased brain concentrations of β-glucuronidase, β-galactosidase, and alkaline phosphatase have also been seen (Dewar, 1981; Dewar and Moffett, 1979).
Carcinogenicity
Chronic application of most pyrethroids tested, including permethrin, rarely and inconsistently caused cancer (DuPont de Nemours Corp., 1989; Extoxnet, 1994; Hallenbeck and Cunningham-Burns, 1985; Ray, 1991; US EPA, 1988, 1989; Waters et al., 1982). One exception is cypermethrin, which was reported to cause benign lung tumors in female mice (US EPA, 1989). Shukla and colleagues (2001) demonstrated that deltamethrin has tumor initiating, but not tumor promoting, activity for skin tumorigenesis in mice.
Mutagenicity and Genotoxicity
One type II pyrethroid, cypermethrin, was tested and found to have no mutagenic or genotoxic effects (FAO/WHO, 1980; Pluijmen et al., 1984; Waters et al., 1982). It was reported, however, to increase polychromatic red cells with micronuclei in bone marrow in
mice (Amer and Aboul-ela, 1985); the clinical significance of this finding is unknown. No mutagenic activity was detected in three in vitro assays for decamethrin (Kavlock et al., 1979). Pluijmen and colleagues (1984) found a lack of mutagenic activity of cypermethrin, permethrin, deltamethrin, bioresmethrin, resmethrin, cismethrin and fenvalerate in S. typhimurium strains. Herrera and Laborda (1988) found no mutagenic activity in S. typhimurium with resmethrin, permethrin and fenvalerate, but did not find mutagenic activity with allethrin in some S. typhimurium strains with or without metabolic activation.
Reproductive and Developmental Effects
In an in vitro test system, none of the eight pyrethroids tested showed substantial estrogenic or antiestrogenic effects at 100–10,000 nM. In vitro tests also showed that they did not alter human estrogen-receptor α-mediated mechanisms (Saito et al., 2000; Sumida et al., 2001).
In general, chronic administration of pyrethroids to female rats does not effect the development of their offspring (Extoxnet, 1994). Pyrethroids are not gonadotoxic, embryotoxic, or teratogenic (Hallenbeck and Cunningham-Burns, 1985; Kaloianova and El Batawi, 1991; Kidd and James, 1991; Litchfield, 1985; Miyamoto, 1976; Polakova and Vargova, 1983; Ray, 1991; US EPA, 1983). High oral doses (250 mg/kg per day) of permethrin during gestational days 6–15, however, reduced the number of offspring (Wauchope et al., 1992). A three-generation study of resmethrin reported slight increases in premature stillbirths and a decrease in pup weight (Ray, 1991). Taken together, the data suggest that pyrethroids are unlikely to cause reproductive and teratogenic effects (Kamrin, 1997).
Immunotoxic Effects
Although detailed studies of the effects of pyrethrins and pyrethroids on immune measures have not been conducted, permethrin has been reported to suppress immune responses in animals. The toxicological significance of these effects to human exposures, however, is difficult to determine because few studies have been conducted in humans and because of interindividual variability (ATSDR, 2001b). Exposure of chickens to a low dose of permethrin in the diet (0.1 ppm) for 3–6 weeks after hatching reduced their immune responsiveness (McCorkle et al., 1980). The response of mouse lymphocytes to mitogens in vitro was suppressed by permethrin at 20 µM (Stelzer and Gordon, 1984). That suppression showed little or no stereospecificity and was caused by both type I and type II pyrethroids, so it was suggested that it resulted from a nonspecific effect on membrane lipids. Permethrin also inhibited the mitogenic response of human lymphocytes to phytohemagglutinin in vitro (Diel et al., 1998). Feeding animals pyrethroids at high concentrations in long-term studies, however, did not cause an increase in infections (Ray, 1991). Cypermethrin has been reported to suppress humoral and cell-mediated immune responses in rats and rabbits directly (Dési et al., 1985). It was suggested that changes in the immune system constitute an early sign of mild exposure and could be an important part of early diagnosis (Dési et al., 1990).
Topical permethrin exposure might produce systemic immune effects. Thymic weight was reduced and splenic weight was increased with no change in body weight in mice by permethrin (at 0.5, 1.0, or 5.0 µL/day, which corresponds to topical insecticide application at about 22–220 mg/kg per day) applied to the shaved dorsal interscapular region
daily for 10 or 30 consecutive days or every other day for seven or 14 exposures. Cell-surface-antigen expression did not change on thymocytes, splenocytes, or bone marrow cells, but the contact-hypersensitivity response was inhibited (Punareewattana et al., 2000). The splenic macrophage-chemiluminescent response was depressed 2 and 10 days after exposure. Phagocytic ability of macrophages was not inhibited. Antibody production, as demonstrated by a plaque-forming cell assay, decreased after 10 consecutive days of exposure (Punareewattana et al., 2001).
Hepatotoxic Effects
Granulomatous changes and giant cell infiltration in the liver have been observed in rodents and dogs experiencing acute fenvalerate intoxication (Okuno et al., 1986; Parker et al., 1984). Those changes, however, are thought to reflect a foreign-body response to deposition of crystals of a type of cholesterol, 2-(4-chlorophenyl)-isovalerate ester, in the liver (Miyamoto et al., 1986). The crystals are formed by microsomal carboxylesterases, and their formation is highly specific for fenvalerate (Kaneko et al., 1986, 1988).
Long-term treatment of rats and mice with pyrethroids produced a variety of liver changes, including increased liver weights (US EPA, 1988; Hallenbeck and Cunningham-Burns, 1985; Lehman, 1965); and microgranulomatous lesions (Kaneko et al., 1986; Okuno et al., 1986; Parker et al., 1983; Ray, 1991).
Other Health Effects
Blood glucose concentrations were increased by acute pyrethroid administration to rats; type II pyrethroids were more potent than type I pyrethroids (Cremer and Seville, 1982; Ray and Cremer, 1979). Thyroid weight was increased by chronic administration of resmethrin (US EPA, 1988). In addition, behavioral changes, decreased blood glucose concentrations, decreased body weights, and increased serum urea concentrations were noted in a 90-day resmethrin inhalation experiment with rats (US EPA, 1988).
Interactions with Other Agents
Piperonyl butoxide is a microsomal mixed-function oxidase inhibitor and has been used to increase the potency of pyrethroids by decreasing their metabolism (O’Brien, 1967). Inasmuch as some of the organophosphorous insecticides inhibit esterases and pyrethroids are hydrolyzed by esterases, synergism of the actions of pyrethroids and organophosphorous insecticides might pose a hazard.
Interactions have been seen between pyrethroids and DEET. Permethrin (15, 30 or 60 mg/kg), pyridostigmine (10 mg/kg), and DEET (50, 200 or 500 mg/kg) had little or no effect on the locomotor behavior of rats when given individually (Hoy et al., 2000a). Behavioral changes were seen, however, when two of them were applied concurrently. In males, the combination of permethrin and pyridostigmine affected speed, and permethrin and DEET affected speed and thigmotaxis. No effects were seen in female rats (Hoy et al., 2000a). Hoy and colleagues (2000b) also investigated open field locomotor activity. In males pyridostigmine and DEET decreased locomotor activity, but DEET in combination with permethrin increased locomotor activity. Females treated with pyridostigmine in combination with permethrin spent more time in the center zone of the open field locomotor activity area, indicating a decrease in thigmotaxis (Hoy et al., 2000b).
The effects of concurrent exposure to pyridostigmine, permethrin, and DEET on multiple fixed-ratio and fixed-interval schedules of reinforcement in rats have also been studied (van Haaren et al., 2001). Exposure to either pyridostigmine or permethrin decreased fixed-ratio and fixed-interval response rates, and DEET decreased both response rates but only at the highest dose. Synergistic effects were observed only on fixed-interval response rate. Those rates were decreased after concurrent exposure to half the dose of pyridostigmine and half the dose of permethrin that alone affected fixed-interval response rate. The rates were also decreased after concurrent exposure to half the effective dose of pyridostigmine and half the effective dose of DEET. In addition, the permeability of the blood-brain barrier of the rat was unaffected by permethrin and slightly decreased by DEET but substantially reduced by a combination of the two (Abou-Donia et al., 2001).
LINDANE
Lindane belongs to a class of insecticides known as the organochlorines. Lindane is the γ-isomer of 1,2,3,4,5,6-hexachlorocyclohexane (HCH). The insecticidal activity of HCH was discovered in 1942.
Use of most organochlorine compounds has been banned since the 1970s in many countries, including the United States, because of their environmental persistence (such as DDT), their ability to bioconcentrate, and their biomagnification (Ecobichon, 2001). Lindane is used for treatment of ectoparasites on humans and animals, and it is used by humans as a lotion and a shampoo (Kwell®) (Facts and Comparisons, 2001; Woolley et al., 1985).
Chemistry
Technical HCH is comprised of four major isomers, α-, β-, γ-, and δ-HCH, in which the concentration of lindane (the γ-isomer) can range from 12 to 99%. Lindane is the most toxic of the isomers; its insecticidal activity is 28–10,000 times higher than that of the others (Ullmann, 1972). Although it is a misnomer, “benzene hexachloride” is a common name used in the United States for the mixture of HCH isomers. The structure of lindane is shown in Figure 3.4.
FIGURE 3.4 Structure of lindane.
Toxicokinetics
Lindane is rapidly absorbed from the gastrointestinal tract. It is much more slowly absorbed dermally (Dick et al., 1997). Once absorbed, it can be metabolized by dehydrogenation, dehydrochlorination, and hydroxylation in the liver. The metabolites of lindane are less acutely toxic than lindane, so those metabolic pathways are considered detoxification pathways (Fitzloff et al., 1982). It is excreted as glucuronide, sulfate, and mercapturic acid conjugates (Copeland et al., 1986).
Mechanism of Action
In the cockroach, lindane evokes synaptic after-discharges and excessive release of acetylcholine (Shankland, 1979; Uchida et al., 1975). However, the major target site of lindane in mammals and insects is the γ-aminobutyric acid (GABA) receptor. GABA is the major inhibitory neurotransmitter in the brain. Activation of the GABA receptor opens chloride-ion channels in neurons. The GABA-induced chloride uptake that results from the channel’s opening is inhibited by lindane (Ghiasuddin and Matsumura, 1982; Ogata et al., 1988). Lindane apparently interferes with binding to one subtype of GABA receptors, the GABAA receptors. Enhanced monoaminergic turnover has also been reported (Rivera et al., 1998), and central monoaminergic systems seem to have a role in lindane intoxication (Llorens et al., 1991).
Acute Human Exposures
Lindane has been used by humans to control scabies caused by mites and to combat lice in cream, ointment, emulsion, and aerosol formulations. Prescription products (Kwell® lotion and Kwell® shampoo) are available for human use (Facts and Comparions, 2001). Not many studies regarding lindane intoxication in humans have been published. A few cases of lindane intoxication following use for control of scabies were due largely to gross disregard of directions; it was applied to the entire body for many days or taken orally (Smith, 1991). A few deaths have been caused by accidental lindane ingestion, and there are many reports of nonfatal intoxication.
A large proportion of fatal and nonfatal lindane poisonings have been in children (Joslin et al., 1960; Savage et al., 1971). Accidental poisoning with lindane occurred when 11 people drank coffee prepared with lindane in place of sugar (Smith, 1991). Initial symptoms included malaise, faintness, and dizziness and were followed by collapse and convulsions, which were sometimes preceded by screaming and accompanied by foaming at the mouth and biting of the tongue. Nausea and vomiting occurred in many cases. The patients were unconscious during convulsions, and loss of consciousness lasted for 15 min to 3 h. Nine patients had retrograde amnesia. Most of the patients were discharged the next day.
Poisoning with lindane is often associated with the use of vaporizing devices. In two such cases, headache, nausea, and irritation of eyes, nose, and throat occurred shortly after exposure to vapors; the symptoms were reversible (AMA, 1952). Secondary effects seen following inhalation of lindane include blood dyscrasias, such as anemia, leukopenia, leukocytosis, granulocytopenia, granulocytosis, eosinophilia, thrombocytopenia, increased bone marrow megakaryocytes, and decreased bone marrow megaloblastoid of the erythroid series (Berry et al., 1987; Morgan et al., 1980), although Morgan and colleagues (1980) seriously questioned the role of lindane in blood dyscrasias.
Experimental Data
Neurotoxic Effects
Acute exposure of animals to lindane causes CNS stimulation, motor impairment, excitation, clonic (intermittent) and tonic (continuous) convulsions, increased respiratory rate or respiratory failure, pulmonary edema, and dermatitis (Drummer and Woolley, 1991;
Smith, 1991). Appearance of tremors has not been reported. Lindane caused hypothermia and anorexia in rats (Aldegunde Villar et al., 1981; Camon et al., 1988; Woolley et al., 1985).
Repeated exposure of rats to subconvulsant doses of lindane led to persistent alterations in the CNS as evidenced by increased susceptibility to experimentally induced seizures (Gilbert, 1995). Repeated exposure to lindane also increased the number of errors in a food-reinforced maze; this suggested lindane interference with learning (Dési, 1974). Nonconvulsant doses of lindane were reported to interfere with the ability to acquire and use new information (Tilson et al., 1987).
Rivera and colleagues (1998) investigated the persistent effects of lindane (single or repeated dose) on motor activity. Rat pups were treated with lindane in a single dose (20 mg/kg) or for 7 days (10 mg/kg per day). Motor activity was tested on postnatal day 15. Acute lindane administration decreased motor activity, whereas repeated lindane administration increased motor activity. The authors suggested that the effect was due to an imbalance of the central monoaminergic and GABAA neurotransmitter system (Rivera et al., 1998).
Carcinogenicity
There is not complete agreement on the carcinogenicity of the chlorinated hydrocarbon insecticides, including lindane (Smith, 1991). A number of studies have been conducted on the carcinogenicity of lindane and technical-grade HCH, which contained lindane. Liver tumors and lung tumors were detected in mice fed diets that contained lindane at 12.5–600 ppm for 24–110 weeks (Hanada et al., 1973; Herbst et al., 1975; Ito et al., 1973a; Thorpe and Walker, 1973; Weisse and Herbst, 1977). Other researchers, however, reported no liver tumors in mice fed diets that contained lindane (100–500 ppm) for shorter periods (24–32 weeks) (Ito et al., 1973a,b; Nagasaki et al., 1972). Studies in rats fed lindane-containing diets did not detect an increase in tumors of any type, including liver tumors (Fitzhugh et al., 1950; Ito et al., 1975; NCI, 1977).
Mutagenicity
Data generally indicate that lindane and HCH are not mutagenic (Buselmaier et al., 1973; Lawlor et al., 1979; Probst al., 1981; Shahin and Von Borstel, 1977; Shirasu et al., 1976; Tsushimoto et al., 1983; Wildemauwe et al., 1983).
Reproductive and Developmental Effects
Reproductive studies of lindane have had mixed results. Although a low dose (5 mg/kg/day) of lindane had no effect on reproduction in rats, long-term administration of low doses (10 mg/kg/day for 138 days) of lindane reduced fecundity and litter size in one study (Trifonova et al., 1970). Dzierzawski (1977) reported an increase by a factor of 2–20 in the number of resorbed fetuses in hamsters, rabbits, and rats. In another study, a very low dose (0.5 mg/kg/day) of lindane increased the duration of diestrus, shortened the duration of estrus, lengthened the gestation period, decreased the number of fetuses, increased the number of dead fetuses, and decreased the growth of the young (Nayshteyn and Leybovich, 1971). Lindane given to rats by gavage on days 6–15 of gestation had no effect on the numbers of pregnancies, abortions, and dead or resorbed fetuses (Khera et al., 1979).
Lindane also has been reported to cause the testes of rats to become atrophied. Seminiferous tubules and Leydig cells degenerated completely over a 10-day treatment period (8 mg/kg/day) (Chowdhury et al., 1987). The decreases in sperm production in mice treated with lindane for 8 months were reversible (Smith, 1991). Lindane also reduced “reproductive behavior” in young rams (Beard et al., 1999).
A number of studies indicate that lindane is not teratogenic. No birth defects were noted in the pups of beagles given lindane (7.5 or 15 mg/kg per day) from gestation day 5 through the end of gestation (US National Library of Medicine, 1995). A three-generation study of rats (Palmer et al., 1978) and experiments with mice (Chernoff and Kavlock, 1983; Gray and Kavlock, 1984) also did not demonstrate any teratogenic effects. Rat offspring exposed to lindane via lactation were reported to have deficiencies that appeared in adulthood; the deficiencies included decreases in testicular weight, lowered number of sperm, and reduced testosterone production (Dalsenter et al., 1997). In utero exposure of rats to lindane reduced [35S]t-butyl bicylcophosphorothionate (TBPS) binding in the brainstem, indicating a decrease in expression of GABAA receptors, but the clinical significance is unknown (Brannen et al., 1998).
Immunotoxic Effects
Weanling rats fed high doses of HCH for 90 days showed hypertrophy of the adrenals, reduction of steroidogenic enzymes, and accumulation of lipids that contain cholesterol (Shivanandappa et al., 1982). Immunosuppression was also reported (Descotes, 1986). Lindane given to weanling rats at low doses suppressed the humoral immune response to typhoid-paratyphoid vaccine (Dewan et al., 1980). Lindane in rats also suppressed the formation of an antibody against human serum albumin and at higher doses inhibited the uptake and lysis of bacteria by phagocytes (Rosival et al., 1974). Reduction of titers in an agglutination test was reported in cats that inhaled lindane and in rabbits that received it in their food (Burkatzkaya, 1963).
N,N-DIETHYL-3-METHYLBENZAMIDE (DEET)
N,N-Diethyl-3-methylbenzamide (N,N-diethyl-m-toluamide; DEET) has been on the worldwide market since the 1950s, and has proved to be an effective, broad-spectrum insect repellent. The compound was developed and patented by the US Army in 1946 for use by military personnel and was registered by US EPA in 1957 (US EPA, 1998). DEET can be applied directly to human skin and to clothing and other materials. It is also used on pets and livestock. It has been estimated that more than 30% of the US population uses DEET during insect-biting seasons. Its use reduces the incidence of vectorborne disease transmission, especially in tropical regions, and of insect bites.
Chemistry
The chemical structure of DEET is shown in Figure 3.5. The DEET in registered formulations must be at least 95% meta-isomer (Figure 3.5), but small amounts of the more toxic ortho-isomer and the less toxic para-isomer are permitted (Robbins and Cherniack, 1986). Formulations vary widely, but most products contain 10–25% DEET in alcohol.
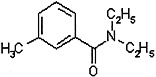
FIGURE 3.5 Structure of DEET.
There are reports that military personnel are issued formulations containing up to 75% DEET.
Toxicokinetics
Topical application of DEET formulations results in accumulation of the repellent in skin layers. Penetration of the skin varies with the formulation, but studies in human volunteers suggest that less than 10% of an applied dose is absorbed. A much wider range of absorption rates has been reported in experimental animals than humans (Qiu et al., 1998). As with most organic compounds, percutaneous absorption would be expected to be greater in areas of skin damage.
DEET is detectable in human blood within 2 h of application to the adult forearm and was found to persist in the blood for at least 4 h (Selim et al., 1995). In rats, peak blood concentrations were achieved 1 h after skin application, and elimination was complete in 2–3 days (Schoenig et al., 1996). The compound and its metabolites are widely distributed in the body. Metabolites include carboxylic acids and hydroxymethyl compounds resulting from oxidation of the methyl group and loss of ethyl groups from the amide side chain (Qiu et al., 1998).
Genetic Polymorphisms and Susceptibility
No information specific to DEET is available on genetic polymorphisms.
Mechanism of Action
Little is known about the toxic mechanism of action of DEET.
Acute Human Exposures
DEET’s overall safety record is good when it is used as directed in the labeling, but three deaths and 10 cases of encephalopathy (all cases except one were in children 8 years old and younger) have been reported to be associated with heavy, and sometimes even short-term, use of DEET formulations (Qiu et al., 1998). There have also been reports of seizures, mostly in children, from exposure to DEET. The national DEET registry (from 1960 to 1997) contains 14 cases of seizures potentially related to DEET for which other more likely causes have not been identified and another 32 cases under review to determine whether there are other more likely causes. In a summary prepared for the reregistration of DEET, US EPA (1998) estimated, on the basis of those data, an incidence of seizures of about one per 100 million users. Reports of acute manic psychosis, cardiovascular toxicity, and anaphylaxis are limited to single, isolated cases, but dermatitis, urticaria, and other skin effects have been more commonly reported (Qiu et al., 1998). Veltri and colleagues (1994) summarized 9086 cases of human exposures to DEET products that had been reported to poison control centers in 1985–1989. The largest fraction (32%) of the cases were related to ocular symptoms, including corneal abrasion, resulting from accidental spraying of products into the eyes. It was possible to follow the course of injury in 24.8% of cases; in 98% of the cases that were followed, there were no long-term sequelae (Veltri et al., 1994).
No other important adverse effects resulting from DEET have been reported in humans. One study of 249 US Gulf War veterans, however, provides evidence of an association between the frequency and amount of skin application of a repellent containing 75% DEET and the risk of a set of symptoms characterized as “arthro-myo-neuropathy” (Haley and Kurt, 1997). Contact dermatitis and urticaria were also reported in some people who used that formulation (Haley and Kurt, 1997).
Experimental Data
Neurotoxic Effects
A single dose of DEET at 1 or 3 g/kg produced neurotoxicity and neuropathy in rats. Many animals recovered fully, a few remained ataxic, others failed to recover muscle tone or righting reflex over 24 h, and some died. Some animals were killed between 2 h and 8 days after treatment for pathologic assessment. Intramyelinic edema, evident from the earliest time, evolved into a patchy and reversible spongiform myelinopathy largely in the cerebellar roof nuclei. Animals that had long-lasting prostration and partially controlled motor seizures displayed scattered neurons with edematous clefts of uncertain origin and significance (Verschoyle et al., 1992). Those findings provide insight into the possible target or mechanism underlying acute DEET-associated encephalopathy.
In subchronic and chronic repeat-dose studies involving dietary administration of DEET to mice, rats, and rabbits, the only consistent effects observed were decreased body weight gain; increased relative weights of testes, liver, and spleen; and increased serum cholesterol (Schoenig et al., 1999). No evidence of an effect on animal survival or abnormal findings of hematology, urinalysis, or gross or microscopic pathology were reported in any dose group. In dogs, tremor, hyperactivity, emesis, excessive salivation, and slightly reduced hemoglobin and hematocrit were reported at oral doses of 400 mg/kg per day given for 1 year; no effects were seen at 100 mg/kg per day (Schoenig et al., 1999).
Schoenig and colleagues (1993) investigated the neurotoxicity of DEET in rats following acute gavage exposure and following multigenerational dietary exposure. Effects on a functional observational battery (FOB) and motor activity measurements were studied following the acute exposure. In addition to the FOB and motor activity tests, an M-maze, passive avoidance acquisition and retention tests, acoustic-startle habituation, and histologic outcomes were also assessed in the multigenerational study. Other than a slight decrease in motor activity at 5000 ppm in the feeding study (rats taken from the second generation of a two-generation study), no evidence of adverse effects was seen.
Reproductive and Developmental Effects
An early study (Gleiberman et al., 1975) reported increased prenatal mortality and some effects on reproductive measures in rats at a maternal dose of 1000 mg/kg per day (dermal). Little evidence of effects on reproduction or development, however, was seen in studies in rats (two studies by the dermal route, one by gavage) and in rabbits (one study by the dermal route, one by gavage) (Schoenig et al., 1994). Another more recent study also did not confirm the findings of Gleiberman and co-workers (1975).
Interactions with Other Agents
The toxicity of DEET coadministered with organophosphorous compounds or permethrin has been investigated to understand toxicosis in domestic cats treated for fleas and ticks. Such research has also been conducted to explore hypotheses related to unexplained illness among US forces in the Gulf War, many of whom used the anticholinesterase drug pyridostigmine bromide (PB) prophylactically against possible nerve-agent exposure in addition to DEET.
Dermal exposures to DEET and at least one pyrethroid have demonstrated unexpected toxicity in cats. Hypersalivation, ataxia, depression, seizures, and death occurred in cats within 4–6 h of dermal application of DEET and fenvalerate (Dorman et al., 1990).
Two laboratories have reported studies in hens and rats demonstrating that various binary and tertiary combinations of PB, DEET, permethrin, and chlorpyrifos produced greater neurotoxicity than the same compounds administered individually at comparable doses (Abou-Donia et al., 1996; van Haaren et al., 2001). Effects on locomotion and thigmotaxis have been reported for combinations of DEET with PB or permethrin. All the studies that showed increased toxicity involved subcutaneous or intravenous (rather than dermal) administration of at least one of the compounds involved.
Increased permeability of the blood-brain barrier and the blood-testis barrier has been reported in rats exposed dermally to DEET and permethrin for 60 days (Abou-Donia et al., 2001). Milder effects were reported for DEET alone, but not for permethrin alone. It is not possible to evaluate the relevance of those experimental findings to humans except to note toxicity may be increased by some combinations of the substances under the specific experimental conditions used.
REFERENCES
Abou-Donia MB. 1995. Organophosphorous pesticides. In: Chang LW, Dyer RS, eds. Handbook of Neurotoxicology. New York: Marcel Dekker, Inc. Pp. 419–473.
Abou-Donia MB, Wilmarth KR, Abdel-Rahman AA, Jensen KF, Oehme FW, Kurt TL. 1996. Increased neurotoxicity following concurrent exposure to pyridostigmine bromide, DEET, and chlorpyrifos. Fundamental and Applied Toxicology 34(2):201–222.
Abou-Donia MB, Goldstein LB, Dechovskaia A, Bullman S, Jones KH, Herrick EA, Abdel-Rahman AA, Khan WA. 2001. Effects of daily dermal application of DEET and permethrin, alone and in combination, on sensorimotor performance, blood-brain barrier, and blood-testes barrier in rats. Journal of Toxicology and Environmental Health 62(7):523–541.
Abu-Qare AW, Abou-Donia AB. 2001. Combined exposure to sarin and pyridostigmine bromide increased levels of rat urinary 3-nitrotyrosine and 8-hydroxy-2'-deoxyguanosine, biomarkers of oxidative stress. Toxicology Letters 123(1):51–8.
Aiuto LA, Pavlakis SG, Boxer RA. 1993. Life-threatening organophosphate-induced delayed polyneuropathy in a child after accidental chlorpyrifos ingestion. Journal of Pediatrics 122(4):658–660.
Aldegunde Villar M, Martin Fargueiro I, Miguez Besada I, Fernandez Otero MP. 1981. Study of the mechanism of the hypothermic action of γ-hexachlorocyclohexane. Acta Cient Compostelana 18:145–154.
Aldridge WN. 1980. Mode of action of pyrethroids in mammals: Summary of toxicity and histological neurophysiological and biochemical studies. In: Mattieu J. ed. Pyrethroid Insecticides: Chemistry and Action. Table Ronde Rouseel UCLAF 37. p. 45.
Aldridge WN. 1990. An assessment of the toxicological properties of pyrethroids and their neurotoxicity. Critical Reviews in Toxicology 21:89–104.
Alexander FE, Patheal SL, Biondi A, Brandalise S, Cabrera ME, Chan LC, Chen Z, Cimino G, Cordoba JC, Gu LJ, Hussein H, Ishii E, Kamel AM, Labra S, Magalhaes IQ, Mizutani S, Petridou E, de Oliveria MP, Yuen P, Wiemels JL, Greaves MF. 2001. Transplacental chemical exposure and risk of infant leukemia with MLL gene fusion. Cancer Research 61(6):2542–2546.
Altenkirch H, Hopmann D, Brockmeier B, Walter G. 1996. Neurological investigations in 23 cases of pyrethroid intoxication reported to the German Federal Health Office. Neurotoxicology 17(3–4):645–651.
AMA (American Medical Association). 1952. American Medical Association Council on Pharmacy and Chemistry report on: Health hazards of electric vaporizing devices for insecticides. Journal of the American Medical Association 149:367–369.
Amer SM, Aboul-ela EI. 1985. Cytogenic effects of pesticides. III. Induction of micronuclei in mouse bone marrow by the insecticides cypermethrin and rotenone. Mutation Research 155(3):135–142.
Asakawa F, Jitsunari F, Miki K, Choi JO, Takeda N, Kitamado T, Suna S, Manabe Y. 1996. Agricultural worker exposure to and absorption of permethrin applied to cabbage. Bulletin of Environmental Contamination and Toxicology 56(1):42–49.
Asmatullah S, Mufti A, Cheema AM, Iqbal J. 1993. Embryotoxicity and teratogenicity of malathion in mice. Punjab University Journal of Zoology 8:53–61.
ATSDR (Agency for Toxic Substances and Disease Registry). 1997. Toxicological Profile for Dichlorvos. Alanta, GA: ATSDR.
ATSDR. 2001a. Draft Toxicological Profile for Malathion. Atlanta, GA: ATSDR.
ATSDR. 2001b. Draft Toxicological Profile for Pyrethrins and Pyrethroids. Atlanta, GA: ATSDR.
Aziz MH, Agrawal AK, Adhami VM, Shukla Y, Seth PK. 2001. Neurodevelopment consequences of gestational exposure (GD14–GD20) to low dose deltamethrin in rats. Neuroscience Letters 300(3):161–165.
Bainova A. 1987. Synthetic pyrethroids—a new group of plant protective drugs. Savr Med 38:3.
Bainova A, Mihovski M, Ismirova N, Benchev I. 1986. Specific Dermal Irritation after Exposure to Synthetic Pyrethroid. 4th Congress of Dermatology, Varna, Bulgaria 1986.
Baker SR, Wilkinson CF. 1990. The Effect of Pesticides on Human Health. Princeton, NJ: Princeton Scientific Publishing Co., Inc.
Ballantyne B, Marrs TC. 1992. Clinical and Experimental Toxicology of Organophosphates and Carbamates. Oxford: Butterworth-Heinemann Ltd.
Baron RL. 1991. Carbamate insecticides. In: Hayes WJ Jr, Laws ER Jr, eds. Handbook of Pesticide Toxicology. Vol. 3. Classes of Pesticides. San Diego: Academic Press Inc. Pp. 1125–1189.
Baynes RE, Halling KB, Riviere JE. 1997. The influence of diethyl-m-toluamide (DEET) on the percutaneous absorption of permethrin and carbaryl. Toxicology and Applied Pharmacology 144(2):332–339.
Beard AP, Bartlewski PM, Chandolia RK, Honaramooz A, Rawlings NC. 1999. Reproductive and endocrine function in rams exposed to the organochlorine pesticides lindane and pentachlorophenol from conception. Journal of Reproduction and Fertility 115(2):303–314.
Berger CW, Sultatos L. 1997. The effects of the phosphorothioate insecticide fenitrothion on mammalian cytochrome P450-dependent metabolism of estradiol. Fundamental and Applied Toxicology 37(2):150–157.
Berry DH, Brewster MA, Watson R, Neuberg RW. 1987. Untoward effects associated with lindane abuse (letter). American Journal of Diseases of Children 141(2):125–126.
Blair D, Dix KM, Hunt PF, Thorpe E, Stevenson DE, Walker AI. 1976. Dichlorvos: A 2-year inhalation carcinogenesis study in rats. Archives of Toxicology 35(4):281–284.
Bradbury SP, Coats JR. 1989. Comparative toxicology of the pyrethroid insecticides. Reviews of Environmental Contamination and Toxicology 108:133–177.
Brannen KC, Devaud LL, Liu J, Lauder JM. 1998. Prenatal exposure to neurotoxicants dieldrin or lindane alters tertbutylbicyclophosphorothionate binding to GABA(A) receptors in fetal rat brainstem. Developmental Neuroscience 20(1):34–41.
Breslin WJ, Liberacki AB, Dittenber DA, Quast JF. 1996. Evaluation of the developmental and reproductive toxicity of chlorpyrifos in the rat. Fundamental and Applied Toxicology 29(1):119–130.
Brophy VH, Hastings MD, Clendenning JB, Richter RJ, Jarvik GP, Furlong CE. 2001. Polymorphisms in the human paraoxonase (PON1) promoter. Pharmacogenetics 11(1):77–84.
Brown MA, Brix KA. 1998. Review of health consequences from high-, intermediate- and low-level exposure to organophosphorous nerve agents. Journal of Applied Toxicology 18(6):383–408.
Burkatzkaya EN. 1963. The effect of hexachlorocyclohexane γ-isomer on the immunobiological reactivity of the body. Gigiena i Sanitariia 28:29–33. As cited in: Hayes WJ Jr, Laws ER Jr. eds. 1991. Handbook of Pesticide Toxicology. Vol. 2, Classes of Pesticides. San Diego: Academic Press, Inc.
Buselmaier W, Röerborn G, Propping P. 1973. Comparative investigations on the mutagenicity of pesticides in mammalian test systems. Mutation Research 21(1):25–26.
Bushnell PJ, Pope CN, Padilla S. 1993. Behavioral and neurochemical effects of acute chlorpyrifos in rats: Tolerance to prolonged inhibition of cholinesterase. Journal of Pharmacology and Experimental Therapeutics 266(2):1007–1017.
Camon L, Martinez E, Artigas F, Sola C, Rodriguez-Farré E. 1988. The effect of non-convulsant doses of lindane on temperature and body weight. Toxicology 49(2–3):389–394.
Carpenter CP, Weil CS, Palm PE, Woodside MW, Nair JH, III, Smyth HF Jr. 1961. Mammalian toxicity of l-naphthyl-N-methylcarbamate (Sevin insecticide). Journal of Agricultural and Food Chemistry 9:30–39.
Carr RL, Chambers HW, Guarisco JA, Richardson JR, Tang J, Chambers JE. 2001. Effects of repeated oral postnatal exposure to chlorpyrifos on open-field behavior in juvenile rats. Toxicological Sciences 59(2):260–267.
Casida JE, Gammon DW, Glickman AH, Lawrence LJ. 1983. Mechanisms of selective action of pyrethroid insecticides. Annual Review of Pharmacology and Toxicology 23:413–438.
Cecchine G, Golomb BA, Hilborne LH, Spektor DM, Anthony CR. 2000. A Review of the Scientific Literature as it Pertains to Gulf War Illness. Arlington, VA: RAND (National Defense Research Institute).
Chambers JE, Levi PE, eds. 1992. Organophosphates, Chemistry, Fate, and Effects. San Diego: Academic Press.
Chan PC, Huff J, Haseman JK, Alison R, Prejean JD. 1991. Carcinogenesis studies of dichlorvos in Fischer rats and B6C3F1 mice. Japanese Journal of Cancer Research 82(2):157–164.
Chanda SM, Pope CN. 1996. Neurochemical and neurobehavioral effects of repeated gestational exposure to chlorpyrifos in maternal and developing rats. Pharmacology, Biochemistry, and Behavior 53(4):771–776.
Chernoff N, Kavlock RJ. 1983. A teratology test system which utilizes postnatal growth and viability in the mouse. Environmental Science Research 27:417–427.
Chowdhury AR, Venkatakrishna-Bhatt H, Guatam AK. 1987. Testicular changes of rats under lindane treatment. Bulletin of Environmental Contamination and Toxicology 38:154–156.
Collins TFX, Hansen WH, Keeler HV. 1971. The effect of carbaryl (Sevin) on reproduction of the rat and gerbil. Toxicology and Applied Pharmacology 19(2):202–216.
Copeland MF, Chadwick RW, Cooke N, Whitehouse DA, Hill DM. 1986. Use of γ-hexachlorocyclohexane (lindane) to determine the ontogeny of metabolism in the developing rat. Journal of Toxicology and Environmental Health 18(4):527–542.
Coulston F, Rosenblum I, Dougherty WJ. 1974. Teratogenic Evaluation of Carbaryl in the Rhesus Monkey (Macaca mulatta). Albany, NY. International Center of Environmental Safety and Albany Medical College. As cited in: Baron RL. 1991. Carbamate insecticides. In: Hayes WJ Jr, Laws ER Jr, eds. Handbook of Pesticide Toxicology. Vol. 3. Classes of Pesticides. San Diego: Academic Press Inc. Pp. 1125–1189.
Cowan J, Sinton CM, Varley AW, Wians FH, Haley RW, Munford RS. 2001. Gene therapy to prevent organophosphate intoxication. Toxicology and Applied Pharmacology 173(1):1–6.
Cranmer MF. 1986. Carbaryl. A toxicological review and risk analysis. Neurotoxicology 7(1):247–328.
Cremer JE, Seville MP. 1982. Comparative effects of two pyrethroids, deltamethrin and cismethrin, on plasma catecholamines and on blood glucose and lactate. Toxicology and Applied Pharmacology 66(1):124–133.
Cullen MR, ed. 1987. The worker with multiple chemical sensitivities: An overview. In: Occupational Medicine: State of the Art Reviews 2(4)655–661.
Dalsenter PR, Faqi AS, Webb J, Merker HJ, Chahoud I. 1997. Reproductive toxicity and toxicokinetics of lindane in the male offspring of rats exposed during lactation. Human & Experimental Toxicology 16(3)146–153.
Daly I. 1996. A 24-month Oral Toxicity/oncogenicity Study of Malathion in the Rat via Dietary Administration. Final report: Lab project No: 90–3641: J-11 90–3641. Unpublished study prepared by Huntington Life Sciences. MRID 43942901. As cited in: ATSDR. 2001. Draft Toxicological Profile for Malathion. Atlanta, GA: ATSDR.
Dam K, Seidler FJ, Slotkin TA. 1998. Developmental neurotoxicity of chlorpyrifos: Delayed targeting of DNA synthesis after repeated administration. Developmental Brain Research 108(1–2):39–45.
Denison MS, Phelan D, Winter GM, Ziccardi MH. 1998. Carbaryl, a carbamate insecticide, is a ligand for the hepatic Ah (dioxin) receptor. Toxicology and Applied Pharmacology 152(2):406–414.
Descotes J. 1986. Immunotoxicology of Drugs and Chemicals. Amsterdam: Elsevier.
Dési I. 1974. Neurotoxicological effects of small quantities of lindane: Animal studies. Internationales Archiv für Arbeitsmedizin 33(2):153–162.
Dési I, Gonczi L, Simon G, Farkas I, Kneffel Z. 1974. Neurotoxicological studies of two carbamate pesticides in subacute animal experiments. Toxicology and Applied Pharmacology 27(3):465–476.
Dési I, Varga L, Dobrony I, Szklenarik G. 1985. Immunotoxicological investigation of the effects of a pesticide; cypermethrin. Archives of Toxicology (Suppl 8):305–309.
Dési I, Nehéz M, Palotás M, Tempfli A, Hogye A, Vetró G. 1990. Experience of health status surveillance of pesticide workers in Hungary. La Medicina del Lavoro 81(6):517–523.
Dewan A, Gupta SK, Jani JP, Kashyap SK. 1980. Effect of lindane on antibody response to typhoid vaccine in weanling rats. Journal of Environmental Science and Health [Part B] B15:395–402.
Dewar AJ. 1981. Neurotoxicity testing with particular reference to biochemical methods. In: Gorrod J, ed. Testing for Toxicity. London: Taylor & Francis, p. 119.
Dewar AJ, Moffett BJ. 1979. Biochemical methods for detecting neurotoxicity—short review. Pharmacology & Therapeutics [Part B] 5(1–3):545–562.
Dick IP, Blain PG, Williams FM. 1997. The percutaneous absorption and skin distribution of lindane in man I. In vivo studies. Human & Experimental Toxicology 16(11):645–651.
Diel F, Detscher M, Borck H, Schrimpf D, Diel E, Hoppe HW. 1998. Effects of permethrin on human basophils and lymphocytes in vitro. Inflammation Research 47:S11–S12.
Dorman DC, Buck WB, Trammel HL, Jones RD, Beasley VR. 1990. Fenvalerate/N,N-diethyl-m-toluamide (DEET) toxicosis in two cats. Journal of the American Veterinary Medical Association 96:100–102.
Drummer HL, Woolley DE. 1991. Toxicokinetics of Ro 5–4864, lindane and picrotoxin compared. Pharmacology, Biochemistry, and Behavior 38(2):235–242.
Dulout FN, Pastori MC, Olivero OA, Gonjzales CM, Loria D, Matos E, Sobel N, deBujan EC, Albiano N. 1985. Sister-chromatid exchanges and chromosomal aberrations in a population exposed to pesticides. Mutation Research 143(4):237–244.
DuPont de Nemours Corp. 1989. Asana XL Technical Bulletin. Wilmington, DE: Dupont.
Dzierzawski A. 1977. Embryotoxicity studies of lindane in the golden hamster, rat and rabbit. Bulletin of the Veterinary Institute in Pulawy 21:85–93.
Ecobichon DJ. 2001. Toxic effects of pesticides. In: Klaassen CD, ed. Casarett and Doull’s Toxicology: The Basic Science of Poisons. 6th ed. New York: McGraw-Hill. Pp. 763–810.
Ecobichon DJ, Joy RM, eds. 1994. Pesticides and Neurological Diseases. 2nd ed. Boca Raton, FL: CRC Press.
Ecobichon DJ, Davies JE, Doull J, Ehrich M, Joy R, McMillan D, MacPhail R, Reiter LW, Slikker W Jr, Tilson H. 1990. Neurotoxic effects of pesticides. Advances in Modern Environmental Toxicology 18:131–199.
Ehrich M, Jortner BS. 2001. Organophosphorous-induced delayed neuropathy. In: Krieger RI, ed. Handbook of Pesticide Toxicology. Vol. 2, Agents. 2nd ed. San Diego: Academic Press. Pp. 997–1012.
Elliott M. 1976. EPA Properties and applications of pyrethroids. Environmental Health Perspectives 14:3–13.
Elliott M. 1977. Synthetic pyrethroids. In: Elliott M, ed. Synthetic Pyrethroids. American Chemical Society Symposium Series 42. Washington, DC: American Chemical Society. Pp. 1–28.
Eriksson P, Fredriksson A. 1991. Neurotoxic effects of two different pyrethroids, bioallethrin and deltamethrin on immature and adult mice: changes in behavioral and muscarinic receptor variables. Toxicology and Applied Pharmacology 108:78–85.
Eriksson P, Talts U. 2000. Neonatal exposure to neurotoxic pesticides increases adult susceptibility: A review of current findings. Neurotoxicology 21(2):37–47.
Extoxnet. 1994. Pyrethrins and Pyrethroids. Available: http://ace.orst.edu/info/extoxnet/pips/pyrethri.htm [accessed July 2002].
Eyer P. 1995. Neuropsychopathological changes by organophosphorous compounds—A review. Human & Experimental Toxicology 14(11):857–864.
Facts and Comparisons. 2001. Drug Facts and Comparisons. St. Louis, MO: Facts and Comparisons.
FAO/WHO (Food and Agriculture Organization/World Health Organization). 1980. FAO Plant Production and Protection Paper 20 (Supplement). Rome: FAO.
Feldman RG. 1999. Occupational and Environmental Neurotoxicology. Philadelphia: Lippincott-Raven.
Fitzhugh OG, Nelson AA, Frawley JP. 1950. The chronic toxicities of technical benzene hexachloride and its alpha, beta and gamma isomers. Journal of Pharmacology and Experimental Therapeutics 100:59–66.
Fitzloff JF, Portig J, Stein K. 1982. Lindane metabolism by human and rat liver microsomes. Xenobiotica 12(3):197–202.
Flannigan SA, Tucker SB. 1985. Variation in cutaneous sensation between synthetic pyrethroid insecticides. Contact Dermatitis 13(3):140–147.
Forshaw PJ, Bradbury JE. 1983. Pharmacological effects of pyrethroids on the cardiovascular system of the rat. European Journal of Pharmacology 91(2–3):207–213.
Fuortes L. 1999. Urticaria due to airborne permethrin exposure. Veterinary and Human Toxicology 41(2):92–93.
Furlong CE, Li WF, Brophy VH, Jarvik GP, Richter RJ, Shih DM, Lusis AJ, Costa LG. 2000. The PON1 gene and detoxication. Neurotoxicology 21(4):581–587.
Gaines TB. 1969. Acute toxicity of pesticides. Toxicology and Applied Pharmacology 14(3):515–534.
Gallo MA, Lawryk NJ. 1991. Organic phosphorus pesticides. In: Hayes WJ Jr, Laws ER Jr, eds. Handbook of Pesticide Toxicology. San Diego: Academic Press. Pp. 917–1123.
Ghiasuddin SM, Matsumura F. 1982. Inhibition of gamma-aminobutyric acid (GABA)-induced chloride uptake by gamma-BHC and heptachlor epoxide. Comparative Biochemistry Physiology C 73(1):141–144.
Gilbert ME. 1995. Repeated exposure to lindane leads to behavioral sensitization and facilitates electrical kindling. Neurotoxicology and Teratology 17(2):131–141.
Gleiberman SE, Volkova AP, Nikolaev GM, Zhukova V. 1975. A study on embryotoxic properties of the repellent diethyltoluamide. Farmakologiya i Toksikologiya 38:202–205
Gollapudi BB, Mendrala AL, Linscombe VA. 1995. Evaluation of the genetic toxicity of the organophosphate insecticide chlorpyrifos. Mutation Research 342(1–2):25–36.
Goncharova NI. 1968. Morphological changes in the blood of animals in sevin poisoning. Veterinariia 45(2):82–83.
Gray LE Jr, Kavlock RJ. 1984. An extended evaluation of an in vivo teratology screen utilizing postnatal growth and viability in the mouse. Teratogenesis, Carcinogenesis, and Mutagenesis 4(5):403–426.
Haley RW, Kurt TL. 1997. Self-reported exposure to neurotoxic chemical combinations in the Gulf War: A cross-sectional epidemiologic study. Journal of the American Medical Association 227(3):231–237.
Haley RW, Billecke S, LaDu BN. 1999. Association of low PON1 type Q (Type A) arylesterase activity with neurologic symptom complexes in Gulf War veterans. Toxicology and Applied Pharmacology 157(3):227–233.
Hallenbeck WH, Cunningham-Burns KM. 1985. Pesticides and Human Health. New York: Springer-Verlag.
Hanada M, Yutani C, Miyaji T. 1973. Induction of hepatoma in mice by benzene hexachloride. Gann 64(5):511–513.
Hayes WJ Jr, Laws ER Jr. eds. 1991. Handbook of Pesticide Toxicology. Vol. 2, Classes of Pesticides. San Diego:Academic Press, Inc.
He F, Sun J, Han K, Wu Y, Yao P, Wang S, Liu L. 1988. Effects of pyrethroid insecticides on subjects involved in packaging pyrethroids. British Journal of Industrial Medicine 45(8):548–551.
He F, Wang S, Liu L, Chen S, Zhang Z, Sun J. 1989. Clinical manifestations and diagnosis of acute pyrethroid poisoning. Archives of Toxicology 63(1):54–58.
He F, Xu H, Qin F, Xu L, Huang J, He X. 1998. Intermediate myasthenia syndrome following acute organophosphates poisoning—an analysis of 21 cases. Human and Experimental Toxicology 17(1):40–45.
Heise GA, Hudson JD. 1985a. Effects of pesticides and drugs on working memory in rats: Continuous delayed response. Pharmacology, Biochemistry, and Behavior 23(4):591–598.
Heise GA, Hudson JD. 1985b. Effects of pesticides and drugs on working memory in rats: Continuous non-match. Pharmacology, Biochemistry, and Behavior 23:599–606.
Herbst M, Weisse I, Koellmer H. 1975. A contribution to the question of the possible hepatocarcinogenic effects of lindane. Toxicology 4(1):91–96.
Hernandez AF, Gonzalvo MC, Gil F, Rodrigo L, Villanueva E, Pla A. 1999. Distribution profiles on paraoxonase and cholinesterase phenotypes in a Spanish population. Chemico-Biological Interactions 119–120:201–209.
Herrera A, Laborda E. 1988. Mutagenic activity of synthetic pyrethroids in Salmonella typhimurium. Mutagenesis 3(6):509–514.
Horn KH, Teichmann B, Schramm T. 1987. Investigation of dichlorvos (DDVP). I. Testing of dichlorvos for carcinogenic activity in mice. Arch. Geschwulfstforsch 57:353–360. As cited in: IARC. 1991. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 53, Occupational Exposures in Insecticide Application, and Some Pesticides. Lyon, France: IARC.
Horn KH, Teichmann B, Schramm T, Nischan P. 1988. Studies on dichlorvos (DDVP). II. Testing of dichlorvos for carcinogenic activity in rats. Arch Geschwulstforsch 58(1):1–10. As cited in: IARC. 1991. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 53, Occupational Exposures in Insecticide Application, and Some Pesticides. Lyon, France: IARC
Hoy JB, Cornell JA, Karlix JL, Schmidt CJ, Tebbett IR, van Haaren F. 2000a. Interactions of pyridostigmine bromide, DEET and permethrin alter locomotor behavior of rats. Veterinary and Human Toxicology 42(2):65–71.
Hoy JB, Cornell JA, Karlix JL, Tebbett IR, van Haaren F. 2000b. Repeated coadministrations of pyridostigmine bromide, DEET, and permethrin alter locomotor behavior of rats. Veterinary and Human Toxicology 42(2):72–76.
Huff JE, Bates R, Eustis SL, Haseman JK, McConnell EE. 1985. Malation and malaoxon: Histopathlogy reexamination of the National Cancer Institute’s carcinogenesis studies. Environmental Research 37(1):154–173.
Husain R, Malaviya M, Seth PK, Husain R. 1994. Effect of deltamethrin on regional brain polyamines and behaviour in young rats. Pharmacology & Toxicology 74(4–5):211–215.
IARC (International Agency for Research on Cancer). 1983. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 30, Malathion. Lyon, France: IARC. Pp. 103–129.
IARC. 1991. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 53, Occupational Exposures in Insecticide Application, and Some Pesticides. Lyon, France: IARC.
Imming RJ, Shaffer BC, Woodward G. 1969. SEVIN®. Safety Evaluation by Feeding to Female Beagles from Day One of Gestation through Weaning of the Offspring. Report from Woodward Research Corporation to Union Carbide Agricultural Products Company, Inc. Research Triangle Park, NC (as cited in Baron RL. 1991. Carbamate insecticides. In: Hayes WJ Jr, Laws ER Jr, eds. Handbook of Pesticide Toxicology. Vol. 3. Classes of Pesticides. San Diego: Academic Press Inc. Pp. 1125–1189).
IOM (Institute of Medicine). 2000. Gulf War and Health, Voume 1: Depleted Uranium, Pyridostigmine Bromide, Sarin, Vaccines. Washington, DC: National Academy Press.
Isshiki K, Miyata K, Matsui S, Tsutsumi M, Watanabe T. 1983. Effects of post-harvest fungicides and piperonyl butoxide on the acute toxicity of pesticides in mice. Safety evaluation for intake of food additives. III. Journal of the Food and Hygiene Society of Japan 24:268–274.
Ito N, Nagasaki H, Arai M, Sugihara S, Makiura S. 1973a. Histologic and ultrastructural studies on the hepatocarcinogenicity of benzene hexachloride in mice. Journal of the National Cancer Institute 51(3):817–826.
Ito N, Nagasaki H, Arai M, Makiura S, Sugihara S, Hirao K. 1973b. Histopathologic studies on liver tumorigenesis induced in mice by technical polychlorinated biphenyls and its promoting effect on liver tumors induced by benzene hexachloride. Journal of the National Cancer Institute 51(5):1637–1646.
Ito N, Nagasaki H, Aoe H, Sugihara S, Miyata Y, Arai M, Shirai T. 1975. Brief communication: Development of hepatocellular carcinomas in rats treated with benzene hexachloride. Journal of the National Cancer Institute 54(3):801–805.
Jamal GA. 1997. Neurological syndromes of organophosphorous compounds. Adverse Drug Reactions and Toxicological Reviews 16(3):133–170.
Jett DA, Navoa RV, Beckles RA, McLemore, GL. 2001. Cognitive function and cholinergic neurochemistry in weanling rats exposed to chlorpyrifos. Toxicology and Applied Pharmacology 174(2):89–98.
Johnson MK, Glynn P. 2001. Neuropathy target esterase. In: Krieger RI, ed. Handbook of Pesticide Toxicology. Vol. 2: Agents. 2nd ed. San Diego: Academic Press. Pp. 953–965.
Joslin EF, Forney RL, Huntington RW Jr, Hayes WJ Jr. 1960. A fatal case of lindane poisoning. In: Proceedings of the National Association of Coroners Seminars, 1958, 1959. Cleveland, Ohio: S.R.Gerber. Pp. 53–57.
Kaloianova FP, El Batawi MA. 1991. Human Toxicology of Pesticides. Boca Raton, FL: CRC Press.
Kamrin MA. 1997. Pesticide Profiles, Toxicity, Environmental Impact, and Fate. Boca Raton: CRC Lewis Publishers.
Kaneko H, Matsuo H, Miyamoto J. 1986. Differential metabolism of fenvalerate and granuloma formation. I. Identification of a cholesterol ester derived from a specific chiral isomer of fenvalerate. Toxicology and Applied Pharmacology 83(1):148–156.
Kaneko H, Takamatsu Y, Okuno Y, Abiko J, Yoshitake A, Miyamoto J. 1988. Substrate specificity for formation of cholesterol ester conjugates from fenvalerate analogues and for granuloma formation. Xenobiotica 18:11–19.
Kavlock R, Chernoff N, Baron R, Linder R, Rogers E, Carver B, Dilley J, Simmon V. 1979. Toxicity studies with decamethrin, a synthetic pyrethroid insecticide. Journal of Environmental Pathology and Toxicology 2(3):751–765.
Khera KS, Whalen C, Trivett G, Angers G. 1979. Teratogenicity studies on pesticidal formulations of dimethoate, diuron and lindane in rats. Bulletin of Environmental Contamination and Toxicology 22(4–5):522–529.
Kidd H, James DR, eds. 1991. The Agrochemicals Handbook. 3rd ed. Cambridge, UK: Royal Society of Chemistry Information Services.
Kitagawa K, Wakakura M, Ishikawa S. 1977. Light microscopic study of endocrine organs of rats treated by carbamate pesticide. Journal of Toxicological Sciences 2:53–60.
Klotz DM, Arnold SF, McLachlan JA. 1997. Inhibition of 17 beta-estradiol and progesterone activity in human breast and endometrial cancer cells by carbamate insecticides. Life Sciences 60(17):1467–1475.
Knox JM 2nd, Tucker SB, Flannigan SA. 1984. Paresthesia from cutaneous exposure to a synthetic pyrethroid insecticide. Archives of Dermatology 120(6):744–746.
Kolmodin-Hedman B, Swensson A, Akerblom M. 1982. Occupational exposure to some synthetic pyrethroids (permethrin and fenvalerate). Archives of Toxicology 50(1):27–33.
Kurt TL. 1998. Epidemiological association in US veterans between Gulf War illness and exposures to anticholinesterases. Toxicology Letters 102–103:523–526.
La Du BN, Aviram M, Billecke S, Navab M, Primo-Parmo S, Sorenson RC, Standford TJ. 1999. On the physiological role(s) of the paraoxonases. Chemico-Biological Interactions 119–120:379–388.
Lawlor T, Haworth SR, Voytek P. 1979. Evaluation of the genetic activity of nine chlorinated phenols, seven chlorinated benzenes and three chlorinated hexanes. Environmental Mutagenesis 1:143.
Lawrence LJ, Casida JE. 1982. Pyrethroid toxicology: Mouse intracerebral structure-toxicity relationships. Pesticide Biochemisrty and Physiology 18(1):9–14.
Lazarini CA, Florio JC, Lemonica IP, Bernardi MM. 2001. Effects of prenatal exposure to deltamethrin on forced swimming behavior, motor activity, and striatal dopamine levels in male and female rats. Neurotoxicology and Teratology 23:655–673.
Lechner DW, Abdel-Rahman MS. 1986. Kinetics of carbaryl and malathion in combination in the rat. Journal of Toxicology and Environmental Health 18(2):241–256.
Ledirac N, Delesculse C, deSousa G, Pralavorio M, Lesca P, Amichot M, Berge JB, Rahmani R. 1997. Carbaryl induces CYP1A1 gene expression in HepG2 and HaCaT cells but is not a ligand of the human hepatic Ah receptor. Toxicology and Applied Pharmacology 144(1):177–182.
Lehman AJ. 1965. Summaries of Pesticide Toxicity. Topeka, Kansas: Association of Food and Drug Officials of the U.S.
LeQuesne PM, Maxwell IC, Butterworth STG. 1981. Transient facial sensory symptoms following exposure to synthetic pyrethroids: A clinical and electrophysiological assessment. Neurotoxicology 2(1):1–12.
Levin ED, Addy N, Nakajima A, Christopher NC, Seidler FJ, Slotkin TA. 2001. Persistent behavioral consequences of neonatal chlorpyrifos exposure in rats. Developmental Brain Research Reviews 130(1):83–89.
Li WF, Costa LG, Richter RJ, Hagen T, Shih DM, Tward A, Lusis AJ, Furlong CE. 2000. Catalytic efficiency determines the in vivo efficacy of PON1 for detoxifying organophosphorous compounds. Pharmacogenetics 10(9):767–779.
Litchfield MH. 1985. Toxicity to mammals. In: Leahey JP, ed. The Pyrethroid Insecticides. London: Taylor & Francis. Pp. 99–150.
Llewellyn DM, Brazier A, Brown R, Cocker J, Evans ML, Hampton J, Nutley BP, White J. 1996. Occupational exposure to permethrin during its use as a public hygiene insecticide. Annals of Occupational Hygiene 40(5):499–509.
Llorens J, Sunol C, Tusell JM, Rodriguez-Farre E. 1991. Evidence for acute tolerance to the behavioral effects of lindane: Concomitant changes in regional monoamine status. Neurotoxicology 12(4):697–705.
Loewenstein-Lichtenstein Y, Schwarz M, Glick D, Norgaard-Pedersen B, Zakut H, Soreq H. 1995. Genetic predisposition to adverse consequences of anti-cholinesterases in atypical BCHE carriers. Nature Medicine 1(10):1082–1085.
Lotti M. 2001. Clinical toxicology of anticholinesterase agents in humans. In: Krieger RI, ed. Handbook of Pesticide Toxicology. Vol. 2, Agents. 2nd ed. San Diego: Academic Press. Pp. 1043–1085.
Lotti M, Moretto A, Zoppellari R, Dainese R, Rizzuto N, Barusco G. 1986. Inhibition of lymphocytic neuropathy target esterase predicts the development of organophosphate-induced delayed polyneuropathy. Archives of Toxicology 59(3):176–179.
Luca D, Balan M. 1987. Sperm abnormality assay in the evaluation of the genotoxic potential of carbaryl in rats. Morphologie et Embryologie 33(1):19–22.
Lucier GW, McDaniel OS, Williams C, Klein R. 1972. Effects of chlordane and methylmercury on the metabolism of carbaryl and carbofuran in rats. Pesticide Biochemistry and Physiology 2:244.
Marrs TC. 1996. Organophosphate anticholinesterase poisoning. Toxic Substance Mechanisms 15:357–368.
Martinez-Chuecos J, del Camen Jurado M, Paz Gimenez M, Martinez D, Menendez M. 1992. Experience with hemoperfusion for organophosphate poisoning. Critical Care Medicine 20(11):1538–1543.
Matsumura F. 1985. Toxicology of Insecticides. 2nd ed. New York: Plenum Press.
Mattsson JL, Wilmer JW, Shankar MR, Berdasco NM, Crissman JW, Maurissen JP, Bond DM. 1996. Single-dose and 13-week repeated-dose neurotoxicity screening studies of chlorpyrifos insecticide. Food and Chemical Toxicology 34(4):393–405.
Mattsson JL, Maurissen JPJ, Nolan RJ, Brzak KA. 2000. Lack of differential sensitivity to cholinesterase inhibition in fetuses and neonates compared to dams treated perinatally with chlorpyrifos. Toxicological Sciences 53(2):438–446.
Maurissen JP, Shankar MR, Mattsson, JL. 2000. Chlorpyrifos: Lack of cognitive effects in adult Long-Evans rats. Neurotoxicology and Teratology 22(2):237–246.
May DG, Naukam RJ, Kambam JR, Branch RA. 1992. Cimetidine-carbaryl interaction in humans: Evidence for an active metabolite of carbaryl. Journal of Pharmacology and Experimental Therapeutics 262(3):1057–1061.
McCorkle F, Taylor R, Martin D, Glick B. 1980. The effect of permethrin on the immune response of chickens. Poultry Science 59(7):1568.
Miyamoto J. 1976. Degradation, metabolism and toxicity of synthetic pyrethroids. Environmental Health Perspectives 14:15–28.
Miyamoto J, Kaneko H, Takamatsu Y. 1986. Stereoselective formation of a cholesterol ester conjugate from fenvalerate by mouse microsomal carboxyesterase(s). Journal of Biochemical Toxicology 1(2):79–93.
Morgan DP, Stockdale EM, Roberts RJ, Walter AW. 1980. Anemia associated with exposure to lindane. Archives of Environmental Health 35(5):307–310.
Moser VC. 1995. Comparisons of the acute effects of cholinesterase inhibitors using a neurobehavioral screening battery in rats. Neurotoxicology and Teratology 17(6):617–625.
Nagasaki H, Tomii S, Mega T, Marugami M, Ito N. 1972. Hepatocarcinogenic effect of α-, β-, γ, and δ-isomers of hexachloride in mice. Gann 63(3):393.
Narahashi T. 1996. Neuronal ion channels as the target sites of insecticides. Pharmacology and Toxicology 79(1):1–14.
Narahashi T. 2001. Neurophysiological effects of insecticides. In: Krieger RI, ed. Handbook of Pesticide Toxicology. Vol. 1. 2nd ed. San Diego: Academic Press. Pp. 335–351.
Nayshteyn SY, Leybovich DL. 1971. Low doses of DDT, γ-HCCH and mixtures of these: Effect on sexual function and embryogenesis in rats. Gigiena i Sanitariia 36(5):19–22 [In Russian]. As cited in: Hayes WJ Jr, Laws ER Jr. eds. 1991. Handbook of Pesticide Toxicology. Vol. 2, Classes of Pesticides. San Diego: Academic Press, Inc.
NCI (National Cancer Institute). 1977. Bioassay of Lindane for Possible Carcinogenicity. CAS No. 58–89–9. Springfield, VA: NTIS PB-273480.
NCI. 1978. Bioassay of Malathion for Possible Carcinogenicity. Washington, DC: US Department of Commerce. NCI-CG-TR-24. Available from: National Technical Information Service; PB-278–257.
NCI. 1979a. Bioassay of Malathion for Possible Carcinogenicity. Washington, DC: US Department of Commerce. NCI-CG-TR-192. Available from: National Technical Information Service; PB-300 301.
NCI. 1979b. Bioassay of Malaoxon for Possible Carcinogenicity. Washington, DC: US Department of Commerce. NCI-CG-TR-135. Available from: National Technical Information Service; PB-299 858.
Neskovic NK, Terzic M, Vitorovic S. 1978. Acute toxicity of carbaryl and propoxur in mice previously treated with Phenobarbital and SKF 525-A. Arhiv za Higijenu Rada i Toksikologiju 29(3):251–256.
NTP (National Toxicology Program). 1977. Bioassay of Dichlorvos for Possible Carcinogenicty (CAS No. 62–73–7) TR-10. NTIS#PB90–198508.
NTP. 1989. Toxicology and Carcinogenesis Studies of Dichlorvos (CAS No. 62–73–7) in F344/N Rats and B6C3F1 Mice (Gavage Studies) TR-342. NTIS# PB90–198508.
O’Brien RD. 1967. Pyrethroids. In: O’Brien RD, ed. Insecticides. Action and Metabolism. New York: Academic Press. Pp. 164–172.
Ogata N, Vogel SM, Narahashi T. 1988. Lindane but not deltamethrin blocks a component of GABA-activated chloride channels. FASEB Journal 2(13):2895–2900.
Okuno Y, Ito S, Seki T, Hiromori T, Murakami M, Kadota T, Miyamoto J. 1986. Fenvalerate-induced granulomatous changes in rats and mice. Journal of Toxicological Sciences 11(1):53–66.
Palmer AK, Cozens DD, Spicer EJF, Worden AN. 1978. Effects of lindane upon reproductive function in a 3-generation study in rats. Toxicology 10(1):45–54.
Pant N, Srivastava SC, Prasad AK, Shankar R, Srivastava SP. 1995. Effects of carbaryl on the rat’s male reproductive system. Veterinary and Human Toxicology 37(5):421–425.
Pant N, Shankar R, Srivastava SP. 1996. Spermatotoxic effects of carbaryl in rats. Human & Experimental Toxicology 15(9):736–738.
Parker CM, McCullough CB, Gellatly JBM, Johnston CD. 1983. Toxicologic and carcinogenic evaluation of fenvalerate in B6C3F1 mouse. Fundamental and Applied Toxicology 3(2):114–120.
Parker CM, Piccirillo VJ, Kurtz SL, Garner FM, Gardiner TH, Van Gelder GA. 1984. Six month feeding study of fenvalerate in dogs. Fundamental and Applied Toxicology 4(4):577–586.
Parker CM, Albot JR, Van Gelder GA, Paterson DR, Taylor JL. 1985. Neuropharmacologic and neuropathologic effect of fenvalerate in mice and rats. Fundamental and Applied Toxicology 5(2):278–286.
Petrelli G, Siepi G, Miligi L, Vineis P. 1993. Solvents in pesticides. Scandanavian. Journal of Work, Environment and Health 19(1):63–65.
Pluijmen M, Drevon C, Montesano R, Malaveille C, Hautefeuille A, Bartsch H. 1984. Lack of mutagenicity of synthetic pyrethroids in Salmonella typhimurium strains and in V-79 Chinese hamster cells. Mutation Research 137(1):7–15.
Polakova H, Vargova M. 1983. Evaluation of the mutagenic effects of decamethrin: Cytogenetic analysis of bone marrow. Mutation Research 120(2–3):167–171.
Pope C, Liu J. 2001. Nonesterase actions of anticholinesterase insecticides. In: Massaro E, ed. Neurotoxicology Handbook. Vol. 1. Totowa, NJ: Humana Press. Pp. 29–43.
Prendergast MA, Terry AV, Buccafusco JJ. 1997. Chronic, low-level exposure to diisopropylfluorophosphate causes protracted impairment of spatial navigation learning. Psychopharmacology 129(2):183–191.
Prendergast MA, Terry AV, Buccafusco JJ. 1998. Effects of chronic, low-level organophosphate exposure on delayed recall, discrimination, and spatial learning in monkeys and rats. Neurotoxicology and Teratology 20(2):115–122.
Probst GS, McMahon RE, Hill LE, Thompson CZ, Epp JK, Neal SB. 1981. Chemically-induced unscheduled DNA synthesis in primary rat hepatocyte cultures: A comparison with bacterial mutagenicity using 218 compounds. Environmental Mutagenesis 391(1):11–32.
Punareewattana K, Smith BJ, Blaylock BL, Robertson JL, Gogal RM Jr., Prater MR, Longstreth J, Snodgrass HL, Holladay SD. 2000. Topical permethrin exposure causes thymic atrophy and persistent inhibition of the contact hypersensitivity response in C57B1/6 mice. International Journal of Toxicology 19:383–389.
Punareewattana K, Smith BJ, Blaylock BL, Longstreth J, Snodgrass HL, Gogal RM Jr, Prater RM, Holladay SD. 2001. Topical permethrin exposure inhibits antibody production and macrophage function in C57B1/6N mice. Food and Chemical Toxicology 39(2):133–139.
Qiu H, Won Jun HW, McCall JW. 1998. Pharmacokinetics, formulation, and safety of insect repellent N,N-diethyl-3-methylbenzamide (DEET): A review. Journal of the American Mosquito Control Association
Ray DE. 1991. Pesticides derived from plants and other organisms. In: Hayes WJ Jr, Laws ER, Jr, eds. Handbook of Pesticide Toxicology. San Diego: Academic Press.
Ray DE, Cremer JE. 1979. The action of decamethrin (a synthetic pyrethroid) in the rat. Pesticide Biochemistry and Physiology 10(3):333–340.
Ray SK, Poddar MK. 1985. Central cholinergic-dopaminergic interaction in carbaryl-induced tremor. European Journal of Pharmacology 119(3):251–253.
Ray DE, Forshaw. 2000. Pyrethroid insecticides: poisoning syndromes, synergies, and therapy. Clinical Toxicology 38(2):95–101.
Richardson RJ. 1995. Assessment of the neurotoxic potential of chlorpyrifos relative to other organophosphorous compounds: A critical review of the literature. Journal of Toxicology and Environmental Health 44(2):135–165.
Rivera S, Rosa R, Martinez E, Sunol C, Serrano MT, Vendrell M, Rodriguez-Farre E, Sanfeliu C. 1998. Behavioral and monoaminergic changes after lindane exposure in developing rats. Neurotoxicology and Teratology 20(2):155–160.
Robbins PJ, Cherniack MG. 1986. Review of the biodistribution and toxicity of the insect repellent N,N-diethyl-m-toluamide (DEET). Journal of Toxicology and Environmental Health 18(4):503–525.
Rodgers KE. 2001. Immunotoxicity of pesticides. In: Handbook of Pesticide Toxicology. Vol. 1. 2nd ed. San Diego: Academic Press. Pp. 769–782.
Rose GP, Dewar AJ. 1983. Intoxication with four synthetic pyrethroids fails to show any correlation between neuromuscular dysfunction and neurobiochemical abnormalities in rats. Archives of Toxicology 53(4):297–316.
Rosival L, Barlogova S, Grunt Yu. 1974. Effect of lindane on certain immunological reactions in rats. Gigienatruda i Professional nye Zabolevaniia 6:53–55 [In Russian]. As cited in: Hayes WJ Jr, Laws ER Jr. eds. 1991. Handbook of Pesticide Toxicology. Vol. 2, Classes of Pesticides. San Diego: Academic Press, Inc.
Rybakova MN. 1966. On the toxic effect of sevin on animals. Gigiena i Sanitariia 31(9):42–47 [In Russian].
Saito K, Tomigahara Y, Ohe N, Isobe N, Nakatsuka I, Kaneko H. 2000. Lack of significant estrogenic or antiestrogenic activity of pyrethroid insecticides in three in vitro assays based on classic estrogen receptor alpha-mediated mechanisms. Toxicological Sciences 57(1):54–60.
Savage EP, Bagby JR Jr, Mounce L, Williams LP Jr, Cholas PH, Cholas G. 1971. Pesticide poisonings in rural Colorado. Rocky Mountain Medical Journal 68(4):29–33.
Schoenig GP, Hartnagel RE Jr, Schardein JL, Vorhees CV. 1993. Neurotoxicity evaluation of N,N-diethyl-m-toluamide (DEET) in rats. Fundamental and Applied Toxicology 21(3):355–365.
Schoenig GP, Neeper-Bradley TL, Fisher LC, Hartnagel RE Jr. 1994. Teratologic evaluations of N N-diethyl-m-toluamide (DEET) in rats and rabbits. Fundamental and Applied Toxicology 23(1):63–69.
Schoenig GP, Hartnagel RE, Osimitz TG, Llanso S. 1996. Absorption, distribution, metabolism, and excretion of N, N-diethyl-m-toluamide in the rat. Drug Metabolism and Disposition 24(2):156–163.
Schoenig GP, Osimitz TG, Gabriel KL, Hartnagel R, Gill MW, Goldenthal EI. 1999. Evaluation of the chronic toxicity and oncogenicity of N, N-diethyl-m-toluamide (DEET). Toxicological Sciences 47(1):99–109.
Schreck CE, Kline DL. 1989. Personal protection afforded by controlled-release topical repellents and permethrin-treated clothing against natural populations of Aedes taeniorhynchus. Journal of the American Mosquito Control Association 5(1):77–80.
Schreck CE, Snoddy EL, Spielman A. 1986. Pressurized sprays of permethrin or DEET on military clothing for personal protection against Ixodes dammini (Acari:Ixodidae). Journal of Medical Entomology 23(4):396–399.
Selim GP, Hartnagel RE Jr., Osimitz TG, Gabriel KL, Schoenig GP. 1995. Absorption, metabolism, and excretion of N,N-diethyl-m-toluamide following dermal application to human volunteers. Fundamental and Applied Toxicology 25:95–100.
Shahin MM, Von Borstel RC. 1977. Mutagenic and lethal effects of a-benzene hexachloride, dibutyl phthalate and trichloroethylene in Saccharomyces cerevisiae. Mutation Research 48(2):173–180.
Shailesh KK, Pais P, Vengamma B, Muthane U. 1994. Clinical and electrophysiological study of intermediate syndrome in patients with organophosphorous poisoning. Journal of the Association of Physicians of India 42(6):451–453.
Shankland DL. 1979. Action of dieldrin and related compounds on synaptic transmission. In: Narahashi T, ed. Neurotoxicology of Insecticides and Pheromones. New York: Plenum Press. Pp. 139–153.
Sheets LP. 2000. A consideration of age-dependent differences in susceptibility to organophosphorous and pyrethroid insecticides. Neurotoxicology 21(1–2):57–63.
Shirasu Y, Moriya M, Kato K, Furuhashi A, Kada T. 1976. Mutagenicity screening of pesticides in the microbial system. Mutation Research 40(1):19–30.
Shivanandappa T, Krishnakumari MK, Majumder SK. 1982. Inhibition of steroidogenic activity in the adrenal cortex of rats fed benzene hexachloride (hexachlorocyclohexane). Experientia 38(10):1251–1253.
Sholdt LL, Rogers EJ Jr, Gerberg EJ, Schreck CE. 1989. Effectiveness of permethrin-treated military uniform fabric against human body lice. Military Medicine 154(2):90–93.
Shukla Y, Arora A, Singh A. 2001. Tumourigenic studies on deltamethrin in Swiss albino mice. Toxicology 163(1):1–9.
Sideroff SI, Santolucito JA. 1972. Behavioral and physiological effects of the cholinesterase inhibitor carbaryl. Physiology & Behavior 9(3):459–462.
Singh JM. 1973. Decreased performance behavior with carbaryl—an indication of clinical toxicity. Clinical Toxicology 6(1):97–108.
Slauter R. 1994. 18-Month oral (dietary) oncogenicity study in mice: Malathion. Lab project No. 668–001. Unpublished study prepared by International Research and Development Corp. MRID 43407201 As cited in: ATSDR 2001a).
Smalley HE. 1970. Diagnosis and treatment of carbaryl poisoning in swine. Journal of the American Veterinary Medical Association 156(3):339–344.
Smalley HE, Curtis JM, Earl FL. 1968. Teratogenic action of carbaryl in beagle dogs. Toxicology and Applied Pharmacology 13(3):392–403.
Smalley HE, O’Hara PJ, Bridges CH, Radeleff RD. 1969. The effects of chronic carbaryl administration on the neuromuscular system of swine. Toxicology and Applied Pharmacology 14(3):409–419.
Smallridge RC, Carr FE, Fein HG. 1991. Diisopropylfluorophosphate (DFP) reduces serum prolactin, thyrotropin, luteinizing hormone, and growth hormone and increases adrenocorticotropin and corticosterone in rats: Involvement of dopaminergic and somatostatinergic as well as cholinergic pathways. Toxicology and Applied Pharmacology 108(2):284–295.
Smith AG. 1991. Chlorinated hydrocarbon insecticides. In: Hayes WJ Jr, Laws ER Jr, eds. Handbook of Pesticide Toxicology. Vol. 2. New York: Academic Press.
Soderlund DM, Clark JM, Sheets LP, Mullin LS, Piccirillo VJ, Sargent D, Stevens JT, Weiner ML. 2002. Mechanisms of pyrethroid neurotoxicity: Implications for cumulative risk assessment. Toxicology
Somkuti SG, Lapadula DM, Chapin RE, Abou-Donia MB. 1991. Light and electron microscopic evidence of tri-o-cresyl phosphate (TOCP)-mediated testicular toxicity in Fischer 344 rats. Toxicology and Applied Pharmacology 107(1):35–46.
Song JH, Narahashi T. 1996. Modulation of sodium channels of rat cerebellar Purkinje neurons by the pyrethroid tetramethrin. Journal of Pharmacology and Experimental Therapeutics 277(1):445–453.
Song X, Seidler FJ, Saleh JL, Zhang J, Padilla S, Slotkin TA. 1997. Cellular mechanisms for developmental toxicity of chlorpyrifos: Targeting the adenylyl cyclase signaling cascade. Toxicology and Applied Pharmacology 145(1):158–174.
Stelzer KJ, Gordon MA. 1984. Effects of pyrethroids on lymphocyte mitogenic responsiveness. Research Communications in Chemical Pathology and Pharmacology 46(1):137–150.
Street JC, Sharma RP. 1975. Alteration of induced cellular and humoral immune respones by pesticides and chemicals of environmental concern: Quantitative studies of immunosuppression by DDT, Aroclor 1254, carbaryl, carbofuran, and methylparathion. Toxicology and Applied Pharmacology 32(3):587–602.
Sumida K, Saito K, Ooe N, Isobe N, Kaneko H, Nakatsuka I. 2001. Evaluation of in vitro methods for detecting the effects of various chemicals on the human progesterone receptor, with a focus on pyrethroid insecticides. Toxicology Letters 118(3):147–155.
Tamura H, Maness SC, Reischmann K, Dorman DC, Gray LE, Gaido KW. 2001. Androgen receptor antagonism by the organophosphate insecticide fenitrothion. Toxicological Sciences 60(1):56–62.
Tang J, Carr RL, Chambers JE. 1999. Changes in rat brain cholinesterase activity and muscarinic receptor density during and after repeated oral exposure to chlorpyrifos in early postnatal development. Toxicological Sciences 51(2):265–272.
Thomson WT. 1985. Insecticides. In: Agricultural Chemicals: Book I. Davis, CA: Thomson Publication.
Thorpe E, Walker AI. 1973. Toxicology of dieldrin (HEOD). II. Comparative long-term oral toxicity studies in mice with dieldrin, DDT, phenobarbitone, β-BHC and γ-BHC. Food and Cosmetics Toxicology 11(3):433–442.
Tilson HA, Shaw S, McLamb RL. 1987. The effect of lindane, DDT and chlordecone on avoidance responding and seizure activity. Toxicology and Applied Pharmacology 88(1):57–65.
Tomova LM. 1982. Study of Occupational Dermatoses Among Field Workers, Referat (Ph.D. thesis), Sofia.
Trifonova TK, Gladenko IN, Shulyak VD. 1970. Effect of hexachlorocyclohexane gamma-isomer and sevin on sexual function. Veterinariia (Kiev) 6:91–93 [In Russian]. As cited in: Hayes WJ Jr, Laws ER Jr. eds. 1991. Handbook of Pesticide Toxicology. Vol. 2, Classes of Pesticides. San Diego: Academic Press, Inc.
Tsushimoto G, Chang CC, Trosko JE, Matsumura F. 1983. Cytotoxic, mutagenic, and cell-cell communication inhibitory properties of DDT, lindane, and chlordane on Chinese hamster cells in vitro. Archives of Environmental Contamination and Toxicology 12(6):721–729.
Tucker SB, Flannigan SA. 1983. Cutaneous effects from occupational exposure to fenvalerate. Archives of Toxicology 54(3):195–202.
Uchida M, Irie Y, Kurihara N, Fujita T, Nakajima M. 1975. The neuroexcitatory, convulsive and lethal effects of lindane analogs on Periplaneta americana (L.). Pesticide Biochemistry and Physiology 5:258–264.
Ullmann E. 1972. Lindane. Monograph of an Insecticide. Verlag K.Schillinger, Freiburg im Breisgau, Germany. Pp. 383.
US EPA (US Environmental Protection Agency). 1983. Tolerances in food administered by EPA: Pemethrin. Federal Register 48:36246–36247.
US EPA. 1988. Pesticide Fact Sheet Number 193: Resmethrin. Washington, DC: Office of Pesticides and Toxic Substances.
US EPA. 1989. Pesticide Fact Sheet Number 199: Cypermethrin. Washington, DC: Office of Pesticides and Toxic Substances.
US EPA. 1998. R.E.D. (Reregistration Eligiblity Decisions) FACTS: DEET. EPA-738-F-95–010. Available: http://www.epa.gov/REDs/factsheets/0002fact.pdf [accessed July 2002].
US EPA. 2000. Malathion: Human Health Risk Assessment for the Reregistration Eligibility Decision (RED) Document. Washington, DC: Office of Prevention, Pesticides and Toxic Substances.
US National Library of Medicine. 1995. Hazardous Substances Data Bank. Bethesda, MD: U.S. National Library of Medicine.
van Haaren F, Haworth SC, Bennett SM, Cody BA, Hoy JB, Karlix JL, Tebbett IR. 2001. The effects of pyridostigmine bromide, permethrin, and DEET alone, or in combination, on fixed-ratio and fixed-interval behavior in male and female rats. Pharmacology Biochemistry and Behavior 69(1–2):23–33.
Van Maele-Fabry G, Laurent C, Willems JL. 2000. Dichlorvos and carcinogenicity: A systematic approach to a regulatory decision. Regulatory Toxicology and Pharmacology 31(1):13–21.
Vasilescu C, Florescu A. 1980. Clinical and electrophysiological study of neuropathy after organophosphorous compounds poisoning. Archives of Toxicology 43(4):305–315.
Veltri JC, Osimitz TG, Bradford DC, Page BC. 1994. Retrospective analysis of calls to poison control centers resulting from exposure to the insect repellent N, N-diethyl-m-toluamide (DEET) from 1985 to 1989. Clinical Toxicology 32(1):1–16.
Verschoyle RD, Brown AW, Nolan C, Ray DE, Lister T. 1992. A comparison of the acute toxicity, neuropathology, and electrophysiology of N,N-diethyl-m-toluamide and N,N-dimethyl-2,2-diphenylacetamide in rats. Fundamental and Applied Toxicology 18(1):79–88.
Vijverberg HPM, van den Bercken J. 1990. Neurotoxicological effects and the mode of action of pyrethroid insecticides. Critical Reviews in Toxicology 21(2):105–126.
Ward SA, May DG, Heath AJ, Branch RA. 1988. Carbaryl metabolism is inhibited by cimetidine in the isolated perfused rat liver and in man. Journal of Toxicology, Clinical Toxicology 26(5–6):269–281.
Ware GW. 1989. The Pesticide Book. 3rd ed. Fresno, CA: Thomson Publications.
Warmke JW, Reenan RA, Wang P, Qian S, Arena JP, Wang J, Wunderler D, Liu K, Kaczorowski GJ, Van der Ploeg LH, Ganetzky B, Cohen CJ. 1997. Functional expression of Drosophila para sodium channels. Modulation by the membrane protein TipE and toxin pharmacology. Journal of General Physiology 110(2):119–133.
Waters MD, Sandhu SS, Simon VF, Mortelmans KE, Mitchell AD, Jorgenson TA, Joens DC, Valencia R, Garrett NE. 1982. Study of pesticide genotoxicity. Basic Life Sciences 21:275–326.
Wauchope RD, Buttler TM, Hornsby AG, Augustijn-Beckers PWM, Burt JP. 1992. SCS/ARS/CES pesticide properties database for environmental decision making. Reviews of Environmental Contamination and Toxicology 123:1–155.
Weiss LR, Orzel RA. 1967. Enhancement of toxicity of anticholinesterases by central depressant drugs in rats. Toxicology and Applied Pharmacology 10(2):344–349.
Weisse I, Herbst M. 1977. Carcinogenicity study of lindane in the mouse. Toxicology 7(2):233–238.
Whitney KD, Seidler FJ, Slotkin TA. 1995. Developmental neurotoxicity of chlorpyrifos: Cellular mechanisms. Toxicology and Applied Pharmacology 134(1):53–62.
WHO (World Health Organization). 1986. Environmental Health Criteria for Carbamate Pesticides: A General Introduction. Geneva: World Health Organization.
Wildemauwe C, Lontie JF, Schoofs L, van Larebeke N. 1983. The mutagenicity in procaryocytes of insecticides, acaricides, and nematicides. Residue Reviews 89:129–178.
Woolley D, Zimmer L, Dodge D, Swanson K. 1985. Effects of lindane-type insecticides in mammals: Unsolved problems. Neurotoxicology 6(2):165–192.
Wright CDP, Forshaw PJ, Ray DE. 1988. Classification of the actions often pyrethroid insecticides in the rat, using the trigeminal reflex and skeletal muscle as test systems. Pesticide Biochemistry and Physiology 30(1):79–86.
Wright DM, Hardin BD, Goad PW, Chrislip DW. 1992. Reproductive and developmental toxicity of N,N-diethyl-m-toluamide in rats. Fundamental and Applied Toxicology 19(1):33–42.
Wu A, Liu Y. 2000. Apoptotic cell death in rat brain following deltamethrin treatment. Neuroscience Letters 279(2):85–88.
Wu A, Ren T, Hu Q, Liu Y. 2000. Deltamethrin induces altered expression of P53, Bax and Bcl-2 in rat brain. Neuroscience Letters 284(1–2):29–32.
Yakim VS. 1967. Data for substantiating the MAC values for Sevin in the air of the working zone. Gigiena i Sanitariia 4:23–33. [In Russian]. As cited in: International Programme on Chemical Safety/WHO. 1994. Environmental Health Criteria 153: Carbaryl. Geneva: World Health Organization.











































