
4
SOLVENT TOXICOLOGY
Solvents are a heterogeneous group of thousands of chemical compounds that can dissolve other chemicals (Rosenberg et al., 1997). That property makes them useful in occupational and home settings for a variety of purposes, including cleaning and degreasing; a few hundred of them have found use as commercial products, many as degreasing agents in industrial settings. Water is the most common solvent, and many solvents are water-based. Non-water-based, or organic, solvents are hydrocarbon-based and lipophilic and therefore are able to extract, dissolve, or suspend fats, oils, and waxes.
Although Congress identified the specific insecticides for study in the legislation on Gulf War exposures, it did not identify the solvents that should be examined by the committee. The committee itself identified a large number of solvents sent to the Gulf War (Chapter 1 and Appendix D), many of which have been extensively studied experimentally.
In light of the large number of solvents sent to the Gulf War and because the toxicity data are used only for background information and supportive evidence (Chapter 2), an extensive review of toxicologic studies of solvents is not appropriate here. Instead, this chapter begins with a review of the general chemistry and toxicokinetics of solvents, the effects of exposure to solvents as a general class, and general interactions that might occur between solvents and other chemicals. Those sections are followed by information on some groups of organic solvents that have unique toxicologic properties or effects or that warrant special mention because of their use in the Gulf War: the aromatic hydrocarbons, halogenated hydrocarbons, alcohols, glycols, glycol ethers, esters, ketones, and petroleum distillates.
It should be noted that data on the solvents discussed here have been reviewed elsewhere—for example, by the Agency for Toxic Substances and Disease Registry, the World Health Organization, the International Agency for Research on Cancer, and the Environmental Protection Agency and in chapters in toxicology textbooks (e.g., Bruckner and Warren, 2001). The reader is referred to those sources for more detailed reviews of the experimental and human data on the various solvents.
GENERAL SOLVENT INFORMATION
Toxicokinetics
The lipophilicity of many organic solvents facilitates their absorption after inhalation or oral or dermal exposure (Bruckner and Warren, 2001). Because of their volatility, the major route of occupational exposure to solvents is inhalation, although dermal exposure can be important in some cases. Once it is absorbed, the disposition and metabolism of a solvent are affected by the route of exposure and by its chemical and physical properties. After ingestion, metabolism and elimination can occur in the first pass through the liver and lungs, before the compound reaches the systemic circulation. Following inhalation exposure most solvents are absorbed rapidly and extensively into the pulmonary and arterial circulation. Some solvents are metabolized to less toxic compounds, others to more toxic compounds or toxic intermediates. Regardless of the route of exposure, unmetabolized lipophilic solvents are distributed predominantly to fatty tissues. Water-soluble solvents, such as alcohols are an exception; they are distributed with body water. Generally, the solvents used in the Gulf War are not highly persistent in the body and are eliminated in a matter of days once exposure ceases.
Toxicity
The central nervous system (CNS) is a major target of organic solvents (Bruckner and Warren, 2001). The CNS-depressing effects of acute, high-dose exposure to solvents are well established in humans and animals, and exposure to most solvents can lead to narcosis if the dose is high enough. In addition, most organic solvents are mucous-membrane irritants and dermal irritants, but only following repeated or prolonged dermal exposure to high concentrations. Permanent CNS damage can occur in humans who chronically abuse solvents, but the effects of low-dose exposure to solvents are less clear. The data on low-dose exposures to solvents come primarily from the epidemiologic literature, which is discussed in the health-outcomes chapters, Chapters 5–9.
In addition to the general chronic solvent-induced CNS effects, some solvents (for example, n-hexane, carbon disulfide, and methyl n-butyl ketone) cause peripheral neuropathy (Graham et al., 1995); symptoms of subchronic and chronic exposure, appear insidiously over weeks to months. The specific solvents known to produce peripheral neuropathy are not believed to have been sent to the Gulf War and are not examined in Chapters 5–9. The clinical features of the neuropathy include sensory loss, distal weakness, and areflexia with a slowing of motor conduction velocity. Nerve biopsy shows axonal degeneration with neurofilamentous axonal swelling and axonal atrophy. Symptoms can progress for months after exposure stops (Huang et al., 1989). In milder cases, symptoms eventually resolve; in more severe cases, residual disability persists over the long term (Arlien-Sobørg, 1992; Feldman, 1998). To judge by the onset and the clinical course, peripheral neuropathy from solvent exposure is a long-term effect. Such peripheral neuropathy was first recognized in humans exposed in the occupational setting and later verified in experimental animals. Peripheral neuropathy is also seen in persons who abuse solvent products containing n-hexane or methyl-n-butyl ketone. N-hexane, carbon disulfide, and methyl-n-butylketone produce identical pathologic and clinical changes. The neurotoxicity of n-hexane and methyl-n-butylketone was found to be mediated by a common
metabolite, 2,5-hexanedione. In contrast, carbon disulfide does not share that metabolite and does not require metabolism for its toxicity. Review of the solvents sent to the Gulf War provides no evidence that military personnel were exposed to n-hexane, methyl-n-butyl ketone, or carbon disulfide. Furthermore, none of the solvents used in the Gulf War is expected to be neurotoxic through a mechanism similar to that of n-hexane, because none of them has a γ-diketone metabolite thought to be required to produce an “n-hexane-like” neuropathy.
Interactions
In most settings, exposures occur to mixtures of solvents (Bruckner and Warren, 2001). Because many solvents have a common mechanism of action for some outcomes (for example, CNS depression), there can be additive effects. Some solvents (such as ethanol) can induce metabolic enzymes. Those enzymes can catalyze the metabolic activation of some solvents and the metabolic detoxification of others. Therefore, the induction of those enzymes can lead to a potentiation of the toxicity of some solvents and reduce the magnitude and duration of the toxicity of other solvents. Interactions can also occur between compounds that have similar toxic outcomes (for example, carbon tetrachloride and chloroform and their effects on the liver).
Genetic Polymorphisms and Individual Susceptibility
Numerous genetic polymorphisms in metabolic enzymes can alter the toxicokinetics, and consequently the toxicity, of solvents that they metabolized (Bruckner and Warren, 2001; Löf and Johanson, 1998). People who take medications or are exposed to other chemicals that affect those enzymes might have altered susceptibility to some solvents, and people with such conditions as liver disease or kidney disease might be more susceptible to the toxicity of solvents.
AROMATIC HYDROCARBONS
Aromatic hydrocarbons are a class of compounds that contain an unsaturated ring structure (Bruckner and Warren, 2001). Benzene, toluene, and xylenes are aromatic hydrocarbons that were or are widely used as solvents and that are thought to have been sent to the Gulf War (see Figure 4.1 for structures). Although no longer used widely as a general-purpose solvent because of concerns about its toxicity, benzene is still used in the synthesis of other chemicals and is a component of gasoline and JP-8 jet fuel. The metabolism of benzene, which occurs in the liver and to a lesser extent in the bone marrow, plays an important role in its toxicity. Benzene is metabolized to benzene oxide, an epoxide, through an oxidation reaction catalyzed primarily by cytochrome P450 2E1. Cytochrome P450 2E11/6 also participates in benzene biotransformation. Benzene oxide can then be metabolized to various compounds, including o-benzoquinone and p-benzoquinone, which are thought to be the two main metabolites that mediate the toxicity of benzene.
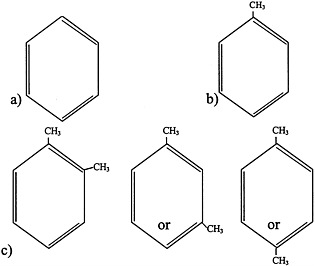
FIGURE 4.1 Structure of a) benzene, b) toluene, and c) xylenes.
Data from laboratory animals and humans show that benzene affects the bone marrow in a dose-dependent manner, causing anemia, leukopenia, and thrombocytopenia; continued exposure causes aplasia and pancytopenia (Bruckner and Warren, 2001). Benzene also has carcinogenic properties. In experimental animals, an increased incidence of malignant lymphomas and some solid tumors have been seen after exposure to high doses of benzene. As discussed in Chapter 6, benzene has also been associated with some types of leukemia in humans. Differences in cancer sites among species indicate that species differences in the carcinogenicity of benzene exist.
Because of the health concerns associated with benzene, it has been replaced in many uses with other solvents, especially toluene and xylenes (Bruckner and Warren, 2001). Toluene and xylenes are widely used in the production of other chemicals and, like benzene, are components of gasoline. Toluene is also present in paints, thinners, cleaning agents, and glue and is widely abused as an inhalant. Toluene and xylenes have the same general toxicity as many solvents, but animal data do not indicate that they are hematopoetic toxicants; the tumors associated with exposure to benzene do not appear to be associated with exposure to toluene and xylenes. Auditory toxicity has been demonstrated in animals following toluene exposure. In rats, intermediate exposures (1000 and 1200 ppm for 14 hours/day for 2 to 9 weeks) resulted in permanent loss of hearing in the high frequency range. A loss of hair cells has been seen following exposure to auditory-toxic concentrations of toluene and could be involved in the underlying mechanism (ATSDR, 2000). Toluene and xylenes are methylated benzenes and, unlike benzene, are metabolized at the methyl group(s) and then readily eliminated.
HALOGENATED HYDROCARBONS
Halogenated hydrocarbons contain at least one halogen atom (such as chlorine, bromine or fluorine). Four halogenated hydrocarbons are discussed in this section: tetrachloroethylene, trichloroethylene, methylene chloride, and chloroform.
Tetrachloroethylene
Tetrachloroethylene (perchloroethylene) is one of the most widely used chlorinated solvents. It is used in dry cleaning and as a degreaser, and it is a component of water repellents, heat-exchange fluids, grain fumigants, and typewriter correction fluids (Bruckner and Warren, 2001).
There is evidence that tetrachloroethylene is carcinogenic in animals (Bruckner and Warren, 2001). An increased incidence of mononuclear-cell leukemia was observed in male and female Fischer 344 rats exposed to tetrachloroethylene. Renal tubular cell adenomas and adenocarcinomas, which are rare in untreated male rats, were observed in male rats exposed to tetrachloroethylene. Oral exposure of male and female mice to tetrachloroethylene over a lifetime resulted in a dose-dependent increase in the incidence of hepatocellular carcinomas.
Tetrachloroethylene’s ability to cause liver tumors in mice appears to be mediated by trichloroacetic acid, its major metabolite (ATSDR, 1997a). Trichloroacetic acid can induce peroxisome proliferation in mice, which can apparently play a role in liver cancer in B6C3F1 mice (Bull, 2000). Trichloroacetic acid induces peroxisome proliferation to a much lesser extent in rats and does not lead to liver cancer in rats (Bull, 2000). Data indicate that humans are relatively insensitive to peroxisome proliferators (Cattley et al., 1998) and produce only very small amounts of trichloroacetic acid after tetrachloroethylene exposure (ATSDR, 1997a). Therefore, that mechanism of liver carcinogenesis in rodents might not be relevant to humans exposed to tetrachloroethylene (Cattley et al., 1998). Another mechanism proposed to underlie the toxicity of tetrachloroethylene is the production of a reactive epoxide intermediate in the metabolism of tetrachloroethylene to trichloroacetic acid (ATSDR, 1997a).
The renal carcinogenesis observed in a low incidence in male rats may occur, in part, from the conjugation of tetrachloroethylene with glutathione in the liver, followed by the formation of genotoxic metabolites in the kidney by β-lyase (Bruckner and Warren, 2001). That glutathione metabolite is formed in substantially smaller amounts in humans (Volkel et al., 1998). It could, nevertheless, play a role in renal carcinogenesis in humans exposed chronically to very high concentrations of tetrachloroethylene. Another mechanism that might underlie the renal toxicity of tetrachloroethylene in male rats, the accumulation of α-2µ-globulin, is also not relevant to humans, because humans do not produce α-2µ-globulin or related proteins in quantities of concern (Green et al., 1990).
Trichloroethylene
Trichloroethylene is a common environmental contaminant that is used as a degreaser, in textile processing, and as an extraction solvent. Trichloroethylene is found in common consumer products, such as typewriter correction fluids, paint removers and strippers, adhesives, spot removers, and rug-cleaning fluid (Bruckner and Warren, 2001).
Most absorbed trichloroethylene is oxidized by hepatic cytochrome P450s to chloral hydrate, trichloroethanol, and trichloroacetic acid (Bruckner and Warren, 2001). Trichloroethylene when inhaled or ingested has been shown to induce liver cancer in B6C3F1 mice but not in rats. That species difference is thought to be due largely to the greater oxidative metabolism of trichloroethylene in mice than in rats. The carcinogenicity of trichloroethylene is mediated largely by trichloroacetic acid and dichloroacetic acid (a minor metabolite), which are thought to contribute to an increase in liver cancers by
inducting peroxisome proliferation. As discussed previously, that mechanism of carcinogenesis might not be relevant to humans (Cattley et al., 1998). In addition, trichloroacetic acid and dichloroacetic acid decrease the rate of normal hepatocyte replication, conferring a selective growth advantage to initiated tumorigenic cells (an example of “negative selection”). Another effect of trichloroacetic acid and dichloroacetic acid that might be related to liver carcinogenicity is the stimulation of DNA replication in hepatocytes. That effect, however, manifests only at very high concentrations (Bull, 2000). Trichloroethylene at low doses also increases renal tubular cell adenomas in male rats.
Inhaled trichloroethylene is a respiratory carcinogen in mice but not in rats (Bruckner and Warren, 2001). Chloral hydrate is the metabolite primarily responsible for pulmonary tumor formation. Clara cells, which have high P450 activity to metabolize trichloroethylene, are much more numerous in the mouse lung than in the lungs of rats and other animals. The human lung has very few Clara cells. They are the primary target of trichloroethylene in the mouse lung (Green, 2000). Toxicity to Clara cells is characterized by vacuolization, necrosis and an increase in cell replication. Inhalation of trichloroethylene in rats has also resulted in increased leukemias and Leydig cell tumors. Male rats exhibited a dose-related increase in Leydig cell tumors and a slight increase in tubular renal adenocarcinomas.
Methylene Chloride
Methylene chloride is used as an extractant for fats and paraffins in the food and pharmaceutical industries and as a postharvest fumigant for grains and strawberries. It was used in hair sprays until 1989 (FDA, 1989) and was used to remove caffeine from coffee.
Exposure to methylene chloride is of concern primarily because of its potential as a human carcinogen, in light of its carcinogenicity in rodents. Evidence suggests that oral exposure to methylene chloride increases the incidence of liver cancer. Acute exposure to methylene chloride induces ornithine decarboxylase in laboratory animals, which is indicative of the promotion of liver cancer (Kitchin and Brown, 1989). Chronic high exposure to methylene chloride in laboratory animals, particularly mice, results in an increase in lung adenomas or carcinomas. Increased numbers of salivary gland sarcomas have been seen in male rats (Burek et al., 1984) and benign mammary tumors in female rats (Burek et al., 1984; Mennear et al., 1988; Nitschke et al., 1988) after exposure to high concentrations of methylene chloride vapors. An increased incidence of lung tumors, but not liver tumors, was seen.
Genotoxic end points have been observed in mice and rats and their tissues after methylene chloride exposure. There has been a great deal of research to define mechanisms underlying the carcinogenicity of methylene chloride, so that the relevance of the murine tumors to humans can be understood (Green, 1997). Liver and lung tumors in mice do not seem to be associated with overt cytotoxicity or increased replicative DNA synthesis (Maronpot et al., 1995). Induction of the tumors is generally believed to be due to a reactive intermediate generated by the glutathione-S-transferase (GST) pathway (Andersen et al., 1987). The theta class GST isozyme GST T1–1 catalyzes the conversion of methylene chloride to S-chloromethylglutathione in the liver and lung, which appears to break down rapidly to glutathione and formaldehyde. The formation of this unstable intermediate occurs in the proximity of DNA in mice and results in single-strand DNA breaks that could play a role in the development of cancer.
An alternative mechanism has been suggested to underlie the carcinogenicity of methylene chloride. Casanova and colleagues (1997) proposed that formaldehyde, a metabolite of methylene chloride, causes the formation of DNA-protein cross-links. When hepatocytes from humans and from several other species were incubated with methylene chloride, DNA-protein crosslinks were found only in the mouse samples. That suggests that mice are more susceptible than other species to the carcinogenic effects of methylene chloride.
Chloroform
Chloroform was introduced in 1847 as an inhalation anesthetic (Bruckner and Warren, 2001). It is no longer used as an anesthetic in humans, but it is used in some endodontic procedures and in the administration of drugs for the treatment of some diseases. It is used in the production of chemicals, as an extraction solvent, as a heat-transfer medium in fire extinguishers, and as an intermediate in the preparation of dyes and pesticides.
The metabolism of chloroform (Figure 4.2) is important to its toxicity, because its toxicity is mediated by reactive metabolites that bind to macromolecules (lipids and proteins) of the endoplasmic reticulum. Most chloroform metabolism occurs by oxidation.
A primary target of chloroform is the liver, and liver necrosis is one of the major toxic effects that has been observed in humans and animals after inhalation and oral exposure to high doses of chloroform (Bruckner and Warren, 2001). Leakage of cytoplasmic liver enzymes into the bloodstream, increases in liver triglycerides, and decreases in liver reduced-glutathione levels have been seen in rodents after exposure to chloroform. Other effects seen in rats and mice are centrilobular hepatocyte necrosis, vascular degeneration in midzonal and periportal portions of the liver lobule, and a decrease in the eosinophilia of the centrilobular and midzonal hepatocyte cytoplasm, acute hepatitis, increased liver weights, and diffuse centrilobular swelling.
The kidneys are another major target of chloroform after both inhalation and oral exposure (Bruckner and Warren, 2001). Tubular necrosis, increased kidney weight, increased cell proliferation, and epithelial cell lesions in the proximal convoluted tubules have been seen in mice after inhalation exposure to chloroform. Tubular necrosis, tubular swelling, and increased kidney weight have occurred in rats after oral exposure.
Liver and kidney tumors have also been seen in animals after exposure to chloroform; the tumors depend on the species, strain, and sex of the animal and on the dosage of chloroform (Bruckner and Warren, 2001). Inhalation exposure to chloroform in BDF1 male mice resulted in renal tubular tumors (Nagano et al., 1998). Exposure to chloroform in drinking water resulted in increased hepatic neoplastic nodules in female Wistar rats and adenofibromas in male and female Wistar rats (Tumasonis et al., 1987). Oral exposure studies indicate a dose-related increase in hepatocellular carcinomas in both male and female mice receiving chloroform in corn oil by gavage (NCI, 1976). An increase in renal tumors was seen in male rats receiving chloroform by gavage (tubular adenoma and carcinoma) (NCI, 1976) and in drinking water (renal tubular adenomas and adenocarcinomas) (Jorgenson et al., 1985). Male mice, but not female mice or male and female rats, demonstrated an increase in renal tumors following exposure to chloroform in a toothpaste base (Palmer et al., 1979; Roe et al., 1979). Those carcinogenic effects, however, occur only at high doses. The nonlinearity of the relationship between chloroform dose and tumor formation is consistent with the evidence that chloroform is not genotoxic. The
carcinogenic effects are thought to be associated with regenerative hyperplasia that is in response to cell death and the data suggest that human exposures do not cause hyperplasia, and therefore would not result in carcinogenesis (Bruckner and Warren, 2001).
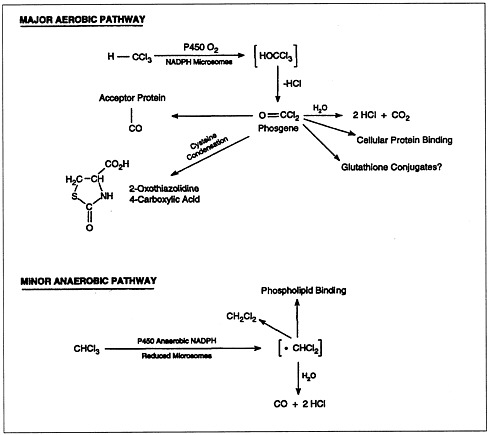
FIGURE 4.2 Metabolic pathways of chloroform biotransformation.
SOURCE: ATSDR, 1997b.
ALCOHOLS
Alcohols are widely used solvents that elicit the general solvent effects discussed earlier. Figure 4.3 shows the chemical structures of four representative alcohols: two primary aliphatic alcohols, methanol and ethanol; one secondary aliphatic alcohol, isopropanol; and one cyclic secondary alcohol, cyclohexanol.
In addition to CNS depression, methanol exposure in primates results in serious effects mediated by metabolites. Methanol can produce severe acidosis and retinal damage
in primates by the production of its metabolite formate at exposures that do not produce substantial CNS depression (ATSDR, 1993; Tephly, 1991); these effects occur after high-dose exposure. Considerable differences exist among species in the capacity of their folate-dependent one-carbon pool to metabolize formate. In nonprimates, formate is metabolized rapidly; in primates, it is metabolized slowly and accumulates, leading to metabolic acidosis and ocular damage. The relative deficit in formate metabolism in primates has been attributed to lower tissue folate concentrations. It is consistent with that hypothesis that nonprimate laboratory animals fed a folate-deficient diet demonstrate sensitivities to methanol that are similar to those seen in primates (Tephly, 1991; Valentine, 1990). Methanol-induced ocular lesions are characterized by a loss of retinal ganglion cells, retinal edema, and demyelination of the temporal retina. Necrosis of cells with or without hemorrhage in the basal ganglion and widespread hypoxia/ischemic damage are also associated with methanol intoxication. At low doses, however, the formate detoxification pathway is not overwhelmed, and formate does not build up.
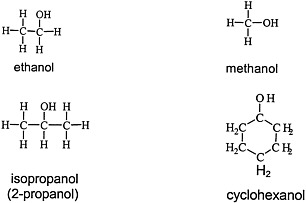
FIGURE 4.3 Structure of various alcohols.
Administration of cyclohexanol to male gerbils and rats by subcutaneous injection (15 mg/kg/day) for 21 and 37 days, respectively, was associated with reproductive toxicity (Tyagi et al., 1979). The adverse effects observed included decreased weights of the testes, epididymis, seminal vesicles, and ventral prostate and degenerative changes in spermatozoa and spermatozoa precursors. Similar changes in the testes were seen in rabbits given cyclohexanol by gavage (25 mg/kg/day) for 40 days (Dixit et al., 1980). In contrast, no change in testicular weights was observed in Sprague Dawley rats given cyclohexanol by gavage (455 mg/kg/day) for 7 days (Lake et al., 1982).
Ethanol can elicit a wide array of serious adverse effects when ingested, but it is unlikely that such effects are associated with the internal doses likely to result from inhalation.
GLYCOLS
Glycols, which are characterized by the presence of two hydroxyl groups (see Figure 4.4 for structures), are major constituents of antifreeze, airplane deicers and anti-icers, brake fluids, and heat exchangers. Propylene glycol is used in cosmetics and foods and as a vehicle for drug delivery.
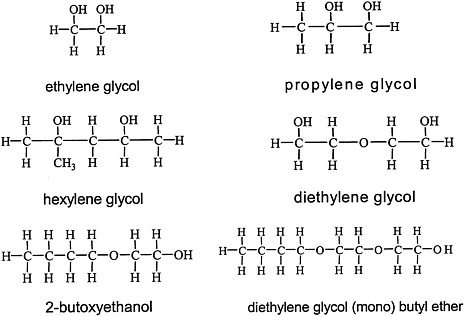
FIGURE 4.4 Structure of various glycols.
Ethylene glycol, propylene glycol, diethylene glycol, and hexylene glycol differ greatly in their potential to produce acute or chronic toxicity (ATSDR, 1997c; BIBRA, 1991, 1993; Lakind et al., 1999; Ruddick, 1972; Snyder and Andrews, 1996). Glycols are metabolized through successive oxidative steps, and the resulting metabolites account for the observed differences in toxicity among the glycols (ATSDR, 1997c; Lakind et al., 1999; Ruddick, 1972; Snyder and Andrews, 1996). Propylene glycol, which is metabolized by alcohol dehydrogenase and aldehyde dehydrogenase to lactate, that can be used in gluconeogenesis, has low toxicity. The principal metabolic pathway for ethylene glycol proceeds through glycoaldehyde, glycolic acid, glyoxylic acid, and oxalic acid. The rate-limiting step in this pathway is the oxidation of glycolic acid to glyoxylic acid. As a result, substantial concentrations of glycolic acid accumulate. The buildup of glycolic acid is thought to be a major factor in the metabolic acidosis involved in the toxicity of ethylene glycol.
Renal toxicity of ethylene glycol or diethylene glycol has been observed in experimental animals (including rodents, primates, canines, and felines) after oral, dermal, and inhalation exposure (ATSDR, 1997c; Lakind et al., 1999; Snyder and Andrews, 1996). The renal lesions produced are similar among species, but humans and cats are 2–5 times more sensitive than rodents and dogs. The lesions observed include renal tubular epithelial degeneration and necrosis. The clinical signs associated with the renal toxicity include polyuria, anuria, crystaluria, and oliguria.
The mechanism of renal toxicity is a matter of debate, and several contributing mechanisms have been proposed (Lakind et al., 1999). The oxalic acid metabolite chelates calcium, generating calcium oxalate, which precipitates in numerous organs. Hypocalcemia
may ensue, with the potential to exacerbate cardiovascular effects produced by the metabolic acidosis. Deposits of calcium oxalate crystals in the renal interstitium have been proposed as contributing to renal dysfunction through physical damage and blockage of the tubules. Evidence has also been presented that other acid metabolites, including glycolic acid and glyoxylic acid, contribute to renal toxicity through direct cytotoxic effects on the renal tubular epithelium. Frantz and colleagues (1996) demonstrated that differences in the metabolism and kinetics of ethylene glycol between mice and rats are related to the differential susceptibility of the two species to ethylene glycol. Metabolic acidosis and an increased osmolal gap may also contribute to renal toxicity by altering intracellular osmotic pressure.
Diethylene glycol shares with ethylene glycol the ability to produce renal toxicity (BIBRA, 1993; Snyder and Andrews, 1996). In contrast with ethylene glycol, however, it does not produce metabolic acidosis or calcium oxalate crystals. The observation that diethylene glycol produces identical lesions and is a more potent nephrotoxicant has been presented to support the contribution of non-oxalic acid metabolites to glycol-mediated nephrotoxicity.
GLYCOL ETHERS
Glycol ethers are used extensively in consumer products, including paints and textile dyes; as an anti-icer in brake fluids; and as a gasoline additive (Bruckner and Warren, 2001). This section discusses ethylene glycol monomethyl ether (EM), ethylene glycol monoethyl ether (EE), propylene glycol monomethyl ether (PM), and ethylene glycol monobutyl ether (EB) (see Figure 4.5 for structures).
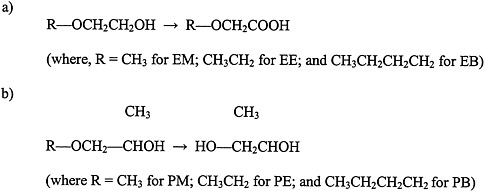
FIGURE 4.5 Structure of glycol ethers and their metabolites.
a) ethylene glycol ethers, and b) propylene glycol ethers. Abbreviations: EM, ethylene glycol monomethyl ether; EB, ethylene glycol monobutyl ether; PM, propylene glycol monomethyl ether; PE, propylene glycol monoethyl ether; PB, propylene glycol monobutyl ether.
Although ingestion of glycol ethers is not acutely hazardous, reproductive toxicity and teratogenicity are of concern in connection with some of them (Bruckner and Warren,
2001). High exposures to EM and EE cause testicular atrophy and a decrease in white blood cells in mice. Treatment of rabbits and rats for 13 weeks with EM caused degeneration of testicular germinal epithelium and infertility; partial recovery was seen after 13 weeks. Exposure of pregnant rats to EE for 7 h/day on gestational days 7–15 caused fetal death with no signs of maternal toxicity. Cardiovascular and skeletal malformations were seen. Cardiovascular malformations have also been seen in the offspring of pregnant rabbits exposed to EE.
Exposure to PM, however, is not associated with reproductive or teratogenic effects (Bruckner and Warren, 2001). The differences in toxicity between PM and the other glycol ethers appear to be due to differences in their metabolism. Evidence indicates that the toxic effects of EM, EE, and EB are mediated by alkoxyacid metabolites (for example, methoxyacetic acid is a metabolite of EM). In contrast, the nontoxic PM does not have an alkoxyacid metabolite; it is metabolized to propylene glycol.
ESTERS
Organic esters are produced by the condensation of an alcohol and carboxylic acid with removal of water (see Figure 4.6 for structures of esters). The chemical properties of the ester depend on the structure of its alcohol and acid moieties. The two esters of potential concern in this document, because of their use in the Gulf War, are butyl acetate, an aliphatic ester, and 1-methoxy-2-propanol acetate, a propylene glycol ether ester. Butyl acetate is used as a flavoring agent and a solvent for cosmetics, lacquers, and adhesives. 1-Methoxy-2-propanol ether acetate (propylene glycol monomethyl ether acetate, or PGMEA) is an ester of acetic acid and 1-methoxy-2-propanol. Because it contains both an ester and an ether linkage, PGMEA has unique solvent properties and is valued as a solvent for oils, gums, and resins.
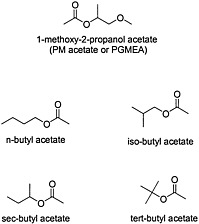
FIGURE 4.6 Structure of various esters.
Both butyl acetate and PGMEA are considered to have low toxicity in humans. Although hepatotoxicity has been seen in workers exposed to a mixture of solvents that included butyl acetate, no direct evidence supports butyl acetate as the agent responsible for the hepatotoxicity (Franco et al., 1986).
Only mild reproductive effects have been reported for butyl acetate in experimental animals. Increased testicular weights were reported in rats exposed to n-butyl acetate by inhalation over 13 weeks, but no testicular lesions accompanied the change in weight. n-Butyl acetate had no adverse effect on female fertility and fetal development (David et al., 2001). Another study found that pregnant rats exposed to n-butyl acetate had fetuses with lower weights than controls, but this was attributed to decreased food consumption by dams. No significant differences were seen in developmental defects in the fetuses of the exposure group, relative to controls, so the
authors concluded that n-butyl acetate is not teratogenic (Elder, 1989). Unlike the ethylene glycol ethers, propylene glycol ethers and acetates, such as PGMEA, do not appear to have significant teratologic or reproductive toxicity effects. However, the PGMEA isomer 2-methoxy-1-propanol acetate, found as an impurity in commercial-grade PGMEA, has been reported to be teratogenic in both rats and rabbits. Because 2-methyoxy-1-propanol acetate can comprise as much as 5% of commercial PGMEA, the reproductive toxicity of this isomer may have implications for exposures to commercial-grade PGMEA.
KETONES
Ketones, organic compounds that contain a carbonyl functional group (C=O), are widely used as solvents and in chemical manufacturing (Rosenstock and Cullen, 1986). The general ketone structure is shown in Figure 4.7.
In general, ketones are not considered to be acutely toxic, and acetone, the most widely used ketone, is considered relatively nontoxic (Bruckner and Warren, 2001). As previously mentioned, some ketones produce a specific type of peripheral neuropathy that is mediated by their common metabolite, 2,5-hexanedione. There is no evidence that any of the
FIGURE 4.7 Basic structure of ketones.
ketones thought to have been sent to the Gulf War cause that peripheral neuropathy or have 2,5-hexanedione as a metabolite. There is evidence, however, that coexposure to some of the compounds sent to the Gulf War (specifically, methyl ethyl ketone and methyl isobutyl ketone) can potentiate peripheral neuropathies induced by n-hexane and methyl-n-butyl ketone (ATSDR, 1999; Ichihara et al., 1998). Although the exact mechanism of that potentiation is not clear, urinary concentrations of 2,5-hexanedione increased with potentiation of n-hexane neurotoxicity by methyl-n-butyl ketone (Ichihara et al., 1998).
PETROLEUM DISTILLATES
There was the potential for exposure to at least two refined-petroleum mixtures in the Gulf War: Stoddard solvent (dry-cleaning safety solvent) and naphtha. Stoddard solvent has many uses as a fuel or fuel additive, lubricant, dry-cleaning agent, chemical intermediate, and general cleaner and degreaser. Naphtha is used in paints, insecticide formulations, and solvent extraction processes and as a component in gasoline blending. Many fuels, including jet fuels, were used in the Gulf War. Those compounds are not reviewed in this volume of Gulf War and Health, however, because they will be reviewed in the next volume of Gulf War and Health.
Stoddard solvent is composed of a mixture of hydrocarbons (usually seven to 10 carbons long) that are produced during the refinement of crude oil. It is a colorless, flammable liquid that is insoluble in water. Naphtha is a general term for petroleum distillates that contain predominantly C5−C13 aliphatic hydrocarbons.
The relative percentage of individual components affects the toxicokinetics of Stoddard solvent. A hydrocarbon’s blood:air partition coefficient is a key factor in the rate
and extent of its systemic absorption in the lungs. Aromatic compounds are absorbed more rapidly and completely through the lungs than are the long-chain aliphatic components, which have low solubility in the blood (Klaassen and Rozman, 1991). Nevertheless, the long-chain aliphatic components are also well absorbed. The internal doses of the individual components, therefore, will differ from their magnitudes of exposure.
Similarly, the toxicity of Stoddard solvent and naphtha is not usually determined by a single component, although the contribution of the individual hydrocarbons can influence the potential hazards associated with those compounds. 140°C Flash Stoddard solvent and naphtha contain the volatile hydrocarbons pentane through octane. This range of hydrocarbons includes hexane, a recognized neurotoxicant that causes a distal neurofilamentous axonopathy. n-Hexane’s concentration in Stoddard solvent is low enough that it is not expected to pose a risk of induced peripheral neuropathy (Chang, 1987). No evidence of this type of injury has been identified in experimental animals after exposure to Stoddard solvent. Although one study in rats has shown decreased nerve conduction velocities in the tail axon, as well as axonal prenodal swelling and demyelinated foci after dermal exposure to white spirit formulations (Stoddard solvent is a type of white spirit) the swellings were mild and not of the neurofilamentous type (Verkkala et al., 1984). Other investigations have generally not corroborated those findings.
Hydrocarbon-induced renal toxicity has been observed in male rats after inhalation exposure. The renal toxicity is thought to arise from an α2µ-globulin interaction, which results in the pathologic changes characteristic of the hydrocarbon-induced nephrotoxicity seen in male rats (Alden, 1986). Because α2µ-globulin is peculiar to male rats, a similar renal toxicity is not expected to occur in humans. Renal toxicity has not been reported in humans, rabbits, guinea pigs, dogs, or monkeys.
REFERENCES
Alden CL. 1986. A review of unique male rat hydrocarbon nephropathy. Toxicologic Pathology 14(1):109–111.
Andersen ME, Clewell HJ, Gargas ML, Smith FA, Reitz RH. 1987. Physiologically based pharmacokinetics and the risk assessment process for methylene chloride. Toxicology and Applied Pharmacology 87(2):185–205.
Arlien-Søborg P. 1992. Solvent Neurotoxicity. Boca Raton, FL: CRC Press.
ATSDR (Agency for Toxic Substances and Disease Registry). 1993. Methanol toxicity. American Family Physician 48:163–171.
ATSDR. 1997a. Toxicological Profile for Tetrachloroethylene. Atlanta, GA: ATSDR.
ATSDR. 1997b. Toxicological Profile for Chloroform. Atlanta, GA: ATSDR.
ATSDR. 1997c. Toxicological Profile for Ethylene Glycol and Propylene Glycol. Atlanta, GA: ATSDR.
ATSDR. 1999. Toxicological Profile for n-Hexane. Atlanta, GA: ATSDR.
ATSDR. 2000. Toxicological Profile for Toluene. Atlanta, GA: ATSDR.
BIBRA Working Group. 1991. Toxicity profile of hexylene glycol. Carshalton, UK: TNO BIBRA International Ltd.
BIBRA Working Group. 1993. Toxicity profile of diethylene glycol. Carshalton, UK: TNO BIBRA International Ltd.
Bruckner JV, Warren DA. 2001. Toxic effects of solvents and vapors. In: Klaassen CD, ed. Cassarett and Doull’s Toxicology: The Basic Science of Poisons. 6th ed. New York: McGraw-Hill. Pp. 763–810.
Bull RJ. 2000. Mode of action of liver tumor induction by trichloroethylene and its metabolites, trichloroacetate and dichloroacetate. Environmental Health Perspectives 108(Suppl. 2):241–259.
Burek JD, Nitschke KD, Bell TJ, Wackerle DL, Childs RC, Beyer JE, Dittenber DA, Rampy LW, McKenna MJ. 1984. Methylene chloride: A two-year inhalation toxicity and oncogenicity study in rats and hamsters. Fundamental and Applied Toxicology 4(1):30–47.
Casanova M, Bell DA, Heck HA. 1997. Dichloromethane metabolism to formaldehyde and reaction of formaldehyde with nucleic acids in hepatocytes of rodents and humans with and without glutathione S-transferase T1 and M1 genes. Fundamental and Applied Toxicology 37:168–180.
Cattley RC, DeLuca J, Elcombe C, Fenner-Crisp P, Lake BG, Marsman DS, Pastoor TA, Popp JA, Robinson DE, Schwetz B, Tugwood J, and Wahli W. 1998. Do peroxisome proliferating compounds pose a hepatocarcinogenic hazard to humans? Regulatory Toxicology and Pharmacology 27(1 Pt 1):47–60.
Chang YC. 1987. Neurotoxic effects of n-hexane on the human central nervous system: Evoked potential abnormalities in n-hexane polyneuropathy. Journal of Neurology, Neurosurgery, and Psychiatry 50(3):269–274.
David RM, Tyler TR, Ouellette R, Faber WD, Banton MI. 2001. Evaluation of subchronic toxicity of n-butyl acetate vapor. Food and Chemical Toxicology 39:877–886.
Dixit VP, Gupta RS, Kumar S, Joshi BC. 1980. Reversible chemical sterilization: Effects of cyclohexanol administration on the testes and epididymes of male rabbit. Indian Journal of Physiology and Pharmacology 24(4):278–286.
Elder RL. 1989. Final report on the safety assessment of ethyl acetate and butyl acetate. Journal of the American College of Toxicology 8(4):681–705.
FDA (Food and Drug Administration). 1989. Cosmetics; ban on the use of methylene chloride as an ingredient of cosmetic products. Federal Register 54(124):27328–27342.
Feldman R. 1998. Occupational & Environmental Neurotoxicology. Philadelphia: Lippincott-Raven.
Franco G, Fonte R, Tempini G, Candura F. 1986. Serum bile acid concentrations as a liver function test in workers occupationally exposed to organic solvents. International Archives of Occupational and Environmental Health 58(2):157–164.
Frantz SW, Beskitt JL, Grosse CM, Tallant MJ, Dietz FK, Ballantyne B. 1996. Pharmacokinetics of ethylene glycol. II. Tissue distribution, dose- dependent elimination, and identification of urinary metabolites following single intravenous, peroral or percutaneous doses in female Sprague-Dawley rats and CD-1 mice. Xenobiotica 26(11):1195–1220.
Graham, DG, Amarnath V, Valentine WM, Pyle SJ, Anthony DC. 1995. Pathogenic studies of hexane and carbon disulfide neurotoxicity. Critical Reviews in Toxicology 25:91–112.
Green T. 1997. Methylene chloride induced mouse liver and lung tumours: An overview of the role of mechanistic studies in human safety assessment. Human and Experimental Toxicology 16(1):3–13.
Green T. 2000. Pulmonary toxicity and carcinogenicity of trichloroethylene: Species differences and modes of action. Environmental Health Perspectives 108(Suppl 2):261–264.
Green T, Odum J, Nash JA, Foster JR. 1990. Perchloroethylene-induced rat kidney tumors: An investigation of the mechanisms involved and their relevance to humans. Toxicology and Applied Pharmacology 103(1):77–89.
Huang CC, Chu NS, Cheng SY, Shin TS. 1989. Biphasic recovery in n-hexane polyneuropathy. A clinical and electrophysiological study. Acta Neurologica Scandinavia 80(6):610–615.
Ichihara G, Saito I, Kamijima M, Yu X, Shibata E, Toida M, Kakeuchi Y. 1998. Urinary 2,5-hexanedione increases with potentiation of neurotoxicity in chronic coexposure to n-hexane and methyl ethyl ketone. International Archives of Occupational and Environmental Health 71(2):100–104.
Jorgenson TA, Meierhenry EF, Rushbrook CJ, Bull RJ, Robinson M. 1985. Carcinogenicity of chloroform in drinking water to male Osborne-Mendel rats and female B6C3F1 mice. Fundamental and Applied Toxicology 5(4):760–769.
Kitchin KT, Brown JL. 1989. Biochemical effects of three carcinogenic chlorinated methanes in rat liver. Teratogenesis, Carcinogenesis, and Mutagenesis 9(1):61–69.
Klaassen CD, Rozman K. 1991. Distribution, excretion, and absorption of toxicants. In: Klaassen CD, Amdur MO, Doull J, eds. Casarett and Doull’s Toxicology: The Basic Science of Poisons. 4th ed. New York: Pergamon Press. Pp. 50–57.
Lake BG, Foster JR, Collins MA, Stubberfield CR, Gangolli SD, Srivastava SP. 1982. Studies on the effects of orally administered dicyclohexyl phthalate in the rat. Acta Pharmacologica et Toxicologica (Copenhagen) 51(3):217–226.
Lakind JS, McKenna EA, Hubner RP, Tardiff RG. 1999. A review of the comparative mammalian toxicity of ethylene glycol and propylene glycol. Critical Reviews in Toxicology 29(4):331–365.
Löf A, Johanson G. 1998. Toxicokinetics of organic solvents: A review of modifying factors. Critical Reviews in Toxicolog 28(6):571–650.
Maronpot RR, Devereux TR, Hegi M, Foley JF, Kanno J, Wiseman R, Anderson MW. 1995. Hepatic and pulmonary carcinogenicity of methylene chloride in mice: A search for mechanisms. Toxicology 102(1–2):73–81.
Mennear JH, McConnell EE, Huff JE, Renne RA, Giddens E. 1988. Inhalation toxicity and carcinogenesis studies of methylene chloride (dichloromethane) in F344/N rats and B6C3F1 mice. Annals of the New York Academy of Sciences 534:343–351.
Nagano K, Nishizawa S, Yamamoto S et al. 1998. Inhalation carcinogenesis studies of six halogenated hydrocarbons in rats and mice. In: Chiyotani K, Hosoda Y, Aizawa Y eds. Advances in the Prevention of Occupational Respiratory Diseases,. Elsevier Science B.V. As cited in US EPA (US Environmental Protection Agency) 2001. Toxicological Review of Chloroform. IRIS (Integrated Risk Information System). Available: http://www.epa.gov/iris/toxreviews/index.html [accessed July 2002]
NCI (National Cancer Institute). 1976. Report on Carcinogenesis Bioassay of Chloroform. Bethesda, MD: Carcinogenesis Program, National Cancer Institute
Nitschke KD, Burek JD, Bell TJ, Kociba RJ, Rampy LW, McKenna MJ. 1988. Methylene chloride: A 2-year inhalation toxicity and oncogenicity study in rats. Fundamental and Applied Toxicology 11(1):48–59.
Palmer AK, Street AE, Roe FJ, Worden AN, Van Abbe NJ. 1979. Safety evaluation of toothpaste containing chloroform. II. Long term studies in rats. Journal of Environmental Pathology and Toxicology 2(3):821–833.
Roe FJ, Palmer AK, Worden AN, Van Abbe NJ. 1979. Safety evaluation of toothpaste containing chloroform. I. Long-term studies in mice. Journal of Environmental Pathology and Toxicology 2(3):799–819.
Rosenberg J, Cone JE, Katz EZ. 1997. Solvents. In: LaDou J, ed. Occupational and Environmental Medicine, 2nd ed. New York: Lange Medical Books/McGraw-Hill.
Rosenstock L, Cullen MR. 1986. Organic solvents and related substances. In: Clinical Occupational Medicine. Philadelphia: Saunders. Pp. 214–225.
Ruddick JA. 1972. Toxicology, metabolism and biochemistry of 1,2-propanediol. Toxicology and Applied Pharmacology 21(1):102–111.
Snyder R, Andrews LS. 1996. Toxic effects of solvents and vapors. In: Klaassen CD, ed. Cassarett and Doull’s Toxicology: The Basic Science of Poisons. 5th ed. New York: McGraw-Hill. Pp. 737–771.
Tephly TR. 1991. The toxicity of methanol. Life Sciences 48(11):1031–1041.
Tumasonis CF, McMartin DN, Bush B. 1987. Toxicity of chloroform and bromodichloromethane when administered over a lifetime in rats. Journal of Environmental Pathology, Toxicology and Oncology 7(4):55–63.
Tyagi A, Joshi BC, Kumar S, Dixit VP. 1979. Antispermatogenic activity of cyclohexanol in gerbil (Meriones hurriane Jerdon) and house rat (Rattus rattus rufescens). Indian Journal of Experimental Biology 17(12):1305–1307.
Valentine WM. 1990. Toxicology of selected pesticides, drugs, and chemicals. Short-chain alcohols. Veterinary Clinics of North America: Small Animal Practice 20(2):515–523.
Verkkala E, Pfaffli P, Savolainen H. 1984. Comparison of local neurotoxicity of three white spirit formulations by percutaneous exposure of rat tail nerve. Toxicology Letters 21(3):293–299.
Volkel W, Friedewald M, Lederer E, Pahler A, Parker J, Dekant W. 1998. Biotransformation of perchloroethene: Dose-dependent excretion of trichloroacetic acid, dichloroacetic acid, and N-acetyl-S-(trichlorovinyl)-L-cysteine in rats and humans after inhalation. Toxicology and Applied Pharmacology 153(1):20–27.
















