
3
The Framework
Under the provisions of the Dietary Supplement Health and Education Act (DSHEA), dietary supplements are to be considered as foods and assumed safe unless the Food and Drug Administration (FDA) has evidence that the supplement or one of its ingredients presents “a significant or unreasonable risk of illness or injury” when used as directed on the label or under normal conditions of use. Since the FDA is not authorized to require or impose premarket safety evaluations for dietary supplement ingredients marketed for use in the United States before October 15, 1994, FDA itself must monitor safety data and gather and assess existing information on safety to determine if a significant or unreasonable risk is present.
Thus the purpose of the Framework1 described in this chapter is to provide a process for FDA to translate the results of their scientific review into a decision regarding regulatory action needed to protect the health of the public.
CONSIDERATIONS IN DESIGNING THE FRAMEWORK
The Framework consists of two components: (1) a process for prioritizing, evaluating, and describing available information to establish risk of
harm, and (2) a set of science-based principles that serve as guidelines for evaluating risk to human health.
For the Framework to be useful, FDA must have adequate resources for implementation. To be credible, it must be scientifically based and include guidelines for obtaining and integrating the totality of the information from many areas of science. The Framework should allow FDA to react to information, as well as to proactively gather information. It needs to be efficient and provide a system for updating information as new information becomes available. In providing a scientific infrastructure for the evaluation of the safety of dietary supplement ingredients, the framework must facilitate decision-making regarding a dietary supplement’s potential to cause harm when uncertainty exists. Adequate staff with appropriate expertise must be available within FDA to administer the process and evaluate the information.
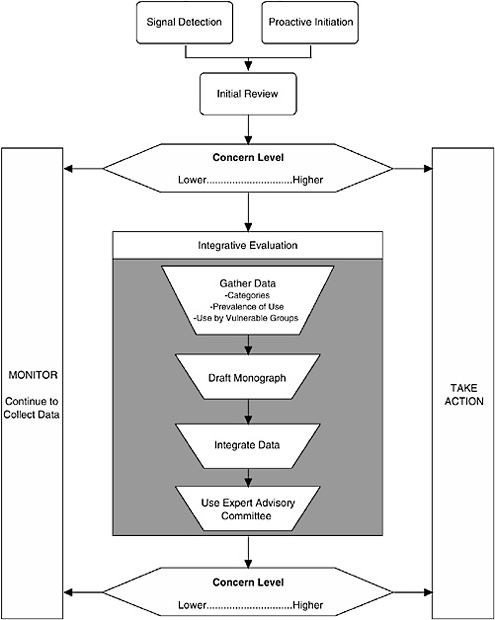
The Framework described here characterizes the nature of the scientific evidence that FDA is likely to encounter and describes a process for organizing this evidence to assess where a dietary supplement ingredient lies on a spectrum of concern2 (see Figure 3-1). As the level of concern increases, so does the potential for a “significant or unreasonable risk,” the standard warranting regulation under the Food, Drug, and Cosmetic Act (FDCA), as amended by DSHEA.
I. THE PROCESS
The process comprises three major components:
-
Signal detection,
-
Initial review of the signal, and
-
Integrative evaluation.
Signal Detection
According to the DSHEA, it is assumed that dietary supplements are generally safe; given the large number of dietary supplement ingredients, it is unlikely that FDA will have the resources or the need to evaluate each ingredient uniformly to determine if it presents an unreasonable risk of illness or injury. Thus, at least initially, it is assumed that some “signal” will indicate that an ingredient’s safety may need to be reviewed. When a signal is detected, it is up to FDA to decide the next step once the credibility of the
signal is evaluated and to determine the possibility that the ingredient caused the adverse effect noted (using the guiding principles outlined later in this chapter and discussed in detail in the chapters that follow).
What Constitutes a Signal?
FDA is likely to receive or become aware of a variety of signals suggesting potential risks to human health with the use of a dietary supplement ingredient. Signals may come to FDA’s attention and thus be “detected” through notice of regulatory action taken by other countries regarding a specific dietary supplement, through routine monitoring of medical and scientific literature, directly through it’s own Special Nutrition/Adverse Event Monitoring System, or through consultation with experts. FDA may also become aware of signals indirectly through reports in the media, through new data from animal experiments suggesting a specific risk, or through information provided by consumer protection advocacy groups. Signals can thus come from many sources and originate from many different types of scientific data. Given the significant number of dietary supplement ingredients, FDA’s attention should focus on signals that indicate a serious3health problem may result from ingestion of a dietary supplement ingredient.
Quality of the Signal
In this first component little is done to evaluate the quality of the data because the focus is simply on signal detection. While some signals may result from concerns expressed by other expert bodies, such as those described in Chapter 2, or by case reports of adverse effects, the quality of the signal is not reviewed until the second component of the process (initial review of available information). The quality of the information behind the signals detected will be highly variable and in some cases may provide only weak evidence or be of little use or credence. Nonetheless, detecting these signals requires the attention of qualified professional staff at FDA and will result in a reaction by FDA (even if the reaction is only to consider the signal of little importance, as described in the next component).
Proactive Initiation of Review
In contrast to reaction based on detecting a signal, FDA may decide to proactively initiate a review of a dietary supplement ingredient due to high prevalence of use in the general population, high prevalence of use by a particularly vulnerable population, or other factors. More than likely, however, a signal indicating possible concerns will be the instigating factor in further review of a substance.
Prioritization for Review
One of the requirements of the study was to develop a framework that would include criteria for how the review of safety of dietary supplements and ingredients should be prioritized. It was suggested that a scheme to initially identify dietary supplement ingredients considered of higher priority for subsequent review based on only one criterion, such as end-organ toxicity in animal studies or the structure of one or more known compounds present in the ingredient be devised and applied to all dietary supplement ingredients. However, given the wide variety of dietary supplement ingredients available, the multiple forms of a specific ingredient that are sold, the voluntary and thus varying nature of the data available on an ingredient, and the wide variety of adverse effects that are possible for dietary supplements and the dependence of such effects on exposure levels, such a scheme is not feasible nor scientifically defensible.
This is not to say that the availability of data from only one category is not enough to determine a higher level of concern. As emphasized in the following chapters on the various types of data, any one category of information can raise concern to a level that requires action by FDA. A hierarchy of adverse effects that warrant greater concern than others based on collective judgment is provided in Chapters 4 through 7; however, a formulaic or algorithmic approach that considers all the important variables—such as the dose at which such effects may occur, the relevance of the information, the information available suggesting the ingredient may be safe—is not useful given the multidimensional matrix that would be needed.
The signal detection step, followed by an initial review of the information, should serve to identify those dietary supplement ingredients that are in need of further review and evaluation via an integrated evaluation.
Initial Review of Available Information
The second component of the Framework is to conduct an initial review of available information. First, the nature of the information generating the signal is examined to determine the appropriate level of concern
regarding a risk to human health. This component is not envisioned as a detailed analysis of data, but rather as an assessment of the concern level warranted by the nature of the evidence (e.g., quality of the report, applicability to humans, route of exposure) and whether the information raises questions that require further examination.
Second, some effort can be made to gather easily available data to place the detected signal in context; such additional information may come from many sources, including other categories of data. Thus this initial review of the signal information need not be limited to reviewing only the information associated with the signal. For example, if the signal is a case report suggesting a possible problem in an elderly woman and clinical trials of the ingredient exist, these should be considered during the initial review.
Level of Concern
The outcome of the initial review is a determination of the initial overall level of concern to decide if an integrative evaluation is needed. Higher concerns warrant an integrative evaluation; lower concerns do not.4 A decision about an ingredient with a moderate concern level should be made after a review of other information to see if other signals are apparent; for example, if the initial signal is animal data that warrant moderate concern, a cursory literature search on the substance or a review of FDA’s adverse event monitoring system could be conducted to determine if other data about the ingredient raise concerns as well, leading to the need for further evaluation.
Assuming that sufficient evidence may not be available from just one type or category of data to cause a higher level of concern, it is important for FDA to consider data from other categories to determine if a higher level of concern may exist.
Decisions Possible Based on Initial Review
When the initial review of the nature of the evidence available indicates a higher level of concern, FDA would then initiate an integrative evaluation process or possibly decide to take immediate action, if the concern is serious enough and the data are strong. If the level of concern is categorized as relatively low, FDA would continue to monitor signals and incorporate the
information obtained into a monitoring database for future use if new data regarding the ingredient become available.
Maintaining a database of specific issues to monitor would allow FDA staff familiar with the criteria outlined in Chapters 4 through 8 to systematically look for information that may address the data gaps. Similarly, data collected should be saved in case a decision is made to move to an integrative evaluation. If a decision is made to conduct an integrative evaluation, but a monograph is not subsequently prepared, then information and a summary of the thought processes involved in the integrative evaluation should be noted and filed for future consideration. Making data gaps and unanswered questions available to other interested parties such as the National Toxicology Program of the National Institute of Environmental Health Sciences (NIEHS) or the Office of Dietary Supplements, both part of the National Institutes of Health (NIH), Department of Health and Human Services, would allow them to incorporate these data needs on specific dietary supplement ingredients into their programs of work.
In summary, once the initial level of concern based on the initial review of the signal is determined, FDA might decide that continued routine monitoring is needed, or it could decide to proceed with an integrative evaluation. This depends on the level of concern raised by the signal: ingredients provoking higher concern should proceed to the integrative evaluation; ingredients resulting in lower levels of concern would generally not proceed; and ingredients with moderate concerns might proceed after considering additional information not necessarily related to the initial signal, such as prevalence of use or concern related to a specific vulnerable population group. Since it is assumed by the DSHEA that dietary supplements are safe, there should be relatively few dietary supplement ingredients that will be categorized as of higher concern after the initial review and thus warrant further examination. This allows FDA to focus its efforts on dietary supplement ingredients that are strong candidates for regulation.
Integrative Evaluation
The third component of the Framework is to conduct an integrative evaluation for those dietary supplement ingredients that are deemed to warrant further investigation, based on the preliminary data reviewed in the second component and the resulting relative placement on the spectra of concern continuums. There are four aspects to the Integrative Evaluation component (see Figure 3-1): in-depth literature search and review, drafting a safety monograph based on this information, integrating the available data into an analysis to complete the monograph, and possibly referring the draft monograph and accompanying information to an expert committee for additional input prior to determining whether to take regulatory action.
Reviewing the Literature
A critical review of the literature is a three-part process. First, multiple databases are searched for information on the dietary supplement ingredient and other substances with similar taxonomical, structural, or functional properties. Such searches are broad-based, and include information on safety and biological activity of the ingredients, including human data, animal data, and in vitro data.
Second, each primary research paper is reviewed for internal consistency; for example, are proper methodologies used? Do data fit the conclusions? Are the associations real? Is appropriate information included? Are there chance, bias, confounding variables, a lack of coherence, or other significant internal issues or limitations that should be taken into account?
Third, the external consistency of the research papers must be judged as a group. Are the studies coherent as a whole? Is there strength in the associations, general agreement, etc.? Studies can then be sorted into those that suggest that there is little risk of illness or injury when consuming the supplement ingredient, those that indicate a relevant concern for risk of illness or injury, and those that have equivocal results. Each should then be examined for flaws and strengths in accordance with the principles and concepts discussed in the subsequent chapters on each general category of data (Chapters 4 through 7).
Focused Versus Broad-Based Evaluation. An integrative evaluation might be reactive to the signal and focused in nature, in that it is conducted to examine a specific moderate- or high-level concern about an ingredient, or it might be more proactive and broad-based, in that it is looking for any risk associated with use of the dietary supplement ingredient. As described above in the description of the signal detection component, a proactive integrative evaluation might be initiated simply because a large percentage of the population is using the ingredient, rather than as a reaction to a particular safety concern.
The amount of information gathered depends on the nature of the harmful effect that is the focus of concern. If a focused evaluation is conducted, it is assumed that less information will be reviewed. However, the relative importance of an individual study is established in conjunction with an evaluation of other relevant literature. Clearly, data or information outside the primary safety concern may include information that has a direct bearing on the overall evaluation of the safety concern identified in the signal component. Thus a comprehensive review can provide information that may raise concerns in other areas not relevant to the focus, but which should not be ignored in a safety monograph.
Relevancy of Data. Gathering data and reviewing it for relevance provides the scientific base upon which FDA can substantiate its conclusions. Data that are not relevant to safety or to the concern in a focused integrated evaluation need not be incorporated into the report; however, acknowledging that it was considered and deemed irrelevant will be helpful if the information has been characterized by others as substantiating safety.
Drafting a Safety Monograph
In most cases, the integrative evaluation will be documented in a monograph5 that summarizes the categories of data available and their use in drawing conclusions about the potential risk associated with use of the ingredient. Evidence obtained either from only one category of data or from integrating all the categories that results in an increased level of concern should result in a higher priority for development of a safety monograph.
A monograph need not be developed for every dietary supplement ingredient, as it is assumed that only those ingredients with moderate or higher concern levels following the initial review will be subject to an integrative evaluation (essential nutrients represent a special case; see Annex 3-1). The monograph may not need to cover every concern, in which case a focused integrative evaluation and resulting monograph would be completed. In a few cases, where available information obtained in the initial review results in a highly significant level of concern, it may be necessary to undertake regulatory action prior to or without developing a monograph. The development of a monograph may be resource and time intensive, especially when initiated proactively and thus with a much broader focus.
However, the development of a monograph provides a method to document in a systematic format the evidence on which FDA can base a regulatory decision. The science-based guiding principles described in the following sections of this chapter, and explained in detail in the following chapters, should be used to reach a decision regarding whether there is an unreasonable risk of illness or injury.
The general types of information to be collected and used in the integrative evaluation and thus collated in a monograph are listed in Box 3-1 and include a description of the ingredient (e.g., constituents, different types of preparations, typical intake amount and duration, historical use) and available information about toxicities and safety (human data, animal data, data describing risks associated with related substances, and in vitro data). In most cases, this information will be gathered from the medical and
|
BOX 3-1
|
scientific literature. However, additional information may be obtained by requesting information from clinical investigators who have published reports about the particular ingredient, as well as by requesting information from industry (e.g., distributors and manufacturers) and other stakeholders. The collected information should be collated into a draft safety monograph. The monograph should be prepared using a standard format to summarize all the data collected on the ingredient (see Annex 3-2 to this chapter for a more detailed discussion of monograph preparation).
Integrating the Data to Determine Risk
The data evaluation component of the integrative evaluation should be conducted on the initial assumption that consumption of the supplement should not present an unreasonable risk of illness or injury, as is assumed in DSHEA. To overturn this assumption, the end result of the review should demonstrate that there is an unreasonable risk of illness or injury to the consumer.
When evidence on a dietary supplement ingredient presents a moderate or higher level of concern relative to this risk, data from other categories should be considered to evaluate biological plausibility and consistency. Integration within and across the other categories of data will help determine if an unreasonable risk exists by looking at the overall picture. Such an analysis can be represented by creating a causal model diagram—a tool to organize the data to visualize how the different types of available data link together to establish risk (described in Chapter 10). For example, in reviewing the potential for concern in the use of saw palmetto for the prototype monograph described in this report, data from all categories were integrated to make a conclusion about risk (see Chapter 11 and Appendix H).
The principles described for considering the various types of data and modifying factors (Chapters 4 through 9), as well as the principles described for how to integrate among and within categories of data (Chapter 10), are applied in the integrative evaluation. They should be followed in assessing and weighing the different types of evidence that enter into the decision. They are summarized in the conclusions in the safety monograph. The conclusions should describe:
-
The relevance of the evidence;
-
How the dose, manner of use, and product affect conclusions about risk;
-
The seriousness of the potential harm suggested by the evidence; and
-
The quality and strength of the evidence.
Review of the information does not need to prove toxicity, only that there is an unreasonable risk of its occurrence (see Box 3-2). Such an analysis is captured in the monograph. The evaluation of the totality of the scientific evidence is thus summarized in conclusions about risk based on the high level of concern resulting from the in-depth review and analysis of the available information.
To guide those making conclusions about risk as a result of the integrative evaluation component, it might be possible to develop a taxonomy of levels of risk—such as “no basis for concern,” “some grounds for further monitoring,” “some basis for concern about risk,” or “presents a risk that warrants regulation under the FDCA as amended by DSHEA.” This was not done in this report because the definitions might become too prescriptive given the variety of information and types of dietary supplement ingre-
|
BOX 3-2 It has been said that the “… dose differentiates a poison from a remedy.” Even essential substances for humans, such as oxygen and water, can be toxic in high concentration or if imbibed in large amounts. Thus no substance is completely “safe.” Safety is a qualitative term that is applied to a variety of situations or environmental factors and is related to the context in which it is evaluated. What is safe in one situation (e.g., driving 50 mph) might be considered unsafe in another. In relation to ingested substances, in some cases it is possible that concerns related to adverse effects resulting from consumption may be mitigated by benefits derived from the substance when ingested. For drugs and medical devices, safety is evaluated as a measure of potential harm relative to benefit (see Chapter 1). For food additives, safety is defined as the reasonable certainty of no harm, without consideration of benefit beyond that of improving the functional characteristics of the resulting food product, such as retarding microbial growth or maintaining texture. The DSHEA classifies dietary supplements similarly to food, and therefore supplements are considered, like conventional foods, to be reasonably safe. While dietary supplements are biologically active substances that may have desirable health benefits, they may also cause adverse health outcomes. DSHEA requires that the FDA determine that a dietary supplement ingredient is unsafe (i.e., consumption results in unreasonable risk of illness or injury at recommended intake levels) rather than requiring that a manufacturer provide data supporting its safety, as it does for food additives, drugs, and medical devices. Since FDA’s authority is limited to evaluating a dietary supplement ingredient for potential to cause illness or injury, but it cannot take into account possible beneficial effects on health, any safety framework for a dietary supplement ingredient must depend on (1) the accumulation of evidence indicating potential for harm and (2) the determination of when this accumulated evidence raises concern to a point that a significant or unreasonable risk exists. |
dients. Thus this Framework, while qualitatively providing descriptions of points on a continuum of relative concern about risk derived for the various types of data, does not include a metric for categorization of risk.
Referring Review to an External Advisory Committee
After considering the conclusions about risk in the draft monograph, FDA should make a decision to (1) take regulatory action, (2) not take regulatory action and continue to monitor for new data regarding safety, or (3) refer the dietary supplement ingredient to an advisory committee of multidisciplinary experts for a safety review.
It is expected that FDA may want further input from an advisory committee on many of the dietary supplement ingredients undergoing an integrative evaluation because only ingredients with significant potential for concern are likely to reach this stage, and outside evaluation may be critical to ensure that all relevant information was reviewed. Also, in cases where FDA does not have internal scientists with the appropriate expertise, it may be cost-effective to create an external advisory committee to provide further input on the safety of the dietary supplement ingredient. This could be an activity under the existing Food Advisory Committee of the Center for Food Safety and Applied Nutrition of FDA, or it could be an additional committee, either standing or ad hoc, depending on the ingredients to be reviewed. See Annex 3-2 for additional discussion of the composition of an advisory committee.
Where the data and thus conclusions are not clear-cut, an external advisory committee would thus be constituted for the following reasons:
-
While there may be credible evidence that the ingredient may cause harm, further review may be needed by consultants with specific knowledge about the ingredient, as well as by consultants with specific knowledge about the safety issues raised, to interpret the totality of the data and derive conclusions and recommendations.
-
The available evidence may be of questionable scientific basis or it may be difficult to interpret.
-
Insufficient data may be available to allow the rationale for the decision to be clearly established.
-
It provides a mechanism for public input.
These reasons are only examples, as many other circumstances may trigger the need for external advisory committee review (See Annex 3-3 for committee composition).
After reviewing the information collected in the draft monograph and in the public information sessions envisioned as part of their deliberations,
the external advisory committee should provide input to FDA regarding revisions in the draft monograph, as needed, to create as complete a picture of the available scientific information on safety as possible, within the resources made available to FDA. The advisory committee should evaluate the ingredient based on the weight of the scientific evidence as described in the previous section.
The advisory committee’s report should include comments about the risks and hazards that may be associated with use by the general population, as well as risks that may be particular to subgroups of the population. As much as possible, the advisory committee should describe how its review of the safety depends on how the ingredient is used—the dose, manner, and form.
The advisory committee may conclude that there is inadequate evidence within the available information to suspect a hazard to the public when the ingredient is used at the levels recommended on the label or at levels that might reasonably be expected. If current use does not demonstrate a hazard, the advisory committee may decide to comment on whether it is possible to foresee that a significant increase in consumption would constitute a hazard. If there is not enough information available to conduct a scientific evaluation of the safety of the dietary supplement, the advisory committee should indicate this.
In cases where the data are insufficient to determine whether a hazard exists, conclusions should also be accompanied by a brief description of additional research that would be most useful in forming science based decisions.
Decision to Take Action
After the advisory committee’s review is shared with FDA, the completed monograph and the advisory committee’s comments should be posted on FDA’s website. One of the important components of DSHEA was that the public should be educated about dietary supplements. FDA thus has a responsibility to educate consumers about the safety of supplement ingredients, and the public availability of the completed monographs can be an important aspect of the educational process. The monographs can provide the public with a reputable summary of the available information and scientific uncertainties about the inherent safety of the supplement ingredient whose safety has been questioned.6 Importantly, public access to infor-
mation from an advisory committee will add to the quality and strength of the available scientific literature.
The decision to refer a dietary supplement ingredient to an external advisory committee rests with FDA. As with other federal advisory committees, while the external advisory committee opinions or conclusions should be based on the information and data presented, the decision on whether to follow the determinations of the external advisory committee rests with FDA, as it alone possesses regulatory authority in these matters. If FDA decided to take an action, it would initiate a judicial enforcement proceeding, such as a seizure, suit for injunction, or prosecution, designed to elicit a court ruling that the supplement was unsafe. In order to justify the use of FDA’s resources to the extent envisioned by the Framework, the results of the integrative evaluation should play a pivotal role in establishing that a supplement ingredient is unsafe.
As a result of the integrative evaluation, it is quite possible that FDA will decide to take action, declaring that a dietary supplement ingredient presents a significant or unreasonable risk of illness or injury. It is also possible that, more selectively, concerns related to the use of a supplement by a vulnerable group within the population may be highlighted, so that specific action related to the use by specific groups is possible where warranted, even though the general population may not be at the same level of risk.
An added benefit of making monographs easily available to the public is that industry and publicly funded scientists may choose to conduct studies that address the concerns raised, thereby increasing the knowledge base regarding the safety of dietary supplements. The general public, as well as industry, pharmacists, health care providers, and distributors, will benefit from the publicly available information and individually can decide whether to use, sell, or recommend the dietary supplement ingredient in question, regardless of whether FDA decides to take action or not.
Decision to Continue to Monitor
When the review of information, either at the initial review step or as a result of an integrative evaluation, indicates a lower level of concern, FDA should continue to monitor information it receives relative to the dietary supplement ingredient. Monitoring consists of either passively watching for new signals of other concerns about the ingredient or developing search strategies to routinely search the scientific literature for new data to address specific concerns. (See Chapter 12 for how monitoring might be approached for some of the dietary supplement ingredients reviewed in the prototypes.) Monitoring might also include working with the National Toxicology Program at NIEHS or the Office of Dietary Supplements at the NIH to initiate
research addressing unanswered questions relative to some of the signals detected.
II. APPLYING SCIENCE-BASED PRINCIPLES TO ESTABLISH RISK
In outlining the task, FDA requested that the Framework include a method based on safety concerns to categorize and prioritize dietary supplement ingredients sold in the United States. Given the variety of types of information that are likely to be available, the Framework classifies scientific information into four broad categories for use in determining the potential for serious harm for a specific dietary supplement ingredient. These categories of data include:
-
Human data,
-
Animal studies,
-
In vitro experiments, and
-
Information on related substances.
Subsequent chapters describe the types of information that may be available in each category of data and the strengths and weaknesses of these different data sources in evaluating the potential of a dietary supplement ingredient to cause harm (Chapters 4 through 7). Also described are how to consider the potential for dietary supplement interactions with drugs and other xenobiotics7 (Chapter 8), important considerations that should be factored into evaluations when vulnerable populations consume dietary supplements or when supplements are widely consumed (Chapter 9), and considerations for integrating the available data from various sources to determine an overall level of concern (Chapter 10) using a causal model diagram. The level of concern appropriate for a specific piece of information within a particular data category (i.e., human, animal, in vitro, or related substances information) is summarized in diagrams that relate the available evidence to show the level of concern when consuming a dietary supplement ingredient.
Evidence that results in a higher level of concern indicates a more immediate priority for further investigation to determine if an unreasonable risk to public health exists. In contrast, a single piece of information resulting in a lower level of concern may suggest that continued routine monitoring for new evidence is warranted—evidence that might elevate the level of concern and thus its priority for increased scrutiny.
Although each chapter strives to describe all types of information that may be available, it is important to recognize that for most dietary supplement ingredients, it will be difficult, if not impossible, to find useful information from all data categories. The following section provides an overview of the types of information that may be encountered and summarizes general scientific guidelines for assessing the relevance and quality of the available information from each data category (Box 3-3). More specific information and details are provided in Chapters 4 through 9.
Spectra of Concern
As briefly outlined in the process description earlier in this chapter, included in the Framework is a qualitative method to evaluate the nature of the evidence for a specific piece of information within a particular data category (i.e., human, animal, in vitro, or information about related substances). Distinguishing characteristics of evidence determine where a piece of information falls on the continuum of lower to higher level of concern. This is summarized in diagrams referred to as spectra of concern. Evidence that results in a higher level of concern indicates a more immediate priority for investigating further whether an unreasonable risk to public health exists, because a higher level of concern suggests a potential risk to public health. In contrast, a single piece of information resulting in a lower level of concern may suggest continued routine monitoring for new evidence that might elevate the level of concern and thus initiate increased scrutiny.
Human Data
Information about human use of dietary supplement ingredients may be in the form of formal studies, such as clinical studies or trials and epidemiological studies; in the form of spontaneously reported adverse event reports or literature case reports; or in the form of information about historical use of the ingredient. Because there is no requirement that dietary supplement ingredients undergo formal studies prior to marketing, formal study data on a dietary supplement ingredient will be less commonly available than spontaneous adverse event reports and information about historical use. The lack of such data, however, does not diminish their importance.
Data about human intake can be useful either as indicators of possible risk or, conversely, as mitigators of concerns raised by other categories of data. Within each type of human data, questions can be asked about the nature and quality of the scientific information to determine whether the information raises the level of concern regarding the probability to cause harm. While discussed in detail in Chapter 4, the general spectra of concern
|
BOX 3-3
|
related to human data are identified in Tables 3-1 through 3-5. An important concept in Chapter 4 that is not captured in the spectra of concern tables is that historical use information becomes less relevant as difference from traditional use increases. Changes in historical versus modern use may arise from new methods of preparation (e.g., plant part used or extraction process) or new patterns of use (e.g., higher intake level, route of administration, duration and frequency of consumption, indication for use).
Animal Data
Information about animal exposure to dietary supplement ingredients may be in the form of formal studies, such as traditional toxicity studies, safety pharmacology data, or observations from clinical veterinary medicine. Because dietary supplement ingredients are not required to undergo formal animal toxicity testing before marketing, extensive toxicity studies are uncommon, but limited amounts of animal data for a number of dietary supplement ingredients are available in the scientific literature. Despite the challenges of dealing with incomplete data, available animal data warrant attention when assessing risk of dietary supplement ingredients.
Animal studies are powerful because controlled studies can be conducted to predict effects that might not be detected from customary use by humans until they result in overt harmful effects. Animal studies are especially useful in detecting effects of chronic exposures and effects on reproductive and developmental processes because epidemiological methods of studying humans are especially problematic in these areas. The ability to administer agents to animals during their entire lifespan enables scientists to ascertain the potential toxic effects that may arise for long-term (chronic) exposure. Animal studies thus serve as important hypothesis generators and may be sufficient to indicate potentially unreasonable risk to human health, which justifies their use in evaluating the risks of dietary supplement ingredients to humans.
In general, adverse effects observed in well-designed and conducted animal studies should be treated as if they occur in at least some members of the human population, assuming humans receive a sufficiently high dose. With some notable and important exceptions, the biological factors affecting the capacity of chemical substances to cause toxicity are broadly similar across mammalian species. While discussed in detail in Chapter 5, the general spectrum of concern related to animal data is described in Table 3-6, based on the relative seriousness of adverse effects seen in animal studies (Box 3-4).
TABLE 3-1 Relative Spectrum of Concern for Individual Spontaneous Adverse Event Reports
|
Increasing Concern |
||
|
|
||
|
Describes a serious adverse event with less information than would justify moderate or strong concern, and/or with prominent confounding factors (e.g., multiple concomitant substances and/or conditions) |
Describes a serious adverse event with some, but not all, characteristics associated with strong concern |
Describes a well-documented serious adverse event with plasma levels (if available) at a relevant range and demonstrates dechallenge and rechallenge (if possible), temporality, and strong attribution |
TABLE 3-2 Relative Spectrum of Concern for Case Series of Spontaneous Adverse Event Reports
|
Increasing Concern |
||
|
|
||
|
Describes a series of serious adverse events, with less information than would justify moderate or strong concern, and/or prominent confounding factors (e.g., multiple concomitant substances and/or conditions) |
Describes a series of serious adverse events, with some, but not all, characteristics associated with strong concern |
Describes a series of well-documented cases demonstrating consistent serious adverse events and clinical findings, and dechallenge (if possible), temporality, and strong attribution |
TABLE 3-3 Relative Spectrum of Concern Raised by Historical Evidence of Toxicity
|
Increasing Concern |
||
|
|
||
|
Traditional cautions (contraindications) exist regarding use in certain populations or circumstances |
Traditional cautions (contraindications) exist regarding use in certain populations or circumstances that, if ignored, might be associated with a serious adverse effect (e.g., do not use during pregnancy) |
There is clear evidence that traditional use causes conditions considered to be serious adverse events (e.g., hallucination, lethal poisoning) |
TABLE 3-4 Spectrum of Relative Concerns with Clinical Studies Data
|
Increasing Concern |
||
|
|
||
|
Describes a serious adverse event, but with less information than would justify moderate or strong concern, and/or the interpretation of the clinical study is hampered by the presence of prominent confounding factors (e.g., multiple concomitant substances and/or conditions) that could not be controlled by balancing AND/OR Prominent methodological concerns (e.g., unexplained high level of dropouts, lack of control groups) |
Nonsignificant, but clinically important, trend of a higher rate of a serious adverse event OR Abnormalities in clinical laboratory values OR Other abnormalities, such as electrocardiographic findings in the dietary supplement ingredient group |
A significantly higher incidence of a serious adverse event OR Other potentially dangerous abnormalities, such as in clinical laboratory values that are associated with risk of serious adverse events OR Other abnormalities, such as electrocardiographic findings in the dietary supplement ingredient group |
TABLE 3-5 Spectrum of Relative Concerns with Epidemiological Data
|
Increasing Concern |
||
|
|
||
|
Case-control or cohort study (including registries), with small,a but statistically significant, relative risk or odds ratio of a serious adverse event OR Large relative risk or odds ratio of a serious adverse event that is not statistically significant OR Poorly conducted studies with large or significant effects |
Case-control or cohort study (including registries), with moderate, statistically significant relative risk or odds ratio of a serious adverse event OR Moderate relative risk or odds ratio of a serious adverse event that is not statistically significant but that implies a trend |
Well-conducted case-control or cohort study (including registries), with large, statistically significant relative risk or odds ratio of a serious adverse event |
|
a In short, 2 or less is generally considered weak association, and 3 or more is considered strong, but this is only a very general “rule of thumb” guidance, which is somewhat debatable. |
||
TABLE 3-6 Relative Spectrum of Concern: Guidelines for Types of Evidence from Animal Studiesa
|
Increasing Concern |
||
|
|
||
|
At least one acceptable, quality study showing effects of Category A at Dose > 1,000× Human Intake |
At least one acceptable, quality study showing effects of Category A at Dose > 100 to < 1,000× Human Intake |
At least one acceptable, quality study showing effects of Category A at Dose < 100× Human Intake |
|
OR |
OR |
OR |
|
At least one acceptable, quality study showing effects of Category B at Dose > 100× Human Intake |
At least one acceptable, quality study showing effects of Category B at Dose > 10 to < 100× Human Intake |
At least one acceptable, quality study showing effects of Category B at Dose < 10× Human Intake |
|
OR |
OR |
OR |
|
At least one acceptable, quality study showing effects of Category C at Dose > 10× Human Intake OR Studies showing adverse effects, but which cannot be interpreted because of deficiencies in design, conduct, or reporting OR Acceptable, quality non-oral studies indicating adverse effect from Category A, B, or C |
At least one acceptable, quality study showing effects of Category C at Dose > 1 to < 10× Human Intake |
At least one acceptable, quality study showing effects of Category C at Dose ≤ 1× Human Intake |
|
a Categories A, B, and C refer to relative seriousness of a variety of adverse effects identified in animal studies, ranging from reproductive failure (A) to reduced food consumption (C). See Box 3-4 for further examples. |
||
Information About Related Substances
Information about substances related to the dietary supplement ingredient of interest may be useful when predicting risk to human health. Such substances may be related to dietary supplement ingredients in one of several ways, such as:
-
Chemical relatedness—the ingredient or constituent of an ingredient is similar to known toxic chemicals, or is known to contain chemicals similar in structure to known toxicophores;8
|
BOX 3-4 Category A (most serious)
Category B (moderately serious)
Category C (less serious)
|
-
Taxonomic relatedness—the ingredient is from the same classification as a known toxic plant species, genera, or family;
-
Functional relatedness—the ingredient or chemical constituent shares a common biological target or mechanism of action that is clearly tied to a toxic effect demonstrated with another substance. This includes endogenous substances and mimetics of endogenous substances, when the effect of increasing the amount of an endogenous substance is linked to an adverse health effect.
Taken together, the value and utility of this information to predict risk depends on the type of dietary supplement ingredient that is being considered. A concern may be raised about a botanical dietary supplement based on information about risk associated with known chemical constituents, as well as information about risk associated with related toxic plants. Similarly, pure single chemical compounds may be of concern based on comparison to known risk-associated chemical compounds and chemical moi-
eties (toxicophores) that raise concern of safety. Substances that are normally present in the human body (endogenous substances) may be of concern based on knowledge of what the substances do in the body at normal concentrations, and an understanding of what might occur if the normal concentrations are exceeded. For dietary supplements for which the chemical composition is undefined,9 but for which information about biological activity is available, it may be helpful and it is appropriate to consider whether the exhibited biological activity is the basis for safety concerns of other substances that are considered potentially harmful. While discussed in detail in Chapter 6, the general spectrum of concern regarding related substances is described in Table 3-7, based on knowledge of the relative seriousness of adverse effects seen with ingestion of related substances.
In Vitro Studies
A range of in vitro experimental systems are used to gain insight into the risk of adverse effects of compounds. These systems include isolated organs, isolated cells, microorganisms, subcellular organelles, and molecular entities such as enzymes, receptors, transport proteins, isolated membranes, and genes or gene fragments. A primary advantage of conducting in vitro studies is that their reductionist (non-whole-organism) approach allows insight into a compound’s mechanisms of action that might be more difficult to obtain in a whole-animal study. The control possible with in vitro experiments enables examination of the effect on the target process or structure in isolation from confounding factors. For example, control over the concentration of the chemical of interest or of one or more of its metabolites enables the interactions among chemicals or metabolites to be studied. In vitro experiments are also generally more rapid and less expensive to conduct than in vivo studies; thus, in vitro studies are more likely than in vivo studies to be available for assessment of dietary supplement safety.
Some experiments are specifically designed to examine safety endpoints while the information provided by other experiments is less specific about an ingredient’s biological activity. Because no battery of tests is required on dietary supplement ingredients, results from safety tests common to other chemicals are not widely available. When they are available, these “validated” in vitro assays—assays that are accepted for use in predicting effects on whole organisms—can be of significant use. In such studies, the serious-
ness of harm can be predicted by a given assay. While discussed in detail in Chapter 7, the general spectrum of concern regarding in vitro data is described in Table 3-8 and is based on the predictability that adverse effects may occur in vivo.
Interactions
One of the major concerns about the safety of dietary supplement ingredients is that interactions between a supplement and other ingested substances (drugs, other dietary supplement ingredients, conventional foods) will result in adverse clinical outcomes due to an increase or decrease in the level of the dietary supplement in the organism, an increase or decrease in the level of other xenobiotics, or combined toxicities.
Interactions can be detected with human, animal, or in vitro studies or predicted on the basis of how related substances behave. There are numerous mechanisms for interactions among xenobiotics, but most can be categorized as direct chemical-chemical, pharmacodynamic, or pharmacokinetic interactions. In direct chemical-chemical interactions, the action of one or both chemicals is modified by taking them within a relatively short time of each other. With pharmacodynamic interactions, there is a change in response to either the dietary supplement ingredient or the xenobiotic, but with no change in plasma concentration in either. Pharmacokinetic interactions, which occur when one substance affects the absorption, distribution, metabolism, or excretion of the other, result in altered levels of one of the substances or its metabolites. In vitro and in vivo experimental methods for identifying ingredients that may cause such interactions are available. While discussed in detail in Chapter 8, the general spectrum of concern regarding interactions among dietary supplement ingredients and other dietary supplements, foods, or drugs is described in Table 3-9, based on prediction of serious adverse events. It should be noted that the potential seriousness of these interactions varies.
Prevalence of Use and Vulnerable Groups in the Population
The scientific bases for evaluating the safety of dietary supplement ingredients described in this section are critical in determining which dietary supplement ingredient warrants the most immediate attention (i.e., in setting priorities). However, it is also appropriate to take other information into consideration when setting priorities. That is, given similar degrees of concern about risk, attention from FDA is more appropriately directed towards a supplement that is being used by a greater portion of the population. It is also important to consider the safety of the most sensitive groups. These two factors are discussed in detail in Chapter 9.
TABLE 3-7 Relative Spectrum of Concern for Relatedness Information
|
Type of Information |
Increasing Concern |
||
|
|
|||
|
Botanical chemical constituents of concern |
Plant contains constituents that are known to be toxic to humans, but the constituents are commonly consumed in similar amounts in conventional food products |
Plant contains constituents that are known to be toxic to humans at low concentration of these substances in the plant part used for the supplement is not characterized OR Plant part used for the supplement contains constituents that are toxic to humans, but there is credible reason to believe that the constituent may not cause serious adverse effects at the amount typically ingested |
Plant contains constituents that are known to be toxic to humans at low concentrations and the concentration concentrations, but the of substances found in the plant part used for formulating the supplement has been characterized |
|
Taxonomic relationship to other botanicals of concern |
In a plant family that contains known toxic genera, but the supplement is not in a genus known to be toxic AND |
Same genus known to be toxic to humans or animals OR In a plant family that is known to contain toxic plants but |
Same species as a known toxic plant that is not ingested as a food |
|
|
It has a history of use as a food in a preparation and method similar to its current use |
not in a genus known as a food plant OR In a plant family that is known to contain toxic plants, and in a genus that may have a history of food use, but supplement is either a concentrated extract or from a different plant part than is ingested as a food |
|
|
Chemical structure |
Structurally similar to, or likely contains, a chemical compound known to be toxic to humans or animals when ingested in high doses |
Supplement contains chemical constituent known to be toxic to humans or animals |
Supplement contains chemical constituent known to be toxic at very low doses to humans |
|
Endogenous substances or mimetics of endogenous substances |
May result in tissue concentrations that would be expected to cause biological effects (either because homeostasis is disrupted or because the substance has potent biological activities), but the seriousness of the biological effect is not definite |
Results seen in tissue concentrations that would be expected to cause biological effects (either because homeostasis is disrupted or because the substance has potent biological activities), but the seriousness of the biological effect is not definite |
Results seen in tissue concentrations that would be expected to cause biological effects (either because homeostasis is disrupted or because the substance has potent biological activities) that are considered serious |
TABLE 3-8 Relative Spectrum of Concern for In Vitro Data
|
Increasing Concern |
||
|
|
||
|
Standardizeda subcellular and cellular assays validated for the purpose of establishing in vivo toxic effect |
Standardized subcellular and cellular assays validated for the purpose of establishing in vivo toxic effect |
Standardized subcellular and cellular assays validated for the purpose of establishing in vivo toxic effect |
|
AND |
AND |
AND |
|
Multiple different assays suggesting the same pathological condition or endpoint |
Multiple different assays suggesting the same pathological condition or endpoint |
Multiple different assays suggesting the same pathological condition or endpoint |
|
AND |
AND |
AND |
|
Poor consistency/reproducibility in response AND No knowledge about concentration of toxicant in blood or tissue |
Consistency in response AND No knowledge about concentration of toxicant in blood or tissue |
Knowledge of presence of toxicant in blood or tissue enhanced by knowledge of concentrations comparable with those causing toxicity in vitro |
|
OR |
OR |
OR |
|
Standardized assays validated for the purpose of establishing organ toxicity |
Standardized assays validated for the purpose of establishing organ toxicity |
Standardized assays validated for the purpose of establishing organ toxicity |
|
AND |
AND |
AND |
|
Multiple different assays suggesting the same pathological condition or endpoint |
Multiple different assays suggesting the same pathological condition or endpoint |
Multiple different assays suggesting the same pathological condition or endpoint |
Vulnerable subpopulations can be defined as groups of individuals who are more likely to experience an adverse event related to the use of a particular dietary supplement ingredient or individuals in whom the specific adverse effects identified are more likely to be serious in comparison with the general population. Characteristics that contribute to such vulnerability may be physiological (including genetic predisposition) and include age, developmental stage (e.g., pregnancy or fetal period), presence of other diseases, or concurrent use of medications or other therapeutic practices.
When evaluating risk and reviewing data, it is important to ask if ingredients are more likely to cause harmful effects to particular subgroups of the population, especially if those subgroups are known to consume the particular ingredient of concern. Vulnerability of a population subgroup is
|
Increasing Concern |
||
|
|
||
|
AND |
AND |
AND |
|
Poor consistency/ reproducibility in response AND No knowledge about concentration of toxicant in blood or tissue OR Results obtained with nonstandardized, nonvalidated assays OR Results from microarray experiments show a gene expression pattern predictive of dangerous compoundsb |
Consistency in response AND No knowledge about concentration of toxicant in blood or tissue |
Knowledge of presence of toxicant in blood or tissue at concentrations comparable with those causing toxicity in vitro |
|
a Standardized in this context means that the assay is performed consistently across laboratories and often is officially promulgated by a standardization body, such as AOAC International (formerly the Association of Official Analytical Chemists), or the protocol is specified by a regulatory agency. b Toxicogenomics is a relatively new field, the impact of which is not possible to predict at this point. However, these types of data may become more important as the field progresses. If the value of genomics, proteomics, and other new technologies in identifying dangerous compounds is demonstrated in the future, then such results may warrant more concern than is indicated in this figure. |
||
TABLE 3-9 Spectrum of Concern for Interactions
|
Increasing Concern |
||
|
|
||
|
Pharmacokinetic and/or pharmacodynamic data suggesting a supplement-drug/food/other dietary supplement interaction that might lead to a serious adverse event and/or identifying a population at risk for a serious adverse event |
Pharmacokinetic and/or pharmacodynamic data documenting a supplement-drug/food/ other dietary supplement interaction that might leads to a serious adverse event and/or identifying a population at risk for a serious adverse event |
Pharmacokinetic and/or pharmacodynamic data documenting a supplement-drug/food/other dietary supplement interaction that lead to a serious adverse event |
described as a modifying factor, in that whether identifiable subpopulations are particularly susceptible to harm should always be taken into consideration when setting priorities for review.
GENERAL PRINCIPLES AND CONCEPTS WHEN CONSIDERING DATA
The principles for evaluating specific types of information are described above, but some concepts are more global in nature, because they are applicable to all types of data or because they are principles for integrating different types of data that may or may not be consistent.
Concentration of Substances at the Site of Action
A critical factor in determining toxicity of a compound in a dietary supplement is not necessarily the ingested amount, but rather the unbound (free) concentration of an active ingredient at its receptor site. Once absorbed, distribution of the ingredient is via the systemic circulation to its receptor site.
Bioavailability (i.e., the rate and extent, or fraction, of delivery of a compound to the systemic circulation) has a significant effect on the concentration achieved. Bioavailability is greatly affected by the composition of the dosage form, first pass metabolism in the intestines and liver, and physiological factors, such as the rate of gastric emptying. Bioavailability and the rates of metabolism and excretion are the major determinants of serum concentration of a given dose of product.
Knowing the concentration of the unbound fraction of a compound in plasma will assist in assessing the relevance of in vitro data. Also, the plasma concentration can assist in comparing data across animal species (note that the concentration of the parent compound and/or any active metabolite is frequently used when the unbound fraction is unknown). Knowing the concentration of the unbound compound in plasma may be used as a surrogate marker for toxicity potential if a relationship has been established between the concentration and toxicity. For example, studies evaluating barbiturate sleep time illustrate a similar effect for a given plasma concentration across animal species; barbiturate sleep times may vary among species, but each species appears to awaken at approximately the same barbiturate plasma concentration (Gillette, 1976).
When judging whether the concentration will reach levels of concern in humans in the absence of information relating dose to systemic concentration, conservative assumptions should be used. In the absence of specific data about an ingredient in humans, one should assume rapid absorption and 100 percent bioavailability and divide the dose administered by the
plasma volume10 to estimate the maximum achievable concentration from a single dose. These assumptions may not be accurate, but they do provide a reasonable basis for making decisions.
Bioavailability is further discussed in Chapter 5 as it relates to route of exposure when considering exposure of animals through non-oral routes. It is also discussed in Chapter 6, when looking at concentrations of substances that are similar to endogenous substances. In Chapter 10, the concept is discussed in terms of integrating data that may appear inconsistent.
Proof of Harm
To evaluate the safety of dietary supplements, it is necessary to determine if an unreasonable or significant risk exists—not to have complete evidence that a dietary supplement ingredient causes a serious adverse event—which is a lower standard than conclusive proof. The difference between proof of harm and risk of harm should be considered when judgment rather than strict interpretation of facts needs to be made.
Absence of Evidence
Absence of evidence of risk does not indicate that there is no risk. In some cases, some data will indicate a risk, while other data will not suggest the risk exists, producing what could be interpreted as an inconsistency. Even if a study showing lack of adverse effects is reported, if the study is not adequately designed to identify risk (e.g., not sufficiently powered, incompletely reported, does not include positive controls, or otherwise has inadequate mechanisms for detecting adverse events), it is not scientifically valid to use such information to mitigate suggested risk from other sources. This concept is discussed in Chapter 4, as it relates to comparing different types of human data. It is also discussed in Chapter 10, as it relates to comparing human and animal data.
Considering Consistency and Biological Plausibility
In many circumstances, data will need to be collated within the same category or across several categories to determine the appropriate overall level of concern. In integrating observations across categories of data, consistency and evidence of biological plausibility should raise the level of
concern. This weaving together of available information can be facilitated, and conceptually illustrated, by the use of causal evidence models. A causal evidence model (see Figure 3-2) provides a structure to help interpret available data from a number of sources in order to address a specific safety question, and is explained in detail in Chapter 10. The model describes the relationship among a dietary supplement ingredient, potential adverse health effects, and biological effects by depicting the relationship as linkages illustrated with arrows. The type of arrow illustrates the type of evidence: convincing data are depicted by solid arrows, and weak or less conclusive data are depicted by dashed arrows. The path between a dietary supplement ingredient and an adverse health effect illustrates the strength of their potential relationship. When the available information is integrated, multiple links between the dietary supplement ingredient and a given health outcome are illustrated by multiple arrows. Evidence from all types of study designs may form linkages to aid in determining the extent of association between dietary supplement exposure and adverse health effects or outcomes. Causal models are useful when a single type of evidence is weak or does not illustrate a relationship, but other related information is available, as may often be the case with dietary supplement ingredients.
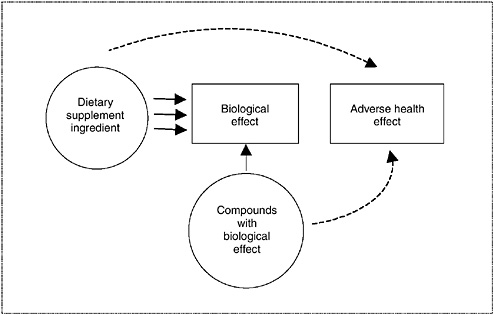
FIGURE 3-2 Diagram of causal model relating information to an adverse health effect.
UTILIZING THE FRAMEWORK
The request from FDA to develop a framework for evaluating the safety of dietary supplement ingredients also included a request that prototype monographs for six dietary supplement ingredients be developed as examples of how the Framework should be applied. Chapter 11 provides case studies of how the available evidence for six dietary supplement ingredients could be evaluated using the spectra of concern discussed in this chapter and described in detail in the following chapters.
Given the fact that these are prototype monographs, they should not be considered as representing findings related to these six dietary supplement ingredients. Rather, they are examples of how to approach reviewing and evaluating the various types of available information on dietary ingredients. Appendixes D through I contain summaries of the six prototype monographs. (The full prototype monographs are available on the web, at www.iom.edu/fnb.) Appendixes J and K contains examples of two focused prototype monographs to show how FDA could focus on determining a level of concern related to one specific adverse effect or outcome when identified. Conducting a broad-based comprehensive assessment would typically identify all data about the dietary supplement ingredient and would lead to a description and evaluation of other adverse effects—which would be a resource-intensive process.
SUMMARY
This chapter outlines a system for conducting a review of the safety of dietary supplement ingredients. Conducting the safety evaluation consists of three components: signal detection, an initial review of the available information, and, when needed, an integrative evaluation. Based on detection of a signal or a proactively initiated review of a dietary supplement ingredient, FDA evaluates the detected signal by conducting a brief initial review of readily available information to determine whether there is a need for a comprehensive review, termed an integrative evaluation. When an integrative evaluation is undertaken, FDA, or a contractor of FDA, prepares the initial draft monograph that is a collection and review of available safety information. In some cases, the integrative evaluation process, during which a draft monograph is developed, may provide sufficient evidence for FDA to decide on a course of action without use of an external advisory committee or public input. However, it is expected that when the data are not sufficiently definitive for FDA to make a decision about whether to take action, it will request the assistance of an advisory committee to review the information.
The external advisory committee, if constituted, reviews the draft monograph, determines if additional information should be collected, and holds
sessions for input from the public. It then recommends modifications to the draft monograph as appropriate and summarizes concerns based on the evidence. The completed monograph, with input from the external advisory committee, is then made public in an easily accessible format.
In any scientific evaluation, at least four categories of information can be considered informative for evaluating the risk of ingesting chemicals, including dietary supplements: human data, animal data, in vitro data, and information about related substances. Evidence in any one of these four major categories can provide considerable guidance regarding the ingredient’s safety. The chapters that follow provide detailed information on the use of this information, and how to integrate the available data to determine the extent to which an unreasonable risk of illness or injury from ingestion of a dietary supplement exists.
ANNEX 3-1 APPLYING THE SAFETY FRAMEWORK TO REQUIRED NUTRIENTS
Essential nutrients (i.e., vitamins and mineral elements) are unique compared to many other categories of dietary supplements in that much more data regarding adverse effects of overconsumption in humans are available. Structures of the vitamins have been elucidated, relative activity of closely related compounds determined, and, at least at physiologic doses, biological activities for these compounds are reasonably well characterized. Most have been characterized in terms of potency in standards such as those produced by the U.S. Pharmacopoeia, as is required by law when monograph standards are available for them. Several nutrients are regulated as generally recognized as safe substances, approved food additives, and as over-the-counter and prescription drugs. It is for this reason that the essential nutrients are considered differently within the Framework than other types of dietary supplement ingredients.
As summarized in Chapter 2, a system for reviewing data about the safety of vitamins and essential elements already exists. Since 1940, the Food and Nutrition Board of the National Academies has been commissioned by federal agencies to set Recommended Dietary Allowances (RDAs) for nutrients; more recently this was expanded to include other reference intake levels for nutrients, now collectively termed Dietary Reference Intakes (DRIs) (IOM, 1994, 1997). With the exception of only a few nutrients, scientific data on all vitamins and mineral elements, and some other nutrients, have been reviewed recently through the DRI process (IOM, 1997, 1998b, 2000, 2001, 2002, 2004).
While the RDAs are designed to be recommendations for intake to ensure that the needs of almost all apparently healthy individuals in a
population group (such as women over 70 years of age or adolescent boys) are met in order to avoid nutrient deficiencies and to decrease risk of chronic disease, other reference values included as part of the new DRIs provide upper levels of intake for vitamins and mineral elements that, if consumed below the specified level on a continuing basis, should not cause specific identified adverse effects of overconsumption (IOM, 1998a). These upper levels of intake are called “tolerable upper intake levels,” or ULs.
For the vitamins and the mineral elements that have been established as required by humans, a considerable amount of primary data relating to animal or experimental studies, human studies, and in vitro studies are available, and these data have been reviewed as part of the DRI consideration of the UL. The DRI review can thus provide substantial data and background information for an FDA evaluation; the process for incorporating this information is provided in the following sections.
Signal Detection for Nutrients
When nutrients present in dietary supplements are suggested for use at levels greater than established ULs, it is appropriate to be concerned, and thus this is an initial “signal” analogous to that described for other dietary supplement ingredients. However, the initial review in response to the signal should be focused on the DRI review of the serious adverse effects that were identified as potentially occurring at high intake levels, recognizing that there has been an uncertainty factor applied to ensure that few, if any, individuals will be adversely affected at the UL level. New information (post-DRI review) on adverse effects of consuming a nutrient also serves as an initial signal. In summary, it is appropriate to a priori consider any marketing of nutrients as dietary supplements above the UL to be potentially of some risk, but whether the risk is unreasonable will depend on the data available. Nonetheless, vitamins and mineral elements are not innocuous substances. Consumption at high levels of some nutrients is associated with illnesses and death as documented in the DRI reports (IOM, 1997, 1998b, 2000, 2001, 2002, 2004).
Initial Review for a Nutrient
Whenever the safety of a vitamin or mineral is considered, the first step should be to consult the results of the DRI UL process. It is possible that a UL was not established for a nutrient when the data were reviewed, but the review process and the data considered are included in the specific DRI report and serve as a credible, nonbiased review of the scientific data available at the time of review. If a UL was established for the vitamin or mineral under question, it is important to consider the following:
-
Was the information suggesting concern available before the publication of the most recent DRI review, and were these data considered in the evaluation of ULs?
One limitation is that the reviews are conducted at specified intervals, so it is possible that safety issues relating to dietary supplements that are essential nutrients might be newly identified within the time period after the last DRI review and prior to an upcoming updated review.
-
Is the ingredient in use the same formulation or is the dosage outside the ranges previously reviewed, and thus beyond the coverage of the most recent DRI review?
-
Was the intake level under question addressed in the DRI report? There is increasing use of very high doses of nutrients in dietary supplements, so various biological activities, toxicities, and adverse effects may have been incompletely addressed (or not verified) in the most recent DRI review. These reviews necessarily must cover issues related to intakes over a wide range, looking for the lowest intake level at which adverse effects are noted, so that the issues at the highest levels of intake are not the sole (or primary) focus of the reviews.
-
What was the critical endpoint or adverse effect used to set the UL? Was it a benign reversible adverse event or a serious and irreversible condition?
-
Is concern about consumption above the UL directed primarily at specific populations, or does it apply widely? If a higher level of concern is directed primarily at specific populations, it may be appropriate for the integrative evaluation to focus on that concern as it relates to that population group.
It is necessary to consider the basis for the UL, with less concern being warranted for non-life-threatening or self-limiting effects, as well as the recommended dose of the supplement to determine the level of concern. The conclusions of the DRI review should be given much greater weight than other data available at the time of the review but not considered in it. This allows an immediate determination of the level of concern and thus a fairly rapid determination of the need to go forward with an integrated evaluation, following similar procedures to that of other dietary supplement ingredients (as described in earlier sections of this chapter).
Integrative Evaluation for a Nutrient
It is quite possible that situations will arise where, due to information which becomes available after the DRI review on the severity of the adverse effect, or the vulnerability of a population at risk, there will be a higher level of concern and thus a need to go further than the DRI review to
determine if a significant or unreasonable risk of illness or injury exists when consuming a nutrient as a dietary supplement at its suggested level of intake. This is the integrative evaluation component.
An integrative evaluation should use as its basis the DRI review and analysis, recognizing that for some nutrients (i.e., vitamin K, β-carotene, arsenic, chromium, silicon, thiamin, riboflavin, vitamin B12, pantothenic acid, biotin, potassium, and sulfate) the available data, while reviewed in the DRI series, were deemed insufficient to develop a UL based on requirements of the model of risk assessment used. This does not indicate that high intakes pose no risk of adverse effects (IOM, 1998a, 2000, 2001, 2004), but that a thorough review by an expert group could not identify dose- response evidence from chronic intakes that would provide a basis for establishing the level at which adverse effects might occur.
If a UL was not established for the vitamin or mineral element under question, it is important to consider the following:
-
Was the substance reviewed by the DRI process, even if a UL was not established?
-
Is there new evidence suggesting risk that was not available at the time of the DRI review?
-
Was significant concern about serious harm expressed in the DRI review, even if a DRI could not be established because of limits in the data or acuteness of the adverse effect (e.g., arsenic)?
It is assumed that the integrative evaluation for a nutrient would contain the same general components as for other dietary supplement ingredients, with the exception that significant information is already captured by virtue of the DRI process and its reports. In situations where new data or information indicate that higher concern is warranted, the nutrient would enter the evaluation in the same manner as other supplement ingredients. Nutrients are usually well-characterized chemically and thus there is less concern about active ingredient identification and function.
ANNEX 3-2 MONOGRAPH PREPARATION AND PUBLIC ACCESS
Preparation of Draft Monographs
In evaluating evidence indicating that an ingredient may present an unreasonable risk to human health, a comprehensive examination of the literature is required, recognizing that not all studies are relevant to ascertaining the safety of a dietary supplement ingredient. The initial step in
preparing a monograph is to gather as much information as possible from the published literature and other sources regarding the potential hazards of consuming the supplement. Multiple comprehensive databases, such as MEDLINE (NLM, 2003a), TOXLINE (NLM, 2003b), and EMBASE should be searched (Elsevier, 2003). In addition, NAPRALERT (Farnsworth, 2003) can be searched if the supplement is a natural product or botanical. To search for potential ingredient-drug or ingredient-ingredient interactions, the Metabolism and Transport Drug Interaction Database (UW, 2003) is a useful tool.
Abstracts and titles should be reviewed for relevance to the adverse event or harmful effect of concern. Although review articles may be useful for the purpose of providing literature references and an overview of the data, review of the original articles from the peer-reviewed literature is essential to obviate any bias or unsubstantiated opinion of the authors of the review. Information in other non-peer-reviewed literature that raises concerns about adverse effects should not be ignored. In order to get as much information as possible, FDA should request the voluntary submission of safety data information from industry and other stakeholders. This request may be made through notice in the Federal Register and through the FDA website. FDA should also request information directly from manufacturers and distributors of the ingredient under consideration, if they are known.
Collecting descriptive and safety information and organizing and summarizing the information into a draft safety monograph will require significant expertise and resources. Time and other resources required for completion of the draft monographs are likely to vary, depending on the amount and complexity of safety-related information available for the ingredient under consideration, as well as the focus of the monograph. FDA may choose to prepare monographs internally, or it may choose to contract the work out to organizations, individuals, or both.
The extent of time and effort devoted to preparation of monographs on dietary supplement ingredients will depend on FDA’s prioritization of need. FDA could set priorities and develop a complete list of substances warranting monographs first. Alternatively, it could retain one or more individuals or groups to develop monographs and determine the need for individual monographs on an ongoing basis as priority setting proceeds or as new needs emerge. The former approach may be more cost effective to implement, given that the latter approach might not provide continuity in workload. However, preparing monographs for substances considered of high priority will require more resources up front if given to contractors and will be dependent on information available at the time high-priority substances are identified.
As discussed previously, monographs, whether prepared internally or
by a contract organization, should be evaluated to determine if the conclusions could be improved by input from additional expert judgment. The decision to undertake a monograph internally or by contract to an outside group will depend on FDA’s resources and internal expertise.
The monographs developed should not be considered static documents. New information should be added as it becomes available, and an organized process for adding information should be developed. The process should also include periodic reviews of monographs to determine if additional external reviews are appropriate.
Public Access to the Monograph
After the advisory committee’s summary is shared with FDA, the revised monograph and the advisory committee’s summary should be posted on FDA’s website. One of the important components of DSHEA is that the public should be educated about dietary supplements. FDA thus has a responsibility to educate consumers about the safety of dietary supplement ingredients, and the public availability of the final monographs can be an important aspect of the educational process. The monographs can provide the public with a reputable summary of the available information and scientific uncertainties about the inherent safety of the supplement ingredient. Importantly, public access to information from an advisory committee will add to the quality and strength of the available scientific literature.
ANNEX 3-3 THE USE OF AN EXTERNAL ADVISORY COMMITTEE
The decision to refer a dietary supplement ingredient to an external advisory committee rests with FDA, which has the authority to refer to such a committee for any reason deemed necessary, as discussed in the text.
The external advisory committee needs to include experts in critical key disciplines. FDA has significant experience in establishing advisory committees and already has rules regarding membership (e.g., conflict of interest) in place. Possible approaches that FDA may wish to consider include:
-
A standing committee of about seven persons, with the option to add one or two scientists with special expertise, as needed, for the review of individual substances.
-
A standing committee of five scientists representing core disciplines, and the addition of three or four special experts depending on the nature of the ingredient, the data to be evaluated, and whether a focused or broad-based evaluation was required. The presence of individuals with expertise in either the ingredient under review or the purported adverse effect of the
-
ingredient is critical to providing a well-documented review of the literature where the data are equivocal.
To ensure that the critical evaluation of the information contained in the monograph and related information is as free of conflict of interest and as objective as possible, the external advisory committee should be composed of expert scientists who have appropriate training, education, and experience. Whether as a result of the appointment of a committee by FDA or by contract with a scientifically based, nonprofit organization, examples of expertise to be included are toxicology, preferably with expertise in safety evaluation; pharmacognosy; clinical pharmacology; nutritional science; epidemiology; biostatistics; clinical trials; medicinal chemistry and structure–activity relationships; pharmacokinetics; consumer behavior related to dietary supplement use; and public health, as well as ad hoc consultants with expertise in specific fields on an as-needed basis (e.g., specialists needed to evaluate particular ingredients, such as experts on oriental medicine, herbalists, veterinary toxicologists, or clinicians with relevant experience). Advisory committee members should be selected based on their disciplinary expertise rather than as representatives of stakeholder viewpoints, and they should not have a financial stake in the outcome of the process or otherwise have a real or perceived conflict of interest.
After the external advisory committee is assembled, a draft monograph should be released, and the public should be provided with an opportunity to comment on the completeness of the data included, as well as on the strength and relevance to humans of the different types of evidence. Industry and other stakeholders should be given time during meetings of the external advisory committee to provide input into the process. The external advisory committee should provide advice on the further refinement of the draft monograph as it reviews all the data and summarizes its conclusions.
REFERENCES
Elsevier. 2003. EMBASE (Excerpta Medica Database). Online. Available at http://www.elsevier.nl/homepage/sah/spd/site/locate_embase.html. Accessed March 13, 2004.
Farnsworth NR. 2003. NAPRALERT (Natural Products ALERT) Database. Chicago: University of Illinois at Chicago.
Gillette JR. 1976. Application of pharmacokinetic principles in the extrapolation of animal data to humans. Clin Toxicol 9:709–722.
IOM (Institute of Medicine). 1994. How Should the Recommended Dietary Allowances be Revised? Washington, DC: National Academy Press.
IOM. 1997. Dietary Reference Intakes for Calcium, Phosphorus, Magnesium, Vitamin D, and Fluoride. Washington, DC: National Academy Press.
IOM. 1998a. Dietary Reference Intakes: A Risk Assessment Model for Establishing Upper Intake Levels for Nutrients. Washington, DC: National Academy Press.
IOM. 1998b. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: National Academy Press.
IOM. 2000. Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids. Washington, DC: National Academy Press.
IOM. 2001. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc. Washington, DC: National Academy Press.
IOM. 2002. Dietary Reference Intakes for Energy, Carbohydrate, Fiber, Fat, Fatty Acids, Cholesterol, Protein, and Amino Acids. Washington, DC: National Academy Press.
IOM. 2004. Dietary Reference Intakes for Water, Sodium, Potassium, Chloride, and Sulfate. Washington, DC: The National Academies Press.
Merriam-Webster. 2001. Merriam-Webster’s Collegiate Dictionary. 10th ed. Springfield, MA: Merriam-Webster.
NLM (National Library of Medicine). 2003a. PubMed. Online. Available at http://www.ncbi.nlm.nih.gov/entrez/query.fcgi. Accessed March 13, 2004.
NLM 2003b. Toxline. Online. Available at http://toxnet.nlm.nih.gov/cgi-bin/sis/htmlgen?TOXLINE. Accessed March 13, 2004.
UW (University of Washington). 2003. Metabolism and Transport Drug Interaction Database. Online. Available at http://depts.washington.edu/didbase. Accessed September 24, 2004.









































