
7
Selecting an In-Market Surveillance Plan
ABSTRACT
In-market surveillance is an essential element of regulating the safety of ingredients new to infant formulas and should be included in new safety assessment guidelines. There are two components of in-market surveillance. The first, the “monitoring component,” involves procedures to detect adverse effects upon infants after a formula has been put on the market. The second, the “follow-up component,” concentrates on possible long-term adverse effects after the period of maximum formula usage. Although formal regulatory guidelines for in-market surveillance do not exist in the United States or Canada, infant formula manufacturers routinely conduct passive surveillance via toll-free calls, contact with health care professionals, and reports from their field sales force.
Satisfactory completion of the appropriate preclinical and clinical studies diminishes the likelihood of systematic adverse reactions; however the risks for adverse reactions cannot be ignored because adverse effects may not be detected in preclinical studies if the wrong animal model was chosen, if the assessment instrument chosen measured a function other than the one adversely affected by the new ingredient, or if a subpopulation of individuals who are highly sensitive to the new ingredient added to infant formula was not sufficiently represented in clinical studies.
The committee believes that there is a crucial need for follow-up strategies to ensure safety and normal development of the infant population (e.g., brain areas that are adversely affected by new ingredients may not functionally become apparent until later in development, or early exposure to a toxin may increase susceptibility to later exposure to toxins). The committee recommends that a systematic plan for continued in-market monitoring and long-term surveillance become an essential part of all submissions for regulatory agency review seeking to add a new ingredient to infant formula. The in-market surveillance strategies the committee proposes are specifically designed for evaluation of the safety of new ingredients added to infant
formulas and do not necessarily apply to the safety of new ingredients not intended for infant formulas.
Infant formula manufacturers should select in-market strategies by implementing a hierarchy of three levels of assessment, including passive surveillance (level 1 assessment), expert panel reviews of the literature (level 2 assessment), and active surveillance (level 3 assessment). Active surveillance is the most complex assessment and includes options such as studying specific populations, conducting retrospective studies, using the Pediatric Research in Office Settings program of the American Academy of Pediatrics, and conducting clinical follow-up studies of the original study populations. Selection of the appropriate level of assessment is based upon conditions under which potential adverse effects of a new ingredient added to infant formulas might have been missed in preclinical or clinical studies. The length of the follow-up period will depend on the targeted area (organ system), preclinical and clinical studies, and the nature of the added ingredient.
BACKGROUND
The Importance of Systematic In-Market Surveillance
Satisfactory completion of the appropriate preclinical and clinical studies diminishes the likelihood of adverse reactions to infant formulas that contain new ingredients once they have been placed on the market. However completion of those studies does not ensure absolute safety, and the probability of adverse reactions cannot be overlooked for several reasons.
First, adverse effects may not be detected in preclinical studies if the wrong animal model is chosen to map onto a function at the human level (see Chapter 5). Second, in clinical studies a less-sensitive instrument may fail to detect an adverse reaction due to poor initial outcome measurement (Clarke and Clarke, 1981; see Chapter 6). Third, adverse effects could be missed during clinical studies if the instrument chosen, even if highly sensitive, was measuring a function other than the one adversely affected by the new ingredient. A similar point holds in regard to predictor measures. Analytical procedures have been developed that are capable of measuring compounds at concentrations as low as picograms per liter. Concurrent appreciation of potential biological changes has not kept pace with this degree of laboratory sophistication. Finally, the sampling design of clinical trials could have been inappropriate. Given what is known about variability in individual or subpopulation sensitivities to exposure to dietary ingredients (Beaton, 1986; Rutter and Pickles, 1991) or toxic substances (Bellinger, 1995; Ruff, 1999), if a subpopulation of individuals who are highly sensitive to the new ingredient added to an infant formula are not sufficiently represented in the clinical studies, adverse effects may not be detected until the formula is marketed to a wider population.
These reasons support the need for a systematic plan to include both types of in-market surveillance into every submission to the regulatory agency for the addition of an ingredient new to infant formulas. The two types of in-market surveillance are discussed below.
RECOMMENDATION: A systematic plan for continued in-market monitoring and long-term surveillance should be an essential part of submissions for regulatory agency review in assessing the safety of ingredients new to infant formulas.
Monitoring
The first component of in-market surveillance, the monitoring component, involves procedures to detect adverse effects to infants after a formula has been put on the market. As discussed above, the probability that negative effects will emerge during in-market monitoring is likely to be quite small if the appropriate preclinical and clinical studies detected no negative effects associated with the introduction of a new ingredient to an existing formula. However the number of infants enrolled in clinical trials is small in relation to in-market use, and the trials may not detect a full range of variations. For this reason it is important to integrate in-market monitoring procedures into the evaluation process to judge the safety of new ingredients introduced into infant formulas.
Follow-up
The second component of in-market surveillance, the follow-up component, is one that is less often considered during clinical trials to assess the safety of new ingredients added to infant formulas. In contrast to in-market monitoring, which focuses on adverse effects occurring during the period of maximum formula usage, in-market follow-up concentrates on possible long-term adverse effects after the period of maximum formula usage. The length of a follow-up study will depend upon the nature of the ingredient, so the expert panel should define it on a case-by-case basis. As described later in this chapter, higher levels of assessments should be performed when the ingredient may affect slow-developing brain regions, hormone or neurotransmitter function, or behavior. In addition, follow-up during critical life transitions, such as entry into school or onset of puberty, should be emphasized.
Most clinical studies are confined to a short amount of time for the period of maximum exposure to the formulas, and they track adverse patterns only during the time of maximum exposure. However there is also the possibility that the new ingredient’s negative effects on the growth and development of children may have delayed onset and only appear later in life. Evidence from a number of sources supports the validity of this statement. First, there are both preclinical and clinical studies that document long-term cognitive and behavioral effects of early exposure to toxic substances (Galler and Tonkiss, 1998; Jacobson and Jacobson, 2000; Leech et al., 1999; Leviton et al., 1993; Richardson, 1998; Richardson et al., 1996; Romano and Harvey, 1998; Wasserman et al., 2000).
More critically, although the overall pattern of evidence is not totally consistent, there are a number of examples from both the clinical and preclinical research literature where negative effects of early exposure to toxic substances were not found upon initial testing, but did appear during follow-up assessment (Weiss, 1995; Winneke, 1990). Toxic substances that do not show their effects until well after the period of exposure to the substance have been labeled as chemical or neurobehavioral “time bombs” (Russell, 1990; Spencer, 1990). Delayed effects have been shown for prenatal or early postnatal exposure to drugs, alcohol (Griffith et al., 1994; Singer et al., 2002), and lead (Bellinger et al., 1991). Although not directly related to toxic exposure, epidemiological studies have also indicated that there may be delayed long-term adult biomedical consequences as a result of the quality of very early nutrition (Barker et al., 1993; Jackson, 2000) or of the level of morbidity in the first year of life (Bengtsson and Lindstrom, 2000).
There are a number of mechanisms through which a delayed impact of early exposure to toxic substances might occur. One involves cumulative effects. Biologically, cumulating intake of a low-level toxin can result in the gradual replacement of a specific neurotransmit-
ter by a less efficient molecular substitute; only over time will the detrimental impact of the substitute become manifest (Russell, 1990). Another example is the long-chain polyunsaturated fatty acids (LC-PUFAs) that have been determined as GRAS for addition to infant formulas. These LC-PUFAs are derived from genetically selected algae that produce triglycerides with a high content of either arachidonic acid or docosahexaenoic acid onto two or three of the fatty acid chains of the triacylglycerol molecule. This contrasts to the triacylglycerol makeup of human milk, where LC-PUFAs rarely form two-thirds of the fatty acids of a single triacylglycerol molecule. In this case, diglycerides resulting from hydrolysis of the algae triglycerides in infant formula would be different than those produced from human milk and different biological effects could occur (e.g., specific diglycerides have very different effects in cell signaling pathways and could have different “triggering” effects on certain cellular pathways).
Such a cumulative effect scenario is particularly likely to occur when a toxic substance is contained in formula, which may be the main nutrient source for an infant over an extended period of time. A cumulative effect process, wherein initial small deficits cumulate and become significant only over time, appears to be a particularly likely scenario when the critical mechanisms are biobehavioral in nature (Wachs, 2000). Such a process could occur when toxic exposure leads to changes in infant temperament characteristics, such as an increased level of irritability or a reduced level of responsivity. Toxin-driven changes in temperament could adversely impact upon critical influences on later development, such as patterns of parent-child relations, which over time can lead to long-term developmental-behavioral deficits (Bendersky et al., 1996; Chasnoff et al., 1987; Fried, 1989; O’Connor et al., 1993; Schneider et al., 1999).
Alternatively, the area of the brain that is damaged by a toxic substance may be one in which the functions mediated by this area become apparent only later in development, or it could be one in which connections go from a damaged area to a later-developing area (Lyon and Gadisseux, 1991; Weiss, 1995). In these cases, the consequences of early exposure to toxic substances could occur only after sufficient time has elapsed for the brain-mediated function to appear in a normal developmental sequence. For example, early damage to areas of the brain associated with motor control may not be seen until that time period when infants would be expected to acquire voluntary movement sequences (Lyon and Gaddisseux, 1991).
Finally, there is evidence from both preclinical (Spear et al., 1998) and clinical studies (Mayes et al., 1998) that early exposure to toxic substances may increase the organism’s sensitivity or vulnerability to later biological or psychosocial stressors. A similar argument for greater susceptibility has also been made in regard to mechanisms underlying the long-term biomedical consequences of early nutritional deficiencies (Waterland and Garza, 1999). If increased susceptibility to later stresses occurs, fewer developmental-behavioral consequences of early exposure to toxins would appear during the relatively sheltered years of infancy, with increased developmental risk when environmental demands increase as the child gets older. Intraventricular hemorrhage in infancy is one example of increased susceptibility to later stress. The impact of intraventricular hemorrhage on cognitive performance seems to fade through the preschool years, but reappears when the child enters grade school (e.g., a high-demand situation) (Sostek, 1992).
The available evidence supports the importance of a program of systematic and continued monitoring of potential consequences of the addition of new ingredients to infant formulas and systematic in-market follow-up past the infancy period. Such a program is especially important for ingredients with putative or potential biological effects.
Limitations of In-Market Surveillance
Although there is ample justification for a systematic program of in-market monitoring and follow-up surveillance, many factors make such a program difficult to implement, and there are many questions and issues that need to be considered before setting up such a program. There are both logistical problems (e.g., tracking enrolled subjects, continuity of the research team, record retention in follow-up studies, knowing or anticipating what elements to track) and methodological problems (e.g., length of follow-up, confounding factors, reconstructing causality in monitoring studies, having sufficient statistical power to detect what may be subtle, yet clinically significant, adverse effects).
One major issue is the problem of confounding factors. It is obviously difficult, years later, to differentiate effects related to new ingredients from effects due to later exposure to other risk factors. Even with longitudinal studies there are significant logistical problems with associating a single added ingredient in infant formula with any observed outcome. Small differences in the amount or type of a carbohydrate, protein, or fat source may not have the implications of the addition of a bioactive substance completely new to infant formulas.
Throughout life the infant, then the child, and finally the adult, is exposed to a wide variety of food substances with potentially unknown composition. The recent explosion in the use of dietary supplements is an example. Study participants can have difficulty with retrospective recall of prior events, as in the case of whether there was a concurrent use of breastfeeding, even if partial, along with formula feeding. Lifestyle factors may not be obvious, and perhaps not admitted, by the participant. Developmental risk factors not related to infant formulas, such as family stress, also can result in long-term deficits. The longer the follow-up interval, the greater the influence of these possible confounding factors.
The length of time that investigators should study children who were placed on a formula containing a new ingredient presents another set of problems. The length of a study may depend on the nature of the substance added. Formulas and solid infant foods may change over the years of the study so that different cohorts may ingest different nonstudy foods, making comparison difficult.
Defining an adverse event may also be difficult in conducting long-term studies (e.g., beyond 1 year). Investigators must define the duration (permanent or not) and nature of an adverse event. As noted in Chapter 6, there also is a question of what functional domains would need to be assessed when conducting in-market surveillance. Ideally the type of compound added to the formula would dictate what form of follow-up would be most important. However sufficient data may not be available to precisely specify associations between a compound and an affected function.
Finally, as outlined in Chapter 2, sample sizes must be large enough to ensure sufficient statistical power in follow-up studies. As noted in Chapter 6, analysis indicated that the majority of studies investigating the cognitive effects of the addition of LC-PUFAs to infant formulas had insufficient statistical power. The problem of sufficient statistical power to detect adverse consequences is particularly critical when follow-up studies are conducted years after the child has ceased consumption of infant formula. In this situation adverse consequences linked to prior consumption of infant formula may be indirect rather than direct, and thus more subtle, so larger sample sizes will be needed to ensure that there is sufficient statistical power. For example, using a statistical power level of 0.80 as the goal and comparing two types of formula with new ingredients versus a control group (standard formula or breastfed), a sample size of 52 children per group will be required to detect a moderate-effect size difference. In contrast, under the same conditions, a sample size of 140
children per group will be required to detect an intermediate-effect size (i.e., between small and moderate). Unless there are compelling reasons to do otherwise, the committee recommends having sufficient power to detect differences between groups of 0.20 standard deviations or less when estimating sample-size needs in follow-up studies. As is noted later in this chapter, at a population level, even effect sizes of this magnitude can have important clinical implications.
The cost to ensure sufficient power and demonstrate statistical significance of long-term follow-up studies may be a factor to consider when designing such studies. It is beyond the scope of the committee’s charge to set cost limits or recommendations.
Current Regulatory Guidelines
As discussed in Chapter 4, formal regulatory guidelines for in-market surveillance do not exist for infant formulas. Section 412 of the Federal Food, Drug and Cosmetic Act states that manufacturers must retain “… all complaints and the maintenance of files with respect to, and the review of, complaints concerning infant formulas which may reveal the possible existence of a hazard to health.”
As a matter of current practice, infant formula manufacturers routinely conduct passive surveillance via toll-free calls, contact with health care professionals, and reports from their field sales force. Since infants are unable to verbally communicate, any adverse effects must be observed and reported through the parents or caregiver, thus special attention must be paid to detect adverse or unusual reactions when feeding infant formulas containing new ingredients.
Similarly, in Canada there are no explicit guidelines or requirements for in-market surveillance of infant formulas specified under Canada’s Food and Drug Regulations in Divisions 16 (Food Additives), 25 (Infant Formula), or 28 (Novel Foods).
OVERVIEW OF RECOMMENDED LEVELS OF ASSESSMENT
RECOMMENDATION: Determine in-market surveillance strategies by implementing a hierarchy of three levels of assessment:
-
Level 1 assessment: passive surveillance. Toll-free or Internet reporting to formula manufacturers or to the regulatory agency. For example, in August 2002 the U.S. Food and Drug Administration (FDA) established a passive surveillance system for food, cosmetics, and dietary supplements (OSAS, 2002). This system, the Adverse Events Reporting System, will be part of the MedWatch program. Formula manufacturers could add this Internet web site’s address to the labeling on each can of formula next to the toll-free reporting number.
-
Level 2 assessment: panel review. In-market panels to review existing data (both published and proprietary). Issues with regard to the selection and composition of such panels have been discussed extensively in Chapter 4. The same selection and composition recommendations presented earlier also hold with regard to in-market panels.
-
Level 3 assessment: active surveillance. The following are examples of three different active surveillance options:
-
Use of follow-up surveillance populations where there is known use of the formula containing the new ingredient or ingredient source (e.g., by using databases
-
-
from the Special Supplemental Nutrition Program for Women, Infants and Children or health maintenance organizations).
-
Use of existing pediatric networks as a data source (e.g., Pediatric Research in Office Settings Program of the American Academy of Pediatrics).
-
Clinical follow-up of the original study populations. Such studies would be part of the original study design—not an add-on study.
Based upon testimony to the committee, companies that market infant formulas often rely on level 1 assessment for in-market surveillance. However there are inherent flaws in this assessment level. One such flaw is the risk of underestimating actual negative occurrences given that not all caregivers whose infants experience problems will call in a report. Underestimation is an even greater problem for long-term follow-up since caregivers are not likely to link a child’s current problems to intake of infant formula years ago, even when such a linkage may occur. Because the committee does not believe that passive surveillance (level 1 assessment) is sufficient for monitoring all ingredients that might be added to infant formulas, a hierarchy of the options was developed.
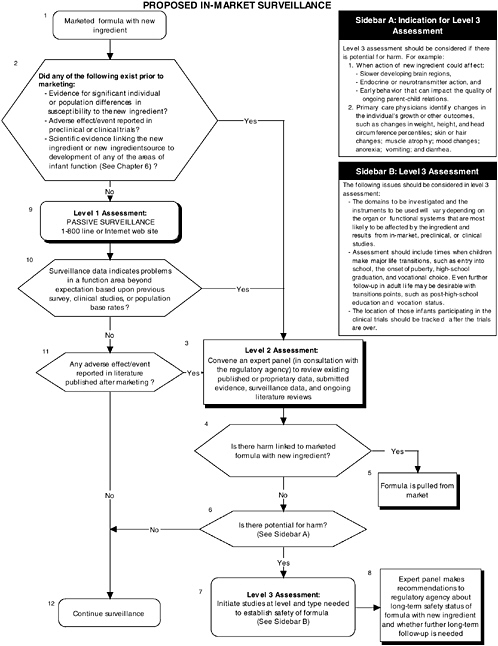
Level 1, 2, and 3A assessments would seem most appropriate for in-market monitoring studies, whereas level 2 and 3 assessments would be appropriate for in-market follow-up studies. These levels should not be considered as either-or alternatives. In some cases results from one level could lead to application of a higher level of assessment, as in the case when potential negative effects emerging from in-market panels would suggest that more data be gathered using methods from the higher levels. Figure 7-1 provides an overview of the decision-making process.
In addition to surveillance, a level 2 review of all pertinent data, both published and unpublished, available since the submission to the regulatory agency should be conducted approximately 2 to 4 years after introduction into the market of the product containing the new ingredient.
CRITERIA AND METHODS FOR IN-MARKET SURVEILLANCE
In-Market Monitoring Programs in the First Year
The criteria used to determine the choice of level of assessment and strategies to be utilized are based on the previous discussion in this chapter of conditions under which potential adverse effects of a new ingredient added to infant formula might have been missed in preclinical or clinical trials. Regardless of the level that is chosen, it is expected that systematic data collection procedures (level 1 assessment) or systematic review procedures (level 2 assessment) will ensure that in-market monitoring information will be assessed for each area of function reviewed in Chapter 6.
The choice of level 1 assessment for in-market monitoring is recommended only when all of the following conditions occur:
-
There is no evidence (presented in the submission) that indicates significant individual or population differences in susceptibility to the ingredient, metabolites, secondary effectors, or source.
-
There is no evidence of adverse effects in preclinical or clinical studies, including adverse effects with potentially plausible alternative explanations (e.g., the effects are viewed as the result of random chance or the reviewers believe that there may be methodological or
-
statistical problems in the studies). This means that even if potentially plausible alternative explanations are offered to explain adverse findings, level 1 in-market monitoring would not be warranted since adverse effects were reported.
-
A review of the relevant scientific literature indicates that there is no link between the ingredient, metabolites, secondary effectors, or source, and the development of any of the areas of infant function described in Chapter 6.
The choice of level 2 assessment for in-market monitoring must occur when any one of the following conditions occurs:
-
There is existing evidence for significant individual or population differences in susceptibility to the ingredient, metabolites, secondary effectors, or source.
-
There is evidence of some type of adverse effect detected during preclinical or clinical studies. This means that even if potentially plausible alternative explanations are offered to explain adverse findings, level 2 in-market monitoring is warranted since adverse effects were reported.
-
A review of the relevant scientific literature indicates there is a link between the ingredient, metabolites, secondary effectors, or source and the development of any of the areas of infant function described in Chapter 6.
Level 2 assessments also would be automatically activated if level 1 assessments (in-market toll-free or Internet reporting strategies) are utilized and information indicates problems in a function area over and above what might be expected based upon previous survey or clinical information or population base rates. If the level 2 assessment panel review indicates that existing published or proprietary data show deviations over and above what might be expected based on population base rates, then a study using one or more of the databases in the level 3 assessment would be required. A systematic re-evaluation of safety issues (level 3 assessment studies) is necessary even when individual risk levels are of small effect size, given that even small individual deficits can be magnified when cumulated at a population level (Lester et al., 1998; Rice, 1990; Scott et al., 1994, 1999).
The choice of domains investigated during such a study would depend on the conclusions of the review panel. As noted in Chapter 4, it is essential that each review panel include qualified scientists from all of the domain areas discussed in Chapter 6. If the potential detrimental effects appear to be restricted to a specific organ or functional system, then a detailed investigation of this system with screening assessments of other systems or functions potentially linked to the functioning of the specific organ system would be appropriate. For example, growth could be easily measured during physical examinations of children in surveillance populations by plotting height, weight, head circumference, and perhaps body composition, especially body fat. If the organ or function system involved had known direct links to other organs or functions, then a detailed investigation of the linked systems would be warranted (e.g., given known links between brain and cognitive function, the potential impact of a new ingredient on brain development would require detailed assessment of both brain and cognitive function using the level 3 instruments described in Chapter 6).
In-Market Follow-up Assessments
Potential long-term adverse consequences associated with the addition of ingredients new to infant formulas may not be detected in clinical studies. Therefore, for every new ingredient to be added to an infant formula, a plan for a systematic approach for in-market
follow-up of the new formula should be required as part of the submission to the regulatory agency. The studies should specially look for effects at the times when children make major life transitions, such as entry into school. If evidence from the first transition period indicated adverse effects, future studies would be warranted at later transition periods, such as the onset of puberty, high-school graduation, post-high-school education, and vocational choice.
The level of follow-up studies would depend on a number of specific criteria. For example, the level of follow-up surveillance may be influenced by the type of substance added to formulas. For ingredients that change only the flavor, color, or texture, minimal concern may exist for long-term changes, especially if such compounds have a long history of use in food. In contrast, a greater concern may be warranted in the case of nutritional substances. For example, soy-based infant formulas are widely used and account for approximately 40 percent of formula sales in the United States. Most studies of growth and development of infants consuming soy formula follow the infants only to age 1 year. Mendez and coworkers (2002) emphasize the need for follow-up of these infants in the areas of reproduction, immune function, thyroid function, visual acuity, and cognitive function. In addition, evidence indicating that exposure to toxins may increase the organism’s responsivity to stress suggests that existing follow-up data should be evaluated or new follow-up data should be collected at time periods when children make life transitions that would increase the level of stress or demand upon the child, such as entry into school.
Based on the evidence cited earlier in this chapter, higher levels of follow-up assessment levels would be particularly critical when any one of the following situations occurs:
-
The action of the new ingredient relates to the development of slower developing brain regions.
-
The action of the new ingredient could affect endocrine or neurotransmitter action.
-
The action of the new ingredient may have affected early child behavioral changes (e.g., temperament) that can impact upon the quality of ongoing parent-child relations (see Chapter 6).
-
Primary-care physicians identify changes in the individual’s growth or other outcomes, such as changes in weight, height, and head circumference percentiles; skin or hair changes; muscle atrophy; mood changes; anorexia; vomiting; or diarrhea.
-
The action of the new ingredient may have an effect only when individuals are exposed to excess calories or other specific situations.
In the submission to the regulatory agency, the following criteria should be referred to when justifying the strategy proposed for in-market follow-up of new ingredients added to infant formulas.
The choice of level 2 assessment (review panel) for in-market follow-up is recommended when any one of the following conditions occurs:
-
In-market monitoring reveals adverse effects reported for the new ingredient or ingredient source.
-
There is existing evidence for significant individual or population differences in susceptibility to the ingredient, metabolites, secondary effectors, or source.
-
There is evidence of adverse effects in preclinical or clinical studies, including adverse effects with potentially plausible alternative explanations (e.g., the effects are viewed as the result of random chance or the reviewers believe that there may be methodological or
-
statistical problems in the studies). This means that even if potentially plausible alternative explanations are offered to explain adverse findings, level 2 in-market monitoring would not be warranted since adverse effects were reported.
-
A review of the relevant scientific literature indicates that there is existing evidence linking the new ingredient, metabolites, secondary effectors, or source to the growth and development of organ systems whose functions become apparent after the period of maximum exposure to infant formula, have known long-term direct consequences, or impact upon developmental outcomes that could result in cumulative adverse effects over time.
The choice of level 3 assessments (options A through C) for in-market follow-up must occur when any one of the following conditions occurs:
-
In-market monitoring reveals a greater than expected number of adverse effects reported for the new ingredient or ingredient source above what might be expected based on population base rates, and an expert panel (level 2 assessment) concludes that there is potential harm for the population. A systematic re-evaluation of long-term safety issues (level 3 assessment studies) is necessary even when individual risk levels are of small effect size, given that even small individual deficits can be magnified when cumulated at a population level (Lester et al., 1998; Rice, 1990; Scott et al., 1994, 1999).
-
There is evidence of some type of adverse effect detected during preclinical or clinical trials and an expert panel concludes that there is potential for harm. This means that even if potentially plausible alternative explanations are offered to explain adverse findings, level 2 in-market monitoring is not warranted since adverse effects were reported.
-
A review of the relevant scientific literature by an expert panel (level 2 assessment) indicates that new evidence exists linking the ingredient, metabolites, secondary effectors, or source to the growth and development of organ systems whose functions become apparent after the period of maximum exposure to infant formula, have known long-term direct consequences, or a link to developmental outcomes that could result in cumulative adverse effects over time.
The choice between options A through C in level 3 assessment (see earlier section, “Recommended Levels of Assessment”) will depend on which populations with known intakes of the formula under question are most available. However the committee recommends as a matter of procedure that efforts be made to keep track of the location of individuals in the original clinical trials (option C), at least through the grade-school years, since this level offers the highest probability of accurately assessing intake of the formula under question. One implication of this recommendation is the importance of ensuring that there is a sufficient sample size in the original clinical trials so that subject attrition does not compromise the level of statistical power in follow-up studies using the original trial population. As discussed earlier in this chapter, sufficient statistical power is especially crucial in follow-up studies since effect sizes may be smaller than in the original clinical trials.
Regardless of which option is chosen, the choice of domains to be investigated in follow-up evaluations and the instruments to be used for each domain will depend on a variety of factors. If the potential detrimental effects appear to be restricted to a specific organ or functional system, then a detailed investigation of this system with screening assessments of other systems or functions potentially linked to the functioning of the specific organ system would be appropriate. For example, anti-infective ingredients (probiotics, prebiotics, lactoferrin) may need follow-up of intestinal function and flora, while immune modulators may
be assessed with both T-cell and B-cell function. As discussed earlier, if the organ or function system involved had known direct links to other organs or functions, then a detailed investigation of the linked systems would be warranted. It also is essential to include qualified scientists from all of the domain areas discussed in Chapter 6 throughout the decision-making process, including the level of follow-up assessment used and the domains where more detailed follow-up will be needed.
Specific instruments chosen must be age appropriate, have documented sensitivity to toxic exposure, and require the least level of invasiveness that still allows sufficient sensitivity. For example, Delaney-Black and colleagues (1998) used routine achievement testing conducted by the child’s school system as measures of cognitive functioning in their investigation of the long-term effects upon school-age children who had been exposed to cocaine in the prenatal period. Measures of child behavior were based on parent and teacher responses to standardized rating scales that assessed different dimensions of child adjustment. Follow-up assessments could also look for evidence of referrals to special education, medical services for achievement-related disorders (e.g., attention deficit hyperactivity disorder or learning disabilities), or mental health treatment for associated disorders (e.g., conduct disorder). These sorts of evaluations will need to address confidentiality issues in the evaluation protocol. Again, the decision of which specific measures should be used will require the participation of expert scientists in the domain under consideration (see Chapter 6).
Existing Models for In-Market Surveillance
Despite the difficulties inherent in drawing useful conclusions from long-term, in-market surveillance studies, possible research models for such studies exist. For example, an opportunity to study long-term effects of nutritional changes may be possible with the beginning of the National Children’s Study, authorized by Congress in 2000. This study proposes to follow 100,000 children from birth to age 21, with a specific focus on environmental influences on growth and development (National Children’s Study, 2003). Nutrition will obviously be a major focus of this study. Data from this type of longitudinal follow-up study might be used to determine if there were increases in specific adverse events, over what would be expected at a population level, after a formula containing a new ingredient was introduced to the general marketplace.
In terms of actual research models, a recent analysis of the outcomes of infants placed on soy formula is an example of extending what was originally a short-term study of growth of infants between the age of 9 days and 16 weeks (Strom et al., 2001). The original study was performed from 1965 to 1978. The investigators returned to their original population to investigate possible long-term endocrinological and reproductive outcomes of infants who had received soy formula containing phytoestrogens. Of the 952 subjects in the original cohort, the investigators had to reject 48 subjects because of exposure to formulas other than soy. Of the remaining 904 subjects, 51 were lost to follow-up and 42 refused to participate. Overall, 90 percent of the original cohort was located and data were available from 85 percent. This study was unable to identify any effect on endocrinological or general health. While this type of study is difficult to conduct because both patients and investigators move, it shows that formula follow-up studies are possible when there is tenacity and continuity of investigators.
Other research models also exist. An efficacy and safety study of an infant formula with added LC-PUFAs was carried out for 18 months (Lucas et al., 1999). Another study of the addition of LC-PUFAs with a follow-up period of 14 months (Auestad et al., 2001) was extended, with the original study group of patients, to a follow-up period of 39 months
(Auestad et al., 2003). In 1978 and 1979, two soy-based formulas were released that contained markedly decreased chloride content. The result was a large number of cases of what was termed the “chloride deficiency syndrome” diagnosed between 1 and 6 months of age (Roy, 1984). These infants presented with failure to thrive, lethargy, muscular weakness, and loss of appetite. Laboratory analysis revealed metabolic alkalosis, hypochloremia, hypokalemia, and hyponatremia. Nine- and 10-year follow-up of some of these infants showed no measurable deficits in cognitive development (Willoughby et al., 1990).
The Strom and colleagues (2001) soy study, as well as other longitudinal studies, such as the Framingham Heart Study (NHLBI, 2002) and the Harvard Physicians Health Study (Steering Committee of the Physicians’ Health Study Research Group, 1989), demonstrates that drawing valid conclusions from long-term surveillance studies can be accomplished, even considering the multiple methodological problems inherent in such studies.
SUMMARY
The two components of in-market surveillance include monitoring for adverse effects in infants after a formula has been introduced and long-term follow-up to ensure that there are no delayed effects. There are a variety of logistical and methodological problems associated with in-market surveillance, especially in regard to long-term follow-up (e.g., tracking subjects, reconstructing causality). However there is a crucial need for such long-term surveillance (e.g., brain areas that are adversely affected by new ingredients may not functionally become apparent until later in development, or early exposure to a toxin may increase susceptibility to later exposure to toxins). The committee recommends a systematic plan for both continued in-market monitoring and long-term surveillance as an essential part of each safety evaluation seeking to add new ingredients to infant formulas. The committee provides three possible levels of assessment for in-market surveillance, along with criteria to decide which surveillance level is appropriate for a new ingredient.
REFERENCES
Auestad N, Halter R, Hall RT, Blatter M, Bogle ML, Burks W, Erickson JR, Fitzgerald KM, Dobson V, Innis SM, Singer LT, Montalto MB, Jacobs JR, Qiu W, Bornstein MH. 2001. Growth and development in term infants fed long-chain polyunsaturated fatty acids: A double-masked, randomized, parallel, prospective, multivariate study. Pediatrics 108:372–381.
Auestad N, Scott DT, Janowsky JS, Jacobsen C, Carroll RE, Montalto MB, Halter R, Qiu W, Jacobs JR, Connor WE, Connor SL, Taylor JA, Neuringer M, Fitzgerald DM, Hall RT. 2003. Visual, cognitive, and language assessments at 39 months: A follow-up study of children fed formulas containing long-chain polyunsaturated fatty acids to 1 year of age. Pediatrics 112:e177–e183.
Barker DJ, Gluckman PD, Godfrey KM, Harding JE, Owens JA, Robinson JS. 1993. Fetal nutrition and cardiovascular disease in adult life. Lancet 341:938–941.
Beaton GH. 1986. 1986 EV McCollum International Lectureship in Nutrition. Toward harmonization of dietary, biochemical, and clinical assessments: The meanings of nutritional status and requirements. Nutr Rev 44:349–358.
Bellinger DC. 1995. Interpreting the literature on lead and child development: The neglected role of the “experimental system.” Neurotoxicol Teratol 17:201–212.
Bellinger D, Sloman J, Leviton A, Rabinowitz M, Needleman HL, Waternaux C. 1991. Low-level lead exposure and children’s cognitive function in the preschool years. Pediatrics 87:219–227.
Bendersky M, Alessandri SM, Lewis M. 1996. Emotions in cocaine-exposed infants. In: Lewis M, Sullivan MW, eds. Emotional Development in Atypical Children. Hillsdale, NJ: Lawrence Erlbaum Associates. Pp. 89–108.
Bengtsson T, Lindstrom M. 2000. Childhood misery and disease in later life: The effects on mortality in old age of hazards experienced in early life, southern Sweden, 1760–1894. Popul Stud (Camb) 54:263–277.
Chasnoff IJ, Burns KA, Burns WJ. 1987. Cocaine use in pregnancy: Perinatal morbidity and mortality. Neurotoxicol Teratol 9:291–293.
Clarke ADB, Clarke AM. 1981. “Sleeper effects” in development: Fact or artifact? Dev Rev 1:344–360.
Delaney-Black V, Covington C, Templin T, Ager J, Martier S, Compton S, Sokol R. 1998. Prenatal coke: What’s behind the smoke? Ann N Y Acad Sci 846:277–288.
Fried PA. 1989. Cigarettes and marijuana: Are there measurable long-term neurobehavioral teratogenic effects? Neurotoxicology 10:577–584.
Galler JR, Tonkiss J. 1998. The effects of prenatal protein malnutrition and cocaine on the development of the rat. Ann N Y Acad Sci 846:29–39.
Griffith DR, Azuma SD, Chasnoff IJ. 1994. Three-year outcome of children exposed prenatally to drugs. J Am Acad Child Adolesc Psychiatry 33:20–28.
Jackson AA. 2000. Nutrients, growth, and the development of programmed metabolic function. Adv Exp Med Biol 478:41–55.
Jacobson SW, Jacobson JL. 2000. Teratogenic insult and neurobehavioral function in infancy and childhood. In: Nelson CA, ed. The Minnesota Symposia on Child Psychology. Vol 31: The Effects of Early Adversity on Neurobehavioral Development. Mahwah, NJ: Lawrence Erlbaum Associates. Pp. 61–112.
Leech SL, Richardson GA, Goldschmidt L, Day NL. 1999. Prenatal substance exposure: Effects on attention and impulsivity of 6-year-olds. Neurotoxicol Teratol 21:109–118.
Lester BM, LaGasse LL, Seifer R. 1998. Cocaine exposure and children: The meaning of subtle effects. Science 282:633–634.
Leviton A, Bellinger D, Allred EN, Rabinowitz M, Needleman H, Schoenbaum S. 1993. Pre- and postnatal low-level lead exposure and children’s dysfunction in school. Environ Res 60:30–43.
Lucas A, Stafford M, Morley R, Abbott R, Stephenson T, MacFadyen U, Elias-Jones A, Clements H. 1999. Efficacy and safety of long-chain polyunsaturated fatty acid supplementation of infant-formula milk: A randomised trial. Lancet 354:1948–1954.
Lyon G, Gadisseux J-F. 1991. Structural abnormalities of the brain in developmental disorders. In: Rutter M, Casaer P, eds. Biological Risk Factors for Psychosocial Disorders. New York: Cambridge University Press. Pp. 1–19.
Mayes LC, Grillon C, Granger R, Schottenfield R. 1998. Regulation of arousal and attention in preschool children exposed to cocaine prenatally. Ann N Y Acad Sci 846:126–143.
Mendez MA, Anthony MS, Arab L. 2002. Soy-based formula and infant growth and development: A review. J Nutr 132:2127–2130.
National Children’s Study. 2003. What is the National Children’s Study? Available at http://nationalchildrensstudy.gov/about/overview.cfm. Accessed November 21, 2003.
NHLBI (National Heart, Lung, and Blood Institute). 2002. Framingham Heart Study. 50 Years of Research Success. Online. Available at http://www.nhlbi.nih.gov/about/framingham/index.html. Accessed July 1, 2002.
O’Connor MJ, Sigman M, Kasari C. 1993. Interactional model for the association among maternal alcohol use, mother-infant interaction, and infant cognitive development. Inf Behav Dev 16:177–192.
OSAS (Office of Scientific Analysis and Support). 2002. Letter to Stakeholders. Announcing CAERS—The CFSAN Adverse Event Reporting System. Online. Center for Food Safety and Applied Nutrition, Food and Drug Administration. Available at http://www.cfsan.fda.gov/~dms/caersltr.html. Accessed January 21, 2003.
Rice DC. 1990. The health effects of environmental lead exposure: Closing Pandora’s box. In: Russell RW, Flattau PE, Pope AM, eds. Behavioral Measures of Neurotoxicity: Report of a Symposium. Washington, DC: National Academy Press. Pp. 243–267.
Richardson GA. 1998. Prenatal cocaine exposure. A longitudinal study of development. Ann N Y Acad Sci 846:144–152.
Richardson GA, Conroy ML, Day NL. 1996. Prenatal cocaine exposure: Effects on the development of school-age children. Neurotoxicol Teratol 18:627–634.
Romano AG, Harvey JA. 1998. Prenatal cocaine exposure: Long-term deficits in learning and motor performance. Ann N Y Acad Sci 846:89–108.
Roy S III. 1984. The chloride depletion syndrome. Adv Pediatr 31:235–257.
Ruff HA. 1999. Population-based data and the development of individual children: The case of low to moderate lead levels and intelligence. J Dev Behav Pediatr 20:42–49.
Russell RW. 1990. Neurobehavioral time bombs: Their nature and their mechanisms. In: Russell RW, Flattau PE, Pope AM, eds. Behavioral Measures of Neurotoxicity: Report of a Symposium. Washington, DC: National Academy Press. Pp. 206–225.
Rutter M, Pickles A. 1991. Person-environment interactions: Concepts, mechanisms, and implications for data analysis. In: Wachs TD, Plomin R, eds. Conceptualization and Measurement of Organism Environment Interaction. Washington, DC: American Psychological Association. Pp. 105–141.
Schneider ML, Roughton EC, Koehler AJ, Lubach GR. 1999. Growth and development following prenatal stress exposure in primates: An examination of ontogenetic vulnerability. Child Dev 70:263–274.
Scott KG, Shaw KH, Urbano JC. 1994. Developmental epidemiology. In: Friedman SL, Haywood HC, eds. Developmental Follow-up. Concepts, Domains, and Methods. San Diego: Academic Press. Pp. 351–374.
Scott KG, Avchen RN, Hollomon HA. 1999. Epidemiology of child developmental problems: The extent of the problems of poor development in children from deprived backgrounds. Food Nutr Bull 20:34–44.
Singer LT, Arendt R, Minnes S, Farkas K, Salvator A, Kirchner HL, Kliegman R. 2002. Cognitive and motor outcomes of cocaine-exposed infants. J Am Med Assoc 287:1952–1960.
Sostek AM. 1992. Prematurity as well as intraventricular hemorrhage influence development outcome at 5 years. In: Friedman SL, Sigman MD, Sigel IE, eds. The Psychological Development of Low-Birthweight Children. Annual Advances in Applied Developmental Psychology. Vol 6. Norwood, NJ: Ablex Publishing. Pp. 259–274.
Spear LP, Campbell J, Snyder K, Silveri M, Katovic N. 1998. Animal behavior models. Increased sensitivity to stressors and other environmental experiences after prenatal cocaine exposure. Ann N Y Acad Sci 846:76–88.
Spencer PS. 1990. Chemical time bombs: Environmental causes of neurodegenerative diseases. In: Russell RW, Flattau PE, Pope AM, eds. Behavioral Measures of Neurotoxicity: Report of a Symposium. Washington, DC: National Academy Press. Pp. 268–284.
Steering Committee of the Physicians’ Health Study Research Group. 1989. Final report on the aspirin component of the ongoing Physicians’ Health Study. N Engl J Med 321:129–135.
Strom BL, Schinnar R, Ziegler EE, Barnhart KT, Sammel MD, Macones GA, Stallings VA, Drulis JM, Nelson SE, Hanson SA. 2001. Exposure to soy-based formula in infancy and endocrinological and reproductive outcomes in young adulthood. J Am Med Assoc 286:807–814.
Wachs TD. 2000. Necessary but Not Sufficient. The Respective Roles of Single and Multiple Influences on Individual Development. Washington, DC: American Psychological Association.
Wasserman GA, Liu X, Popovac D, Factor-Litvak P, Kline J, Waternaux C, LoIacono N, Graziano JH. 2000. The Yugoslavia Prospective Lead Study: Contributions of prenatal and postnatal lead exposure to early intelligence. Neurotoxicol Teratol 22:811–818.
Waterland RA, Garza C. 1999. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr 69:179–197.
Weiss B. 1995. Incipient hazards of cocaine: Lessons from environmental toxicology. In: Lewis M, Bendersky M, eds. Mothers, Babies, and Cocaine: The Role of Toxins in Development. Hillsdale, NJ: Lawrence Erlbaum Associates. Pp. 41–55.
Willoughby A, Graubard BI, Hocker A, Storr C, Vietze P, Thackaberry JM, Gerry MA, McCarthy M, Gist NF, Magenheim M, Berendes H, Rhoads GG. 1990. Population-based study of the developmental outcome of children exposed to chloride-deficient infant formula. Pediatrics 85:485–490.
Winneke G. 1990. Neurobehavioral toxicity of selected environmental chemicals: Clinical and subclinical aspects. In: Russell RW, Flattau PE, Pope AM, eds. Behavioral Measures of Neurotoxicity: Report of a Symposium. Washington, DC: National Academy Press. Pp. 226–242.















