
4
Defining, Interpreting, and Applying Concepts of Risk and Benefit in Clinical Research Involving Children
In research involving human subjects, risk is a central organizing principle, a filter through which protocols must pass.
National Bioethics Advisory Commission (NBAC), 1998, p. 89
If risk is an important filter for research protocols involving adults, it is an even finer filter for research that involves children. Federal regulations and international guidelines have established criteria for approving and conducting research involving children that are generally more stringent than those that apply to research involving mentally competent adults. This stringency is most notable as it relates to the level of risk to which child research participants can be exposed, particularly when the research does not offer the prospect of direct benefit.
The committee was specifically asked to consider the regulatory definition of “minimal risk.” Because this concept is closely related to several other concepts in the federal regulations on research involving children, the committee also examined certain of these concepts, including “minor increase over minimal risk,” “disorder or condition,” “commensurate experience,” and “vital importance.” In addition, the chapter discusses the regulatory requirements that risks to research participants be minimized and that the risks be reasonable in relation to the anticipated benefits. In this
chapter, as elsewhere in the report, when the text mentions requirements for the approval of research, it is referring to research covered by federal regulations because it is federally supported, conducted, or regulated or because an individual institution has extended the regulations to all of its research.
Although the interpretation and application of risk criteria to research protocols are sometimes straightforward, disagreements between investigators and reviewers of proposed research and disagreements among reviewers are not uncommon. The analysis and recommendations presented in this chapter are intended to encourage greater consistency in regulatory interpretation and promote explicit attention to all the criteria for approving research protocols that include children.
It is important to note at the outset that much research—whether having a prospect of benefit or not—cannot be known to be entirely free of risk. If applied to studies without prospect of benefit, a “no risk” standard for approving research would make impossible much research that can advance the health and well-being of children. The risk categories analyzed in this chapter—involving minimal risk or a minor increase over minimal risk—are intended to allow important research to go forward by permitting children who do not have the potential to benefit directly from research to be exposed to only a small risk of harm. Still, a clear tension can exist between the goal of advancing clinical research that may benefit future children and the goal of limiting the risks to individual children who participate in clinical studies. That tension underscores the importance—as discussed further in chapter 5—of a sound process for explaining to parents and, as appropriate, children the purposes of a study, its potential harms and benefits, and the rights of prospective research participants to refuse participation.
BASIC CONCEPTS OF RISK, HARM, AND BENEFIT
Risk is a complex concept that has different meanings in different contexts. Broadly, risk refers to a potential harm or the potential of an action or event to cause harm. Specific risks can be characterized along several dimensions, including the probability of a given harm as well as its likely severity and duration.
A harm is a hurtful or adverse outcome of an action or event. It makes one’s situation worse, temporarily or permanently. Research harms can occur or be evident close in time to the research intervention, but they also may occur or become apparent long after the research has been completed.
Harms resulting from research participation may be physical (e.g., pain, disability, discomfort, or death), psychological (e.g., fear, anxiety, depression, or embarrassment), or social (e.g., peer disapproval, economic loss, or
legal jeopardy). For research that includes children, investigators and reviewers of research must consider potential harms such as fear and separation from parents that are usually not considered in studies involving adults (AAP, 1995). Discussions of research ethics also identify harms related to the violation of privacy or confidentiality and, more abstractly, to the lack of respect for individuals that occurs if research participants are treated as objects or means to an end (see, e.g., NBAC, 2001a and NHRPAC, 2002).
One difference between the federal regulations and the 1977 report of the National Commission for the Protection of Human Subjects of Biomedical and Behavioral Research (National Commission) is that the regulatory definition of minimal risk refers to harms or discomfort, whereas the latter report’s definition mentions only harm (National Commission, 1977). This could be seen as unimportant because a discomfort is, by definition, a kind of harm, in that it makes a person’s situation worse. A certain amount of discomfort could also be viewed as unimportant in assessments of overall risks of harm to a child. For example, the supplementary discussion in the National Commission report refers to “unusual discomfort” as a harm. The regulatory language, nonetheless, calls the attention of investigators and reviewers of research to harms that children may experience but that are less dramatic than, for example, death, disability, prolonged pain, or other lasting physical, psychological, or social harm. Such discomfort-as-harm covers a range of transitory but unpleasant physical and psychosocial experiences (e.g., pain, nausea, embarrassment, and fear).
A benefit is a positive or valued outcome of an action or event. A potential benefit is a positive but uncertain outcome, for example, the desired result of an experimental intervention. Discussions—and federal regulations as well—often refer to “risks and benefits” when it is appropriate to use the parallel language of “potential harms and potential benefits.”
Potential benefits, like risks or potential harms, have the dimensions of probability, magnitude, and duration. They likewise may be physical (e.g., cure of a disease, slowing of the progression of a disease, or relief from pain), psychological (e.g., relief from depression or improved quality of life), or social (e.g., removal or lessening of a condition that is stigmatizing or that interferes with employment).
Most discussions of risk, harm, and benefit focus on potential harms or potential benefits to the individual research participant. For certain studies involving children, however, federal regulations require consideration of possible benefits to other children. Specifically, as discussed below, when proposed research involves a minor increase over minimal risk and does not offer the prospect of direct benefit, the research must be limited to children with a disorder or condition and must be expected (among other criteria) to generate vital knowledge about the disorder or condition (45 CFR 46.406; 21 CFR 50.53). That is, the child participating in the research is not ex-
pected to benefit directly from the study, but future children may. In addition, studies that are otherwise not approvable under the regulations may be approved by the Secretary of the U.S. Department of Health and Human Services (DHHS) or the Commissioner of the Food and Drug Administration (FDA) if the studies, among other things, present “a reasonable opportunity to further understanding, prevention or alleviation of a serious problem” with children’s health or welfare (45 CFR 46.407; 21 CFR 50.54). Again, any potential benefit is not directly to the child research participant but to other children in the future.
Although federal regulations include, as one element of informed consent, “a description of any benefits to the subject or to others which may reasonably be expected,” the corresponding description of risks cites only risk to the research participant (45 CFR 46.116(a)(3); 21 CFR 50.25(a)(3)) from the research. The regulations require research protocols to include written procedures for promptly reporting of “problems involving risks to subjects or others” (45 CFR 46.103(b)(5); 21 CFR 56.108(b)(1)), but overall, the emphasis in the regulations is on risks to research participants.
Some research clearly presents risks to others. For example, participants in a smallpox vaccine trial may pose a risk of disease transmission to individuals in close contact with them. Certain genetic studies have the potential to cause serious emotional distress to family members and impair family relationships in the short- and long-term. If confidentiality is breached, such studies may present the additional risks of stigmatization or economic harm to the family. Investigators and reviewers of research should normally consider such risks as part of their broader moral responsibility to minimize or prevent harm.
In addition, certain research involving children may pose risks to members of ethnic, cultural, religious, or other communities. Studies may, for example, focus attention on the prevalence of a medical condition or problem (e.g., diabetes or teen alcohol abuse) in certain ethnic or other groups (see, e.g., Arrillaga, 2001 and Stiffman et al., 2002). Such attention may reinforce prejudices about minority groups, stigmatize their members, and promote discrimination (Fisher et al., 2002). Reflecting concerns about stereotyping, discrimination in jobs or insurance, and other short- or long-term negative effects of research, some Indian tribes such as the Navaho Nation and the Cherokee Nation have organized formal institutional review boards (IRBs) to review research protocols (Hillabrant, 2002; see also IHS, 2002). (Whether or not they have organized their own IRBs, tribes must approve research conducted on tribal lands, unless the tribe has delegated review to another entity, such as the Indian Health Service.)
ASSESSING THE LEVEL OF RISK POSED BY A RESEARCH PROTOCOL
Categorizing, evaluating, and weighing the potential harms of proposed research are among the most challenging and subjective tasks for those charged with reviewing pediatric research. In 1998, the National Bioethics Advisory Commission observed that “relatively little progress has been made in describing the criteria for assessing risk by IRBs” (NBAC, 1998, p. 89, emphasis in the original).
Required Determinations
Box 4.1 summarizes the determinations about possible research harms and benefits that must be made before federally funded, sponsored, or regulated research involving children may be approved. As described in Chapter 3, some of these requirements must be met for all covered human research; other provisions are specific to children.
The provisions specific to children establish four basic categories of approvable research involving children (see Box 3.3 in Chapter 3). Figure 4.1 presents an algorithm for using the assessed level of risk, the possibility for direct benefit, and other criteria to determine whether research fits one of these categories. For research that involves a control group of healthy children without a disorder or condition and without prospect of direct benefit from the research, the research procedures for that group would have to involve no more than minimal risk.
Defining Minimal Risk
Subpart A of the DHHS regulations defines minimal risk as meaning “that the probability and magnitude of harm or discomfort anticipated in the research are not greater in and of themselves than those ordinarily encountered in daily life or during the performance of routine physical or psychological examinations or tests” (45 CFR 46.102(i); 21 CFR 50.3(k)). Subpart D of the regulations then explicitly applies this definition to research involving children.
The interpretation of what constitutes minimal risk is important to investigators because several practical consequences may flow from a judgment that a proposal does not exceed that risk threshold. For example, under certain circumstances, as described in Chapter 3, such a proposal may be eligible for expedited IRB review.
|
BOX 4.1 Are risks to participants minimized by using procedures that are consistent with sound research design and that do not unnecessarily expose subjects to risk and, whenever appropriate, by using procedures already being performed on the subjects for diagnostic or treatment purposes? 45 CFR 46.111(a)(1); 21 CFR 56.111(a)(1) Are risks to participants reasonable in relation to anticipated benefits to subjects and to the importance of the knowledge reasonably anticipated from the research? 45 CFR 46.111(a)(2); 21 CFR 56.5111(a)(2) Is the selection of participants equitable, taking into account the purposes of the research, its setting, and the special problems of research involving vulnerable populations, such as children? 45 CFR 46.111(a)(3); 21 CFR 56.111(a)(3) Are safeguards included to protect participants who are likely to be vulnerable to coercion or undue influence, such as children? 45 CFR 46.111(b); 21 CFR 56.111(b) Does research meet the regulatory criteria for research involving children? 45 CFR 46.404-407; 21 CFR 50.51-54 (see Figure 4.1) Are appropriate provisions made for monitoring participant safety? 45 CFR 46.111(a)(6); 21 CFR 56.111(a)(6) Are appropriate provisions made for protecting privacy and confidentiality? 45 CFR 46.111(a)(7); 21 CFR 56.111(a)(7) |
How Investigators and IRBs Interpret Minimal Risk
The committee found few systematic studies documenting how clinical investigators or reviewers of research interpret minimal risk in general or with reference to the risks of either “daily life” or “routine medical or psychological examinations.” The first comparison (daily life) covers a very large array of possible experiences involving a sizeable range of risks, even for people not engaged in clearly high-risk activities such as skydiving. As described by Kopelman, this part of the definition may try “to explain the obscure (what studies have an appropriately low risk to allow participation by child-subjects) with the more obscure (what is the probability and magnitude of risk people normally encounter in daily life) (Kopelman, 2000, p. 751). The concept of “minor increase over minimal risk” builds on the
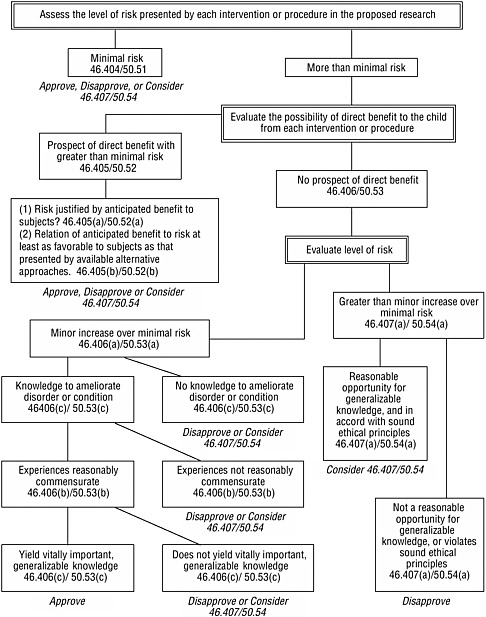
interpretation of minimal risk and, therefore, shares in its uncertainty or confusion.
That the “minimal risk” standard invites variable interpretation has long been clear. A 1981 article reported on a survey of pediatric researchers and department chairs that found variations in how they applied the risk categories in proposed federal regulations to several procedures used in pediatric studies (Janofsky and Starfield, 1981). For example, when asked to assess tympanocentesis (puncturing of the ear drum) in children up to 1 year of age, 46 percent of respondents classified the procedure as involving a minor increment over minimal risk, 40 percent thought that it involved more than a minor increase, but 14 percent thought that it posed minimal risk or less. The level of agreement on a risk category typically was less than 70 percent. An accompanying editorial called for better standards of risk assessment in children’s research and suggested (without results) that a task force of the American Academy of Pediatrics develop consensus opinions about the risks of different procedures (Lascari, 1981).
Some 20 years after the study by Janofsky and Starfield was published, a survey of chairs of IRBs again found variation in the assessments of risk that a variety of research procedures presented to children (Wendler, 2003; Shah et al., 2004). For example, when asked to assess the risk presented by a confidential survey of sexual activity, 29 percent of respondents classified it as presenting more than a minor increase over minimal risk, whereas 44 percent thought that it presented minimal risk. Fifteen percent of respondents classified a blood draw once a week for 6 weeks as minimal risk, but 32 percent classified it as more than a minor increase over minimal risk. Eighty-one percent judged a single blood draw as minimal risk, but 17 percent labeled it as a minor increase over minimal risk. For research involving lumbar puncture without conscious sedation, 32 percent of respondents classified the research as involving a minor increase over minimal risk when it involved ill children and 6 percent classified it as minimal risk; for healthy children, the corresponding figures were 16 and 2 percent. The respondents might have shown greater agreement if they had been reviewing real protocols, and it is not clear whether respondents were giving their own views or the views that they thought others held. Also, the study did not determine the nature and extent of the experience of the IRB chairs with protocols involving children. Nonetheless, the results still point to the considerable subjectivity of risk assessments.
Chapter 8 cites analyses of actual IRB determinations that show similar variability in risk categorization. Most involve determinations reached by multiple IRBs reviewing the same protocol for multicenter trials. This variability in risk categorization raises ethical concerns about whether children are being appropriately protected from research risks. It also presents practical problems for those involved in designing and implementing important
and often complex pediatric studies. The next sections of this chapter offer interpretations of minimal risk and minor increase over minimal risk that the committee believes will reduce the confusion and disagreement surrounding these concepts. Nonetheless, given the lack of relevant data and the subjective aspects of risk assessments, differences in judgments will certainly continue. For that reason, it is important that those designing and developing research explain the evidence and rationales behind their judgments. Likewise, those reviewing a research protocol should explain the bases for their determinations about its approval or disapproval.
Indexing Assessments of Minimal Risk to the Experience of Healthy Children in Daily Life
The regulatory definition of minimal risk, which was first adopted in 1981, departs in some respects from the definitions offered by the National Commission in its 1977 report, Research Involving Children (see Chapter 1). That report defined minimal risk as “the probability and magnitude of physical or psychological harm that is normally encountered in the daily lives, or in the routine medical or psychological examination, of healthy children” (National Commission, 1977, p. xix, emphasis added). In contrast, the definition presented in Subpart A and cited in Subpart D of the federal regulations has no modifying phrase referring to healthy individuals. When what is now Subpart A was first published in the Federal Register in 1981, the preamble—but not the actual language of the regulations—described minimal risk in terms of “those risks encountered in the daily life of the subjects of the research” (DHHS, 1981, online, unpaged, emphasis added). This use of the research participants’ experiences as the point of reference has come to be called the “relativistic interpretation” of the minimal-risk standard.
A relative interpretation theoretically allows high-risk studies to be approved as “minimal-risk” studies if members of target research populations experience high risks in their daily lives, including in their homes, schools, sports activities, or neighborhoods. In addition to such environmental risks, some potential target research populations may, by virtue of their medical condition or its treatment, experience substantial everyday risks, distress, and uncomfortable medical examinations that are, for them, routine but not minimal in burden or discomfort. A relative interpretation of minimal risk would permit comparably high risks in research for these already high-risk children. In contrast, more fortunate research populations that experience low levels of risk in daily life would have a correspondingly low risk threshold for assessing whether a study presented minimal risk.
Consistent with the conclusions of a number of other groups (see, e.g., the National Commission, 1977; NBAC, 2001a; and NHRPAC, 2001), the
committee rejected a relative interpretation of minimal risk, that is, an interpretation that allows the application of different standards or thresholds of minimal risk for different children and permits greater risk for those already disadvantaged by illness, poverty, and other burdens. The relative interpretation of the minimal-risk standard is inconsistent both with an ordinary or commonsense understanding of the concept of minimal risk and with the objective of providing special protections to child participants in research. It misinterprets and undercuts the moral and social purpose behind the minimal-risk standard; namely, to guide judgments about when risks are low enough to safely and ethically enroll children in studies that are not designed to benefit them (Kopelman, 1989, 2000).
Furthermore, allowing a relative interpretation of minimal risk would violate the ethical principle of justice, which requires that the burdens and benefits of research be distributed equitably. As a political and practical matter, it could also create social dissension if those in disadvantaged communities or populations understood that their children could be exposed to a higher risk in research than better-off children (Kopelman, 1989, 2000). Thus, the standard for interpreting risk has moral and social as well as legal and scientific implications.
In rejecting a relative interpretation of minimal risk, the committee agreed with the National Human Research Protections Advisory Committee (NHRPAC) that the interpretation of the concept should be “indexed” to the experiences of the “normal, healthy, average child” (NHRPAC, 2002, p. 3).1 The threshold of minimal risk should thus be the same for healthy and ill children.2 Furthermore, the interpretation should not change for groups of healthy children whose daily experiences involve risks greater than those that most other children experience as part of daily life. Thus, children who live in dangerous environments (e.g., with abusive parents or in unsafe housing) would not be exposed to more research risk than children who live in safer environments.
Interpretations of risk may, however, take into account children’s developmental status or age because the physical or psychological risk of a research procedure can vary for younger and older children. That is, what is minimal risk for an 8-year-old may be high risk for an infant. Also, in some cases, a procedure that is judged to involve minimal risk to healthy children
may present more than minimal risk to children with certain medical conditions. For example, intramuscular injections that are safe for healthy children would be risky for children with hemophilia. The eligibility and screening criteria described in research protocols should be sufficient to exclude such vulnerable children from studies that otherwise present minimal risk.
Determinations about risk should consider the duration and cumulative characteristics of research interventions or procedures, for example, the number of procedures included in a protocol or the number of times that an individual procedure is repeated in a given period of time. Certain studies may include several different procedures that involve minimal risk or burden individually but that taken together present more than minimal risk. Similarly, although a single blood draw by needle stick normally involves minimal harm or discomfort, multiple needle sticks for blood draws in a short period could, depending on the child’s age and other circumstances, present more than minimal risk of harm or discomfort. In other words, risk can be cumulative.
The committee agreed with NHRPAC’s basic argument about the meaning of minimal risk, but it had some concerns about a further statement that interpreted minimal risk as “the socially allowable risks which parents generally permit their children to be exposed to in nonresearch situations” (NHRPAC, 2002, p. 1). The terms socially allowable and generally permit were presumably intended to exclude comparisons with risky actions or situations (e.g., failing to place infants and small children in car seats during automobile trips) that might be permitted in children’s daily lives by irresponsible or ignorant parents. The committee agrees that it is appropriate to discourage comparisons of minimal-risk research to such dangerous situations as “what a child might encounter … while playing in traffic.”3
Nonetheless, that an action has majority social approval and is legal does not necessarily make it an appropriate basis of comparison for the assessment of risks to child participants in research. For example, researchers could conceivably claim that an experiment that involved hitting a child presented only “minimal risk” if state laws or school board policies expressly permitted spanking (see, e.g., AAP, 2001) and public opinion polls in the state showed majority support for the practice as part of school life.
Comparisons with Risks of Routine Examinations for Healthy Children
In addition to the risks of daily life, the federal regulations provide a second standard for assessing minimal risk in research, specifically, the risks “ordinarily encountered … during the performance of routine physical or psychological examinations or tests.” Just as the committee concluded that the assessment of “risks of daily life” standard should be indexed to the experiences of normal, healthy, average children, it likewise concluded that the “routine examination” standard be interpreted with reference to the experiences of normal, healthy, average children. Ill children may routinely undergo much more burdensome and risky examinations. Again, assessments can appropriately take age into account because routine examinations differ for infants, children, and adolescents.
The components of “routine medical examinations” have no precise, universally accepted definition but what is sometimes called a well-child physician visit offers a reasonable basis for comparisons. In addition to a history, such a visit typically includes several routine, age-appropriate physical and psychological examinations or tests, guidance and education (for the child, the parents, or both), and immunizations.
Recommended elements of the physical examination component of a well-child visit are not entirely uniform (see, e.g., USPSTF, 1995; Schuster, 2000; and Behrman et al., 2004), in part because those making recommendations must often rely on clinical experience and judgment rather than solid scientific evidence about the potential benefits and risks of specific assessments or tests. Depending on the child’s age, the physical examination may include measurements of height, weight, and head circumference; assessment of obesity with skin-fold calipers (pincher-like devices used to determine levels of subcutaneous fat); collection of blood; measurement of heart rate and blood pressure; collection of voided urine; testing of fine and gross motor development; and hearing and vision tests. The recommended elements are most extensive for neonates and older infants and most limited for school-age, preadolescent children.
A central objective of the history component of a well-child visit is to identify health risks (e.g., poor diet or a lack of seat belt or car seat use). Because many health risks vary by age, so does the history component of an examination. For an adolescent’s routine medical examination, a full history includes exploration of sexual, smoking, and other behaviors that have health consequences.
Notwithstanding some disagreement about the specifics, the components of a well-child visit appear to be fairly modest in number and, taken individually, are reasonably well-characterized in content. They clearly include a far smaller set of activities than the activities of daily life. They also present a more limited range of risk of harm.
In addition to psychological assessments that may be part of a well-child visit, psychological tests considered routine for healthy children include standardized measures of infant functioning and standardized intelligence tests for children and adolescents. For example, to assess infant behavior, including responses to stress, the Brazelton Neonatal Behavioral Assessment Scale exposes infants to both pleasant stimuli and unpleasant stimuli, such as pinpricks (Brazelton et al., 1987). If the Brazelton assessment is used as a benchmark for determining acceptable research risk, researchers would clearly be able to expose infants to mildly unpleasant stimuli. As another example, the Bayley Scales of Infant Development, which are used for children from age 1 month to 3 1/2 years, include both mental scales and motor scales (Bayley, 1993). In the administration of the Bayley scales, the child is asked to display gross motor skills, such as standing, walking, and jumping, and fine motor skills, such as grasping an object. The child may also be asked to point to pictures, imitate words spoken by the examiner, or imitate the examiner’s actions. The examiner tests a child’s reaction to actions such as shaking a rattle behind the child’s head and placing a mirror in front of the child. The Denver Developmental Screening Test II is used with children from birth to age 6 years (Frankenburg et al., 1992). This test also assesses gross motor and fine motor skills. It also assesses language skills and social skills, for example, by having the child play ball with the examiner and by having the child wash and dry his or her hands.
Intelligence tests, such as the widely-known Stanford-Binet Intelligence Scale, the Wechsler Preschool and Primary Scale of Intelligence, and the Wechsler Intelligence Scale for Children, include questions designed to assess knowledge and reasoning (Thorndike et al., 1986; Wechsler, 1989, 1991). The Wechsler scales also include performance items that require children to perform tasks such as assemble puzzles, arrange pictures in a sequence, and reproduce a design.
For a research procedure or intervention that is not normally part of routine medical or psychological examinations for children, the question is whether it presents a risk that is “equivalent” to that encountered in such examinations (NHRPAC, 2002). In its discussion of procedures involving minimal risk, the National Commission mentioned—in addition to physical examinations and psychological tests—“immunization, modest changes in diet or schedule, … and noninvasive physiological monitoring” (National Commission, 1977, pp. xix-xx). For the most part, assessments of equivalence are likely to be more straightforward for comparisons to routine medical and psychological examinations than for comparisons involving the larger range of risks encountered in daily life.
Recommended Interpretation of Minimal Risk
To recapitulate, the committee appreciated the subjective dimensions involved in interpreting the concept of minimal risk and the frequent lack of data to guide assessments of the risk presented by a particular procedure in the context of a particular study. It recognized that variability in assessments will continue, which makes it important that those designing and reviewing research explain the bases for their judgments of risk. Recommendations later in this chapter call for federal agencies or advisory groups to provide additional guidance for investigators and reviewers about procedures that normally involve minimal risk or a minor increase over minimal risk.
On ethical grounds, the committee rejected a “relative” interpretation of minimal risk; that is, an interpretation that allows the application of higher thresholds of minimal risk for children who experience higher risk in their daily lives as a result of their place of residence, family situations, medical condition, or other burdensome circumstances. Instead, the assessment of risk should be indexed to the experiences of average, normal, healthy children. It may take age into account and should consider the duration of potential harms or discomfort.
Recommendation 4.1: In evaluating the potential harms or discomfort posed by a research protocol that includes children, investigators, and reviewers of research protocols should
-
interpret minimal risk in relation to the normal experiences of average, healthy, normal children;
-
focus on the equivalence of potential harms or discomfort anticipated in research with the harms or discomfort that average, healthy, normal children may encounter in their daily lives or experience in routine physical or psychological examinations or tests;
-
consider the risk of harms or discomfort in relation to the ages of the children to be studied; and
-
assess the duration as well as the probability and magnitude of potential harms or discomfort in determining the level of risk.
Defining Minor Increase over Minimal Risk and Associated Terms
Minor Increase over Minimal Risk
In its 1977 report on research involving children, the National Commission recommended that children with a disorder or condition be allowed to participate in research that does not hold the prospect of directly benefiting them if the research involves no more than a minor increase over
minimal risk and has the prospect of providing knowledge vital for understanding or ameliorating the disorder or condition. The Commission argued (with dissents by 2 of 11 members) that “foreseeable benefit to an identifiable class of children may justify a minor increment of risk to research subjects” (National Commission, 1977, p. 125).
In Section 406 of 45 CFR 46 and Section 53 of 21 CFR 50, federal regulations follow the Commission’s advice. The section thus permits children with disorders or conditions to be exposed to slightly more than minimal risk—without prospect of direct benefit—to gain important knowledge about those disorders or conditions. For research involving adults, federal regulations provide no equivalent risk category. The assumption is that adults can decide for themselves if they wish to consent to research, including research that involves greater than minimal risk absent any prospect of direct benefit.
Whatever disputes exist about the interpretation of the “minimal risk” threshold are compounded in interpreting the “minor increase over minimal risk” threshold. Neither the regulations nor the National Commission’s report provides a definition of “minor increase over minimal risk.” The Council for International Medical Organizations noted that there is no precise, internationally accepted definition of a “slight or minor increase” above the risks encountered in routine medical or psychological examinations (CIOMS, 2002, Guideline 9). In commentary, the National Commission mentioned research that “goes beyond, but only slightly beyond, the minimal risk” (National Commission, 1977, p. 130). NHRPAC referred to risks that are “just a bit more” or “a little more than minimal” (NHRPAC, 2002, p. 3). The committee endorses these interpretations that stress that the increase over minimal risk should be “only slightly beyond” or “just a bit more” than that level.
In its report, NHRPAC concluded that just because children may experience invasive procedures with considerable risk and discomfort while they are being treated for a disease, this “does not justify risks greater than a minor increase over minimal in a research study that provides no prospect of direct benefit to the individual subjects” (NHRPAC, 2002, p. 3). The committee agrees. That is, consistent with the interpretation of minimal risk, what constitutes “a bit more” risk in research involving children is not relative and does not allow a higher threshold for children with high-risk or high-burden conditions than for children with less serious conditions.
Recommendation 4.2: In evaluating the potential harms or discomfort posed by a research protocol that includes children who have a disorder or condition but no prospect of benefiting from participation, investigators and reviewers of research protocols should
-
interpret minor increase over minimal risk to mean a slight increase in the potential for harms or discomfort beyond minimal risk (as defined in relation to the normal experiences of average, healthy, normal children);
-
assess whether the research procedures or interventions present experiences that are commensurate with, that is, reasonably comparable to experiences already familiar to the children being studied on the basis of their past tests or treatments or their knowledge and understanding of the treatments that they might undergo in the future;
-
consider risks of harms or discomfort in relation to the ages of the children to be studied; and
-
assess the duration as well as the probability and magnitude of potential harms or discomfort in determining the level of risk.
Disorder or Condition
Just as federal regulations at 45 CFR 46.406 and 21 CFR 50.53 offer no definition of a minor increase over minimal risk, they likewise offer no definitions of “disorder” or “condition.” Again, this section of the regulations, among other provisions, allows children to be enrolled in studies that involve a minor increase over minimal risk but no prospect of direct benefit. Thus, the approval of much research that may ultimately benefit various groups of children hinges on the definition and interpretation of these terms.
The meaning of the term condition has, in particular, been variably interpreted. Some of this variability, however, probably stems from problems with the interpretation of minimal risk. For example, when IRBs narrowly interpret “minimal risk” and routinely classify research that involves potentially controversial topics (e.g., surveys of adolescent sexual activity or drug use) as involving a minor increase over minimal risk (rather than no more than minimal risk), approval may then depend on whether the target research group is determined to have a condition (e.g., “being an adolescent”).
Stedman’s Medical Dictionary defines disorder as “a disturbance of function, structure, or both, resulting from a genetic or embryonic failure in development or from exogenous factors such poison, trauma, or disease” (Stedman, 2000). This definition encompasses physical and mental health problems arising from such common sources as disease, trauma, congenital anomalies, neurodevelopmental problems, and genetic abnormalities.
The term condition presents more difficulties. Standard medical dictionaries do not define it (except as a verb), and other dictionaries are not helpful in the context of clinical research. In recommending that research involving children be conducted to promote children’s health and well-
being, the National Commission stated in its 1977 report that “it is necessary to learn more about normal development as well as disease states in order to develop methods of diagnosis, treatment and prevention of conditions that jeopardize the health of children, interfere with optimal development, or adversely affect well-being in later years” (National Commission, 1977, p. 1). The National Commission did not, however, explicitly define “condition.”4
Views on the meaning of “condition or disorder” cover a wide spectrum. At one end of the spectrum is the view that “disorder or condition” refers only to an illness, disease, injury, or defect. The committee rejected this view as too narrow, noting that it would reduce flexibility in studying children who are currently healthy but at risk of serious illnesses that could potentially be prevented or mitigated through early interventions.
Others interpret condition so broadly that almost any social, developmental, or other characteristic would qualify as a condition and, thereby, justify exposing a child to a higher level of research risk. This would make virtually meaningless the distinction between research approvable under Section 404 and research approvable under Section 406 of the DHHS regulations. Use of such broad interpretations could also unjustly single out groups of children already burdened by poverty and other social disadvantages for research that would not necessarily benefit them.
Thus, although the committee agreed with NHRPAC that a condition “can be understood more broadly than simply a specific disease or diagnostic category” (NHRPAC, 2002, p. 3), the committee did not fully accept that group’s description of condition as “relating to [1] a specific characteristic which describes a group of children, [2] a physical, social, psychological, or neurodevelopmental condition affecting children, or [3] the risk of certain children developing a disease in the future based on diagnostic testing or physical examination” (NHRPAC, 2002, p. 3, bracketed numbers added for clarity). As examples of conditions, the NHRPAC report cited two periods of childhood (infancy and adolescence) and socioeconomic circumstances, for example, poverty and institutionalization.
The committee recognized that it is important to understand the correlates of health and disease and to identify specific circumstances or conditions—not just correlates—that contribute to children’s poor or good health. For example, what about poverty—for example, persistent poor nutrition—contributes to specific health problems? Nonetheless, children’s social, economic, racial, ethnic, and environmental characteristics or circumstances
do not, in themselves, necessarily justify exposing a child to a higher level of risk in research that is not expected to benefit them directly. Issues of justice must be considered.
If a characteristic of a group of children is to be designated as a condition that allows children to be exposed to a higher level of risk without prospect of benefit, the link between the characteristic and a deficit in health or well-being should be supported by scientific evidence or clinical knowledge. Thus, for this kind of research, investigators who define a research population on the basis of social characteristics or “conditions”—such as ethnicity, family circumstances, or economic status—must present a case that the condition has a negative effect on children’s health and well-being that is relevant to the research question. They must also make the case that the research can be expected to generate vital knowledge about the condition.
Recommendation 4.3: In determining whether proposed research involving a minor increase over minimal risk and no direct benefit can be approved, the term condition should be interpreted as referring to a specific (or a set of specific) physical, psychological, neurodevelopmental, or social characteristic(s) that an established body of scientific evidence or clinical knowledge has shown to negatively affect children’s health and well-being or to increase their risk of developing a health problem in the future.
In the committee’s experience, investigators and reviewers of research sometimes ignore the “disorder” or “condition” criterion in assessing whether research is approvable under 45 CFR 46.406 or 21 CFR 50.53. Investigators and IRBs should be explicit about how this and other criteria are met for research approved under this section.
Recommendation 4.4: For purposes of determining whether proposed research involving a minor increase over minimal risk and no direct benefit can be approved, institutional review boards should make a determination that
-
the children to be included in the research have a disorder or condition;
-
the research is likely to generate vital knowledge about the children’s disorder or condition;
-
the research procedures or interventions present experiences that are commensurate with, that is, reasonably comparable to, experiences already familiar to the children being studied on the basis of their past tests or treatments or on their knowledge and understanding of the treatments that they might undergo in the future; and
-
the research does not unjustly single out or burden any group of children for increased exposure to research risk on the basis of their social circumstances.
Commensurate
In approving research that involves a minor increase over minimal risk, investigators and reviewers of research must also determine that “the intervention or procedure presents experiences to subjects that are reasonably commensurate with those inherent in their actual or expected medical, dental, psychological, social, or educational situations” (45 CFR 46.406(b); 21 CFR 50.53(b)).5 This provision is based on another recommendation in the 1977 report of the National Commission. The Commission suggested that this requirement would help children who have the capacity to agree or assent to research participation “make a knowledgeable decision … based on some familiarity with the intervention or procedure and its effects” (National Commission, 1977, p. 5). NHRPAC observed that the requirement could also help parents make thoughtful judgments about permitting a child’s participation in research.
As a synonym for commensurate, the National Commission referred to research activity that was “reasonably similar” to procedures that prospective research participants ordinarily experience. Dictionary definitions of commensurate or commensurable also emphasize the concepts of sameness, as well as proportionality or correspondence (American Heritage, 1992; Merriam-Webster, 2003). Thus, a child might not have experienced a particular research procedure, but the procedure could still be described to the child as potentially presenting levels of pain, immobility, anxiety, time away from home, or other effects that would be similar to those produced by procedures that they have experienced. In any case, even if a procedure is commensurate with a child’s experiences, it cannot be approved under this category of research if it involves more than a minor increase over minimal risk.
Familiarity with a procedure may make further experiences with that or a similar procedure less frightening or burdensome to an ill child than it would be to a healthy child. In some cases, however, past experience with a procedure could make a child more fearful or anxious. The National
Commission’s explanation of this provision indicates that investigators and reviewers of protocols should not assume that familiarity with a procedure reduces the burden on a child research participant. Rather, investigators should plan to assess the views and concerns of the child and the parents when children’s assent and parent’s permission for a child’s participation in research are discussed. That advice remains appropriate.
Direct Benefit
Under 45 CFR 46.405 and 21 CFR 50.52, child participants in research can be exposed to more than a minor increase over minimal risk if—among other conditions—the research presents the prospect of direct benefit. A direct benefit is a tangible positive outcome (e.g., cure of disease, relief of pain, and increased mobility) that may be experienced by an individual. When a research procedure or intervention has the prospect of directly benefiting child research participants, it can be approved even if it presents more than a minor increase over minimal risk. In addition, the relationship between the anticipated direct benefit and the risk or potential harm should be at least as favorable for the proposed research procedure as for the alternatives available to the children. This follows the recommendation of the National Commission (1977) that a child should not be disadvantaged by being enrolled in a research study (see the discussion below of clinical equipoise).
Research participants may also anticipate collateral, indirect, or side benefits that are not related to the research objectives as such (Churchill et al., 2003). For example, those participating in research may appreciate the opportunity to learn more about their condition or develop social relationships with others in similar circumstances (Churchill et al., 2003). Collateral or indirect benefits should not, however, be considered in assessing a research procedure’s potential benefits in relation to its potential harms or be allowed to make up for shortfalls in the prospect of direct benefit from the research procedure (NBAC, 2001b). Similarly, although research participants may view gifts or payments for research participation as benefits, federal guidance makes clear that such payments should not be included by investigators or IRBs in their risk-benefit assessments (OPRR, 1993; see also Chapter 6).
Ordinarily, research that holds out the prospect of direct benefit evaluates an intervention intended to prevent, diagnose, or treat illness or injury. In addition, the regulations mention monitoring procedures that might contribute to a child’s well-being (45 CFR 46.405; 21 CFR 50.52). For example, a study testing a new method for monitoring blood sugar levels might have the prospect of reducing discomfort or inconvenience for the children involved in the study.
Environmental circumstances can affect assessments of whether research has the potential for direct benefit. For example, in recent discussions of a proposed test of dilute smallpox vaccine in young children, one issue was whether the threat of a terrorist attack involving smallpox was, in the words of one reviewer, “a fantastically remote possibility or a real threat” (Hammerschmidt, 2002, p. 2). That reviewer judged that the prospect of direct benefit was highly speculative and that the arguments for direct benefit were not conclusive. Another reviewer noted, in contrast, that most vaccine research involving common childhood illnesses did have the prospect of directly benefiting children participating in the research and that the risks were low (Halsey, 2002).
As discussed in Chapter 2, the usual phase 1 trial with healthy adult volunteers seeks to determine the maximum tolerated dose and pharmacokinetic characteristics of a drug intended for a condition that these volunteers do not have. Such a trial does not hold out the prospect of direct benefit. In contrast, clinical trials involving children usually follow studies with adults (and possibly special laboratory and animal studies) that have provided information about safety and pharmacokinetics and that have given at least preliminary indications of efficacy. This information can help shape the design of an early-phase pediatric trial, for example, by guiding the selection of a drug dose that will maximize the potential for benefit and reduce the associated potential for harm as much as possible, although the trial may still involve more than a minor increase over minimal risk. Particularly when standard treatment alternatives have been exhausted and the probable outcome for the child without the trial intervention is grim, the prospect of benefit may arguably be considered reasonable in relation to the risks, even when further testing of efficacy is continuing in adult studies (see, e.g., Kodish, 2003c).
Vital Importance and Research Approvable Under Section 407 or Section 54
When proposed pediatric studies involve more than minimal risk and no direct benefit, approval of the research requires that IRBs determine not only that the children included in the study have a disorder or condition but also that the proposed research may produce vitally important scientific knowledge about the disorder or condition. The committee stressed earlier that all these determinations should be an explicit part of the review and approval process.
Although the standard of “vital importance” is required for research to be approved under 45 CFR 46.406 or 21 CFR 50.53, different language is used in 45 CFR 46.407 or 21 CFR 50.54, which allows research that cannot otherwise be approved to be referred to and approved by the Secre-
tary of DHHS or the Commissioner of FDA. Section 407 requires a determination that the research offers “a reasonable opportunity to further the understanding, prevention, or alleviation of a serious problem affecting the health or welfare of children.” Arguably, the latter is a less stringent standard than the standard of vital importance. That is, proposed research might more easily be justified on the ground that it “furthers” knowledge than on the grounds that it is “vital.”
As described in the 1977 report of the National Commission, the referral of proposed research for “national” review should be reserved for “exceptional situations” and research of “major significance.” Given this context, the committee believed that the criterion for judging the potential contribution of research must, ethically, be as stringent for reviews conducted under Section 407 as for those conducted under Section 406. Thus, although it is not required by the regulations, the standard of “vital importance” should be applied by the panels involved in the review of proposals referred to the Secretary of DHHS or the Commissioner of FDA for approval. The Secretary’s Advisory Committee on Human Research Protections (SACHRP) should incorporate this criterion when it considers adjustments in the process for Section 407 reviews (see Chapter 8).
Recommendation for Additional Government Guidance
Ideally, investigators and reviewers would have some data on which to base assessments of the risk presented by common research procedures, whether or not the procedures are part of routine physical examinations. For many routine medical and psychosocial interventions, however, risks may at best have been characterized (e.g., as involving bruising, pain, or anxiety) but not assessed in terms of the frequency, intensity, or duration of harms or discomfort. Although it is reasonable for reviewers of research proposals to seek information on the risks of research procedures or interventions, they may find that no evidence is available. Even when some data are available about the risk of harm, judgments about what is “minimal” or “minor” often have a significant subjective component.
The committee concluded that IRBs should be encouraged to develop written rationales (e.g., in IRB minutes) to explain the basis for their judgments about the risks presented by procedures included in a research protocol. Such a practice should help reviewers of research identify more clearly the evidence or other bases for their judgments.
In addition, the committee concluded that useful guidance can be provided to pediatric investigators and reviewers of pediatric research if continued efforts are made to develop consensus identifications of procedures that present minimal risk or no more than a minor increase over minimal risk to children participating in research (NBAC, 1998; Kopelman, 2000).
The NHRPAC work group engaged in such an effort and produced a table in which they categorized a number of common research procedures according to the level of risk presented by a single use of the procedure. That table is reproduced in Table 4.1.
Although NHRPAC has been disbanded, the successor SACHRP should be encouraged to continue its predecessor’s work to develop consensus assessments about the risk of common research procedures. To provide further guidance for investigators and reviewers of research, these consensus assessments should be accompanied by examples, citations of any relevant data, and explanations of the rationales for the categorization of procedures as involving either minimal risk or a minor increase over minimal risk.
The Office for Human Research Protections (OHRP) can also assist
TABLE 4.1 Common Research Procedures by Category of Risk
investigators and IRBs by developing explicit guidance based on the information and recommendations presented in this report and the work of NHRPAC and, eventually, SACHRP. Such official guidance is essential to direct the attention of IRBs to these resources. In addition to providing guidance about the interpretation of the terms discussed in this chapter, OHRP should provide examples of studies and procedures to clarify the limits of minimal risk and a minor increase over minimal risk. The examples should indicate qualifying factors that may affect research risk; for example, the expertise of those performing a procedure and the adequacy of the facilities (e.g., in case of a research emergency). Although FDA has provided much more detailed guidance on many aspects of research conduct and the interpretation of regulations than OHRP, it should work with OHRP on the development of this guidance.
Recommendation 4.5: The Secretary’s Advisory Committee on Human Research Protections (U.S. Department of Health and Human Services) should continue the work of its predecessor committee by developing additional consensus descriptions of procedures or interventions that present minimal risk or no more than a minor increase over minimal risk. In addition, the Office for Human Research Protections and the Food and Drug Administration should cooperate to develop and disseminate guidance and examples for investigators and institutional review boards to clarify important regulatory concepts and definitions (including definitions of minimal risk, minor increase over minimal risk, condition, and prospect of direct benefit).
OTHER ISSUES RELATED TO THE ASSESSMENT OF RISK
In addition to the issues discussed in the preceding sections, other questions about the assessment of research risks warrant consideration. The following discussion considers three questions. First, should determinations about potential harms and benefits be made individually for each research procedure or intervention included in a study, or is it appropriate to judge the research as a whole or as a package? Second, how should research risks be assessed in relation to the anticipated or hypothesized benefits, given other alternatives available to research participants? Third, how should a research protocol be examined to assess whether it minimizes risks to participants.
Assessing Level of Risk by Protocol Components or as a Whole
Most complex clinical research involves several research procedures or interventions. Certain procedures may hold out the prospect of direct benefit to the research participant; others may not. A 2001 report from NBAC
pointed out that all research involves some procedures or methods that are used “solely for the purpose of answering the research question(s)” (NBAC, 2001b, p. 77). Such procedures may be medical (e.g., collection of blood by venipuncture or via a catheter), statistical (e.g., random assignment of subjects to different arms of a clinical trial), or administrative (e.g., review of medical charts).
For example, to gain additional knowledge about an experimental intervention, a research protocol might include an aspiration of bone marrow that would not offer the prospect of direct benefit to the child participant. To be considered for approval, such a “research-only” procedure must present no more than a minor increase over minimal risk. To meet this requirement and also minimize risk, procedural sedation for the aspiration of bone marrow might be restricted to local anesthesia and intravenous medications (e.g., a narcotic and a benzodiazepine). The purpose of the restriction would be to ensure that the level of sedation was moderate, thus preserving protective airway reflexes.
When a research protocol involves multiple procedures, including some without prospect of direct benefit to the research participant, how should the protocol be assessed to determine whether it meets the regulatory criteria for approval? (For discussions related to this question, see, e.g., National Commission, 1977; Freedman, 1987; NBAC, 2001b; Weijer, 2001; and Nelson, 2003.) One answer is that a research protocol should be assessed and approved as a whole—even if some components do not individually meet the criteria for approval—as long as the level of risk overall is reasonable in relation to the anticipated benefit.6
An alternative view—which the committee adopted—is that not only must the research be considered as a whole, but each intervention or procedure must also be assessed independently against the regulatory criteria for approval. Thus, for pediatric studies, the presence of an intervention or procedure that offers the prospect of direct benefit cannot be used to justify the exposure of a child to other procedures that present more than a minor increase over minimal risk but no prospect of direct benefit. Furthermore, as noted earlier, the cumulative risk or burden of a protocol should be assessed. This is important because research may involve several different
|
6 |
For example, in clarifying the analysis that recently led one IRB to refer a protocol to the Secretary of DHHS for consideration, the IRB stated that federal regulations imply that the IRB should make “a risk determination regarding the collective nature of the research procedures” and argued that making “determinations for individual procedures seems of little help in determining an overall risk assessment” (see questions and answers at http://ohrp.osophs.dhhs.gov/panels/407-04pnl/response.htm). |
procedures that may involve minimal risk or burden individually but that may present more than minimal risk when considered collectively.
In the DHHS regulations, Section 405 (concerning approval of studies presenting more than minimal risk and the prospect of direct benefit) and Section 406 (concerning approval of studies presenting more than minimal risk without the prospect of direct benefit) both refer to the risk presented by an intervention or procedure (rather, referring generally to the risk presented by the research). This regulatory language is consistent with a “component” assessment of risk. If elements are evaluated individually, then one component of a protocol might be approved under Section 405 of the regulations, whereas another might either be approved under Sections 404 or 406 or be judged to be not approvable under any of the three primary regulatory categories. (The corresponding FDA regulations are at 21 CFR 50.51 to 53.)
In its 1977 report, the National Commission stated that in assessing the “overall acceptability” of proposed research, “the risk and anticipated benefit of activities described in a protocol must be evaluated individually as well as collectively, as is done in clinical practice” (National Commission, 1977, p. 4). NBAC likewise recommended that “[i]n general, each component of a study should be evaluated separately” and, further, “[p]otential benefits from one component of a study should not be used to justify risks posed by a separate component of a study” (NBAC, 2001b, p. 77). This committee agrees.
Recommendation 4.6: Institutional review boards should assess the potential harms and benefits of each intervention or procedure in a pediatric protocol to determine whether each conforms to the regulatory criteria for approving research involving children. When some procedures present the prospect of direct benefit and others do not, the potential benefits from one component of the research should not be held to offset or justify the risks presented by another.
Assessing Whether Risks Are Reasonable in Relation to Anticipated Benefits: Clinical Trials and Clinical Equipoise
Current regulations provide that a child should not be exposed to more than a minor increase over minimal risk unless the research intervention or procedure offers a prospect of direct benefit. In its report on children, the National Commission argued further that a child should not be disadvantaged by being enrolled in a research study (National Commission, 1977). Two requirements follow from this argument. First, if more than a minor increase over minimal risk is involved, the risk of harm associated with an intervention must be justified by the prospect of direct benefit to the child.
No similar justification is required for research with adults (45 CFR 46.111; 21 CFR 56.111). Second, the balance of anticipated benefits and the risk of harm must be comparable to that for the alternatives available to the child outside of the research. When these other alternatives are taken into account, a child’s health or welfare should not be placed in jeopardy by the decision to enter the child into a research protocol.
The argument of the National Commission bears some resemblance to the concept of “clinical equipoise” (or “research equipoise”), which, in turn, is related to the notion of a null hypothesis (i.e., that there is no difference between two alternatives). The concept of equipoise has emerged as an important criterion in evaluating the ethical acceptability of a clinical trial and determining the appropriate comparison groups—or arms—for a trial (see, e.g., Freedman, 1987; Weijer et al., 2000; and NBAC, 2001b; see also Sackett, 2000 and Shrier, 2001). As originally defined by Freedman, clinical equipoise exists when “there is genuine uncertainty within the expert medical community—not necessarily on the part of the individual investigator—about the preferred treatment” for a condition (Freedman, 1987, p. 141; see also Tri-Council, 1998).
Thus, at the beginning of a clinical trial with an intervention arm and a control arm, no participant would be assigned to receive care known to be inferior to an alternative. Reflecting their assessment of findings from laboratory, animal, or other human research, some experts might view one option as inferior; but both options should be endorsed by “at least a respectable minority of expert practitioners” (Weijer et al., 2000, p. 757) based on “reasoned” uncertainty about the evidence (Mann and Djulbegovic, 2003). The case that such uncertainty exists can be presented by investigators and assessed by reviewers of research protocols.7
Freedman and colleagues (1996), among others, have argued that the principle of clinical equipoise is founded in part on a physician investigator’s therapeutic obligation to patients and that one situation that can violate this obligation involves the use of placebo controls when an effective, active treatment can serve as a control (Rothman and Michels, 1994). A counterview distinguishes the obligation of clinicians to act in an individual patient’s best interest from the obligations of researchers to create knowledge using ethical, scientifically valid methods, which may—under carefully defined
and limited conditions—include a placebo-controlled trial when an effective treatment exists (see, e.g., Emanuel and Miller, 2001 and Miller and Brody, 2002; see also, Ellenberg and Temple, 2000 and Truog et al., 1999).8 One such condition is an especially meticulous informed consent process that carefully explains what a placebo is and makes clear to prospective research participants that they could be assigned to the placebo arm of the trial.
The use of placebo control groups has long been controversial (see, e.g., Ellenberg and Temple, 2000; NBAC, 2001b; and Emanuel and Miller, 2001). This controversy flared in October 2000 when the World Medical Association (WMA) revised the Declaration of Helsinki (WMA, 2002). One revision (paragraph 29) stated that any new preventive, diagnostic, or therapeutic method should be tested against the best current method and that the use of a placebo (or no treatment) control group should be limited to situations in which there is no proven alternative method. After considerable controversy over the revision, WMA qualified its opposition to state (in a footnote) that a placebo control may be ethical if a compelling scientific case supports the use of a placebo control rather than an active control or if the condition being studied is minor and the risk to the group receiving the placebo is minor (WMA, 2002).
In 2000, the International Conference on Harmonisation adopted a guideline that would allow a placebo control when there is no serious harm of withholding effective therapy (ICH, 2000a; see also FDA, 2001d). A placebo-controlled could also be ethical when the proven effective treatment has such severe toxicity that many patients would refuse treatment or when comparison with an active control treatment would not yield scientifically valid results. Similarly, the recent revision of the Guidelines of the Council of International Medical Organizations stated that placebo controls can be ethical (1) when there is no established effective intervention, (2) when withholding and established effective intervention would result in only temporary discomfort or a delay in the relief of symptoms, and (3) when an active control trial would not yield scientifically reliable results
|
8 |
FDA is perceived by many as conditioning the approval of drugs, in most instances, on the provision of information from placebo-controlled trials (see, e.g., Cowdry, 1997). FDA rules and guidelines do not explicitly require such trials. FDA policies require “adequate and well-controlled studies” and identify five types of studies that may be acceptable under some circumstances: placebo concurrent control, dose-comparison concurrent control (in which at least two different doses of the same drug are compared), no treatment concurrent control, active treatment concurrent control, and historical control (21 CFR 314.26(b)(2)). In responding to comments on the so-called pediatric rule (see Chapter 2), FDA stated that alternatives to placebo-controlled trials should be used if such trials can provide adequate information about the effectiveness of a therapy (FDA, 1998e). |
and the use of placebo would not pose any risk of serious or irreversible harm to the research participants (CIOMS, 2002).
The Committee on Drugs of the American Academy of Pediatrics (AAP) also has a statement on the use of placebo controls. Such use should be limited to situations
-
when there is no commonly accepted therapy for the condition and the agent under study is the first one that may modify the course of the disease process;
-
when the commonly used therapy for the condition is of questionable efficacy;
-
when the commonly used therapy for the condition carries with it a high frequency of undesirable side effects and the risks may be significantly greater than the benefits;
-
when the placebo is used to identify incidence and severity of undesirable side effects produced by adding a new treatment to an established regimen; or
-
when the disease process is characterized by frequent, spontaneous exacerbations and remissions and the efficacy of the therapy has not been demonstrated (AAP, 1995, p. 294).
The AAP statement does not mention the withholding of commonly accepted therapy when that would only result in minor discomfort. Such research would likely be approvable under federal regulations.
Whatever the criteria outlined by national and international organizations, they may still not justify the inclusion of children in a placebocontrolled trial. For research involving children and a placebo control group to be approved by an IRB under federal regulations, either (1) the balance of potential harms and benefits for children in the placebo control arm must be as favorable as those for children receiving the active, standard treatment or (2) the potential harms to which children in the placebo control arm would be exposed are no more than minimal or involve only a minor increase over minimal risk. Although ethical and regulatory principles allow adults to give their informed consent to participate in research involving more risk, they do not allow parents or children to agree to accept such research risk for a child.
Other requirements also apply. As for all research, risks must be minimized (see further discussion below). In addition, the creation of a data and safety monitoring board is typically required for clinical trials (see Chapter 3).
For a placebo-controlled trial, the process of requesting parental permission should make clear that the proposed research could involve foregoing or delaying a known effective therapy and that the child could be
assigned to either a placebo-control group (with no benefit expected) or a group receiving an experimental intervention (with a prospect of benefit). (The same kind of explanation is also necessary when active treatment control groups are employed.) Parents should also be informed if children in the placebo-control group will eventually “cross over” to the experimental intervention. They should likewise be provided clear explanations about why a placebo arm is necessary to answer the research question and what measures that will be taken to ensure the child’s safety and well-being. As discussed in the next chapter, studies indicate that research participants and parents of research participants may not understand these and other aspects of research.
Given the controversy over placebo-controlled trials, more published data are needed on health outcomes for research participants receiving placebos. A common misperception among patients (and even some clinicians) is that research participants assigned to placebo arms are necessarily at greater risk than those assigned to treatment arms. Particularly in early-phase studies, however, the participants in placebo arms may have fewer adverse events. As discussed above, if the condition of research equipoise is indeed met, then no research participants should be exposed to treatment (including administration of a placebo) known to be inferior to an alternative.
Assessing Whether Risk Is Minimized
One ethical and regulatory responsibility of investigators is to minimize the risk that research presents to participants. In assessing whether risks are being minimized, attention often focuses on the risk presented by the procedures or interventions that are being tested for safety or efficacy. Investigators and reviewers must, however, also consider whether risks are minimized for interventions or procedures intended solely to collect information (e.g., blood draws and lumbar punctures). One advantage of separately evaluating each intervention or procedure in a research protocol, as recommended above, is that it encourages attention to risk minimization for all the procedures.
As observed in Chapter 1, poorly designed research will usually fail to answer the research question. One example is research that is designed without adequate attention to the sample size needed to detect a meaningful difference between an experimental intervention and a placebo or control treatment. At a minimum, such research wastes the time of research participants. Depending on its particular faults, poorly designed research can also expose research participants to avoidable harm and can dissipate potential benefits. An important emphasis of specialized education for clinical re-
searchers is techniques of modern research design and data analysis that help minimize the exposure of research participants to such avoidable risks.
Depending on the procedures or interventions involved in a research protocol and the characteristics of the research population, investigators and reviewers of research may consider several questions in determining whether the protocol proposes appropriate steps to minimize risk. Box 4.2 lists a number of these questions. Some—for example, whether the research design is sound—apply to any research, whereas others—for example, whether the research setting is equipped to meet children’s developmental requirements—focus on particular concerns in studies that include infants, children, or adolescents.
The guidelines listed in Box 4.2 include several items related to the qualifications of the research team and the characteristics of the research site that may affect the risk associated with a particular clinical study. The creation of pediatrics as a specialty and the founding of children’s hospitals as institutions were motivated, in part, by perceptions that excellent physical and psychosocial care for children requires specialization. Similarly, the creation of regional pediatric trauma and neonatal intensive care centers reflects a judgment that the concentration of care for patients with complex conditions in designated units will improve outcomes.
Many interventions or procedures would clearly involve unacceptable risks if they were undertaken by generalist physicians in ordinary community hospitals. Even within pediatric centers, research involving high-risk procedures, for example, gross surgical resections and stem cell transplants may involve higher risks if they are performed by staff and institutions that are less experienced with those highly specialized procedures or the management of their side effects in children with specific cancers. Thus, reviewers of research protocols may consider the relevance of specialization in facilities and personnel when making their determinations about risk. In addition, the IRB may determine that information about staff and institution experience should be included in discussions with parents about a child’s research participation.
IRB members may sometimes find it appropriate to examine specific data about the performance of the investigators and the research setting to help them assess the level of risk posed by a research procedure and the extent to which risks have been minimized. Most data available to IRBs are likely to involve serious adverse outcomes (e.g., death, disability, hospital admission or extension of the hospital stay, or the need for a “rescue” procedure). No data may be available to judge outcomes such as pain, fear, or other forms of distress. Ideally, comparative data could be used to assess performance history.
|
BOX 4.2
|
CONCLUSION
Under federal regulations, some research that would be approvable for adults involves more risk than is allowable for children. The regulations recognize children’s greater vulnerability and need for protection. Unfortunately, the concepts of risk on which decisions about approvable research hinge have significant subjective elements. The same applies to several other key concepts in the regulations. Reasonable people may disagree when interpreting these concepts, especially when little or no evidence is available to inform judgments. Nonetheless, if investigators and IRB members apply the definitions presented here, judgments about similar protocols should be more consistent. They should also conform more to the ethical principles underlying the regulations.
In addition, the consistency and quality of evaluations can be improved if investigators and IRB members pay more systematic attention to all the requirements (not just the provisions related to risk) included in the four sections of the DHHS and FDA regulations that set criteria for permissible research involving children. Applications for IRB approval and IRB records should include rationales related to each of the key concepts. The last chapter of this report recommends that IRBs and federal agencies provide clear, easily located guidance that will help both investigators and IRB members understand and fulfill their responsibilities.

































