
3
Obstacles and Opportunities for Framing Future Research
OVERVIEW
Humans exist in complex milieus, and their association with disease is affected both by the environment in which they live and by their genetic susceptibility to particular diseases. People live in concert with microbial agents that may or may not cause disease in particular individuals, depending on their environment and their genetics.
There are substantial obstacles to identifying organisms associated with a particular chronic disease. First, organisms can act in a “hit and run” manner, in which they cause disease initially but then are either resolved due to natural immunity or are successfully eliminated with antibiotics. The damage has been done, however, resulting in chronic disease. For some chronic diseases of this type, such as Reiter’s syndrome, Guillain-Barré syndrome, or rheumatic heart disease, it is very difficult to find a fingerprint of the organism in the disease tissue. Second, organisms can be latent at the time of diagnosis. They may not be actively replicating, so there is no active RNA transcription. Third, chronic latent or recurrent infection may be involved in the pathogenesis so that, again, the organism may not be active at the time of diagnosis. Fourth, organisms may need a particular predisposing environment or a host with a particular genetic susceptibility, so the simple presence or absence of the organism may be misleading.
To address these problems, evidence is assembled in a number of ways: from epidemiological studies, from microbiological assessment of pathogenesis and etiology, from studies that mimic the disease process in vitro or in animals, and from clinical treatment trials.
Patrick Moore described how the discovery of new pathogens will require the talents of multiple disciplines, including epidemiology, clinical medicine, molecular biology, and pathology. Moore used the example of the identification of the virus that causes Kaposi’s sarcoma, which often strikes gay men, to discuss general issues in causality and to illustrate the limits of current approaches for determining causality for a newly discovered agent and disease. The causative agent, named Kaposi’s sarcoma-associated herpesvirus (KSHV), was identified using a genetic technique called representational difference analysis. Once the virus was identified, in 1994, events moved rather quickly—a fact that speaks to the importance of new pathogen discovery. The virus’s genome has been sequenced, serologic tests have been developed, and studies have been initiated to understand its epidemiology and to test possible treatments. Moreover, the virus has since been found to cause at least two other types of disease. Based on this experience, Moore pointed to the need for researchers to move beyond Koch’s postulates or other traditional guidelines in their efforts to determine disease causality for suspect microbes. Researchers are attempting to do this by applying various new techniques emerging from molecular biology and biotechnology. It seems clear as well that epidemiologists developing new criteria for causality will have to incorporate new pathogenic mechanisms that are not accounted for in current disease models.
Mikhail Pletnikov discussed the importance of expanding research to better understand the interplay of genetic and environmental factors in the causation of a number of important developmental behavioral disorders. Among methodological problems of studying the gene-environment interplay is the difficulty in firmly defining environmental factors and making them quantifiable. In this context, virus infections provide a promising research avenue, because of their etiologic connection to several neurodevelopmental disorders, including autism and schizophrenia, and because of the reliability of quantification of viral effects on brain and behavior. In particular, Pletnikov described work using an animal model to study gene-environmental interactions that occur during neonatal infection with Borna disease virus (BDV). Neonatal BDV infection in rats has been shown to produce distinct neuroanatomical, neurochemical, and behavioral abnormalities that resemble pathological and clinical features of some human developmental disorders. The significance of studying neonatal exposure derives from the fact that the effects of many genetic and environmental risk factors are evident either prior to or around the time of birth, and the interaction between them often is apparent well before the onset or diagnosis of the chronic disease condition. Thus, studying the effects of neonatal BDV infection across the entire postnatal period in genetically different strains of rats will aid in understanding the course and time-dependent character of the interaction of genetic background features and the virus infection. In this way, the model system may allow study of some tremendously complex mechanisms relevant to developmental disorders.
David Persing provided an overview of recent research in the area of infec-
tion, cancer, and the immune response. Current evidence suggests that inherited predisposition to cancer probably accounts for only a subset of total cancer patients, and in most models of the development of neoplasia, an underlying assumption is the contribution of an array of intrinsic and extrinsic factors within a multistep process. A basic prerequisite of many models is an increase in the baseline proliferation rates of essentially normal cell populations that leads to dysregulation of normal growth control mechanisms. Since many infectious processes often lead, directly or indirectly, to increased cell turnover and proliferation, certain agents are now widely regarded as carcinogens. Some of the pathogens that have been linked to cancer include human papillomaviruses (cervical cancer and other skin cancers), human T-cell leukemia viruses (adult T-cell leukemias and lymphomas in endemic areas), hepatitis B virus (liver cancer), Epstein-Barr virus (Burkitt’s lymphoma and nasopharyngeal carcinoma), and Helicobacter pylori infection (gastric carcinoma and MALT lymphoma).In addition, new disease associations are being made with respect to previously known pathogens, such as the association of chronic hepatitis C virus infection with non-Hodgkin’s lymphoma in certain populations.
In a separate presentation, Persing described recent and continuing advances in the development and application of techniques for identifying pathogens that cause chronic diseases. Although a paper on these subjects does not appear in this chapter, the following paragraph notes the highlights.
The ability to detect and manipulate nucleic acid molecules in microorganisms has created a powerful means for identifying previously unknown microbial pathogens and for studying the host-pathogen relationship. Although a paper on these subjects does not appear in the ensuing text, the highlights of his presentation are discussed here. Among the new technologies that Persing described is broad-range polymerase chain reaction, which has proved instrumental in linking a growing number of pathogens with chronic diseases, and representational difference analysis, which is an efficient means for finding differences between complex genomes and for identifying specific DNA sequences from the genomes of unknown pathogens. Researchers also are making use of sophisticated new DNA microarrays and biosensors that, among other things, can monitor host response as an indicator of the presence of infection or inflammation. In addition, new methods for generating “libraries” of genetic information from very small amounts of material are making it easier to conduct very specific and sensitive serologic tests. Equipped with these and other advanced tools, researchers are becoming better able to move beyond the limitations of Koch’s postulates and to link infectious agents with chronic diseases more precisely and with greater confidence than ever before.
In the ensuing discussions, participants began to sketch in some of the characteristics of a comprehensive and coordinated effort that would enhance efforts both to identify links between infectious microorganisms and chronic diseases and to develop and implement interventions to minimize their health conse-
quences. The goal was not to set specific priorities, but to identify opportunities. Highlighting a selection of the traits identified may provide a glimpse of the overall picture envisioned.
Participants agreed, for example, on the need to develop standardized definitions of infections and disease, to enable comparisons across studies and conclusions about causality, and on the need to ensure that laboratory assays maintain universally high standards of specificity, sensitivity, and reproducibility. New laboratory technology also is needed that can meet such performance standards while handling high throughput rates, in order to handle analyses of large cohorts in a reasonable amount of time, a trait that likely will be required in many future projects. Comparable efforts are needed to ensure that epidemiological studies are conducted with vigor and in an appropriate manner. One step will involve linking of databases that are designed (or modified) to be compatible. Peer review journals can reinforce performance standards if publication depends on the use of sound laboratory assays and epidemiologic design capable of supporting the conclusions.
Continued studies are needed to define temporal relationships between infections and disease—that is, what stage of infection determines outcome (e.g., first infection, reinfection, persistent infection, coinfection, or subsequent cross-reacting infection). Studies also are needed to clarify at which stage infection must be prevented or treated in order to minimize or eliminate chronic sequelae. It will be important to determine the expected benefit of actions, to ensure that the benefits will outweigh any possible risks. In other words, intervention should decrease chronic disease burden without unduly endangering the people who receive care.
There is a need to better understand the natural history, especially the earliest stages, of chronic diseases of unknown or incompletely known origins. What makes this task especially important is the hit-and-run nature of some diseases in which microbes set adverse events in motion and then disappear, the increased difficulty of imputing causation to microbes detected late in the course of disease, and the increased ease of treating or preventing disease at early time points. Toward this aim, clinicians should be increasingly encouraged to identify patients who have recently developed or seem to be developing various suspect chronic diseases, to collect in an orderly manner a range of clinical specimens, and then to follow the course of the disease in order to identify tell-tale early clinical features.
Calls were made for more effort devoted to developing animal models of chronic diseases, and to teaching health professionals about their value and their limitations. Animal models can be powerful tools when the etiology or pathogenesis of a disorder is unknown. Psychiatric modeling with animals may present an especially ripe area for probing a variety of important questions, yet many practitioners in the field are not accustomed to working with such models.
Increased emphasis should be placed on longitudinal studies, as well as on follow-up studies and “look back” studies of cohorts and surveillance results that have been generated in the past. Longitudinal studies may prove particularly valu-
able given that rapid advances in the field may mean that we might not know today which pieces of evidence will be needed in the future. Human specimen collections, such as the National Children’s Study that will begin in 2004, may be especially important for longitudinal research.
Participants identified a number of specific populations that should receive additional attention. One such group includes people who move from rural areas into cities, both in the developing and the developed world. Studies are needed to see whether they bring new infections with them, or whether they prove to be susceptible to new infections that they previously had not encountered. With the world’s changing demographics, gathering such information may provide a window into pathogenesis of a number of chronic diseases.
Efforts are needed to address problems related to informed consent. Many workshop participants expressed concern that current regulations and guidelines are too complex, too uncertain, or too restrictive to allow for meaningful sharing of data—and sometimes all three. There was general agreement that informed consent is and must remain an important part of research involving human subjects. But participants also agreed that all parties—from government, academia, and private funding agencies—need to work together to develop a more standardized method for gaining patient consent, for gathering identifying information, and for being able to use this identifying information in the future. This may be an opportunity for multiple institutions and multiple governments, domestic and foreign, to cooperate in devising a system of patient consent that operates more smoothly, protects patient rights, and allows for expanded research on infections and chronic diseases.
Given the magnitude of the outstanding scientific questions, and of the health consequences at stake, an increasing share of future research likely will involve groups of investigators representing a variety of disciplines, or groups of institutions working collaboratively. Although there remains a clear role for individual investigators, it is becoming apparent that large multidisciplinary projects often can best marshal the critical mass needed to address the thorniest biological problems. In many cases, these large projects will include a multinational component, in order to ensure that sufficient attention is paid to multiracial, multiethnic, and multicultural differences.
Participants called on the overall scientific community to evaluate whether it is organized and structured properly to address these issues, and whether its various components communicate effectively. The community also should mount a concerted effort to identify gaps in current knowledge about the etiology of chronic diseases, pinpoint what needs to be done to close those gaps, chart the obstacles that stand in the way, and then identify and provide the necessary financial resources (monetary and human) to drive progress.
Government can play an important role by reorienting its funding priorities. Indeed, the time is ripe. The government is now investing nearly $1 billion in rebuilding the nation’s public health system, and part of the money will go to-
ward linking state health departments more closely with local health departments than has historically been the case. At the same time, government research centers are launching major new interdisciplinary projects, and universities, which often have been in competition with one another, are beginning to join in collaborations. Thus, foundations are beginning to be built for bridges linking public health, clinical medicine, and research. But these promising efforts need to be nurtured to ensure continued cooperation.
PATHOGENS AND DISEASE: ISSUES IN DETERMINING CAUSALITY
Patrick S. Moore
University of Pittsburgh
Pittsburgh, PA
Successful new pathogen discovery requires the talents of multiple disciplines, including epidemiology, clinical medicine, molecular biology, and pathology. In this paper, the identification of Kaposi’s sarcoma-associated herpesvirus (KSHV) illustrates general issues in causality and shows the limits on our ability to determine a causality for a newly discovered agent and disease (Moore and Chang, 1998).
The mysterious outbreak of Kaposi’s sarcoma among gay men was the harbinger of the AIDS epidemic. It is now clear, however, that the AIDS-associated Kaposi’s sarcoma epidemic was actually due to the collision of two independent viruses in a susceptible population: HIV and a new virus, KSHV or HHV8, which was found using molecular techniques (O’Brien et al., 1999).
Over 20 different agents had been put forward as the cause of KS before 1994. To look for the “KS agent” (Beral et al., 1990), Yuan Chang used representational difference analysis (RDA) to compare DNA from a Kaposi’s sarcoma lesion to uninvolved, sterile-site tissue from the same patient on the assumption that the two samples would be genetically identical except for the presence of the putative agent’s genome (Chang et al., 1994).
As shown in Figure 3-1, RDA is a subtractive hybridization technique in which PCR adapters are ligated onto digested DNA from the KS tissue (tester) (Lisitsyn et al., 1993). The tester DNA was then rehybridized back to a ten-fold excess of uninvolved sample DNA (driver) that had been identically digested. For human genomic fragments present in the KS sample, 90 percent of these fragments will form pairs with the corresponding antisense strand lacking the adapter from the normal tissue DNA. PCR with a primer specific to this adapter linearly amplifies these common sequences and, of course, there was no amplification of the DNA rehybridized from the driver alone. Sequences that were unique to the KS lesion, however, reanneal to each other and have adaptors on both ends, so that amplification occurs exponentially. The initial PCR products are then
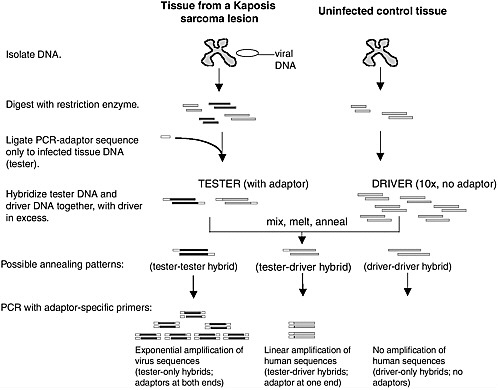
rehybridized again to the adapter-less healthy tissue DNA and the process is repeated, each time selectively enriching for the unique sequences found only in the KS lesion (Gao and Moore, 1996).
Four RDA fragments were generated by this process, two of which were found to be specific for the KS agent. Although these two fragments account for less than 1 percent of the entire 145-kilobase viral genome, the few base-pairs worth of unique information they provided made it possible to develop enough tools to identify the agent.
The two fragments were used as Southern hybridization probes and tested against KS lesions, showing that about three-quarters of the KS lesions were positive for viral DNA. Using internal specific primers from the KS 330 band, a PCR assay was developed that showed 25 out of 27, or 93 percent, of the initial KS lesions tested positive. Moreover, the negative samples were equally telling: one of the two negatives had degraded DNA and was not amplifiable by using cellular primers, and the other one was mislabeled normal human kidney.
KSHV is a gamma herpesvirus belonging to the same class as Epstein-Barr virus (EBV). It is associated with three different major proliferative diseases: Kaposi’s sarcoma, primary effusion lymphoma (PEL) (Cesarman et al., 1995a), a monoclonal B cell lymphoma, and multicentric Castleman’s disease (Soulier et al., 1995), which is a polyclonal hyperplasia caused by a virus-encoded cytokine expressed by KSHV (Parravicini et al., 1997). Nearly all KS and PEL patients have KSHV infection, but only about half of HIV-negative, multicentric Castleman’s disease patients are positive for KSHV infection indicating that this disease has a heterogeneous pathogenesis.
The two aforementioned RDA fragments of the KSHV genome facilitated the identification of infected cell lines to serve as source material for viral DNA and as a reagent for biologic studies (Cesarman et al., 1995b). Genomic library walking was performed using cosmid and lambda libraries from one of these cell lines allowing sequencing of the remainder of the genome (Russo et al., 1996). Using this information, various techniques were used to identify likely antigens and generate serologic tests (Gao et al., 1996a,b; Kedes et al., 1996; Simpson et al., 1996). While identification of high-titered infected cell lines sped up this process, isolating the agent was not essential for developing tools to detect it. Molecular biology has reached the point where it is straightforward to identify a new agent, sequence its genome and develop serologic tests for it without ever having actually purified, living sample of the agent. The virus does not have to be grown in order to apply traditional techniques for determining whether or not an agent is present.
The virus itself is a tremendously interesting scientific problem. It has a long unique coding region containing all of the viral open reading frames. Unlike most viruses, KSHV has pirated cellular genes over its evolution and the viral genes are recognizable homologues to cellular genes of known function. Many of these
genes provide new insights into tumor virology through their control of the cell cycle, prevention of apoptosis, or immune evasion properties.
One might conclude that this virus is completely different from other viruses and not much can be learned from it to extend to other viruses. In fact, the opposite is true. EBV, for example, induces cellular cyclin D2 to drive the cell through the G1/S cell cycle checkpoint; KSHV encodes its own version of a cyclin D with an analogous function. Other examples of functional correspondence between the KSHV homologues and viral genes encoded by even distantly related viruses can be readily seen (Moore and Chang, 1998a, 2001). For this reason KSHV might be considered something like a molecular Rosetta stone because by using it, we can begin to interpret the language of molecular virology in terms of cell biology for many different viruses.
The importance of new pathogen discovery is illustrated by a timeline of KS research. This would be equally true also for hepatocellular carcinoma and hepatitis C or a wide range of diseases where a new pathogen has been found. The point is that things change quite rapidly once the agent is finally found. Moritz Kaposi initially described the disorder in 1873, but not until 70 years later was there a suggestion of an infectious etiology. In 1981, the onset of the AIDS epidemic brought a tremendous increase in scientific interest in this cancer. There was still, however, little known about the pathogenesis of this disorder in 1993 when there were over 200,000 cases of AIDS in the United States. At that time over 20 different agents had been proposed at one time or another as the causal agent for Kaposi’s sarcoma.
The description of KSHV was first published in 1994 and within two years its viral genome was completely sequenced (Neipel et al., 1997; Russo et al., 1996). By that time it was known that the virus was found in all forms of Kaposi’s sarcoma (Boshoff et al., 1995; Chang et al., 1996; Moore and Chang, 1995), serologic tests had been developed (Gao et al., 1996a,b; Kedes et al., 1996; Miller et al., 1996; Simpson et al., 1996) and studies initiated to understand the epidemiology of this virus in KS (Moore et al., 1996; Whitby et al., 1995). Shortly thereafter, studies were performed to see whether ganciclovir, a specific antiviral agent, could be used to treat KS (Martin et al., 1999). At the present, there have been over 2,000 papers published on KSHV and its role in malignancy.
Finding a new pathogen also can benefit other fields. When KSHV was first described, only two other related rhadinoviruses had been described in new world primates. Although we live in North America, humans are still considered old world apes. One was herpesvirus saimiri from squirrel monkeys and the other herpesvirus ateles from spider monkeys. Researchers at the University of Washington began to look for other primate KSHV-like viruses (Rose et al., 1997). Using consensus PCR, two were found in rhesus macaques, and subsequently in all the various branches of the primates, both lower and higher primates. This suggests that the viral ancestor of KSHV evolved with us over time. Even more
interesting, another group found a closely-related but distinctly different virus in rhesus macaques (Desrosiers et al., 1997). It was named rhesus rhadinovirus (RRV) and belongs to a second lineage of rhadinoviruses. RRV was initially only found in the lower primates, but last year an RRV was found in chimps (Greensill et al., 2000; Lacoste et al., 2000; Lacoste et al., 2001). The implication is that there is an ancestral KSHV/RRV-like virus split off in the primate evolution and has followed through with the different primate lineages, probably including humans. Thus, it is almost certain that there is an undiscovered HHV 9.
An issue in new pathogen discovery is making the step from finding a new DNA sequence to determining whether or not it causes a specific disease. Applying Koch’s postulates (Koch, 1942) can elucidate the process:
-
The agent occurs in every case of disease.
-
The agent never occurs as a fortuitous or non-pathogenic strain.
-
The agent can be isolated from the lesion, grown in pure culture, induce disease in a susceptible host and can be re-isolated from an infected susceptible host.
These were postulates that Koch developed for determining the cause of tuberculosis at a time when not much was known of viruses or the carrier state. This was a brilliant attempt to develop a scientific rationale for determining whether an agent is causal for disease or not.
Bradford Hill also developed epidemiologic criteria for causality which are shown here for KSHV and KS (Hill, 1965). Though developed specifically for cigarette smoking, most epidemiologists now use these criteria to determine causality:
-
Is the infection present in cases; do all types of the disease involve infection? Is it reproducible in multiple settings?
-
Does infection precede disease?
-
Is the infection specific to the disease or is it ubiquitous infection among humans?
-
Is the virus localized to the tumor (one interpretation of a biologic gradient)?
-
Do the epidemiologic studies make sense (are they coherent?)?
-
Is it biologically reasonable and do experiments confirm the relationship?
With regard to KSHV, the answers to these questions are largely true. KSHV is present in more than 95 percent of KS lesions. It can be said that the remaining negative 5 percent is probably spurious due to technical difficulties in detection or misdiagnosis, and in fact the virus is absolutely necessary for disease. Though this cannot be proven at present, it can be argued that the situation is very similar to that of papillomavirus and cervical cancer 5 years ago. Is it generalizable? Yes. All types of KS are infected as far as is known. It also appears to have the correct
temporal association in that cohort studies show that patients are infected before developing disease, and not afterwards.
But specificity is an important question. KSHV is not singly associated with Kaposi’s sarcoma. It is also associated with two other diseases. However, the epidemiology of these two diseases makes some sense in terms of Kaposi’s sarcoma, so multiple outcomes are not too worrisome. Depending on the assay that is used, some researchers suggest that the infection rate in the general population for this virus is much higher than alluded to here, but careful studies suggest that less that 5 percent of Americans are infected with KSHV.
Is there a biologic gradient? Yes, there is. Are the epidemiologic findings coherent? Yes, a wide range of epidemiologic studies seem to come to exactly the same set of conclusions. Is it biologically plausible? Yes, there are multiple oncogenes in this virus, related viruses cause cancers, and there are blinded clinical trials which seem to suggest that treatment with ganciclovir prevents the development of Kaposi’s sarcoma.
KSHV and KS was a relatively easy case even though it took two years and seven or eight different studies before these conclusions could be reached. Nonetheless, the case for causality was relatively easy.
Now let’s consider issues where causality is more problematic. First, KSHV has been claimed not only to cause Kaposi’s sarcoma, but also a wide variety of diseases that don’t fit its epidemiologic pattern, such as multiple myeloma and sarcoidosis. Although studies supporting these associations were published in reputable journals, they were based on PCR or had other problems and remain questionable in terms of contemporary epidemiological knowledge. In the age of PCR, it is difficult for the casual observer to sort out what is true and what is not.
Assuming that the problem of poor laboratory technique can be solved, there are three more fundamental problems in determining causality. First, causality is relative and should not be thought of as being cast in stone. Causality depends on pathogenic assumptions. That is where Koch’s postulates fall down and also where Hill’s criteria fall down as well.
For example, if a virus is associated with autoimmune disorders, it can be assumed that one would have an immune response against that virus. In that case the individual may actually clear the virus, so a reverse association would be seen from what would normally be expected following Hill’s criteria. The criteria simply do not apply in this case, even though it is a reasonable possibility.
Second, causality is normative. Researchers can get together and study the data but only a few agree to particular conclusions. When the studies describing KSHV as the cause of KS were completed, it was thought that the issue of causality would be resolved. However, it still required a great deal of interpretation. There were many contradictory studies that were ignored because they were not considered valid. But others might disagree, and this is true for just about any contentious issue. An agent is only considered causal for a disease when a major-
ity of scientists agree and it is passed on as received wisdom to others. By then, few scientists probably know the actual studies that were actually used to determine causality.
Third, no agent causes disease alone. There are some fairly convincing examples, HIV as well as rabies virus. Simply not enough is understood about the epidemiology of rabies virus to know whether or not there are people who have been exposed to it who have not developed disease. But with the possible exception of those two viruses, virtually every other infection can have a symptomatic infection, and disease is determined by other factors than the virus alone.
There are several examples where current methods of determining causality break down. One is the role of EBV in nasopharyngeal carcinoma (NPC). There is extremely strong evidence that EBV is the cause of nasopharyngeal carcinoma, but EBV is a near ubiquitous infection. So Hill’s criteria cannot be used to determine that EBV causes nasopharyngeal carcinoma since it is likely to be a composite risk factor and additional causal factors have to be used in conjunction with EBV infection. These factors are unknown for NPC, but it is easy to see that rather than using EBV infection alone as an exposure variable, it may be more valuable to measure exposure as EBV infection at a certain susceptible age or EBV infection in a cell having a specific mutation.
EBV and NPC shows another problem with Hill’s criteria for causality. RaabTraub developed an assay for EBV terminal repeat monoclonality and using this assay found that in NPC tumors, the precursor cell forming the monoclonal tumor was infected with a monoclonal form of the virus (Raab-Traub and Flynn, 1986). For molecular biologists and virologists, this is overwhelming proof that the virus causes the tumor since the odds of this happening by chance are so small. It is extremely unlikely that a healthy cell was first infected with the virus, and by chance the same cell independently became transformed into an expanding tumor cell population with EBV growing inside of it as a passenger virus. However, this does not neatly fit Hill’s criteria and epidemiologists have no way of weighing the importance of this “overwhelming” piece of molecular evidence, especially in comparison to contradicting evidence such as the ubiquity of EBV infection among humans.
Multicentric Castleman’s disease (MCD) illustrates additional problems related to determining causality for different diseases which look identical. About half of MCD tumors are positive for KSHV and so it seems that there are actually two diseases, not just one, under the label of MCD. KSHV is only considered necessary for the KSHV positive form. Now that it is known what to look for, there may be subtle clinical and pathologic clues that can distinguish the two forms of MCD. Analogous situations can be drawn for hepatocellular carcinoma and hepatitis virus C positive hepatocellular carcinoma, or for meningitis and any of the many causes of meningitis. While hepatitis virus C is not necessary for all liver cancers, it is obviously necessary for hepatitis virus C positive tumors. By defining a subset of diseases associated with a viral infection post-hoc, the cau-
sality argument becomes circular even though we now have good reasons for splitting up a disease manifestation into different diseases with different manifestations.
It is likely that in the future, improved knowledge of pathogenic mechanisms will reveal novel causal relationships. For example, not too long ago the idea that a bacteria could cause stomach ulcers would have been considered laughable. Helicobacter pylori and peptic ulcer disease had a pathogenic mechanism that was poorly understood and thus there was no framework to gauge whether or not a bacteria was the possible cause. In fact, pathologists had seen bacteria associated with ulcers for decades but didn’t remark on them because there was no way to measure their significance.
New ways of determining causality that go beyond Hill’s criteria and Koch’s postulates need to be developed if new and complex mechanisms for disease are to be understood. Researchers have attempted to do this by taking into consideration new techniques of molecular biology (Fredericks and Relman, 1996). It seems clear that epidemiologists developing new criteria for causality will have to incorporate new pathogenic mechanisms that are not currently accounted for.
Unfortunately, no one can predict what new pathogenic mechanisms will be discovered and therefore there are no universal criteria for causality that will not need future revisions. In the end, it cannot be absolutely proved that an agent causes disease, only that it does not. Instead, while criteria such as Hill’s or Koch’s postulates are enormously helpful in guiding our thinking, we should not be constrained by them as has happened in cases like EBV and nasopharyngeal carcinoma. In this case, both science and public health have suffered from rigid adherence to abstract criteria. For cases where established criteria break down, all that can be done is to develop a detailed pathogenic model which can be tested using epidemiologic studies and further modified. In essence, to use the scientific method which is employed by scientists every day.
REFERENCES
Beral V, Peterman TA, Berkelman RL, Jaffe HW. 1990. Kaposi’s sarcoma among persons with AIDS: a sexually transmitted infection? Lancet 335:123–128.
Boshoff C, Whitby D, Hatziioannou T, Fisher C, van der Walt J, Hatzakis A, Weiss R, Schulz T. 1995. Kaposi’s sarcoma-associated herpesvirus in HIV-negative Kaposi’s sarcoma. Lancet 345:1043–1044
Cesarman E, Chang Y, Moore PS, Said JW, Knowles DM. 1995a. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. New England Journal of Medicine 332:1186–1191.
Cesarman E, Moore PS, Rao PH, Inghirami G, Knowles DM, Chang Y. 1995b. In vitro establishment and characterization of two acquired immunodeficiency syndrome-related lymphoma cell lines (BC-1 and BC-2) containing Kaposi’s sarcoma-associated herpesvirus-like (KSHV) DNA sequences. Blood 86:2708–2714.
Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, Moore PS. 1994. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 265:1865–1869.
Chang Y, Ziegler JL, Wabinga H, Katongole-Mbidde E, Boshoff C, Schulz T, Whitby D, Maddalena D, Jaffe HW, Weiss RA, Moore PS. 1996. Kaposi’s sarcoma-associated herpesvirus and Kaposi’s sarcoma in Africa. Archives of Internal Medicine 156:202–204.
Desrosiers RC, Sasseville VG, Czajak SC, Zhang X, Mansfield KG, Kaur A, Johnson RP, Lackner AA, Jung JU. 1997. A herpesvirus of rhesus monkeys related to the human Kaposi sarcomaassociated herpesvirus. Journal of Virology 71:9764–9769.
Fredericks DN and Relman DA. 1996. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clinical Microbiology Reviews 9:18–33.
Gao SJ and Moore PS. 1996. Molecular approaches to the identification of unculturable infectious agents. Emerging Infectious Diseases 2:159–167.
Gao SJ, Kingsley L, Hoover DR, Spira TJ, Rinaldo CR, Saah A, Phair J, Detels R, Parry P, Chang Y, Moore PS. 1996a. Seroconversion to antibodies against Kaposi’s sarcoma-associated herpesvirus-related latent nuclear antigens before the development of Kaposi’s sarcoma. New England Journal of Medicine 335:233–241.
Gao SJ, Kingsley L, Li M, Zheng W, Parravicini C, Ziegler J, Newton R, Rinaldo CR, Saah A, Phair J, Detels R, Chang Y, Moore PS. 1996b. KSHV antibodies among Americans, Italians and Ugandans with and without Kaposi’s sarcoma. Nature Medicine 2:925–928.
Greensill J, Sheldon JA, Murthy KK, Bessonette JS, Beer BE, Schulz TF. 2000. A chimpanzee rhadinovirus sequence related to Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8: increased detection after HIV-1 infection in the absence of disease. AIDS 14:F129–135.
Hill AB. 1965. Environment and disease: association or causation? Proceedings of the Royal Society of Medicine 58:295–300.
Kedes DH, Operskalski E, Busch M, Kohn R, Flood J, Ganem D. 1996. The seroepidemiology of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus): distribution of infection in KS risk groups and evidence for sexual transmission. Nature Medicine 2:918–924.
Koch R. 1942. The aetiology of tuberculosis (translation of Die Aetiologie der Tuberculose [1882]). Pp. 392–406 in Source Book of Medical History, DH Clark, ed. New York: Dover Publications, Inc.
Lacoste V, Mauclere P, Dubreuil G, Lewis J, Georges-Courbot MC, Gessain A. 2000. KSHV-like herpesviruses in chimps and gorillas. Nature 407:151–152.
Lacoste V, Mauclere P, Dubreuil G, Lewis J, Georges-Courbot MC, Gessain A. 2001. A novel gamma 2-herpesvirus of the Rhadinovirus 2 lineage in chimpanzees. Genome Research 11:1511–1519.
Lisitsyn NA, Rosenberg MV, Launer GA, Wagner LL, Potapov VK, Kolesnik TB, Sverdlov ED. 1993. A method for isolation of sequences missing in one of two related genomes. Molekuliarnaia Genetika, Mikrobiologiia, i Virusologiia 3:26–9
Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H, Robinson CA. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. New England Journal of Medicine 340:1063–1070.
Miller G, Rigsby MO, Heston L, Grogan E, Sun R, Metroka C, Levy JA, Gao SJ, Chang Y, Moore P. 1996. Antibodies to butyrate-inducible antigens of Kaposi’s sarcoma-associated herpesvirus in patients with HIV-1 infection. New England Journal of Medicine 334:1292–1297.
Moore PS and Chang Y. 1995. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma lesions from persons with and without HIV infection. New England Journal of Medicine 332:1181–1185.
Moore PS and Chang Y. 1998a. Antiviral activity of tumor-suppressor pathways: clues from molecular piracy by KSHV. Trends in Genetics 14:144–150.
Moore PS and Chang Y. 1998b. The discovery of KSHV (HHV 8). Epstein-Barr Virus Report 5:1–3.
Moore PS and Chang Y. 2001. Kaposi’s sarcoma-associated herpesvirus. Pp. 2803–2833 in Fields Virology, DM Knipe and P Howley, eds. Philadelphia: Lippincott, Williams & Wilkins.
Moore PS, Kingsley LA, Holmberg SD, Spira T, Gupta P, Hoover DR, Parry JP, Conley LJ, Jaffe HW, Chang Y. 1996. Kaposi’s sarcoma-associated herpesvirus infection prior to onset of Kaposi’s sarcoma. AIDS 10:175–180.
Neipel F, Albrecht JC, Fleckenstein B. 1997. Cell-homologous genes in the Kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: determinants of its pathogenicity?. [Review]. Journal of Virology 71:4187–4192.
O’Brien TR, Kedes D, Ganem D, Macrae DR, Rosenberg PS, Molden J, Goedert JJ. 1999. Evidence for concurrent epidemics of human herpesvirus 8 and human immunodeficiency virus type 1 in US homosexual men: rates, risk factors, and relationship to Kaposi’s sarcoma. Journal of Infectious Diseases 180:1010–1017.
Parravinci C, Corbellino M, Paulli M, Magrini U, Lazzarino M, Moore PS, Chang Y. 1997. Expression of a virus-derived cytokine, KSHV vIL-6, in HIV-seronegative Castleman’s disease. American Journal of Pathology 151:1517–1522.
Raab-Traub N and Flynn K. 1986. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell 47:883–889.
Rose TM, Strand KB, Schultz ER, Schaefer G, Rankin GW Jr, Thouless ME, Tsai CC, Bosch ML. 1997. Identification of two homologs of the Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in retroperitoneal fibromatosis of different macaque species. Journal of Virology 71:4138–4144.
Russo JJ, Bohenzky RA, Chien MC, Chen J, Yan M, Maddalena D, Parry JP, Peruzzi D, Edelman IS, Chang Y, Moore PS. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proceedings of the National Academy of Sciences 93:14862–14867.
Simpson GR, Schulz TF, Whitby D, Cook PM, Boshoff C, Rainbow L, Howard MR, Gao SJ, Bohenzky RA, Simmonds P, Lee C, de Ruiter A, Hatzakis A, Tedder RS, Weller IV, Weiss RA, Moore PS. 1996. Prevalence of Kaposi’s sarcoma associated herpesvirus infection measured by antibodies to recombinant capsid protein and latent immunofluorescence antigen. Lancet 348:1133–1138.
Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L. 1995. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 86:1276–1280.
Whitby D, Howard MR, Tenant-Flowers M, Brink NS, Copas A, Boshoff C, Hatzioannou T, Suggett FE, Aldam DM, Denton AS, et al. 1995. Detection of Kaposi’s sarcoma-associated herpesvirus (KSHV) in peripheral blood of HIV-infected individuals predicts progression to Kaposi’s sarcoma. Lancet 364:799–802.
EXPLORING THE GENETIC BACKGROUND–ENVIRONMENT INTERPLAY IN AN ANIMAL MODEL OF NEURODEVELOPMENTAL DISORDERS: A MULTIDISCIPLINARY APPROACH
Mikhail V. Pletnikov, M.D., Ph.D.*
Departments of Psychiatry and Behavioral Sciences, Johns Hopkins University
School of Medicine, Baltimore, MD
and
Laboratory of Pediatric and Respiratory Viral Diseases, Center for Biologics
Evaluation and Research, U.S. Food and Drug Administration, Bethesda, MD
Data from family, twin, and adoption studies convincingly show evidence of a substantial genetic contribution to most neurodevelopmental disorders in humans. Moreover, recent improvements in molecular and genetic technologies have resulted in the implication of genes at several chromosomal loci and a search for candidate genes continues. However, multiple examples of deviation of complex developmental disorders from clear-cut Mendelian transmission cannot be fully explained by incomplete penetrance, variable expressivity, or polygenic etiology. A growing body of evidence suggests an important role of environmental factors in the causation of some developmental behavioral disorders. Unfortunately, environmental studies have been carried out with the same concept in mind, i.e., a search for relevant risk factors that would be self-sufficient to explain the pathogenesis of human conditions. Very little, if any, theoretical or methodological interaction between genetic linkage analysis and environmental (e.g., toxicology or teratology) studies has been undertaken. However, it is the gene-environment interaction that determines variable disease outcomes and responses to treatment and must be addressed in future research approaches.
Although it is clear that there are critical interactions between genes and environment to produce disease phenotypes, this concept has not been a focus of extensive consideration that the important role of environmental factors becomes more apparent in the setting of interaction with genetic determinants. Separating the search for genetic determinants vs. environmental disease etiologies was, in part, based on the assumption that genes and environmental sources are mainly additive in their effects, with the outcome reflecting the sum of their influences. However, the evidence is now clear that genes and environment are interactive as well, and several important issues of the gene-environment interaction are illustrated here with data obtained on the animal model of neurodevelopmental damage in rats neonatally infected with an experimental teratogen, Borna disease virus (BDV).
Among methodological problems of studying the gene-environment interplay is the difficulty in firmly defining environmental factors and making them
quantifiable. In this context, virus infections provide a promising research avenue because of their clear etiologic connection to several human neurodevelopmental disorders and because of reliability of quantification of viral effects on brain and behavior (Johnson, 1998). For example, in the BDV model, neonatal infection of the rat’s brain with this 8.9-kb non-segmented, negative-strand, enveloped RNA virus (Briese et al., 1994; Cubitt et al., 1994) produces distinct neuroanatomical, neurochemical and behavioral abnormalities that resemble pathological and clinical features of human developmental disorders (Carbone et al., 1991; Carbone and Pletnikov, 2000; Bautista et al., 1994, 1995; Dittrich et al., 1989; Pletnikov et al., 1999, 2000, 2001; Hornig et al., 1999; Eisenman et al., 1999; Gonzalez-Dunia et al., 2000; Weissenbock et al., 2000).
Considering gene-environmental interaction may also help us to better understand the nature of some environmental risk factors. For example, in adult infected Lewis rats, BDV-induced brain damage is primarily mediated by T-cell inflammatory response, causing generalized encephalitis and meningitis (Narayan et al., 1983; Hirano et al., 1983). This global damage significantly hampers studies of other pathogenic mechanisms of viral neurotoxicity. In contrast, in different genetic settings (e.g., black hooded rats or tree shrews), adult BDV infection does not appear to evoke significant inflammatory response, providing new insights into the mechanisms of chronic BDV-associated neurobehavioral deficits (Herzog et al., 1991; Sprankel et al., 1978).
Similar methodological advantages are demonstrated by neonatal rat BDV infection serving as an animal model of neurodevelopmental damage, while also emphasizing a neurodevelopmental perspective in studying the gene-environment interaction. The significance of the neurodevelopmental perspective is substantiated by the fact that effects of many genetic and environmental risk factors are evident either prior to or around the time of birth, and the interaction between them is apparent well before the identified onset/diagnosis of the classical, chronic disease condition. For this reason, we study effects of neonatal BDV infection across the entire postnatal period in genetically different strains of rats in order to understand the course and time-dependent character of the interaction of genetic background features and the virus infection.
In the newer view on the gene-environment interaction, wherein environmental factors are considered to have variable effects on individuals with different genotypes, gene-environment interactions result from genetically-mediated differences in sensitivity to environmental factors. This issue clearly underlines a need for searching for genes that mediate susceptibility to environmental factors rather than genes that directly determine specific chronic conditions. From a pathogenesis point of view, if environmental effects are mainly observed in vulnerable individuals, there is also a need in understanding what pathogenic role individual characteristics may play in modulating differential responses to the environment, variable disease outcome, and sensitivity to treatments. Obviously, such studies must be complex and multidisciplinary, simultaneously addressing
alterations in brain structure, chemistry, endocrine function and behaviors in genetically different settings.
Here, using our BDV neonatal rat model of neurodevelopmental damage, we present the data of the developmental analysis of the pathogenic role of baseline (i.e., strain-related) differences in physiology, neurochemistry, and behaviors between two inbred rat strains, Lewis and Fisher344 rats in determining BDV-induced (i) brain damage, (ii) alterations in monoamine systems, (iii) behavioral deficits, and (iv) responses to pharmacological treatment.
Our data show that basic virus infection in both strains is comparable, e.g., similar virus replication and distribution in brain parenchyma in BDV-infected Fisher344 and Lewis rats throughout the postnatal period and similar inhibition of body weight gain in both rat strains. However, the outcome of this virus infection in these two different strains is not identical. Neonatal BDV infection produces a more profound thinning of the neocortex in Fisher344 rats compared to Lewis rats, while a similar reduction in granule cells in the dentate gyrus of the hippocampus and the comparable hypoplasia of the cerebellum was observed in two rat strains (Pletnikov et al., 2002a).
Neurochemical studies indicated regional and strain-specific monoamine alterations in tissue content, turnover and density of post- and presynaptic receptors in developing and adult BDV-infected Lewis and Fisher344 rats at postnatal day (PND) 30 and 120.
The observed strain-specific brain pathology and neurochemical alterations may explain differential behavioral deficits and responses to ameliorative pharmacological treatments in BDV-infected Lewis and Fisher344 rats. For example, when assessed by the prepulse inhibition of the acoustic startle paradigm, neonatal BDV infection impaired sensorimotor gating in Fisher344 but not in Lewis rats at PND 30 and 120 (Pletnikov et al., 2002a). Also, neonatal BDV infection produced greater hyperactivity in Fisher344 rats compared to Lewis rats. The difference in hyperactivity was especially evident at PND 30.
Effects of the interaction of genetic background and environmental insult were further observed in responses of diseased animals to ameliorative pharmacological treatments. Novelty-induced hyperactivity remained unaffected by injections of a serotonin (5-HT) A1 receptors agonist 8-OH-DPAT in BDV-infected Lewis rats, while 8-OH-DPAT significantly decreased novelty-induced hyperactivity in BDV-infected Fisher344 rats. In contrast, novelty-induced hyperactivity was significantly depressed by a selective serotonin reuptake inhibitor (SSRI), fluoxetine, in BDV-infected Lewis rats and remained unaffected in BDV-infected Fisher344 rats (Pletnikov et al., 2002b).
In conclusion, it is likely that the interaction between genetic background and environmental insult contributes to the variability seen in chronic human conditions. The specific mechanisms and processes involved in the genotypeenvironment interaction remain largely unknown and are only just beginning to be explored. In the present work, some theoretical and methodological aspects of
the gene-environmental interaction are discussed and illustrated with the data from the analysis of effects of different genetic background on neurodevelopmental damage and responses to treatment in two rat strains following the neonatal BDV infection.
REFERENCES
Bautista JR, Schwartz GJ, de la Torre JC, Moran TH, Carbone KM. 1994. Early and persistent abnormalities in rats with neonatally acquired Borna disease virus infection. Brain Research Bulletin 34:31–40.
Bautista JR, Rubin SA, Moran TH, Schwartz GJ, Carbone KM. 1995. Developmental injury to the cerebellum following perinatal Borna disease virus infection. Brain Research. Developmental Brain Research 90:45–53.
Briese T, Schneemann A, Lewis AJ, Park YS, Kim S, Ludwig H, Lipkin WI. 1994. Genomic organization of Borna disease virus. Proceedings of the National Academy of Sciences 91:4362–4366.
Carbone K and Pletnikov M. 2000. Borna again, starting from the beginning. Molecular Psychiatry 5:577.
Carbone KM, Park SW, Rubin SA, Waltrip RW II, Vogelsang GB. 1991. Borna disease: association with a maturation defect in the cellular immune response. Journal of Virology 65:6154–6164.
Cubitt B, Oldstone M, de la Torre JC. 1994. Sequence and genome organization of Borna disease virus. Journal of Virology 68:1382–1396.
Dittrich W, Bode L, Kao M, Schneider K. 1989. Learning deficiencies in Borna disease virus-infected but clinically healthy rats. Biological Psychiatry 26:818–828.
Eisenman LM, Brothers R, Tran MH, Kean RB, Dickson GM, Dietzschold B, Hooper DC. 1999. Neonatal Borna disease virus infection in the rat causes a loss of Purkinje cells in the cerebellum. Journal of Neurovirology 5:181–189.
Gonzalez-Dunia DM, Watanabe S, Syan M, Mallory E, de la Torre JC. 2000. Synaptic pathology in Borna disease virus persistent infection. Journal of Virology 74:3341–3448.
Herzog S, Frese K, Rott R. 1991. Studies on the genetic control of resistance of black hooded rats to Borna disease. Journal of General Virology 72:535–540.
Hirano N, Kao M, Ludwig H. 1983. Persistent, tolerant or subacute infection in Borna disease virusinfected rats. Journal of General Virology 64:1521–1530.
Hornig M, Weissenbock H, Horscroft N, Lipkin WI. 1999. An infection-based model of neurodevelopmental damage. Proceedings of the National Academy of Sciences 96:12102–12107.
Johnson RT. 1998. Viral infections of the nervous system. Philadelphia: Lippincott-Raven.
Narayan OS, Herzog K, Frese H, Rott R. 1983. Behavioral disease in rats caused by immunopathological responses to persistent Borna virus in the brain. Science 220:1401–1403.
Pletnikov MV, Rubin SA, Vasudevan K, Moran TH, Carbone KM. 1999. Developmental brain injury associated with abnormal play behavior in neonatally Borna Disease Virus (BDV)-infected Lewis rats: A model of autism. Behavorial Brain Research 100:30–45.
Pletnikov MV, Rubin SA, Schwartz GJ, Carbone KM, Moran TH. 2000. Effects of neonatal rat Borna disease virus (BDV) infection on the postnatal development of monoaminergic brain systems. Brain Research. Developmental Brain Research 119:179–185.
Pletnikov MV, Rubin SA, Carbone KM, Moran TH, Schwartz GJ. 2001. Neonatal Borna disease virus infection (BDV)-induced damage to the cerebellum is associated with sensorimotor deficits in developing Lewis rats infection on the postnatal development of monoaminergic brain systems. Brain Research. Developmental Brain Research 126:1–12.
Pletnikov MV, Rubin SA, Vogel MW, Moran TH, Carbone KM. 2002a. Effects of genetic background on neonatal Borna disease virus infection-induced neurodevelopmental damage. I. Brain pathology and behavioral deficits. Brain Research 944:97–107.
Pletnikov MV, Rubin SA, Vogel MW, Moran TH, Carbone KM. 2002b. Effects of genetic background on neonatal Borna disease virus infection-induced neurodevelopmental damage. II. Neurochemical alterations and responses to pharmacological treatments. Brain Research 944:108–123.
Sprankel H, Richard K, Ludwig H, Rott R. 1978. Behavior alterations in tree shrews (Tupaia glis, Diard 1820) induced by Borna disease virus. Medical Microbiology and Immunology 26:1–18.
Weissenbock H, Hornig M, Hickey WF, Lipkin WI. 2000. Microglia activation and neuronal apoptosis in bornavirus infected neonatal Lewis rats. Brain Pathology 10:260–272.
INFECTION, CANCER, AND THE IMMUNE RESPONSE
David H. Persing, M.D., Ph.D.
Corixa Corporation and the Infectious Disease Research Institute, Seattle, WA
and
Franklyn G. Prendergast, M.D., Ph.D.
Department of Pharmacology and the Mayo Clinic Cancer Center,
Rochester, MN
During the past decade, the scientific community has witnessed a virtual explosion of information regarding the genetic basis of disease, especially of inherited disorders and human cancers. Much of the effort of the Human Genome Initiative has been focused on genetic abnormalities that arise during the development of neoplasia and upon congenital predispositions to cancer that are associated with the inheritance of mutations within tumor suppressor genes and other loci. These studies are of critical importance to our understanding of genetic and cellular processes contributing to neoplasia. However, to date the evidence suggests that inherited predisposition to cancer probably accounts for only a subset of total cancer patients and appears to be insufficient to explain the sporadic cases currently comprising the majority.
In most models of the development of neoplasia, an underlying assumption is the contribution of an array of intrinsic and extrinsic factors within a multi-step process. A basic prerequisite of many models is an increase in the baseline proliferation rates of essentially normal cell populations, accompanied by genotypic and phenotypic alterations leading to dysregulation of normal growth control mechanisms. Accordingly, preneoplastic conditions are often associated with an increase in the proliferation of tissues, and some cancer predisposing conditions result from inherited predispositions toward increased mitotic rate (e.g., familial polyposis). However, virtually any condition leading to increased cellular proliferation, whether by a direct or indirect mechanism, might potentiate the development of malignancy. Since many infectious processes often lead, directly or indirectly, to increased cell turnover and proliferation, certain agents are now widely regarded as carcinogens (Rosenthal and Purtilo, 1997).
In 1991, zur Hausen estimated that a significant fraction of all human cancers worldwide are associated with infections due to viruses, including human papillomaviruses (cervical cancer and other skin cancers), human T-cell leukemia viruses (adult T-cell leukemias and lymphomas in endemic areas), hepatitis B virus (liver cancer), and Epstein-Barr virus (Burkitt’s lymphoma and nasopharyngeal carcinoma) (zur Hausen, 1991). The estimate of the influence of infection may now need to be revised in light of the fact that new viral associations have been discovered and that other, nonviral associations have been uncovered (Rosenthal and Purtilo, 1997). These include a common bacterial pathogen (Helicobacter pylori infection with gastric carcinoma and MALT lymphoma), and new viruses (hepatitis C virus with liver cancer, HHV-6 with non-Hodgkin’s lymphoma, HHV-8 [a.k.a. KSHV]) with Kaposi’s sarcoma, Castleman’s disease, and body cavity lymphomas (Mueller, 1995). In addition, new disease associations are being made with respect to previously known pathogens, such as the association of chronic hepatitis C virus infection with non-Hodgkin’s lymphoma in certain populations (Luppi et al., 1996). The following sections will summarize briefly some of the established and emerging associations between chronic infections and human cancer, as reflected in Table 3-1.
Chronic Bacterial Infections Associated with Human Malignancy
Helicobacter pylori
After many years of unwarranted skepticism, the medical establishment in the United States and worldwide generally now recognizes Helicobacter pylori as the most common cause of diffuse superficial gastritis and gastric and duodenal ulcers (McGowan et al., 1996). Infections caused by H. pylori persist within the gastric mucosa for many years, and the incidence of infection is associated with lower economic status and increasing age. Consistent with the step-wise elucidation of the role of the etiologic agent, the treatment for gastric and duodenal ulcer has evolved from a surgical approach (gastrectomy), to medical management of gastric hyperacidity (H2 blockers), and most recently toward antibiotic therapy directed against H. pylori (McGowan et al., 1996).
Inflammation associated with H. pylori infection may progress to chronic atrophic gastritis, which is a known predisposing condition for the development of gastric carcinoma. Accordingly, H. pylori infection has been linked epidemiologically to gastric adenocarcinoma; many studies involving thousands of participants have now shown an increased risk of gastric cancer in persons with elevated antibodies to Helicobacter pylori and in known H. pylori carriers (Uemura et al., 2001). It is probable that host genetic factors (blood type, HLA type, other immunogenetically determined factors) as well as microbial virulence factors (particularly the presence of the cagA virulence factor) contribute to tissue burden of organisms, persistence of infection, and the nature of the inflammatory response,
TABLE 3-1 Infectious Agents Associated with Malignancies
|
Infectious Agent |
Neoplasm |
|
BACTERIA |
|
|
Helicobacter pylori |
Gastric adenocarcinoma, intestinal type MALT lymphoma, non-Hodgkin’s lymphoma |
|
Fusobacterium fusiforme and Borrelia vincentii |
Squamous cell carcinoma arising from tropical phagedenic ulcer |
|
Vibrio cholerae |
Possible associations with immunoproliferative small intestinal disease (IPSID), non-Hodgkin’s lymphoma |
|
VIRUSES |
|
|
Epstein-Barr virus |
Burkitt’s lymphoma, nasopharyngeal carcinoma, and reversible lymphoproliferative diseases in immunodeficient patients |
|
Human T-lymphotropic viruses I and II |
Adult T-cell leukemia, T-cell lymphoma |
|
Human papillomavirus |
Cutaneous and mucosal papillomas and carcinomas |
|
Hepatitis B and C viruses |
Hepatitis, chronic active hepatitis and hepatocellular carcinoma |
|
Human herpesvirus-8 (KSHV) |
Kaposi’s sarcoma, body cavity lymphoma |
|
Human immunodeficiency virus |
Kaposi’s sarcoma, and non-Hodgkin’s lymphoma, cutaneous and mucosal papillomas and carcinomas |
|
SV-40 |
Possible associations with mesothelioma and ependymoma |
|
PROTOZOA |
|
|
Strongyloides stercoralis |
T-cell leukemia (with HTLV) |
|
Plasmodium falciparuma |
Burkitt’s lymphoma |
|
Schistosoma hematobium |
Squamous cell carcinoma of the urinary bladder |
|
S. mansoni, S. japonicum |
Colonic carcinomas |
|
Clonorchis sinensis |
Cholangiocarcinoma |
|
Opisthorchis viverrini |
Cholangiocarcinoma |
|
aPlasmodium falciparum is a cofactor for development of Burkitt’s lymphoma in endemic regions. |
|
all of which may contribute directly or indirectly to the development of carcinoma (Delchier et al., 2001). The association of H. pylori infection with gastric cancer was an important landmark, because it provided the first definitive link between a chronic bacterial infection, chronic inflammation originating within the target tissue, and the ultimate development of a human cancer.
Perhaps even more provocative has been the association of H. pylori with lymphoproliferative disease. H. pylori infection is associated with a proliferative response of mucosa-associated lymphoid tissues (MALT). This polyclonal or oligoclonal lymphocytic proliferation, which consists mostly of B cells, appears to be predisposed toward the subsequent development of malignant lymphomas of the non-Hodgkin’s type (Delchier et al., 2001). Several studies have now demonstrated regression of early MALT lymphomas by antibiotic treatment directed against H. pylori (Fischbach et al., 1997; Weber et al., 1994). This suggests further that the maintenance of the MALT lesion is directly associated with H. pylori infection, and may in fact be due to an antigen (or perhaps superantigen) and/or cytokine-driven proliferative response. Non-Hodgkin’s lymphomas harboring bcl2 rearrangements may emerge years after regression of the underlying MALT lymphoma by antibiotic therapy directed against H. pylori (Horstmann et al., 1994); it has been proposed that malignant cells may already be present in small numbers within the late-stage MALT lymphoma population prior to antibiotic treatment and may emerge thereafter as an antigen-independent lymphoproliferative disease (Graham et al., 1994; Thijs et al., 1995).
An important implication of these findings is the potential for cancer chemoprevention directed against the underlying infectious agent. Treatment of H. pylori in early stages of infection could result in lower risk of developing complications of chronic infection (Thijs et al., 1995). However, because H. pylori infections are common, and the development of cancer is an uncommon complication of infection, further work should be done to identify at-risk populations in order to better target chemoprevention efforts (Graham et al., 1994).
Other Bacterial Infections
If H. pylori can serve as an infectious trigger of a lymphoproliferative disease, might other chronic bacterial infections also be involved? Small intestinal lymphomas have been described as a complication of chronic infection with Tropheryma whippeli, the Whipple’s disease-associated bacillus, which has been documented to persist for many years (Gillen et al., 1993). Unrecognized cases of Whipple’s disease may be more common than previously suspected, based on findings of several recent studies employing detection methods based on the polymerase chain reaction (PCR) (Ramzan et al., 1997). A disease known as immunoproliferative small intestinal disease (IPSID) has been associated epidemiologically with enteric infection possibly due to Vibrio cholerae, primarily in developing countries (Isaacson, 1994). Hyperplastic lymphoid tissue develops in
the small bowel in association with the disease, converting to malignant lymphoma at later stages. Consistent with the experience of regression of MALT lymphoma in H. pylori-infected patients, tetracycline treatment sometimes causes regression of IPSID lesions in patients with early lesions, and is often used in conjunction with chemotherapy in later stage lesions (Trotman et al., 1999). Finally, tropical phagedenic ulcer is a chronic persistent dermatological infection of developing countries caused by coinfection with Fusobacterium and a spirochete, Borrelia vincentii (Robinson et al., 1988). This infection leads to the development squamous cell carcinoma within the depigmented margins of the ulcerative lesion. Antibiotic treatment is effective for eliminating the bacterial infection and presumably also for reducing cancer risk.
Taken together, it seems that chronic bacterial infections other than H. pylori may also predispose to the development of malignancy, especially non-Hodgkins lymphoma, by virtue of direct or indirect stimuli of target cell proliferation. Given the increasing incidence of non-Hodgkin’s lymphoma in the US, and the fact that the inciting organisms might be detectable in cancer patients at the time of presentation suggests that pathogen discovery efforts aimed at well-defined patient populations might well be productive. More importantly, effective chemoprevention strategies may depend upon the identification of microbial, environmental, and host determinants in the development of neoplasia.
Parasitic Infections Associated with Malignancy
Some infections with protozoa are associated with malignancy in humans. Burkitt’s lymphoma is thought to arise from the convergence of two pathogens in the same host—Epstein-Barr virus and the malarial parasite, Plasmodium falciparum. Clustered cases of Burkitt’s lymphoma match the geographic distribution of holoendemic malaria (Facer and Playfair, 1989). The well-known immunosuppressive effects of chronic malarial infection are thought to predispose the host to the transforming effects of EBV, perhaps by a mechanism similar to that described for EBV-related malignancies in immunosuppressed hosts (Lam et al., 1991). Another possible virus/parasite interaction in the development of malignancy is the possible association of Strongyloides stercoralis in the development of HTLV-associated leukemia which is mentioned below (Plumelle et al., 1997).
Schistosomiasis, a helminth infection with a wide geographic distribution, is associated with carcinoma of the urinary bladder (Rosin et al., 1994). Chronic infection with S. hematobium causes chronic inflammation, fibrosis, and increased proliferation of squamous cells; malignant squamous cell carcinomas usually arise from this premalignant proliferative lesion. In contrast, most malignant tumors of the bladder that arise outside of the context of infection with S. hematobium in endemic areas are transitional cell carcinomas. A similar mechanism of increasing cell turnover may apply to the development of cholangiocarcinoma during
chronic biliary tract infection with Clonorchis sinensis and Opisthorchis viverrini (Shin et al., 1996). Infection with both of these organisms is associated with chronic inflammation and proliferation of the biliary epithelium. Chronic inflammation of the bile duct epithelium due to infection with the newly recognized protozoa Septata intestinalis and Cryptosporidium parvum have been recognized in this country, but it is not yet known whether these infections are associated with malignancy.
Viruses Associated with Human Malignancies
Although malignancies associated with viral infection have been studied in animal models for decades, clear evidence of involvement of viral infection in human malignancies has been lacking until about the past decade. A growing number of viral infections have been associated with various human malignancies including carcinomas and adenomas of the cervix and upper airways, liver cancer, and lymphomas and leukemias. As mentioned above, it is estimated that approximately 15 percent of all malignancies worldwide are associated with known viral infections. This estimate is based on associations with already known viral infections. However, recent history has documented that there are likely to be many viral infections of humans that are still to be uncovered.
Malignancy associated with viral infection has in some cases been attributed to direct effects of viral gene products, as described above for the human papillomavirus, or it may be associated with increased cellular proliferation of a target tissue, as described for the bacterial infections above. In both settings, host immune responses are likely to play an important role in the tolerance of persistent viral infection.
The Gamma-herpesviruses, EBV and KSHV
Since its discovery in the early 1960s in an African Burkitt’s lymphoma cell line, Epstein-Barr virus has become widely regarded as an oncogenic virus, especially in the immunocompromised host. It is also highly prevalent in the US and worldwide, with 80–90 percent seroconversion by young adulthood in most countries. Most childhood infections are clinically silent, but primary EBV infection in older children or young adulthood is the major cause of acute mononucleosis. During primary infection a polyclonal expansion of B cells occurs resulting in the formation of heterophile antibodies as well as EBV-specific cellular and humoral immune responses. Latency is established in a subset of B cells which can be interrupted under certain conditions to produce reactivation of infection (Goldschmidts et al., 1992).
In the mid-1950s, missionary doctor Denis Burkitt described a unique malignant lymphoma of childhood usually involving the jaw and viscera. He later identified the distribution of cases of the lymphoma to areas of Africa that are en-
demic for malaria (the “lymphoma belt”). To this day, Burkitt’s lymphoma remains one of the most dreaded diseases in sub-Saharan Africa. EBV can be found in nearly 100 percent of endemic lymphomas, in contrast to its lower prevalence in non-endemic Burkitt-type malignancies. In both endemic and non-endemic tumor types, translocation of the c-myc cellular oncogene to an expressed locus downstream from the immunoglobulin G promoter (IgG-P) occurs by gene rearrangement. However, the IgG-P/c-myc breakpoints in endemic cases occur within a more tightly clustered region, suggesting stereotypic recombinational patterns in the malaria-associated cases. Whether this pattern of recombination occurs directly in response to malarial infection is currently unknown (Facer and Playfair, 1989), but an attractive hypothesis is that immunoglobulin gene rearrangement driven by expansion of Plasmodium-specific B-cells participates in the development of malignant clones. Differences in EBV subtypes have been observed in endemic and non-endemic cases; endemic cases contain EBV subtype 2, in contrast to non-endemic cases which usually harbor EBV subtype 1 (Magrath et al., 1992). Mechanisms for the possible pathogen interaction between EBV and Plasmodium are poorly understood; the well known immunosuppressive effects of malarial infection have been proposed by several investigators to activate EBV-associated lymphoproliferation (Lam et al., 1991). The combination of immunosuppression with antigen-specific proliferation may, in this model, lead to the development of a unique type of cancer.
EBV is also associated with nasopharyngeal carcinoma (NPC), a malignancy that represents the most common tumor of males in southern China. Additional environmental exposures to salted fish containing nitrosodimethylamines, and perhaps inhaled herbal extracts have been implicated as possible cofactors in the development of this malignancy. Latent forms of the EBV genome can be detected by in situ hybridization. Immunologic predisposition may also contribute, as reflected in relative overrepresentation of certain HLA types in patients with NPC along with evidence of intrafamilial case clustering. In addition, an atypical immune response as determined by the presence of IgA to certain EBV proteins suggests that an aberrant immune response to the virus may underlie cancer risk (Hsu et al., 2001). This EBV-specific IgA response may be an indirect, albeit diagnostically important, indication of EBV infection on mucosal surfaces which itself serves as a proferative stimulus of epithelial cell populations accompanied by lymphocytic infiltration (lymphoepithelioma).
A recent report implicated EBV in the development of human breast cancer (Magrath and Bhatia, 1999); various studies have detected up to a 51 percent prevalence of EBV in breast cancer tissues, depending on the methods used. Since the known EBV-associated carcinomas are lymphoepitheliomas, and since most breast carcinomas are not lymphoepitheliomas, these findings have been controversial and indeed many studies fail to confirm the initial findings (Chu et al., 2001). A critical link to be established here would be the demonstration of unusual immunological responses to EBV proteins as demonstrated for NPC.
In the developed world, the most common EBV-associated tumor is non-Hodgkin’s lymphoma associated with immunosuppression. Most non-Hodgkin’s lymphomas originating in the context of HIV infection are associated with EBV, and CNS lymphomas in AIDS patients are nearly exclusively EBV positive (Knowles, 1996). In transplant recipients, EBV-associated tumors often originate as polyclonal or oligoclonal proliferations, occasionally evolving into monoclonal populations (Ambinder, 1990). Reversal of immunosuppression often leads to regression of polyclonal lymphoproliferative disease, consistent with the control of direct proliferative effects of EBV by cytotoxic T-cell responses. Once the tumor has become predominantly monoclonal, however, reversal of immune suppression often has no effect. The progression of non-Hodgkin’s lymphomas in immunosuppressed patients is reminiscent of the development of MALT lymphoma, in which an initial polyclonal lymphoproliferative response is followed by the evolution of more aggressive tumors that are apparently less subject to immune surveillance.
A new member of the gamma-herpesvirus family was recently discovered by the application of nucleic acid-based pathogen discovery techniques to Kaposi’s sarcoma (KS). KS had long been suspected to have an infectious etiology because of its peculiar epidemiology (Chang and Moore, 1996). The newly recognized virus, which has been called human herpesvirus 8 or KS-associated herpesvirus (KSHV), has been found in HIV-associated cases as well as in non-HIV associated endemic KS in Mediterranean and African men (Moore and Chang, 1995). A similar virus of primates, Herpesvirus samirai, causes an aggressive T-cell lymphoma and polyclonal B-cell proliferation in an experimental model of primate infection (Trimble and Desrosiers, 1991). Current epidemiologic evidence suggests that KSHV is necessary but not sufficient for development of most cases of KS. Defects in immune surveillance may contribute to the evolution of KSHV-associated lesions in a manner similar to its more highly prevalent cousin, EBV; these defects may be acquired or intrinsic in nature.
Recently, KSHV was recognized in dendritic cells of the bone marrow from patients with multiple myeloma and a smaller number of patients with monoclonal gammopathy of unknown significance (MGUS), a paraproteinemia which often predisposes to myeloma (Beksac et al., 2001; Rettig et al., 1997). Multiple myeloma is a malignancy of B-cells which represents the second most frequently diagnosed hematologic malignancy in the US. These findings were and still are controversial, especially from an epidemiologic perspective, and subsequent studies have failed to detect an association with KSHV infection in myeloma patients (Ablashi et al., 2000).
Human Retroviruses
The human retroviruses, human T cell leukemia viruses type I and II, infect millions of persons worldwide and are associated with various neoplastic mani-
festations and immunologic abnormalities. HTLV infects approximately one million people in the Japanese islands and localized populations within the Caribbean basin and South America. HTLV-related lymphomas are common in these areas, and approximately 400 patients develop HTLV associated leukemia yearly in Japan. The fact that only a small subset of HTLV infected patients develop malignant manifestations clearly implicates other factors, either environmental or genetic, that may predispose patients to neoplasia within the multi-step pathway. In the tropics, the high frequency of HTLV-associated malignancies in areas endemic for other protozoal diseases has suggested that coinfection with the latter may play a role in their pathogenesis. Recently, infection with Strongyloides stercoralis was found to be an independent risk factor for development of HTLV-associated leukemia in Jamaica (Plumelle et al., 1997). The increasingly recognized immunosuppressive effects of HTLV infection on the cytotoxic T cell response may also be implicated in the development of other cancers, such as carcinoma of the cervix (Strickler et al., 1995).
Perhaps because it is only rarely associated with lymphomagenesis in the United States, relatively less attention has been paid to this virus compared to its distant cousin, HIV. However, many valuable lessons have been learned about the transforming ability of human retroviruses in the development of HTLV-related neoplasia. Furthermore, with the advent of powerful new pathogen discovery techniques, it is possible that additional human retroviruses will be found and that experience gained from evaluation of HTLV-associated cases could be extrapolated to other diseases. Indeed, given the wide variety of exogenous and endogenous retroviruses found within other higher primate species, this seems likely. Several novel endogenous retroviruses have been isolated and identified in patients with autoimmune diseases and malignancies. In the case of human seminoma, elevated antibody titers to retroviral gag proteins from a novel human endogenous retrovirus (strain K10) that was originally isolated from a seminoma tumor cell line are found in patients and very infrequently in healthy controls (Sauter et al., 1995). Antigens crossreactive with Jaagsiekte virus, a known causative agent in the development of lung adenocarcinoma of sheep, have been detected in human lung carcinoma specimens (Palmarini et al., 1999). Sequences related to known oncogenic retroviruses have also been detected in human breast cancers (Liu et al., 2001). Phylogenetic analyses of other (presumably) exogenous and known endogenous retroviruses have shown that some are related to vertebrate oncoviruses found in mice (mouse mammary tumor virus) as well as to primate endogenous retroviruses. Further work will be necessary to determine whether the sequences of these exogenous and endogenous viruses play a significant role in human cancers or other malignancies.
Malignancies associated with HIV infection include increased susceptibility to cervical dysplasia, squamous cell carcinoma, B cell lymphomas, Kaposi’s sarcoma, and possibly seminomas and testicular cancer. It is interesting that in most examples of increased frequency of malignancy in HIV patients, viral cofactors
have been directly implicated in the progression of neoplasia. Epstein-Barr virus plays an important role in the evolution of B cell lymphomas in these patients, Kaposi’s sarcoma virus participates in the tumorigenesis of Kaposi’s sarcoma, and human papillomavirus infection contributes to the development of carcinomas of the skin and cervix. A reduction in levels of antigen-specific cytotoxic T cell responses are associated with activation of infection or disease progression in all of these cases, and a reduction in the efficiency of immune surveillance has been proposed as a factor in tumorigenesis. Consistent with this hypothesis, patients on immunosuppressive therapy following transplantation appear to be at increased risk for the same spectrum of malignancies with the exception of a lower incidence of KS; accordingly, KSHV appears to be much less prevalent in transplant patients compared to HIV patients, presumably because of the risk factors associated with the predominantly sexual transmission of KSHV (Chang and Moore, 1996).
Hepatitis B Virus
Primary infection with hepatitis B virus usually follows an acute and convalescent course in immunocompetent hosts with ultimate resolution of disease. However, in approximately 5 to 10 percent of adults, and in most infants born to infected carrier mothers, primary infection leads to chronic active hepatitis which may progress to cirrhosis of the liver and hepatocellular carcinoma (Kew, 1996). Continuous proliferation of hepatocytes, which may be present for the life of the host, is a prominent feature of chronic active hepatitis; in such patients, the relative risk of developing hepatocelluar carcinoma is increased 20–40 fold. Because of the large number of carriers of hepatitis B virus (HBV), approximately 200 million persons worldwide, hepatocellular carcinoma is one of the most common cancers in the world and is the most common cancer in HBV-endemic areas of the Far East and sub-Saharan Africa. The development of hepatocellular carcinoma probably depends upon direct effects of certain viral determinants along with chronic persistent proliferation of hepatocytes. HBV-related hepatocellular carcinomas often contain defective HBV genomes expressing one or more viral open reading frames; the viral X gene, which is often expressed in malignant tissues, is a transcriptional transactivator of host promoter sequences including those associated with oncogene expression and integrated viral genomes can activate host proto-oncogenes. There is little doubt that continuous proliferation of hepatocytes is also an important contributor to the development of neoplastic disease, since other disorders associated with chronic hepatitis and hepatocyte turnover (such as chronic alcoholism or hereditary hemochromatosis) may also lead to an increased predisposition to liver cancer, and the effects of alcohol ingestion and viral infection may well be synergistic in this regard (Brechot et al., 1996).
Hepatitis C Virus
A rapidly emerging association of chronic viral infection with malignancy is the development of hepatocellular carcinoma associated with chronic hepatitis C virus infection. In the United States, HCV infection is projected to become the major cause of liver cancer, due in part the large number of chronic carriers in this country, currently estimated at 3.5 million persons. In the case of HCV infection, a major determinant of development of hepatocellular cancer appears to be duration of infection and the degree of persistent liver injury (Zein and Persing, 1996). Several studies have associated certain HCV subtypes with differences in liver disease severity, interferon responsiveness, and duration of infection. Viral subtypes that appear to have been in the U.S. population for longer periods of time are dramatically overrepresented among patients with liver cancer. Specifically, subtype 1b, which is present in 15–20 percent of cases nationwide, is present in 90 percent or more of liver cancer patients in several studies conducted at different centers (Zein et al., 1996). Since presence of genotype 1b appears to be a marker of longer duration of infection in U.S. patients, it is possible that other genotypes will be more commonly implicated in cases of hepatocellular carcinoma in other countries, and that the overrepresentation of genotype 1b in the United States will decline over time. Additional factors such as alcohol consumption may contribute independently to risk of neoplasia, even in patients with more recent infections (Brechot et al., 1996). As for HPV infection, host immunogenetic or other factors may play a role in susceptibility to viral infection as well as in determining the severity of infection (see below).
Recently, chronic HCV infection has been associated with the development of B-cell non-Hodgkin’s lymphomas in an Italian population (Mele et al., 2003) and with the development of cryoglobulinemia and monoclonal gammopathy in the United States (Cacoub et al., 1994). The development monoclonal gammopathy is thought to presage the development of hematologic malignancies in a significant subset of cases; it is not yet known whether HCV-associated monoclonal gammopathy or mixed cryoglobulinemia is a predisposing variable for the development of such malignancies in the United States. One recent study suggests that HCV infection-associated lymphoproliferative disease might include multiple myeloma (Montella et al. 2001). It is possible that MGUS is a marker of an underlying benign, perhaps antigen driven lymphoproliferative process, which may after many years convert to a malignant process via somatic mutation. In this respect, the genesis of neoplasia may be similar to that described for Helicobacter pylori-related MALT lymphoma. The relative disparity in the frequency of non-Hodgkin’s lymphoma associated with HCV infection in the United States compared to that in Europe may conceivably reflect differences in duration of infection within the population, immunogenetic differences, or exposure to other oncoviruses including KHSV.
Human Papillomavirus Infection
In the past decade, human papillomavirus (HPV) has emerged as the single most important risk factor for development of cervical carcinoma, and it may be associated with neoplasia in other tissues as well. To date, more than 80 HPV subtypes have been described in a wide variety of epithelial tissues, with some viral subtypes found exclusively in certain tissues; partial sequences of novel subtypes indicate that over 100 subtypes may exist in humans. It is estimated that approximately 95 percent of cervical cancers worldwide are associated with infection by certain human papillomavirus subtypes, usually types 16 or 18 but occasionally other types. HPV-associated cervical cancers typically harbor at least remnants of the HPV genome in the form of two viral oncogenes which are consistently expressed in malignant tissues. The viral E6 gene product binds to tumor suppressor protein p53, promoting its degradation, and the viral E7 product binds to the retinoblastoma gene product, resulting in a functional inactivation. In addition, the E6 and E7 oncoproteins stimulate cell proliferation by activating cyclins E and A. The combination of these effects is to immortalize keratinocytes in vitro. However, additional mutational events are apparently needed to provide the fully transformed phenotype, since immortalized keratinocytes do not form tumors in vivo (zur Hausen and Rosl, 1994). Nonetheless, the viral E6 and E7 genes and their products may become attractive diagnostic targets for monitoring metastatic disease and for the development of tumor-specific immunotherapeutic protocols.
The association of HPV infection with carcinomas of other tissues has been less clear cut, but is somewhat reminiscent of the early days of research on the association of HPV with cervical cancer in which a fundamental limitation in the detection of the large variety of HPV types led initially to great controversy. On the other hand, the inherent proneness of PCR technology to contamination problems, coupled with its ability to detect extremely small numbers of possibly incidental HPV contaminants, warrants a careful interpretation of the growing number of reports implicating this virus in malignancies of many types. Archetypal “oncogenic” subtypes of HPV appear capable of infecting different anatomical sites, and associations of HPV infection with squamous cell carcinomas of the head and neck are reasonably well established. The involvement of HPV in the pathogenesis of skin cancer, the most common malignancy in the world, was suggested recently and a new family of HPV types has now been described in recurrent squamous cell carcinomas of the skin in transplant patients (Shamanin et al., 1996). This finding seems consistent with the clinical experience in patients who are immunosuppressed by HIV infection, in which cervical dysplasia is highly prevalent (Sun et al., 1995).
The role of HPV infection in epithelial carcinomas of the bladder and lung are also being investigated. Studies of the bovine papilloma virus (BPV) have been shown to have an association with cancer of the bladder in cattle (Olson et
al., 1965). Inoculation of BPV into newborn hamsters or mice has also been demonstrated to induce tumors (zur Hausen, 1996). Some human studies in recent years have suggested that relatively common HPV types can be detected in human transitional cell carcinoma (TCC) of the bladder in up to 34 percent of cases in certain patient populations (Smetana et al., 1995), but the presence of HPV in the neoplastic tissue has been difficult to demonstrate with consistency (Chetsanga et al., 1992). Interestingly, cases of rapidly progressive multifocal TCC of the bladder have been reported in patients following renal and cardiac transplantation, suggesting that HPV may play a more significant role in TCC of the bladder in immunosuppressed patients (Noel et al., 1994). Although smoking history is the leading risk factor in the development of squamous cell carcinoma of the lung, a recent study implicated the presence of HPV type 16 in an unexpectedly high number of cases of well-differentiated squamous cell carcinoma of the lung on the island of Okinawa (Kinoshita et al., 1995). However, although some studies have supported this finding (Hirayasu et al., 1996), others have failed to implicate HPV in lung cancer (Szabo et al., 1994). In this regard, it is interesting to note that risk of cervical cancer is linked epidemiologically to risk of carcinoma of the lung but not uterine or ovarian cancers, and that both of the former are linked to smoking (Anderson et al., 1997). Clearly, additional studies will be necessary which are designed to rule out the effects of incidental virus and DNA contamination, yet also designed to be capable of detection of the widest range of HPV types.
Recent studies of the natural history of HPV infection have suggested that the ability of the genetically heterogeneous papillomaviruses to exploit relative deficiencies in the immune response may represent an important determinant of the risk of developing chronic infection and subsequent neoplastic disease. Attention has focused recently on cytokine production in HPV-specific helper T lymphocyte populations in women with cervical cancer compared to women in whom the disease regresses spontaneously. Several studies have now indicated that failure to mount cytotoxic T cell (CTL) responses to human papillomavirus-infected cells may significantly predispose to the development of cervical cancer (Tsukui et al., 1996); this failure may be virus type-specific, such that the types represented most often in cervical cancer are those most successful at avoidance of CTL activity (Ellis et al., 1995). Conditions associated with reduced CTL activity, such as HIV and HTLV infection, oral contraceptive use, pregnancy, and immune suppression associated with transplantation have likewise been associated with increased HPV viral burden and acceleration of disease progression (Sun et al., 1995). Twin studies have suggested that risk factors for cervical cancer, as for another HPV-associated lesion, epidermoplasia verruciformis, may be inherited (Ahlbom et al., 1997). More recent studies have failed to detect an association with somatic mutations in the gene encoding p53, and have further explored associations with the immunoglobulin gene cluster (Cuzick et al., 2000). Understanding the contribution of the environmental, host somatic, and host im-
munogenetic factors to the development of HPV-related dysplasia is an area of intense research activity which may lead to direct practical benefits to patients in the form of improved immunomodulatory approaches to early and late stage lesions. Since the prevalence of HPV infection is so high, the use of routine testing for HPV is of questionable value (Stoler, 2001), except in patients with Pap smears containing atypical features (ASCUS). Clearly, if additional immunogenetic and other host susceptibility factors can be identified, the positive predictive power of HPV testing in cervical cancer screening programs may increase to acceptable levels (Klug et al., 2001).
The Role of Humoral and Cellular Immune Responses
As illustrated vividly by HIV infection, infection with one pathogen may lead to a predisposition toward other infectious processes as well as cancer; a key feature of the immune suppression associated with HIV infection and pharmacologically-produced immunosuppression following transplantation is a reduction of cytotoxic T-cell (CTL) and related T-helper type 1 responses (Spina and Tirelli, 1992). One important theme that recurs among the above mentioned programs, and in cancer research in general, is the relationship of T cell responses to the evolving tumor. A decline in cytotoxic immunity has been associated with virus-associated tumors in immunosuppressed patients, and relative deficits in the establishment of CTL responses during the initial stages of infection are associated with progression of infection in otherwise immunocompetent hosts. Relative differences in the ability to mount cytotoxic T-cell responses have been noted among mouse strains for several years, and recent evidence suggests that immunogenetic determinants in humans may play similar roles. From a teleological perspective, it is likely that viruses and other pathogens associated with chronic infections in humans have evolved means of avoiding CTL responses in at least some hosts in order to maximize reservoir capacity. An example is the recent documentation of point mutations within a dominant HLA B7-associated CTL epitope of the human papillomavirus E6 gene product; the presence of sequence variation has implications for vaccine design and immunotherapy, and overall, viral diversity must be taken into account in the design of therapeutic strategies.
Just as the suppression of cellular immune responses may relate to development of cancer, the development of humoral immune responses may be extremely useful for determining the history of exposure to the pathogen and for the serologic surveillance necessary to establish the etiologic role of a given pathogen. Serologic studies have been critical to the understanding of the role of H. pylori as an etiologic cofactor in various malignancies; when no such assays are available for a novel infectious agent, surrogate serologic assays can be used as proposed previously (Persing, 1997). Furthermore, since IgG antibody subclasses are generated on the basis of T-cell help, measurement of IgG subclasses may lead to an understanding of the role of T-cell responses (i.e., the proportional Th1
and Th2-type responses) in determining the resolution or persistence of a particular infection (Moro et al., 2001).
Role Reversal: The Cancer Cell as the Ultimate Eukaroytic Parasite
Independent of the etiologic roles of infectious agents in the development of human cancer, in more than one sense, the cancer cell itself represents the ultimate eukaryotic parasite. Cancer cells, like parasitic protozoa, use a variety of mechanisms to avoid immune surveillance including persistence in small numbers, active secretion of immunosuppressive substances, downregulation of MHC class 1-based recognition, and antigenic variation (Buchanan and Nieland-Fisher, 2001). Humoral immune responses to cancer-specific antigens often occur in cancer patients and are undetectable in patients without cancer (Disis et al., 1997), suggesting that immunologic tolerance can be broken and that many types of cancer-specific immune responses, in the form of tissue-specific autoimmune phenomena, can be tolerated in humans. Antibody responses in cancer patients have led to the discovery of many important cancer-specific antigens (Chen, 2000). At the same time, these studies point to the relative lack of efficacy of humoral immune responses in the control of a developing malignancy, much the same way that in chronic carriers of protozoan parasites, immune responses to pathogen-specific antigens happily coexist with the pathogens themselves. Indeed, many of the lessons learned from parasite immunology can be extrapolated to human cancer, especially in the development of new vaccine-based approaches to the treatment of both diseases.
Conclusions
In purely reductionistic terms, infectious diseases can be viewed as horizontally acquired genetic disorders, in which exogenously acquired nucleic acids of a pathogen integrate, either chromosomally, episomally, or extracellularly, with those of the host to disrupt normal cellular processes or produce inflammation. Developing a better understanding of the interactions of human microbial flora with their hosts, along with an understanding of other host and environmental determinants of pathogenicity, represents an increasingly important intersection of infectious disease research with the Human Genome Project. Illustrations of the latter concept are provided by the discovery of resistance to HIV infection by virtue of mutations within a gene encoding an HIV coreceptor (CCR-5) (Samson et al., 1996). Another important example is the inherited susceptibility to Mycobacterium avium-intracellulare infections in families carrying a mutant allele of the interferon-gamma (IFN-γ) receptor (Newport et al., 1996). The identification of such an allele within the human population has far-reaching implications, since it may be associated with a general reduction in the ability to mount cytotoxic T-cell responses during infections of many types. Genetic predisposition to
papillomavirus infection appears to underlie the inherited skin disorder epideromodysplasia verruciformis (Majewski and Jablonska, 1995), in which cytotoxic T-cell responses apparently fail to develop against certain papillomavirus subtypes. Since early generation of such responses appears to be a critical step in the resolution of human papillomavirus infections, patients harboring functionally similar mutations may be uniquely predisposed to the development of cervical and other cancers linked to HPV infection. By focusing on unusually severe outcomes of relatively common infectious diseases, we may be able to identify critical immunogenetic factors in the formation of protective immune responses, and to tailor patient management and cancer chemoprevention efforts (in some cases directed against the pathogens themselves) in appropriate ways.
REFERENCES
Ablashi DV, Chatlynne L, Thomas D, Bourboulia D, Rettig MB, Vescio RA, Viza D, Gill P, Kyle RA, Berenson JR, Whitman JE. 2000. Lack of serologic association of human herpesvirus-8 (KSHV) in patients with monoclonal gammopathy of undetermined significance with and without progression to multiple myeloma. Blood 96:2304–2306.
Ahlbom A, Lichtenstein P, Malmstrom H, Feychting M, Hemminki K, Pedersen NL. 1997. Cancer in twins: genetic and nongenetic familial risk factors. Journal of the National Cancer Institute 89:287–293.
Ambinder RF. 1990. Human lymphotropic viruses associated with lymphoid malignancy: EpsteinBarr and HTLV-1. Hematology Oncology Clinics of North America 4:821–833.
Anderson KE, Woo C, Olson JE, Sellers TA, Zheng W, Kushi LH, Folsom AR. 1997. Association of family history of cervical, ovarian, and uterine cancer with histological categories of lung cancer—the Iowa women’s health study. Cancer Epidemiology, Biomarkers & Prevention 6:401–405.
Beksac M, Ma M, Akyerli C, DerDanielian M, Zhang L, Liu J, Arat M, Konuk N, Koc H, Ozcelik T, Vescio R, Berenson JR. 2001. Frequent demonstration of human herpesvirus 8 (HHV-8) in bone marrow biopsy samples from Turkish patients with multiple myeloma (MM). Leukemia 15:1268–1273.
Brechot C, Nalpas B, Feitelson MA. 1996. Interactions between alcohol and hepatitis viruses in the liver. Clinics in Laboratory Medicine 16:273–287.
Buchanan J and Nieland-Fisher NS. 2001. Role of immune function in human papillomavirus infection. Journal of the American Medical Association 286:1173–1174.
Cacoub P, Fabiani FL, Musset L, Perrin M, Frangeul L, Leger JM, Huraux JM, Piette JC, Godeau P. 1994. Mixed cryoglobulinemia and hepatitis C virus. American Journal of Medicine 96:124–132.
Chang Y and Moore PS. 1996. Kaposi’s Sarcoma (KS)-associated herpesvirus and its role in KS. Infectious Agents & Disease 5:215–222.
Chen YT. 2000. Cancer vaccine: identification of human tumor antigens by SEREX. Cancer Journal 6 (Suppl 3):S208–217.
Chetsanga C, Malmstrom PU, Gyllensten U, Morenolopez J, Dinter Z, Peterson U. 1992. Low incidence of human papillomavirus type 16 DNA in bladder tumour detected by polymerase chain reaction. Cancer 69:1208–1211.
Chu PG, Chang KL, Chen YY, Chen WG, Weiss LM. 2001. No significant association of EpsteinBarr virus infection with invasive breast carcinoma. American Journal of Pathology 159:571–578.
Cuzick J, Terry G, Ho L, Monaghan J, Lopes A, Clarkson P, Duncan I. 2000. Association between high-risk HPV types, HLA DRB1* and DQB1* alleles and cervical cancer in British women. British Journal of Cancer 82:1348–1352.
Delchier JC, Lamarque D, Levy M, Tkoub EM, Copie-Bergman C, Deforges L, Chaumette MT, Haioun C. 2001. Helicobacter pylori and gastric lymphoma: high seroprevalence of CagA in diffuse large B-cell lymphoma but not in low-grade lymphoma of mucosa-associated lymphoid tissue type. American Journal of Gastroenterology 96:2324–2328.
Disis ML, Pupa SM, Gralow JR, Dittadi R, Menard S, Cheever MA. 1997. High-titer HER-2/neu protein-specific antibody can be detected in patients with early-stage breast cancer. Journal of Clinical Oncology 15:3363–3367.
Ellis JR, Keating PJ, Baird J, Hounsell EF, Renouf DV, Rowe M, Hopkins D, Duggan-Keen MF, Bartholomew JS, Young LS et al. 1995. The association of an HPV16 oncogene variant with HLA-B7 has implications for vaccine design in cervical cancer. Nature Medicine 1:464–470.
Facer CA and Playfair JH. 1989. Malaria, Epstein-Barr virus, and the genesis of lymphomas. Advances in Cancer Research 53:33–72.
Fischbach W, Tacke W, Greiner A, Konrad H, Muller H. 1997. Regression of immunoproliferative small intestinal disease after eradication of Helicobacter pylori. Lancet 349:31–32.
Gillen CD, Coddington R, Monteith PG, Taylor RH. 1993. Extraintestinal lymphoma in association with Whipple’s disease. Gut 34:1627–1629.
Goldschmidts WL, Bhatia K, Johnson JF, Akar N, Gutierrez MI, Shibata D, Carolan M, Levine A, Magrath IT. 1992. Epstein-Barr virus genotypes in AIDS-associated lymphomas are similar to those in endemic Burkitt’s lymphomas. Leukemia 6:875–878.
Graham DY, Malaty HM, Go MF. 1994. Are there susceptible hosts to Helicobacter pylori infection? Scandinavian Journal of Gastroenterology Supplement 205:6–10.
Hirayasu T, Iwamasa T, Kamada Y, Koyanagi Y, Usuda H, Genka K. 1996. Human papillomavirus DNA in squamous cell carcinoma of the lung. Journal of Clinical Pathology 49:810–817.
Horstmann M, Erttmann R, Winkler K. 1994. Relapse of MALT lymphoma associated with Helicobacter pylori after antibiotic treatment [letter] [see comments]. Lancet 343:1098–1099.
Hsu MM, Hsu WC, Sheen TS, Kao CL. 2001. Specific IgA antibodies to recombinant early and nuclear antigens of Epstein-Barr virus in nasopharyngeal carcinoma. Clinical Otolaryngology and Allied Sciences 26:334–338.
Isaacson PG. 1994. Gastrointestinal lymphoma. Human Pathology 25:1020–1029.
Kew MC. 1996. Hepatitis B and C viruses and hepatocellular carcinoma. Clinics in Laboratory Medicine 16:395–406.
Kinoshita I, Dosaka-Akita H, Shindoh M, Fujino M, Akie K, Kato M, Fujinaga K, Kawakami Y. 1995. Human papillomavirus type 18 DNA and E6-E7 mRNA are detected in squamous cell carcinoma and adenocarcinoma of the lung. British Journal of Cancer 71:344–349.
Klug SJ, Wilmotte R, Santos C, Almonte M, Herrero R, Guerrero I, Caceres E, Peixoto-Guimaraes D, Lenoir G, Hainaut P, Walboomers JM, Munoz N. 2001. Tp53 polymorphism, hpv infection, and risk of cervical cancer. Cancer Epidemiology Biomarkers and Prevention 10:1009–1012.
Knowles DM. 1996. Etiology and pathogenesis of AIDS-related non-Hodgkin’s lymphoma. Hematology/Oncology Clinics of North America 10:1081–1109.
Lam KM, Syed N, Whittle H, Crawford DH. 1991. Circulating Epstein-Barr virus-carrying B cells in acute malaria. Lancet 337:876–878.
Liu B, Wang Y, Melana SM, Pelisson I, Najfeld V, Holland JF, Pogo BG. 2001. Identification of a proviral structure in human breast cancer. Cancer Research 61:1754–1759.
Luppi M, Grazia Ferrari M, Bonaccorsi G, Longo G, Narni F, Barozzi P, Marasca R, Mussini C, Torelli G. 1996. Hepatitis C virus infection in subsets of neoplastic lymphoproliferations not associated with cryoglobulinemia. Leukemia 10:351–355.
Magrath I and Bhatia K. 1999. Breast cancer: a new Epstein-Barr virus-associated disease? Journal of the National Cancer Institute 91:1349–1350.
Magrath I, Jain V, Bhatia K. 1992. Epstein-Barr virus and Burkitt’s lymphoma. Seminars in Cancer Biology 3:285–295.
Majewski S and Jablonska S. 1995. Epidermodysplasia verruciformis as a model of human papillomavirus-induced genetic cancer of the skin. Archives of Dermatology 131:1312–1318.
McGowan CC, Cover TL, Blaser MJ. 1996. Helicobacter pylori and gastric acid: biological and therapeutic implications. Gastroenterology 110:926–938.
Mele A, Pulsoni A, Bianco E, Musto P, Szklo A, Sanpaolo MG, Iannitto E, De RenzoA, Martino B, Liso V, Andrizzi C, Pusterla S, Dore F, Maresca M, Rapicetta M, Marcucci F, Mandelli F, Franceschi S. 2003. Hepatitis C virus and B-cell non-Hodgkin lymphomas: an Italian multicenter case-control study. Blood. 102(3):996–999.
Montella M, Crispo A, Frigeri F, Ronga D, Tridente V, De Marco M, Fabbrocini G, Spada O, Mettivier V, Tamburini M. 2001. HCV and tumors correlated with immune system: a case-control study in an area of hyperendemicity. Leukemia Research 25:775–781.
Moore PS and Chang Y. 1995. Detection of herpesvirus-like DNA sequences in Kaposi’s sarcoma in patients with and without HIV infection [see comments]. New England Journal of Medicine 332:1181–1185.
Moro MH, Bjornsson J, Marietta EV, Hofmeister EK, Germer JJ, Bruinsma E, David CS, Persing DH. 2001. Gestational attenuation of Lyme arthritis is mediated by progesterone and IL-4. Journal of Immunology 166:7404–7409.
Mueller N. 1995. Overview: viral agents and cancer. Environmental Health Perspectives 103(Suppl 8):259–261.
Newport MJ, Huxley CM, Huston S, Hawrylowicz CM, Oostra BA, Williamson R, Levin M. 1996. A mutation in the interferon-gamma-receptor gene and susceptibility to mycobacterial infection. New England Journal of Medicine 335:1941–1949.
Noel JC, Thiry L, Verhest A, Deschepper N, Peny MO, Sattar AA, Schulman CC, Haot J. 1994. Transitional cell carcinoma of the bladder: evaluation of the role of human papillomaviruses. Urology 44:671–675.
O’Connor F, Buckley M, O’Morain C. 1996. Helicobacter pylori: the cancer link. Journal of the Royal Society of Medicine 89:674–678.
Olson C, Pamucku AM, Brobst DF. 1965. Papillomalike virus from bovine urinary tumours. Cancer Research 25:840–847.
Palmarini M, Sharp JM, de las Heras M, Fan H. 1999. Jaagsiekte sheep retrovirus is necessary and sufficient to induce a contagious lung cancer in sheep. Journal of Virology 73:6964–6972.
Persing DH. 1997. The cold zone: a curious convergence of tick-transmitted diseases. Clinical Infectious Diseases 25(Suppl 1):S35–42.
Plumelle Y, Gonin C, Edouard A, Bucher BJ, Thomas L, Brebion A, Panelatti G. 1997. Effect of Strongyloides stercoralis infection and eosinophilia on age at onset and prognosis of adult T-cell leukemia. American Journal of Clinical Pathology 107:81–87.
Ramzan NN, Loftus E, Burgart LJ, Rooney M, Batts KP, Wiesner RH, Fredricks DN, Relman DA, Persing DH. 1997. Diagnosis and monitoring of Whipple disease by polymerase chain reaction. Annals of Internal Medicine 126(7).
Rettig MB, Ma HJ, Vescio RA, Pold M, Schiller G, Belson D, Savage A, Nishikubo C, Wu C, Fraser J, Said JW, Berenson JR. 1997. Kaposi’s sarcoma-associated herpesvirus infection of bone marrow dendritic cells from multiple myeloma patients. Science 276:1851–1854.
Robinson DC, Adriaans B, Hay RJ, Yesudian P. 1988. The clinical and epidemiologic features of tropical ulcer (tropical phagedenic ulcer). International Journal of Dermatology 27:49–53.
Rosenthal LJ and Purtilo DT. 1997. Neoplasms associated with infectious agents. P. 1707. In Pathology of Infectious Diseases, vol. II, DH Connor, FW Chandler, HJ Manz, DA Schwartz, EE Lack, eds. Stamford, CT: Appleton and Lange.
Rosin MP, Anwar WA, Ward AJ. 1994. Inflammation, chromosomal instability, and cancer: the schistosomiasis model. Cancer Research 54:1929s–1933s.
Samson M, Libert F, Doranz BJ, Rucker J, Liesnard C, Farber CM, Saragosti S, Lapoumeroulie C, Cognaux J, Forceille C, Muyldermans G, Verhofstede C, Burtonboy G, Georges M, Imai T, Rana S, Yi Y, Smyth RJ, Collman RG, Doms RW, Vassart G, Parmentier M. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene [see comments]. Nature 382:722–725.
Sauter M, Schommer S, Kremmer E, Remberger K, Dolken G, Lemm I, Buck M, Best B, NeumannHaefelin D, Mueller-Lantzsch N. 1995. Human endogenous retrovirus K10: expression of Gag protein and detection of antibodies in patients with seminomas. Journal of Virology 69:414–421.
Shamanin V, zur Hausen H, Lavergne D, Proby CM, Leigh IM, Neumann C, Hamm H, Goos M, Haustein UF, Jung EG et al. 1996. Human papillomavirus infections in nonmelanoma skin cancers from renal transplant recipients and nonimmunosuppressed patients [see comments]. Journal of the National Cancer Institute 88:802–811.
Shin HR, Lee CU, Park HJ, Seol SY, JM Chung, Choi HC, Ahn YO, Shigemastu T. 1996. Hepatitis B and C virus, Clonorchis sinensis for the risk of liver cancer: a case-control study in Pusan, Korea. International Journal of Epidemiology 25:933–940.
Smetana Z, Keller T, Leventon-Kriss S, Huszar M, Lindner A, Mitrani-Rosenbaum S, Mendelson E, Smetana S. 1995. Presence of human papilloma virus in transitional cell carcinoma in Jewish population in Israel. Cellular & Molecular Biology 41:1017–1023.
Spina M and Tirelli U. 1992. Human immunodeficiency virus as a risk factor in miscellaneous cancers. Current Opinion in Oncology 4:907–910.
Stoler MH. 2001. HPV for cervical cancer screening: is the era of the molecular Pap smear upon us? Journal of Histochemistry and Cytochemistry 49:1197–1198.
Strickler HD, Rattray C, Escoffery C, Manns A, Schiffman MH, Brown C, Cranston B, Hanchard B, Palefsky JM, Blattner WA. 1995. Human T-cell lymphotropic virus type I and severe neoplasia of the cervix in Jamaica. International Journal of Cancer 61:23–26.
Sun XW, Ellerbrock TV, Lungu O, Chiasson MA, Bush TJ, Wright T Jr. 1995. Human papillomavirus infection in human immunodeficiency virus-seropositive women. Obstetrics & Gynecology 85:680–686.
Szabo I, Sepp R, Nakamoto K, Maeda M, Sakamoto H, Uda H. 1994. Human papillomavirus not found in squamous and large cell lung carcinomas by polymerase chain reaction [see comments]. Cancer 73:2740–2744.
Thijs JC, Kuipers EJ, van Zwet AA, Pena AS, de Graaff J. 1995. Treatment of Helicobacter pylori infections. QJM 88:369–389.
Trimble JJ and Desrosiers RC. 1991. Transformation by herpesvirus saimiri. Advances in Cancer Research 56:335–355.
Trotman BW, Pavlick AC, Igwegbe IC, Goldstein MM. 1999. Immunoproliferative small intestinal disease: case report and literature review. Journal of the Association for Academic Minority Physicians 10:88–93.
Tsukui T, Hildesheim A, Schiffman MH, Lucci JR, Contois D, Lawler P, Rush BB, Lorincz AT, Corrigan A, Burk RD, Qu W, Marshall MA, Mann D, Carrington M, Clerici M, Shearer GM, Carbone DP, Scott DR, Houghten RA, Berzofsky JA. 1996. Interleukin 2 production in vitro by peripheral lymphocytes in response to human papillomavirus-derived peptides: correlation with cervical pathology. Cancer Research 56:3967–3974.
Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. 2001. Helicobacter pylori infection and the development of gastric cancer. New England Journal of Medicine 345:784–789.
Weber DM, Dimopoulos MA, Anandu DP, Pugh WC, Steinbach G. 1994. Regression of gastric lymphoma of mucosa-associated lymphoid tissue with antibiotic therapy for Helicobacter pylori. Gastroenterology 107:1835–1838.
Zein NN and Persing DH. 1996. Hepatitis C genotypes: current trends and future implications. [Review] [50 refs]. Mayo Clinic Proceedings 71:458–462.
Zein NN, Poterucha JJ, Gross J Jr, Wiesner RH, Therneau TM, Gossard AA, Wendt NK, Mitchell PS, Germer JJ, Persing DH. 1996. Increased risk of hepatocellular carcinoma in patients infected with hepatitis C genotype 1b. American Journal of Gastroenterology 91:2560–2562.
zur Hausen H. 1991. Viruses in human cancers. Science 254:1167–1173.
zur Hausen H. 1996. Roots and perspectives of contemporary papillomavirus research. Journal of Cancer Research and Clinical Oncology 122:3–13.
zur Hausen H and Rosl F. 1994. Pathogenesis of cancer of the cervix. Cold Spring Harbor Symposia on Quantitative Biology 59:623–628.







































