
2
Basics of Sound, the Ear, and Hearing
In this chapter we review basic information about sound and about how the human auditory system performs the process called hearing. We describe some fundamental auditory functions that humans perform in their everyday lives, as well as some environmental variables that may complicate the hearing task. We also discuss the types of hearing loss or disorder that can occur and their causes.
INTRODUCTION TO SOUND1
Hearing allows one to identify and recognize objects in the world based on the sound they produce, and hearing makes communication using sound possible. Sound is derived from objects that vibrate producing pressure variations in a sound-transmitting medium, such as air. A pressure wave is propagated outward from the vibrating source. When the pressure wave encounters another object, the vibration can be imparted to that object and the pressure wave will propagate in the medium of the object. The sound wave may also be reflected from the object or it may diffract around the object. Thus, a sound wave propagating outward from a vibrating object can reach the eardrum of a listener causing the eardrum to vibrate and initiate the process of hearing.
Sound waves can be mathematically described in two ways, that is, in two domains. In the time domain, sound is described as a sequence of pressure changes (oscillations) that occur over time. In other words, the time-domain description of a sound wave specifies how the sound pressure increases and decreases over time. In the frequency domain, the spectrum defines sound in terms of the tonal components that make up the sound. A tonal sound has a time-domain description in which sound pressure changes as a regular (sinusoidal) function of time. If one knows the tonal components of sound as defined in the frequency domain, one can calculate the time-domain description of the sound. Using the same analytic tools, the frequency domain representation of a sound can also be calculated from the time-domain description. Thus, the time and frequency domain descriptions of sound are two different ways of measuring the same thing (i.e., the time and frequency domains are functional equivalents). Thus, one can describe sound as temporal fluctuations in pressure, or one can describe sounds in terms of the frequency components that compose the sound.
Largely because tonal (sinusoidal) sounds are the bases of the frequency domain description of sound, a great deal of the study of hearing has dealt with tonal sounds. However, everyday sounds are complex sounds, which are made up of many tonal frequency components. A common complex sound used to study hearing is noise. Noise contains all possible frequency components, and the amplitude of the noise varies randomly over time. A noise is said to be “white noise” if it contains all frequency components each at the same average sound level.
A sound waveform has three basic physical attributes: frequency, amplitude, and temporal variation. Frequency refers to the number of times per second that the vibratory pattern (in the time domain) oscillates. Amplitude refers to sound pressure. There are many aspects to the temporal variation of sound, such as sound duration. Sound pressure is proportional to sound intensity (in units of power or energy), so sound magnitude can be measured in units of pressure, power, and energy. The common measure of sound level is the decibel (dB), in which the decibel is the logarithm of the ratio of two sound intensities or two sound pressures. Frequency is measured in units of hertz (Hz), cycles per second. Measures of time are expressed in various temporal units or can be translated into phase measured in angular degrees. Below are some definitions of terms and measures used to describe sound.
-
Sound pressure (p): sound pressure is equal to the force (F) produced by the vibrating object divided by the area (Ar) over which that force is being applied: p = F/Ar.
-
DekaPascals or daPa; the Système International unit of pressure. One daPa = 100 dynes per cm2, and one atmosphere = 10132.5 daPa.
-
Sound intensity (I): sound intensity is a measure of power. Sound intensity equals sound pressure squared divided by the density (po) of the sound-transmitting medium (e.g., air) times the speed of sound (c): I = p2/poc. Energy is a measure of the ability to do work and is equal to power times the duration of the sound, or E = PT, where P is power and T is time (duration) in seconds.
-
Decibel (dB): dB = 10*log10(I/Iref) or 20*log10(p/pref), where I is sound intensity, p is sound pressure, ref is a referent intensity or pressure, and log10 is the logarithm to the base 10. When pref is 20 micropascals, then the decibel measure is expressed as dB SPL (sound pressure level).
-
Hertz (Hz): hertz is the measure of vibratory frequency in which “n” cycles per second of periodic oscillation is “n” Hz.
-
Phase (angular degrees): one cycle of a periodic change in sound pressure can be expressed in terms of completing the 360 degrees of a circle. Thus, half a cycle is 180 degrees, and so on. Thus, time (t) within a cycle can be expressed in terms of phase (θ, expressed in degrees), θ = 360o(t)(f), where f = frequency in Hz, and t = time in seconds.
-
Tone (a simple sound): a tone is a sound whose amplitude changes as a sinusoidal function of time: Asin(2 πft + θ), where sin is the trigonometric sin function, θ = peak amplitude, f = frequency in Hz, t = time in seconds, and θ = starting phase in degrees.
-
Complex sound: any sound that contains more than one frequency component.
-
Spectrum: the description of the frequency components of sound; amplitude spectrum describes the amplitude of each frequency component; phase spectrum describes the phase of each frequency component.
-
Noise: a complex sound that contains all frequency components, and whose instantaneous amplitude varies randomly.
-
White noise: a noise in which all of the frequency components have the same average level.
The term “noise” can refer to any sound that may be unwanted or may interfere with the detection of a target or signal sound. In some contexts, a speech sound may be the signal or target sound, and another speech sound or a mixture of other speech sounds may be presented as a “noise” to interfere with the auditory processing of the target speech sound. Often a mixture of speech sounds is referred to as “speech babble.”
The Auditory System
The ear is a very efficient transducer (i.e., a device that changes energy from one form to another), changing sound pressure in the air into a
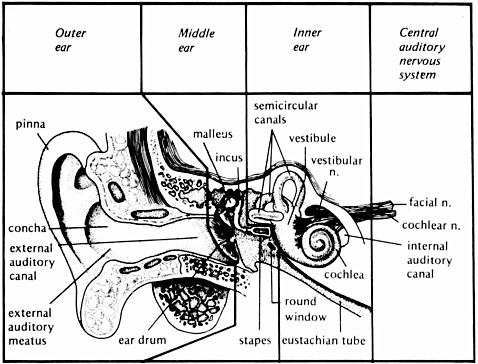
FIGURE 2-1 The anatomy of the auditory system. From Yost (2000, p. 66). Reprinted with permission of author.
neural-electrical signal that is translated by the brain as speech, music, noise, etc. The external ear, middle ear, inner ear, brainstem, and brain each have a specific role in this transformation process (see Figure 2-1).
The external ear includes the pinna, which helps capture sound in the environment. The external ear canal channels sound to the tympanic membrane (eardrum), which separates the external and middle ear. The tympanic membrane and the three middle ear bones, or ossicles (malleus, incus, and stapes), assist in the transfer of sound pressure in air into the fluid- and tissue-filled inner ear. When pressure is transferred from air to a denser medium, such as the inner ear environment, most of the pressure is reflected away. Thus, the inner ear offers impedance to conducting sound pressure to the fluid and tissue of the inner ear. The transfer of pressure in this case is referred to as admittance, while impedance is the restriction of the transfer of pressure. The term “acoustic immittance” is used to describe the transfer process within the middle ear: the word “immittance” combines the words impedance and admittance (im + mittance). As a result of this impedance, there is as much as a 35 dB loss in the transmission of sound pressure to the inner ear. The outer ear, tympanic membrane, and ossicles interact when a sound is present to
focus the sound pressure into the inner ear so that most of that 35 dB impedance loss is overcome. Thus, the fluids and tissues of the inner ear vibrate in response to sound in a very efficient manner.
Sound waves are normally transmitted through the ossicular chain of the middle ear to the stapes footplate. The footplate rocks in the oval window of the inner ear, setting the fluids of the inner ear in motion, with the parameters of that motion being dependent on the intensity, frequency, and temporal properties of the signal. The inner ear contains both the vestibular system (underlying the sense of balance and equilibrium) and the cochlea (underlying the sense of hearing). The cochlea has three separate fluid compartments; two contain perilymph (scala tympani and scala vestibuli), similar to the body’s extracellular fluid, and the other, scala media, contains endolymph, which is similar to intracellular fluids.
The scala media contains the sensorineural hair cells that are stimulated by changes in fluid and tissue vibration. There are two types of hair cells: inner and outer. Inner hair cells are the auditory biotransducers translating sound vibration into neural discharges. The shearing (a type of bending) of the hairs (stereocilia) of the inner hair cells caused by these vibrations induces a neural-electrical potential that activates a neural response in auditory nerve fibers of the eighth cranial nerve that neurally connect the hair cells to the brainstem. The outer hair cells serve a different purpose. When their stereocilia are sheared, the size of the outer hair cells changes due to a biomechanical alteration. The rapid change in outer hair cell size (especially its length) alters the biomechanical coupling within the cochlea.
The structures of the cochlea vibrate in response to sound with a particular vibratory pattern. This vibratory pattern (the traveling wave) allows the inner hair cells and their connections to the auditory nerve to send signals to the brainstem and brain about the sound’s vibration and its frequency content. That is, the traveling wave motion of cochlear vibration helps sort out the frequency content of any sound, so that information about the frequency components of sound is coded in the neural responses being sent to the brainstem and brain.
The fact that the different frequencies of sound are coded by different auditory nerve fibers is referred to as the place theory of frequency processing, and the auditory nerve is said to be “tonotopically” organized in that each nerve fiber carries information to the brainstem and brain about a narrow range of frequencies. In addition, the temporal pattern of neural responses of the auditory nerve fibers responds to the temporal pattern of oscillations of the incoming sound as long as the temporal variations are less than about 5000 Hz.
In general, the more intense the sound is, the greater the number of neural discharges that are being sent by the auditory nerve to the
brainstem and brain. Thus, the cochlea sends neural information to the brainstem and brain via the auditory nerve about the three physical properties of sound: frequency, temporal variation, and level. The biomechanical response of the cochlea is very sensitive to sound, is highly frequency selective, and behaves in a nonlinear manner. A great deal of this sensitivity, frequency selectivity, and nonlinearity is a function of the motility of the outer hair cells.
There are two major consequences of the nonlinear function of the cochlea: (1) neural output is a compressive function of sound level. This means that, at low sound levels, there is a one-to-one relationship between increases in sound level and increases in neural output; however, at higher sound levels, the rate at which the neural output increases with increases in sound level is lower. (2) The cochlea and auditory nerve produce distortion products. For instance, if the sound input contains two frequencies, f1 and f2, distortion products at frequencies equal to 2f1, 2f2, f2-f1, and 2f1-f2 may be produced by the nonlinear function of the cochlea. The distortion product 2f1-f2 (the cubic-difference tone) may be especially strong and this cubic-difference distortion product is used in several measures of auditory function.
At 60 dB SPL the bones of the skull begin to vibrate, bypassing the middle ear system. This direct vibration of the skull can cause the cochlea to vibrate and, thus, the hair cells to shear and to start the process of hearing. This is a very inefficient way of hearing, in that this way of exciting the auditory nervous system represents at least a 60 dB hearing loss.
There are many neural centers in the brainstem and in the brain that process the information provided by the auditory nerve. The primary centers in the auditory brainstem in order of their anatomical location from the cochlea to the cortex are: cochlear nucleus, olivary complex, lateral lemniscus, inferior colliculus, and medial geniculate. The outer, middle, and inner ears along with the auditory nerve make up the peripheral auditory system, and the brainstem and brain constitute the central auditory nervous system. Together the peripheral and central nervous systems are responsible for hearing and auditory perception.
AUDITORY PERCEPTION
In the workplace, hearing may allow a worker to:
-
Communicate using human speech (e.g., communicate with a supervisor who is giving oral instructions);
-
Process information-bearing sounds (e.g., respond to an auditory warning);
-
Locate the spatial position of a sound source (e.g., locate the position of a car based on the sound it produces).
There is a wealth of basic knowledge about how the auditory system allows for communication based on sound, informative sound processing, and sound localization. Listeners can detect the presence of a sound; discriminate changes in frequency, level, and time; recognize different speech sounds; localize the source of a sound; and identify and recognize different sound sources.
The auditory system must often accomplish these workplace tasks when there are many sources producing sound at about the same time, so that the sound from one source may interfere with the ability to “hear” the sound from another source. The interfering sound may make it difficult to detect another sound, to discriminate among different sounds, or to identify a particular sound. A hearing loss may make it difficult to perform one or all of these tasks even in the absence of interfering sounds but especially in the presence of interfering sounds.
Sound Detection
The healthy, young auditory system can detect tones in quiet with frequencies ranging from approximately 20 to 20000 Hz. Figure 2-2 displays the standardized average thresholds for detecting tonal sounds of different frequencies when the sounds are approximately 500 milliseconds (ms) in duration. The sounds to be detected can be presented over calibrated headphones (minimal audible pressure, MAP, measures) or from a loudspeaker in a calibrated free-field environment (minimal audible field, MAF, measures). The headphones can be circumaural, that is, with a headphone cushion that fits around the pinna and the earphone speaker resting against the outside of the outer ear canal, or they can be insert earphones whose earphone loudspeaker fits within the outer ear canal. The thresholds are expressed in terms of decibels of SPL, where zero (0) dB SPL means that the sound pressure level is 20 micropascals (i.e., the referent sound pressure (pref) is 20 micropascals). Upper limits of hearing, indicating the maximum SPL that the auditory system can tolerate, are also indicated in Figure 2-2. Thus, the dynamic range of hearing covers approximately 130 dB in the frequency region in which the human auditory system is most sensitive (between 500 and 4000 Hz). The thresholds for detecting a tonal sound increase as the duration of the sound to be detected decreases at durations shorter than 500 ms, but remain approximately constant as the duration increases above 500 ms.
The detection of tones as characterized by the data of Figure 2-2 is the basis for the primary measure of hearing loss or impairment, the audio-
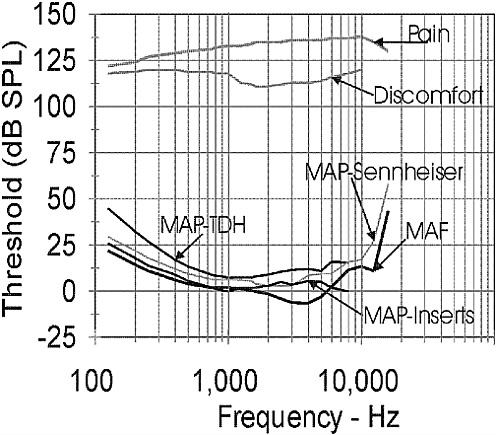
FIGURE 2-2 The thresholds (in dB SPL) of detecting tones and for discomfort and pain are shown as a function of tonal frequency. The MAP thresholds for Sennheiser and TDH are thresholds for two types of circumaural headphones.
gram. The audiogram is a plot of the thresholds of hearing referenced to the appropriate MAP or MAF thresholds shown in the figure. Thus, a person with no hearing loss at all will have a flat audiogram at zero dB HL (dB HL means decibels of hearing level, in which the reference decibel values are the appropriate MAP or MAF dB SPL values shown in Figure 2-2). A person with a 40 dB hearing loss would be said to have a threshold of 40 dB HL. If the tone being detected is 500 Hz, the threshold for detecting the tone in terms of SPL would be 50 dB SPL, since according to Figure 2-2, the threshold for detecting a 500 Hz tone is 10 dB SPL. Thus, the 40 dB hearing loss (40 dB HL) plus the 10 dB SPL threshold yields a threshold of 50 dB SPL.
In many cases, the average threshold (either in dB SPL or dB HL) for several different frequencies may be obtained to provide an estimate of
overall auditory sensitivity or overall hearing loss. In this case, the threshold is referred to as a pure-tone average (PTA) threshold, in which the frequencies used for the threshold averaging are listed in abbreviated form; for example, a PTA threshold obtained at 500 Hz (.5 kHz), 1000 Hz (1 kHz), and 2000 Hz (2 kHz) would be listed as PTA 512, or sometimes PTA (512).
Many of the measurements made in hearing and the equipment used to make these measurements have been standardized by the American National Standards Institute (ANSI, see references). ANSI standards represent documents that have been reviewed by the ANSI process so that the standards represent a consensus of the best and most accurate data, method, or equipment needed to address a particular practical problem or need. Appendix B is a list of the major ANSI standards that are mentioned in this report, with descriptions of each standard. For instance, ANSI S3.6-1996 provides specifications for how audiometers, which are used by audiologists to measure the audiogram, should be built, as well as the standard thresholds of hearing used in the calculation of dB HL. A sample of an audiogram can be seen in Chapter 3.
As the intensity of a tone increases, so does its subjective loudness. Loudness is a subjective indication of the magnitude of sound. One measure of loudness level is the phon. A phon is the subjective loudness of a test sound that is judged equally loud to a standard sound. The standard sound is a 1000 Hz tonal sound presented at some SPL.
For instance, if a test sound is judged to be equally loud to the 1000 Hz standard sound presented at 40 dB SPL, the test sound is said to have a loudness of 40 phons. A sound that has a loudness of 40 phons is usually judged to be very quiet. A standardized method for determining the loudness of complex sounds in phons has also been developed (American National Standards Institute, 2003). The perceived loudness of sound will double for about every 10 dB increase in sound level (e.g., a 60 dB SPL sound may be subjectively twice as loud as a 50 dB SPL sound). People with a hearing loss experience discomfort at about the same SPL as people without hearing loss. Because people with hearing loss have elevated thresholds but have the same upper limit of audibility as people with normal hearing, the change in loudness grows more rapidly as a function of increasing sound level above threshold for a person with a hearing loss than for a person without a hearing loss. This rapid growth of loudness is referred to as loudness recruitment, and it is experienced by almost all people with sensorineural hearing loss.
As the frequency of a sound changes, so does its subjective pitch. Pitch is the subjective attribute of sound that allows one to determine if the sound is high or low along a single perceptual dimension. The pitch of a test sound can be determined by the frequency of a tonal sound that is
judged to be subjectively equal in pitch to the test sound. For instance, a test sound is said to have a pitch of 400 Hz if it is judged equal in pitch to a 400 Hz tone. In addition to being measured in hertz, pitch can also be measured using the 12-note or other musical scale.
While loudness is highly correlated with sound intensity and pitch with frequency, loudness and pitch are subjective attributes of sound that may be correlated with each of the physical attributes of sound: level, frequency, and temporal properties. So for instance, a change in sound frequency may result not only in a change in pitch, but also in a change in loudness.
The presence of another sound (masking sound) presented at the same time as a tone that is to be detected (signal tone) may increase the threshold of the signal tone above that measured in quiet. In this case, the signal tone is being masked by the other sound. Most masking occurs (i.e., thresholds are increased the most) when the masking sound contains the same frequency components as the signal tone. For instance, less masking occurs when a 300 Hz masker masks a 1000 Hz signal than when the masker and signal are both 1000 Hz. Thus, there is a region of frequencies near that of the signal frequency (a band of frequencies with the center of the band being the frequency of the signal) that are critical for masking the signal, and the width of the critical band increases as the frequency of the signal increases. For instance, for a 1000 Hz signal, as long as the masker contains frequencies between approximately 936 and 1069 Hz (a 133-Hz-wide critical band, with 1000 Hz in the geometric center of the band), the masker will be effective in masking the 1000 Hz signal. Maskers with frequencies higher than 1069 Hz or lower than 936 Hz will be less effective in masking the 1000 Hz signal. Thus, a white noise filtered so that the noise contains frequency components between 936 and 1069 Hz will be maximally effective in masking a 1000 Hz tonal signal. If the noise has a bandwidth that is narrower than the critical band, the signal is easier to detect, but if the bandwidth is wider than the critical band, then there is no change in signal detection performance.
For listeners with normal hearing, when the power of the noise in the critical band is equal to the power of the tonal signal, then the signal is usually at its masked threshold. Over a considerable range of frequency, level, and duration, and when the signal and masker occur at the same time, each decibel increase in the level of a masking sound requires approximately a decibel increase in signal level in order for the signal to remain just detectable in the presence of the masker. That is, the signal-to-noise (S/N) ratio required for signal detection remains relatively constant over a large range of frequency, overall level, and duration.
People with hearing loss often have wider critical bands than people with normal hearing, which means that the signal can be masked by
sounds with frequencies farther from the signal frequency. The wider critical band obtained for people with hearing loss is usually found for signals whose frequency content is in the spectral region of their loss. This means that people with hearing loss often require a more intense sound to detect a signal masked by other sounds, especially when the signal contains frequencies that the person with a hearing loss has difficulty detecting in quiet.
The description of masking provided above applies to situations in which the masker and signal are presented at the same time. Masking can occur when the signal is turned off before the masker is turned on (backward masking) and when the masker is turned off before the signal is turned on (forward masking). Less masking occurs for forward and backward masking than for simultaneous masking. There is very little if any masking (i.e., the threshold for detecting a sound is the same as it was in quiet) if the masker and signal are separated by more than about 250 ms. Since people with hearing loss often have difficulty sorting out the temporal properties of sound, they can experience elevated forward and backward masked thresholds, compared with those measured for people with normal hearing.
Thus, a young otologically healthy person in the workplace can detect a signal sound over the frequency range of 20 to 20000 Hz, but the level of the signal sound required for detection depends on such variables as the frequency of the sound, the duration of the sound, and the nature of any other sound that may be present at or near the same time as the signal sound that may mask the signal sound. Masking means the detection threshold of a signal sound has been elevated by the presence of the masking sound. Loudness and pitch refer to subjective attributes of sound that are highly correlated with sound level and frequency, respectively. Sounds that are spectrally similar are more likely to mask each other than are sounds that are not spectrally similar. Signals are most difficult to detect when a masker and signal occur at the same time, but masking can occur when the signals and maskers do not temporally overlap. All of these measures of auditory perception can be adversely affected if a person has a hearing loss.
Sound Discrimination
Over a range of frequencies (approximately 500 to 4000 Hz) and levels (approximately 35 to 80 dB SPL) in which humans are most sensitive, listeners can discriminate a change of about one decibel in sound level and about a half of a percent change in tonal frequency. For instance, a 50 dB SPL sound can be just discriminated from a 51 dB SPL sound, and a 2000 Hz tone can be just discriminated from a 2010 Hz tone. A hearing
loss can lead to elevated level and frequency difference thresholds, making it difficult for the person with a hearing loss to discern the small differences in level and frequency that often accompany changes in the speech waveform.
Long-duration sounds require a larger change in duration for duration discrimination than do shorter duration sounds, although the exact relationship between duration and duration discrimination depends on many factors. Listeners can discriminate a sound whose overall level fluctuates (the sound is amplitude modulated) from a sound whose overall level is steady over time, when the rate of amplitude fluctuation is less than about 50 cycles per second. A sound that is amplitude modulated consists of a carrier sound that has its level varied by a different function, called the modulator. Thus, the level of the carrier sound increases and decreases over time in a manner determined by the modulator.
All of these measures of sound discrimination do not change appreciably as a function of the presence of masking sounds as long as the signal sound is readily detectable. Many people with hearing loss, especially the elderly, have difficulty processing the temporal structure of sounds. These people usually have high temporal difference thresholds, and they require slow rates of amplitude fluctuation to discriminate a fluctuating sound from a steady sound. Thus, people with such losses may not be able to follow some of the rapid fluctuations in sound intensity that are present in many everyday sounds, such as speech and music.
Thus, in the workplace, very small changes in sound level, frequency, and duration can be discriminated even when some masking sounds also exist. As long as the level of a sound does not vary too rapidly, listeners in the workplace should be able to determine that the sound is fluctuating in level (in loudness). People with a hearing loss often perform less well in these auditory discrimination tasks than people with normal hearing.
Sound Identification
Almost all of the research on sound identification has involved speech sounds. The recognition or intelligibility of speech sounds has been studied for a wide range of conditions. These conditions include both alterations of the speech sounds (e.g., whether there is a masking sound present) and aspects of the requirements of the listening task (e.g., the extent to which memory is required). In many speech recognition tasks, listeners are asked to identify phonemes (e.g., vowels), words, nonsense words, or sentences. The recognition task can be open set, in which the listeners are not aware of the set of speech utterances that will be presented, or closed set, in which the speech utterances are known (i.e., come from a list of words or sentences that the listener is aware of). Masking
sounds are usually white noise, speech spectrum noise, or other speech sounds.
Speech recognition or identification is usually measured in one of two ways: the percentage of utterances correctly identified or the level of some stimulus parameter (e.g., the level of a masking noise) yielding a particular percentage-correct speech identification value (e.g., 50 percent correct identification). The speech recognition threshold (SRT) is the level of the speech signal expressed in dB required for a criterion level of performance (e.g., 50 percent correct identification). The term “signal-to-noise ratio” (S/N ratio) is used for the ratio of the speech signal level to masker level (S/N ratio is usually expressed in decibels, and as such S/N ratio is the decibel difference between the level of the speech signal and the masker), when noise is used to mask the ability of listeners to recognize speech and when the levels of the speech and masker are expressed in decibels. In some tasks, another sound (e.g., a brief acoustic click) may be embedded in the speech sound and the detection of the click is used as a measure of how salient different parts of the utterance may be (e.g., if the click is not readily detected, then it may be inferred that the information temporally surrounding the click was crucial for speech processing).
Intelligibility of speech processed in quiet by listeners with normal hearing is somewhat resistant to many forms of physical alterations. Speech can be filtered (allowing only selected frequencies to be presented), speeded up or slowed down, clipped in amplitude, etc., and still be intelligible in a quiet listening environment. However, speech is susceptible to masking or interference from other competing sounds, especially other speech sounds. Several different methods have been proposed to determine the intelligibility of speech in the presence of competing sounds. The articulation index (AI, now called speech intelligibility index, SII), which was devised at Bell Laboratories for developing the telephone system in the 1930s and 1940s, is one method that is currently used (see American National Standards Institute, 2002) to estimate speech intelligibility for situations in which the physical properties (e.g., the spectrum) of the speech and interfering sounds are known. One rule of thumb for listeners with normal hearing is that for a broadband masking stimulus such as a white noise, approximately 50 percent intelligibility occurs when only the speech and noise information is provided and the overall levels of the speech words or syllables and noise are about equal (i.e., when the S/N ratio is zero dB). However, many conditions can alter the relationship between S/N ratio and performance. Listeners with hearing loss often have much more difficulty in recognizing speech that is altered, and their S/N ratio is usually greater than zero dB.
Many different speech tasks and speech utterance lists have been developed to assess the ability of listeners, especially those with hearing
losses, to process speech. These tests allow one to determine more precisely how different components of speech (e.g., vowels versus consonants) are processed or the extent to which familiarity with words influences speech intelligibility. There are many variables that might make it easy or hard to recognize a speech utterance. Speech tests are usually designed to determine if only one or maybe a small number of variables affect the ability of the subject or patient to recognize speech. For instance, many speech tests are intended to determine how much difficulty a person with a high-frequency hearing loss might have in recognizing speech. If the speech test consists of words that the patient is not familiar with, then poor performance on the test might indicate a difficulty with vocabulary rather than a hearing loss. Using test words in a language in which the patient is not fluent could also confound the assessment of hearing loss. Thus, many different speech recognition tests have been developed for the purpose of assessing hearing loss, to help ensure that the results are valid indicators of the relationship between speech recognition and hearing loss. Additional description of several speech intelligibility tests is provided in Chapters 3 and 7.
Processing speech in the workplace can be compromised when competing sounds are present. Sound reproduction systems do not have to be high fidelity to provide for acceptable speech intelligibility in the absence of competing sounds for people with normal hearing, but for people with hearing loss, such high fidelity may be essential for speech communication. However, the higher the fidelity of the reproduction system, the better speech recognition is likely to be when interfering sound sources are present. Hearing loss can lead to a significant loss of speech recognition even with high-quality amplification systems.
Sound Localization
Sound itself has no spatial dimensions, but the source of a sound can be located in three spatial dimensions as a function of the auditory system’s ability to process the sound emanating from a sound source. These dimensions are azimuth—the direction from the listener in the horizontal plane (see Figure 2-3); elevation—the vertical or up-down dimension; and range—distance or the near-far dimension. A different set of cues is used by the auditory system to locate sound sources in each spatial dimension. Sounds from sources located off-center in the azimuth direction arrive at one ear before they arrive at the other ear, and the sound at the near ear is more intense than the sound at the far ear. Thus, interaural differences of time and level are the two cues used for azimuthal (directional) sound localization; interaural time is the major cue for locating low-frequency (below 1500 Hz) sound sources, and interaural
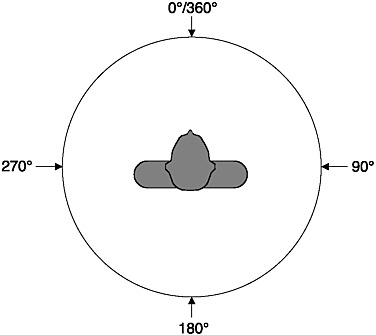
FIGURE 2-3 Azimuth: Overhead view of the listener.
level is the main cue at high frequencies. The interaural level difference results from the fact that the head and body provide an acoustic “shadow” for the ear farther away from the sound source. This “head shadow” produces large interaural level differences when the sound is opposite one ear and is high frequency. Human listeners can discriminate a change in sound source location of about 1-3° angle.
As sound travels from its source to the outer ears of a listener, it passes over and around (is diffracted by) many parts of the body, especially the pinna. These body parts attenuate and slow down the sound wave in a manner that is specific to the frequency of the sound and to the relationship between the location of the sound source and the body, especially the relative vertical location of the source. The head-related transfer function (HRTF) describes the spectral changes that a sound undergoes between the sound source and the outer ear canal. High-frequency sounds are attenuated in a frequency-specific manner that is dependent on the vertical location of the sound source relative to the body. That is, different HRTFs are produced for different vertical sound source locations. In particular, there are spectral regions of low amplitude (spectral notches or valleys) whose spectral loci are vertical-location-specific. Thus, these spectral notches in the HRTF can be a cue for vertical location. The spectral
cues associated with the HRTF are probably also used to help discriminate sounds that come from in front of a listener from those that come from behind. For instance, a sound coming from directly in front of a listener will provide the same interaural time and level differences as a sound coming from directly behind. Spectral cues derived from the HRTF can assist in reducing front-back localization errors.
Faraway sounds are usually softer than near sounds, and this loudness cue can be used to determine the distance of a sound source, assuming the listener has some knowledge about the nature of the source (i.e., some knowledge about how intense the sound is at the source). If there is any reflective surface (e.g., the ground), then the reflection from a near sound source is almost as intense as the sound that arrives at the ears directly from the source, whereas for a faraway sound the reflected to direct sound level ratio is lower. Thus, the ratio of reflected to direct sound level can be a cue for sound source distance perception, and distance perception is poorer in conditions in which there are no reflections.
Locating sound sources can be more difficult for people with hearing loss. This is especially true for listeners with unilateral hearing loss. If a single hearing aid or cochlear prosthesis is used, it may provide only limited assistance for sound localization, since binaural processing is required to locate sounds in the horizontal plane. However, fitting each ear with a hearing aid or cochlear prosthesis does not always assist the patient in sound localization. In most cases, the two aids or prostheses do not preserve all of the acoustic information required by the auditory system to localize a sound source.
In reverberant spaces, such as a room, the sound waveform reflects off the many surfaces, resulting in a complex pattern of sound arriving at the ears of a listener. Listeners are usually not confused about the nature of the actual sound source, including its location, in many reverberant spaces, presumably because the auditory system processes the first sound arriving at the ears and inhibits the information from later-arriving reflected sounds. Since the sound from the source will arrive at the listener before that from any longer-path reflection, auditory processing of the direct sound takes precedence over that of the reflected sound, usually allowing for accurate sound processing even in fairly reverberant environments. Another aspect of sound reflections is that the sound in a reflective space remains in the space after the sound production ends, due to the sound continuing to reflect off the many surfaces. The reverberation time is the time (measured in seconds) that it takes the level of this reverberant or reflected sound to decay by a specified number of decibels, which is usually 60 dB. Rooms that are large and reflective have long reverberation times. People with hearing losses often perform very poorly in reverberant spaces, and the poor performance may persist even when
they use a hearing aid or cochlear prosthesis. That is, people with hearing loss have difficulty recognizing speech signals when the reverberation time is long, especially if the acoustic environment is also noisy.
The detection of a signal sound source at one spatial location in the presence of a masking sound source at another spatial location is improved when the signal and masking sound sources are further apart. That is, the ability to detect a masked signal can be enhanced if the masking sound source is spatially separated from the signal sound source. The improvement in detection threshold as a function of spatial separation is referred to as the spatial masking-level difference. Thus, a variable that could affect speech recognition is the spatial separation of the test signal and other sound sources in the listening environment. Patients fitted with two hearing aids can sometimes take advantage of detecting sounds based on their spatial separation, whereas this becomes more difficult if the patient only uses one hearing aid.
Listening systems that make full use of the spectral information contained in HRTFs of individual listeners can produce sound over headphones that provides a percept as if the sound was emanating from an actual sound source located at some point in space (e.g., in a room). Systems that use HRTF technology can produce a virtual auditory environment for headphone-delivered sounds. Such HRTF-based systems can be used for testing and experimentation, eliminating the need to have specialized calibrated rooms for presenting sounds from different locations.
Thus, in the workplace, listeners can determine the location of sound sources located in all three spatial dimensions. The ability to detect a signal source can be improved if potential masking sound sources are spatially separated from the signal sound source. Having a hearing loss can compromise a person’s ability to locate sounds, and hearing aids may not assist him or her in locating sound sources.
Sound Source Determination
Colin Cherry (1953) pointed out that even at a noisy cocktail party, the human listener is remarkably good at determining many of the sources of sounds (different people speaking or singing, clanging glasses, music from a stereo loudspeaker, a slammed door, etc.), even when most of the sounds from these sources are occurring at approximately the same time. Bregman (1990) referred to this cocktail party effect as “auditory scene analysis.” Since the sounds from many simultaneously presented sound sources arrive at the ears of a listener as a single sound field, it is the auditory system that must determine the various sound sources—that is, determine the auditory scene. Little is known about how the auditory system accomplishes the task of auditory scene analysis, but several po-
tential cues and neural processing strategies have been suggested as ways in which the sources of many sounds can be processed and segregated in a complex, multisource acoustic environment. People with hearing loss often remark that they have problems in noisy situations, such as at a cocktail party, implying that they are not able to determine the auditory scene as well as people without hearing loss.
Thus, listeners with normal hearing can use many potential cues to determine many of the sources of sounds in the workplace, even when the sounds from the sources overlap in time and perhaps in space.
CAUSES OF HEARING LOSS
In general, hearing loss can be caused by heredity (genetics), aging (presbycusis), loud sound exposure, diseases and infections, trauma (accidents), or ototoxic drugs (drugs and chemicals that are poisonous to auditory structures). Hearing loss can categorized into the following ranges based on PTAs (PTA 512):
-
slight (16-25 dB hearing loss)
-
mild (26-40 dB hearing loss)
-
moderate (41-55 dB hearing loss)
-
moderately severe (56-70 dB hearing loss)
-
severe (71-90 dB hearing loss)
-
profound (greater than 90 dB hearing loss)
The loss can be caused by damage to any part of the auditory pathway. Three major types of hearing loss have been defined: conductive, sensorineural, and mixed. Conductive hearing loss refers to damage to the conductive system of the ear—that is, the ear canal, tympanic membrane (eardrum), and ossicles (middle ear bones)—and can include fluid filling the middle ear space. Sensorineural hearing loss indicates a problem in the inner ear, auditory nerve, or higher auditory centers in the brainstem and temporal lobe. Mixed hearing loss designates that the hearing loss has both a conductive and sensorineural component. Treatments for hearing loss involve surgery, hearing aids of various types, cochlear prostheses, medication, and various forms of habilitation and rehabilitation.
Conductive Hearing Loss
If a problem arises in the external or middle ear, a conductive hearing loss occurs that is largely due to the outer and middle ear’s no longer being able to overcome the loss in sound transmission from the outer to
the inner ear. Many conductive hearing losses, due to such causes as a perforation in the tympanic membrane, loss of ossicular continuity, or increased stiffness of the ossicular chain, can be repaired surgically, restoring the conductive hearing loss. During an acute ear infection, fluid can accumulate in the middle ear, resulting in a temporary conductive hearing loss. If the ear develops chronic otorrhea (drainage of purulent fluid), an infected skin cyst (cholesteatoma) may be the cause. A conductive hearing loss without ear pain is the usual course of this disease, but medical attention must be sought to prevent extensive damage to the ossicles and inner ear. Such conductive losses can produce up to a 60 dB hearing loss.
Sensorineural Hearing Loss
Sensorineural hearing loss is caused by problems associated with the neural transduction of sound. Diseases and disorders that damage the cochlea and auditory nerve result in a sensorineural hearing loss. In the past, sensorineural hearing loss was referred to as “nerve deafness”; however, in most instances of sensorineural hearing loss, the auditory nerve is intact and an impairment in the hair cells within the inner ear results in the hearing loss. Loss of hair cells and the neurotrophic factors that they produce eventually lead to nerve cell loss. Because the hair cells and auditory nerve complex relay information in a frequency-specific manner to the brainstem and brain, loss of hair cells in a particular part of the cochlea will cause hearing loss in a particular frequency region. Hair cell damage at the base of the cochlea near the stapes causes high-frequency hearing loss, while hair cell loss away from the base (near the apex) leads to low-frequency hearing loss. Sensorineural hearing losses due to cochlear damage can occur at any frequency and can range from mild to profound.
Presbycusis and noise exposure are the most common causes of adult hearing loss. Both result in an initial high-frequency sensorineural deficit, caused by damage to hair cells at the base of the cochlea. In presbycusis, other cells in the inner ear are also affected in many cases; these include the nerve cells that innervate the hair cells and cells in the structure known as the stria vascularis. In individuals with these conditions, other parts of the inner ear still function, allowing for the normal perception of low-frequency sounds. The primary difficulty for such a person lies in an inability to distinguish high-frequency sounds, such as the consonants of speech that are crucial for human communication. As the conditions progress, middle- and low-frequency hearing can also deteriorate. Traditional acoustic amplification (hearing aids) is often ineffective at making
speech sounds understandable to individuals with high-frequency hearing loss when the loss becomes severe (Ching, Dillon, and Byrne, 1998; Hogan and Turner, 1998). In individuals who have profound sensorineural hearing loss across the frequency range, hearing aids may not be as effective in improving hearing as a cochlear implant. Infections (viral or bacterial), disorders such as Meniere’s disease and autoimmune inner ear disease, hereditary disorders, trauma, and ototoxic drugs are other causes of sensorineural hearing loss.
Acoustic trauma can be a significant problem in the workplace. If the level of sound is intense, especially if the sound lasts for a long time, listeners exposed to such intense sounds may experience either a temporary or a permanent threshold shift—that is, their threshold for detecting sound is either temporarily or permanently elevated above that measured in quiet and before the exposure. A temporary threshold shift (TTS) can recover to normal detection threshold after a few minutes to a few days, depending on the parameters of the exposing sound and their relationship to those of the sound to be detected. Permanent threshold shifts (PTS) never recover and therefore indicate a permanent hearing loss that can range from mild to severe.
There is a trade-off between sound level and duration in terms of producing TTS and PTS. The greater the level or the longer the duration of the exposing sound, the greater the threshold shift and the longer it takes to recover from TTS. Most TTS occurs at frequencies the same or slightly higher than the frequency of the exposing sound. The Occupational Safety and Health Administration (OSHA) and the National Institute of Occupational Safety and Health provide regulations and guidance (e.g., Occupational Safety and Health Administration, 2002) for occupational noise exposure to mitigate its effects in the workplace.
Etiology of Severe to Profound Hearing Loss
It is estimated that 1 person in 1,000 has a severe to profound hearing loss. The number with bilateral (both ears) profound deafness is lower. Loss of hearing has a significant impact on the development of speech and spoken language skills, which are dependent on the age of onset of deafness. Children born with bilateral profound hearing loss or who acquire profound loss before the acquisition of speech and spoken language (approximately age 2 years) are identified as prelingually deafened. Children and adults deafened at any age after developing speech and spoken language are referred to as postlingually deafened.
Children
High-risk factors for congenital hearing loss include a family history of congenital hearing loss or delayed-onset sensory hearing loss of childhood, physical findings (birthweight less than 1,500 grams, craniofacial anomalies, the variable physical signs of Waardenburg’s syndrome), and maternal prenatal infections (cytomegalovirus, syphilis, rubella, herpes).
Prelingual deafness can also arise from severe vital function depression at birth when Apgar scores are in the 0 to 3 range at five minutes. Hyperbilirubinemia severe enough to require exchange transfusion; treatment of postnatal infection with ototoxic drugs, such as gentamicin, tobramycin, kanamycin, and streptomycin; systemic infections including meningitis, congenital syphilis, mumps, and measles all can result in bilateral profound deafness. Closed head trauma and neurodegenerative diseases including Tay-Sachs disease, neurofibromatosis, Gaucher’s disease, Niemann-Pick disease, and myoclonic epilepsy are rare additional etiologies of deafness that manifest themselves in childhood.
Genetic causes for prelingual and postlingual deafness are thought to account for 50 percent of the sensorineural hearing loss in childhood. The remainder are either environmental (about 25 percent) or sporadic idiopathic (about 25 percent). Genetic hearing loss can be congenital or delayed, progressive or stable, unilateral or bilateral, syndromic or nonsyndromic. The majority of genetic hearing losses are thought to be recessive (about 75 percent); 20 percent are attributable to dominant genes and a small percentage are X-linked disorders. Autosomal dominant disorders include Waardenburg’s syndrome (20 percent associated with hearing loss); Stickler’s syndrome (sensorineural or mixed hearing loss, 15 percent); branchiootorenal syndrome; Treacher Collins syndrome (sensorineural or mixed hearing loss); neurofibromatosis II; and dominant progressive hearing loss. Autosomal recessive disorders associated with sensorineural hearing loss are Pendred’s syndrome, Usher syndrome, and Jervell and Lange-Nielsen syndrome. Sex-linked syndromes associated with sensorineural hearing loss are Norie’s syndrome, otopalatodigital syndrome, Wildervaank’s syndrome, and Alport’s syndrome. Most hereditary hearing loss in early childhood is “nonsyndromic,” that is, there are no other apparent abnormalities. While mutations in any of dozens of genes can cause hearing loss, a gene that controls the production of a protein called connexin 26 is responsible for a large proportion of such cases, with studies on various populations reporting differing prevalences (Dahl et al., 2001; Erbe, Harris, Runge-Samuelson, Flanary, and Wackym, 2004; Gurtler et al., 2003; Nance, 2003).
Recently a condition termed “auditory neuropathy” (Starr, Picton, Sininger, Hood, and Berlin, 1996) has been identified in individuals with severe to profound hearing loss. Patients with this condition typically
show severely distorted or absent auditory brainstem response (explained in Chapter 3) with recordings showing prominent cochlear microphonic (CM) components, and normal otoacoustic emissions. The otoacoustic emissions and CM findings in patients with auditory neuropathy usually indicate that the cochlear hair cells, at least the outer hair cells, are functioning normally while the abnormal auditory brainstem response is indicative of disease in the inner hair cells, auditory nerve, or brainstem. Some theorize that the disorder is a specific neuropathy of the auditory nerve; thus the name of the disorder (Starr et al., 1996; Starr, Picton, and Kim, 2001). Starr et al. (2001) have found indirect evidence of peripheral nerve involvement based on sural nerve biopsy or nerve conduction velocity measures. More recently, they have documented specific neuropathy of the auditory nerve in a patient with well-documented clinical signs of auditory neuropathy on audiological tests (Starr et al., 2003). Such histological findings (fair to good hair cell populations along with poor ganglion cell and nerve fiber survival) have been reported previously (Hallpike, Harriman, and Wells, 1980; Merchant et al., 2001; Spoendlin, 1974; see Nadol, 2001, for a review of these pathologies in humans). However, others suggest that there is a “general lack of anatomic foundation for the label” (Rapin and Gravel, 2003, p. 707) because of difficulty in documenting specific peripheral neuropathy in patients, especially at the level of the auditory nerve.
When auditory neuropathy exists, neither the auditory brainstem response nor the otoacoustic emissions can be used to determine the degree of hearing loss. The degree of hearing loss in patients with this condition can be anywhere from none to profound (Sininger and Oba, 2001). The hearing loss of patients with auditory neuropathy can fluctuate dramatically and rapidly, sometimes within a single day (Sininger and Oba, 2001). In rare cases, increases in core temperature from fever can bring on severe to profound hearing loss that will return to prefever levels as the condition resolves (Starr et al., 1998). Recent publications report that cochlear implants have been effective in individuals displaying signs of auditory neuropathy (Peterson et al., 2003; Shallop, Peterson, Facer, Fabry, and Driscoll, 2001). This finding could be due to electrical synchronization of the neural response, or it may suggest that the etiology of profound deafness could be located in the inner hair cells.
An excellent review of sensorineural hearing loss in children can be found in a chapter by Brookhouser (1993).
Adults
Although many diseases and disorders can induce hearing loss, relatively few result in profound sensorineural deafness. Infections, immune-
mediated disorders, trauma, idiopathic and hereditary disorders, and ototoxic agents are the most common etiologies of bilateral profound hearing loss in adults.
The most common infections associated with profound hearing loss are bacterial and viral meningitis. Neural syphilis can produce progressive bilateral fluctuating hearing loss and spells of vertigo. If not treated with long-term antibiotics and steroids, profound hearing loss can occur. Even with treatment, some individuals continue to lose hearing.
Idiopathic disorders of cochlear otosclerosis and Meniere’s disease can produce profound bilateral hearing loss infrequently. Immune-mediated bilateral sensorineural hearing loss has recently been recognized as a cause of profound deafness. A history of rapid progressive hearing loss in both ears over weeks to months is the hallmark of this disease (McCabe, 1979). Fluctuation of hearing and spells of vertigo may occur. Other autoimmune disorders, such as Cogan’s syndrome, Wegener’s granulomatosis, and systemic lupus, also can result in profound hearing loss.
Aminoglycoside antibiotics have long been associated with ototoxicity. These drugs concentrate in perilymph and have a longer half-life in this fluid than in blood. In renal failure their levels are elevated. Aminoglycosides are directly toxic to outer hair cells, but they can also affect ganglion cells. Ototoxicity has been observed within the “safe” limits of nephrotoxicity. The effects can be observed even after discontinuing the drug. The agents associated with the highest degree of toxicity of the inner ear include kanamycin, tobramycin, amikacin, neomycin, and dihydrostreptimycin. (Some families may have a genetic disposition to developing deafness with these drugs.) The only treatment is to discontinue the drug. Antimetabolites such as Cisplatin and nitrogen mustard are also ototoxic. The risk to hearing is related to the amount of a given dose rather than cumulative amount.
TINNITUS AND HYPERACUSIS
Ear disorders cause many different symptoms, including hearing loss, tinnitus, pain, otorrhea (ear drainage), facial nerve paralysis, vertigo, and disequilibrium. Most of these symptoms are only tangentially relevant to the work of the committee, which is limited to the effects of hearing loss. In contrast, tinnitus and a related symptom, hyperacusis, may be considered by some to be “hearing impairments,” although neither tinnitus nor hyperacusis is mentioned in the current SSA regulations covering hearing impairment. We therefore offer a brief discussion in this section, concluding that most people with tinnitus or hyperacusis are not disabled (as that term is defined by SSA) and that the tests audiologists and otolaryngologists use to detect and measure abnormal func-
tion of the ear cannot separate people with tinnitus or hyperacusis who are disabled from those who are not disabled. For these reasons, these symptoms are not discussed in detail in later chapters, and we make no recommendations for procedures SSA might use in evaluating claims for disability based on tinnitus or hyperacusis. More extensive discussions are available in books edited by Tyler (2000) and Snow (2004), and in an earlier NRC report (National Research Council, 1982).
Tinnitus
Tinnitus is a sensation that often is associated with suffering, which may occasionally be severe enough to preclude work. Sensation and suffering are separate aspects of tinnitus and should not be confused.
Tinnitus sensation is the perception of sound when there is no external acoustic stimulus, and it can be either objective or subjective. Objective tinnitus occurs in rare cases when there is an internal acoustic stimulus. For example, turbulent blood flow in an artery close to the ear can make a pulsing sound that is audible not only to the person whose ear is affected, but also to a physician applying a stethoscope to the patient’s head. Subjective tinnitus almost always occurs when there is no acoustic stimulus at all, and only one person can hear the sound. People with subjective tinnitus typically describe their sensations as ringing, buzzing, humming, whistling, or hissing sounds. Most people with tinnitus also have hearing loss that is measurable by audiometry; however, many people with hearing loss do not have tinnitus, and no objective audiological or medical test has been shown to predict whether a person with hearing loss will have subjective tinnitus. In other words, the ears of people with hearing loss and subjective tinnitus are not different, as far as we know, from the ears of people with hearing loss alone.
When asked to match their tinnitus sensations to tones presented from an audiometer, most people select tones that are close to the frequencies they have difficulty hearing. The intensity of an external tone matched in loudness to a person’s tinnitus is usually less than 10 dB above that person’s threshold for the tone. Although there is no objective test that can demonstrate whether a person has tinnitus or not, tinnitus matching results that are repeatable and consistent with the patterns described above can sometimes offer evidence that corroborates a person’s claim to have tinnitus. However, tinnitus matching tests are not well standardized or widely used. Most importantly, they can at best describe tinnitus sensation and say nothing about tinnitus suffering.
The distribution of tinnitus suffering can be described as a pyramid. At its broad base are the majority of people with tinnitus, who find that it does not interfere significantly with their daily lives and never seek medi-
cal attention. The next level includes those who visit a physician, but only to find out whether their tinnitus is a sign of some serious medical problem. A smaller group of people with tinnitus complain of substantial difficulty with activities of daily life, especially sleep disturbance, trouble concentrating, emotional problems (anxiety, depression, etc.), and trouble understanding speech (Tyler and Baker, 1983). Since most people with tinnitus also have audiometrically measurable hearing loss, it is difficult to know whether tinnitus per se interferes with speech understanding. As stated by Stouffer and Tyler (1990), “it seems likely that patients confuse the effects of tinnitus on speech understanding with the effects of hearing loss on speech understanding.” At the narrow top of the pyramid are a very few persons who are severely disabled; cases of suicide have been reported, but almost exclusively in people who have many other risk factors for suicide, such as male sex, advanced age, social isolation, and especially depression (Lewis, Stephens, and McKenna, 1994).
Tinnitus sensation (as measured in the audiology booth using matching tests) and tinnitus suffering (as assessed by self-report) are uncorrelated (Baskill and Coles, 1999). Tinnitus sufferers differ from nonsufferers not in the pitch or loudness of the sounds they hear, but in the nature of their reaction, coping, and adjustment to tinnitus sensations. Patients who go to specialized tinnitus treatment clinics are very frequently found to meet formal psychiatric criteria for diagnosis of major depressive disorder, and about half of these have a history of major depression or anxiety disorder prior to the onset of tinnitus (Sullivan et al., 1988; Zoger, Svedlund, and Holgers, 2002).
Audiologists and otolaryngologists can provide useful information regarding the existence, perceptual qualities, and causation of tinnitus, but a person’s claim to be disabled by tinnitus might best be supported by psychiatric or psychological evidence, as well as by corroborative reports of employers, coworkers, family, and others; audiologists and otolaryngologists cannot provide objective evidence to support or refute such a claim. We note in passing that the American Medical Association’s Guides to the Evaluation of Permanent Impairment (American Medical Association, 2001) permit physicians to add up to 5 percent to a person’s “binaural hearing impairment” score if that person has tinnitus that interferes with activities of daily living. This could increase a person’s “whole person impairment” by no more than 2 percent, unless that person also had impairments in other domains (such as the “mental and behavioral” domain).
Most people with tinnitus do not choose to be treated, in large part because no treatment has been demonstrated to permanently eliminate tinnitus sensation (Dobie, 1999). Masking therapy (the covering up of tinnitus with external sound) can temporarily reduce or eliminate tinnitus
sensation while the masking noise is present. Tinnitus suffering is frequently addressed through psychological counseling and antidepressant or antianxiety drugs (Snow, 2004).
Hyperacusis
The word “hyperacusis” has sometimes been used to refer to “an exceptionally acute sense of hearing” (Dorland, 1974), but it is doubtful that there are actually people who hear so well that their thresholds are distinctly separate from, and better than, the normal distribution of hearing thresholds in young healthy adults. More commonly, clinicians use the term to refer to a relatively rare condition of abnormal intolerance of even moderately loud sounds, such as conversation, traffic, and music (the terms “hyperacusis” and “phonophobia” are often used interchangeably), usually accompanied by avoidance behavior (e.g., wearing earplugs at all times, staying at home, severing social relationships). People who complain of intolerance to everyday sounds usually have bother-some tinnitus as well, but they represent less than 1 percent of tinnitus patients at one national center (Vernon and Meikle, 2000). There is no widely used criterion for the diagnosis of hyperacusis, although some clinicians have used “loudness discomfort levels” (LDLs) for this purpose. People with normal hearing, and most people with hearing loss, report that they can tolerate tones up to 90-105 dB HL in the audiometry booth, while most patients who complain of severe sound intolerance have LDLs below 85 dB HL (Hazell, Sheldrake, and Graham, 2002). Most of these patients have required treatment for psychological problems prior to the onset of hyperacusis (Hazell and Sheldrake, 1991). Treatment of hyperacusis has usually consisted of desensitization to gradually increasing sound levels or treatment of underlying psychiatric disorders.
No objective audiological or medical test can distinguish people who complain of hyperacusis from other people. It is far from clear that hyperacusis should even be considered an ear disorder. As in the case of tinnitus, the best evidence supporting or refuting claims of disability based on hyperacusis is likely to come from psychiatrists and psychologists and from lay persons who can corroborate the claimed disability.
SUMMARY
Adults and children depend on hearing for their ability to function in work, school, and other daily activities, to be able to communicate using speech, and to better process information about objects in their environments. Using hearing to function in the world means detecting, discriminating, localizing, and identifying sound produced by the many sound
sources that constantly surround one. There are many causes of damage to the auditory system that result in a hearing loss that reduces one’s ability to detect, discriminate, localize, or identify sound. These losses can adversely effect an adult’s ability to work and a child’s ability to process sound.



























