
6
DEVELOPING NEW THERAPEUTIC INTERVENTIONS: FROM THE LABORATORY TO THE CLINIC
Moving from an experimental therapy in the laboratory to an approved treatment for patient use involves a prescribed series of steps that validate and ensure the safety and efficacy of the therapeutic intervention (Table 6-1). Although these steps in the drug and device development and approval process are time-consuming and expensive, they are designed and regulated to ensure patient safety.
This chapter discusses the challenges and opportunities involved in developing therapeutic interventions to treat spinal cord injuries. The chapter describes major steps along the research pipeline and discusses the issues involved in translating experimental therapies into clinical practice. Finally, the unique challenges of designing and implementing clinical trials of treatments and therapies for both acute and chronic spinal cord injuries and the tools needed to improve the efficiencies of clinical trials are discussed.
The development of new medications or devices that target a specific complication, such as preventing or reducing neuropathic pain, can take an average of 15 years (Quest for new cures, 2003; PhRMA, 2004a). It often takes at least 3 years, and often upwards of 7 years, for a potential therapeutic compound to be identified and for the preclinical research to be conducted. During this stage, potential therapies are tested, refined, and verified by using multiple in vitro and in vivo assessment assays. Once a likely intervention is identified, it moves through a set of clinical trials that examine the drug’s safety and efficacy in humans (Table 6-1). Using statistical methods, researchers can analyze the results of each phase to identify differences between the outcomes displayed by the test population, which received an experimental therapy, and the outcomes of a control popula-
TABLE 6-1 The Research Pipeline
|
|
|
SOURCE: Adapted from PhRMA (2004b); Quest for new cures (2003). |
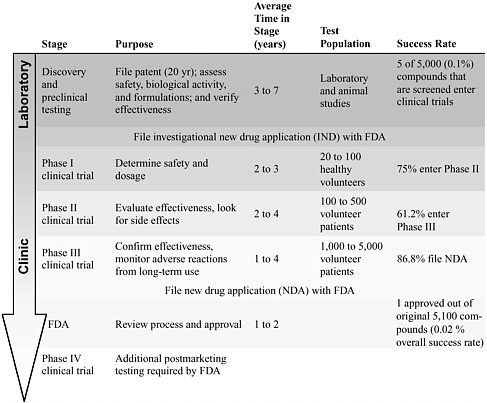
tion, which most often received a sham or a placebo treatment. If a clinical trial is designed and performed correctly, clinicians can use the results obtained with a limited number of participants to guide a treatment for an entire patient population (Matthew, 2001).
Phase I clinical trials are used to determine safety and an appropriate treatment dosage and regimen and, in some cases, are used to perform preliminary analysis of the biological activity of the intervention. Phase II and phase III clinical trials evaluate the efficacy of the new intervention and examine adverse effects in studies with larger populations. In the end, a novel drug that has entered a phase I clinical trial has only an approximately 30 to 40 percent chance of successfully completing a phase III clinical trial and being approved by the Food and Drug Administration (FDA) (Harding, 2004). Phase IV clinical trials are required by the FDA for additional analysis of long-term risks and benefits.
As described in Chapters 4 and 5, a range of approaches relating to the
treatment of spinal cord injuries are being explored, including efforts to prevent or reduce the adverse consequences during the acute phase of the injury and combination strategies designed to remyelinate nerve fibers, promote nerve fiber growth, and prevent cell death. During the development and verification of new therapies, researchers and regulators make decisions regarding when the data are sufficient to indicate that the intervention is efficacious and safe and can move on to the next stage of the process. The challenge is to develop therapies in a timely fashion without undermining future scientific endeavors and, most importantly, without endangering patient safety.
CRITERIA FOR VALIDATING A NOVEL THERAPY
The spinal cord injury research community is making substantial progress in developing novel therapies that may soon be ready for clinical trials. However, many of the alternative therapies that individuals with spinal cord injuries are using are not recommended options or standards of care because they have not been proven to be safe and efficacious (see Appendix F). An overriding concern that arises when a researcher contemplates translating a successful laboratory therapy to the clinical setting is the extent of preclinical data needed to justify proceeding with testing in studies with humans (Ramer et al., 2000; Kleitman, 2004). A coordinated and methodical approach is needed to verify the safety and effectiveness of therapies and treatments that are proceeding through the research pipeline. A 2003 article published in the Journal of Rehabilitation Research & Development describes a set of criteria that should be considered before a treatment can enter into a clinical trial (Table 6-2) (Dietrich, 2003). To meet these criteria, the author recommends that a coordinated effort among the spinal cord injury research community be mobilized to quickly respond to new scientific findings. As discussed in detail in Chapter 7, an enhanced research infrastructure and network is needed to facilitate collaborative research efforts.
Verification of a Therapy’s Preclinical Effectiveness in Replicated Studies with Animals
Preclinical testing provides data on whether a therapeutic intervention holds promise for the treatment of spinal cord injuries in humans. However, because the nature and severity of spinal cord injuries vary between individuals, a wide spectrum of behavioral and functional deficits exist, with no one outcome occurring among those with spinal cord injuries.
An emphasis on the replication of preclinical studies (replication studies) between laboratories is needed. Difficulties in getting replication studies
TABLE 6-2 Criteria for Drug Therapies Entering a Clinical Trial
|
|
SOURCE: Adapted from Dietrich, 2003. |
published and concerns over constraints in meeting the needs of the sponsoring funding agencies may be among the reasons for the lack of replication studies. The spinal cord injury research community needs to embrace and encourage these studies, which can be incorporated into broader studies that not only replicate a previous study but also include novel elements in the experiment. The National Institute of Neurological Disorders and Stroke (NINDS) identified the need for these types of studies by establishing the Facilities of Research Excellence in Spinal Cord Injury (FOR-SCI) funding mechanism in 2002 (NINDS, 2002). The Miami Project to Cure Paralysis is one of the two recipients of a FOR-SCI contract and is conducting studies to review and replicate studies in the areas of neuroprotection and axonal regeneration after a spinal cord injury. The second FOR-SCI contract is with the Reeve-Irvine Research Center and stipulates a focus on interventions to promote regeneration in the chronic setting. Similar contracts should be established for replication studies in other areas of relevant research.
Furthermore, it is critical that research findings, including those from replication studies and studies with negative or inconclusive results, be published in peer-reviewed journals with details about the study design, quantitative end points, and statistical analyses (Dietrich, 2003). Not only should positive study conclusions be presented, but a forum also should be generated to enable peer review of negative conclusions, especially those pertaining to replication studies. These efforts would enable the scientific community to scrutinize the data and would provide information to the spinal cord injury patient population on the results of preclinical and clinical testing of all novel therapies.
Safety Concerns in Moving to Tests with Humans
The safety of human subjects is of paramount concern when decisions are made about when and how to test therapeutic interventions in studies with humans. International codes of ethics, including the Declaration of Helsinki (World Medical Association, 2004), indicate that before an experimental therapy is tried in humans, a high standard of consensus about the appropriateness of a therapy should be established in the scientific community through laboratory and animal experiments (Sugarman, 1999). In 2001, the American Society for Neural Transplantation and Repair developed a set of guidelines that recommend that safety studies be conducted in the best available model—or in the case of spinal cord injuries, multiple models—before the therapy is tested in humans (Redmond et al., 2001). Failure to do so can put the study subjects at undue risk. Specifically, animal models should be used to examine potential toxicities and harmful complications (Dietrich, 2003).
Furthermore, the safety and efficacy of a novel therapy in relation to those of the alternatives available in a particular situation must also be considered (Sugarman, 1999). If no treatments are available and the patient population has a life-threatening condition, bioethicists have argued that “it seems reasonable to pursue experimental alternatives that may be somehow unsafe” (Sugarman, 1999).
Additional bioethical criteria that need to be considered to ensure that it is appropriate to move an experimental therapy from the laboratory bench to clinical trials include the following (Dekkers and Boer, 2001; Sugarman and McKenna, 2003):
-
the study should address an important research question that cannot be answered by use of an alternative study design;
-
the scientific community has reached a consensus about the safety of the proposed experiment on the basis of the findings from preclinical studies;
-
evidence that the intervention might ultimately be beneficial is sufficient;
-
the clinical trial design is based on sound science and has minimal risks and maximal benefits, the outcomes are measurable and meaningful, and the selection of subjects is fair; and
-
valid informed consent is obtained from each participant.
Concerns have been raised about the experimental therapies that are being requested and tried by individuals with spinal cord injuries, even though the interventions have not been assessed for safety and efficacy in
|
BOX 6-1 In recent years, more than 500 individuals with spinal cord injuries (including approximately 100 American patients) have been treated in Beijing, China, with an experimental therapy that involves surgically implanting olfactory ensheathing cells (see Chapter 5) above and below the site of injury (Lev, 2004). These individuals have been willing to spend more than $25,000 each for this procedure (Lev, 2004). Anecdotal reports relate that some individuals appear to regain partial function within days after the surgery, whereas other reports indicate that patients have severe infections shortly after the procedure (Huang et al., 2003; Lev, 2004). The scientific community has not received reports on whether standard preclinical testing has been performed and whether outcomes assessment measures have been collected from every patient, and little is known about whether long-term follow-up is being conducted (Huang et al., 2003). It is therefore unclear to what extent individuals with spinal cord injuries regain function after the procedure. There is general agreement, including from the lead physician performing this procedure, that the improvement occurs too soon after surgery for it to be a direct result of the olfactory ensheathing cells, neuronal regeneration, or remyelination of the remaining neurons. The benefit may be attributable to decompression of the spinal cord, a placebo effect of the surgery, or an as yet unidentified mechanism. The mechanism of recovery needs to be further elucidated. This procedure has raised many issues for individuals with spinal cord injuries and the research community. Individuals with spinal cord injuries argue that they should be able to make decisions regarding their own health. Many scientists and health care professionals are worried, however, that this is an invasive, costly, and potentially dangerous procedure that has not yet been validated (Lev, 2004). A phase I clinical trial of this intervention is under way in Australia and, when it is completed, will provide a better understanding of the safety and efficacy of olfactory ensheathing cell transplants. Until the results of the clinical trials are obtained, however, the spinal cord injury community should be cautioned against receiving such therapies. As Rosenfeld and Gillett have noted, “[s]tem-cell-based technology offers amazing possibilities for the future … but [t]he experimental basis of stem-cell or OEC [olfactory ensheathing cell] transplantation should be sound before these techniques are applied to humans with spinal cord injury” (Rosenfeld and Gillett, 2004). |
clinical trials (Box 6-1). Because of the devastating outcomes of a spinal cord injury, many individuals with such injuries are willing to try therapies that they think may have promise, even if there is the potential for adverse health effects. The challenge for the research community is to better inform individuals about clinical trials and to accelerate the development of interventions while carefully considering patient safety.
LESSONS LEARNED FROM PREVIOUS CLINICAL TRIALS OF SPINAL CORD INJURY INTERVENTIONS
Over the past 5 years, nearly 300 publications in peer-reviewed journals have elucidated the results of clinical trials of interventions for spinal cord injuries (see Appendix G). The majority of these trials are associated with therapies for chronic complications, particularly bowel and bladder problems, exercise and locomotion; spasticity and muscle control; and sexual function; and only a handful have assessed therapies for use during the acute phase of the injury (Figure 6-1). In addition, only a few large-scale phase II and III clinical trials have been conducted, in part because of the absence of therapies that are ready for that stage of the research process (Ellaway et al., 2004).
A 2004 review concluded that many of the most-cited clinical trials of interventions for spinal cord injuries had methodological limitations that hinder interpretation of their results and the ability to generalize their
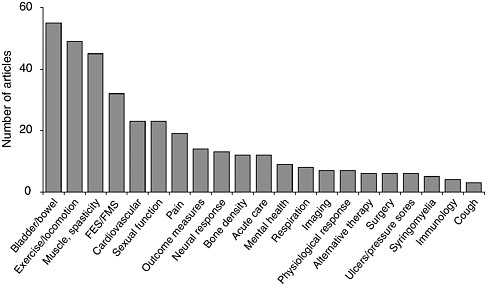
FIGURE 6-1 Publications on clinical trials for interventions for spinal cord injuries (1998 to 2003).
Multiple articles may result from the same clinical trial. A particular article may address multiple secondary conditions. As such, they are counted separately under each category. Abbreviations: FES = functional electrical stimulation; FMS = functional magnetic stimulation.
findings (Dobkin and Havton, 2004). In addition, as noted above, one of the issues raised in discussing clinical trials in this field is the use of alternative therapies that are highly experimental and that are not based on completed clinical trials or that are based on trials whose results are only putative (see Appendix F).
The International Spinal Research Trust (ISRT) recently asserted that continued advances in research laboratories will result in more experimental therapies being ready for clinical trials in the next few years. Furthermore, ISRT concluded that to meet this demand, new well-designed clinical trials that use novel tools to assess the effects of a treatment on the outcomes will need to be developed (Ellaway et al., 2004).
Useful lessons for future trials can be learned from the series of clinical trials that assessed the ability of methylprednisolone to improve the recovery of individuals after a spinal cord injury, including the original three National Acute Spinal Cord Injury Study (NASCIS) trials (Bracken et al., 1984, 1990, 1997) (see Appendix E). Although the authors of the NASCIS trials concluded that methylprednisolone should be a standard treatment for acute spinal cord injuries, many clinicians question its efficacy because of issues regarding the statistical analysis used to assess the improvements to the central nervous system and concern over adverse complications. The limitations of the studies in addressing some of the potential confounding variables may limit the extent to which the results of the studies can be generalized to all individuals with acute spinal cord injuries (Hanigan and Anderson, 1992; Bracken, 2000; Hurlbert, 2000; Dumont et al., 2001). For example, the NASCIS II trial did not include details about other interventions used (e.g., radiology, surgical manipulations, or the extent of rehabilitative therapies) that may have contributed to improvements or recovery (Hanigan and Anderson, 1992). Concerns have also been raised about the robustness of the statistical analysis and the heterogeneity of the injured population used in the studies, which made the baselines of the different populations difficult to compare (Bracken and Holford, 2002). Furthermore, although it was concluded that the NASCIS II and III trials were “well designed and well executed,” post hoc analysis of these trials “failed to demonstrate improvement in primary outcome measures” (motor scores, pinprick scores, and light-touch scores) (Hurlbert, 2000). Consequently, it has been stated that the data describing improved recovery with methylprednisolone treatment are weak and that the improvements observed may represent random events (Hurlbert, 2000). Some have also suggested that it should not be a recommended treatment (Hurlbert, 2000). However, concerns about the potential legal ramifications of not using methylprednisolone may cause some physicians to continue to treat their patients with the medication. To strengthen future trials, it is important to learn from the experiences of the NASCIS studies and other similar studies.
CHALLENGES IN CONDUCTING CLINICAL TRIALS OF SPINAL CORD INJURY INTERVENTIONS
The emergency nature of the incidents that cause spinal cord injuries, the heterogeneity of the resulting injuries and functional limitations, the longitudinal time course, the absence of standardized outcome measures, and the diverse community and the small number of individuals who receive such injuries all contribute to unique challenges in designing and conducting clinical trials of interventions for the treatment of spinal cord injuries.
Obtaining Informed Consent for Acute-Care Clinical Trials
Immediately after a spinal cord injury the body triggers a cascade of responses that can further injure the spinal cord and cause additional complications (see Chapter 2). Many of these changes occur within the first few hours after the injury. For a treatment to be effective at preventing the majority of the complications that arise during the acute stage of the injury, it must be provided to a patient soon after the injury; this period of time is generally thought to be 8 hours (Clifton et al., 2002).
To ensure that a patient is fully aware of the potential benefits and hazards of participating in a clinical trial, federal regulations require that patients provide informed consent before the experimental treatment is initiated. For patients who do not have the capacity to give informed consent, legal proxy can be obtained from a family member or a guardian. Therefore, obtaining informed consent for most clinical trials that examine interventions in the chronic stage of spinal cord injury has not been a major hurdle. However, experimental interventions are being developed that will require clinical trials involving patients in an emergency setting who may be unable to provide informed consent.
However, obtaining informed consent in emergency-care situations has been fraught with challenges. In many cases, interventions need to be administered as soon as possible after the injury and there is no time to wait for a patient to recover sufficiently or for a legal proxy to be obtained (Smithline and Gerstle, 1998). In 1996, revisions to the federal informed-consent regulations were made to allow a limited waiver for informed consent in the case of clinical trials of emergency-care interventions or other similar situations (DHHS, 1996; Smithline and Gerstle, 1998; Clifton et al., 2002). The previous regulations allowed for a waiver of informed consent only if the patient would receive no more than a minimal additional risk by entering into the trial. In the new language, the definition of the risk of the research was changed to “reasonable in relation to what is known about the medical condition of the potential class of subjects” (DHHS, 1996).
This change in language has made a significant difference in the ability of clinical trials of emergency medical treatments to proceed.
An illustration of the need for careful consideration of the design of clinical trials for acute-care interventions is found in the National Acute Brain Injury Study: Hypothermia trial. That study, which was initiated in 1994, before the new waiver consent rules were implemented, examined whether cooling the brain to 33°C within 8 hours of the injury could prevent further secondary damage. No positive effect was found; however, the details of the study design demonstrated the need for timely waiver consent (Clifton et al., 2002). During the first 9 months of the study, on average, it took close to 12 hours to obtain informed consent, initiate the treatment, and achieve the target temperature. However, after the new regulations were passed and the study design was changed to include waiver of consent if a responsible family member could not be located within 1.5 hours after admission of the injured patient to the hospital, the average time dropped to 7.9 hours (Clifton et al., 2002). The authors argue that it would be “impractical and unjust to perform studies of acute brain injury without use of waiver consent when the treatment window is less than six hours” (Clifton et al., 2002). Many similar issues apply to clinical trials for the emergency treatment of spinal cord injuries, and informed-consent waiver guidelines should be developed and standardized for use in clinical trials of interventions for acute care for spinal cord injuries.
Challenges in Patient Recruitment
Clinical trials require an adequate number of participants to ensure that the study groups are representative of the larger patient population and to overcome the variations in results unrelated to the intervention (Pocock, 1983). Furthermore, it is important that the control groups be as homogeneous as possible so that valid comparisons can be made. The combination of these two requirements results in narrow eligibility criteria and, thus, can result in longer patient accrual times (NRC, 1993).
For example, when Acorda was recruiting individuals with spinal cord injuries for a phase III trial of 4-aminopyridine (fampridine), it was estimated that about 1,200 patients would have to be screened to locate 400 patients who met the broad eligibility requirements (inclusion criteria) for the study (Blight, 2004). This process can be much more difficult for trials that have stricter inclusion criteria. The number of potential patients is made even smaller because few individuals enroll in clinical trials. For example, according to the National Cancer Institute, only 2 to 3 percent of cancer patients are ever enrolled in clinical trials (IOM, 1999). It is not totally clear why these numbers are so low, even though the general population is increasingly aware of clinical trials. However, the eligibility re-
quirements are often strict and many patients have concerns about sharing personal information with researchers.
As described throughout this report, individuals with spinal cord injuries are presented with countless challenges in their everyday lives that make it difficult for them to travel. Depending on the severity of the injury and the resources available in their communities, an individual’s ability to travel independently may be further limited. Furthermore, for some individuals the travel necessary for participation in a clinical trial would be a significant expense. These challenges may limit the number of individuals available for a clinical trial, although there are opportunities to use existing regional patient care centers to facilitate access to clinical trials.
To increase the number of individuals with spinal cord injuries as potential clinical trial participants, health care professionals must be aware of ongoing clinical trials so that they can educate their patients about the available options and the benefits of participating in clinical trials. Efforts to increase the dissemination of information to both health care professionals and individuals with spinal cord injuries are needed.
Spontaneous Recovery Can Complicate Interpretation of Results
As discussed in Chapter 1, a small number of patients with incomplete injuries and paraplegia recover some function, particularly bowel and bladder function, usually within the first year after the injury (Maynard et al., 1979; Ditunno et al., 2000). Thus, the potential for the recovery of function that is not due to the intervention complicates the interpretation of clinical trial results and necessitates careful attention to the matching of a control group and the population receiving the intervention.
NEXT STEPS IN DESIGNING AND CONDUCTING CLINICAL TRIALS
Although investigators performing clinical trials of interventions for spinal cord injuries are presented with unique challenges, many tools and techniques can be used to maximize participation and improve data analysis.
Utilize Statistical Methodologies for Addressing Small Numbers of Patients and Heterogeneity of Outcomes
The limited number of individuals who qualify for clinical trials of interventions for spinal cord injuries and the heterogeneity of the nature and the severity of their injuries make it difficult to conduct multiple large-scale, randomized, controlled clinical trials. Furthermore, for studies that
examine cell- or gene-based interventions, standard research designs—which often require large numbers of research participants to achieve adequate statistical power—may not always be possible or appropriate. Therefore, clinical trials may need to be performed with a small sample of patients (small “n” trials). Depending on the clinical end points and the magnitude of the effects desired, a small “n” trial may involve only a few patients or as many as 100 patients. Statistical methodologies (e.g., sequential analysis, hierarchical models, Bayesian analysis, and decision analysis) can be used to provide substantial evidence of efficacy in studies with small sample sizes (Table 6-3) (IOM, 2001). If it is necessary to perform a clinical trial with a small population of participants, a recent Institute of Medicine (IOM) report (2001) recommended that the following steps be followed:
-
clearly define the research question;
-
tailor the study design by giving careful consideration to alternative methods and involve statisticians early in the design process;
-
clarify sample characteristics and methods for reporting the results of clinical trials with small sample sizes;
-
perform corroborative analyses to evaluate the consistency and robustness of the results; and
-
exercise caution in the interpretation of the results before attempting to extrapolate or generalize the findings.
The use of small “n” methodologies can be especially useful for phase I clinical trials that investigate the safety and dosage of new medications or
TABLE 6-3 Appropriate Contexts in Which to Consider a Clinical Trial with a Small Population
|
Context |
Example |
|
A new drug or procedure will be used for the first time in human subjects. |
A traditional phase I study may be used to determine the maximum tolerated dose of a new drug. |
|
The number of subjects available for study is extremely limited. |
The investigator wishes to study changes in bone mineral density in astronauts during extended stays in space. |
|
The study population is small, isolated, or unique. |
The investigator is studying health outcomes unique to a small isolated tribe or in patients with a rare disease. |
|
SOURCE: IOM, 2001. |
|
devices in studies with a small number of patients. Phase II and phase III clinical trials can be performed with small samples of patients, but they usually involve larger sample sizes. As stated in the recent IOM report, “[a] small clinical trial often guides the design of a subsequent trial. Therefore, a key question will be what information from the current trial will be of greatest value in designing the next one?” (IOM, 2001). Many of these methodologies have not been field tested in situations typical of spinal cord injury clinical trials; therefore, it may be appropriate to develop a research initiative to gather experienced clinical trialists to provide recommendations on how small “n” clinical trials could be effectively applied to spinal cord injury clinical trials.
Clinical trials of neuroprostheses provide examples of trials that use a relatively small number of subjects to provide a statistically significant result. These trials are characterized by outcomes that are often clear and immediate. Furthermore, as demonstrated by the examples in Box 6-2, the subject can serve as his or her own control. This allows the outcomes obtained postoperatively with the neuroprosthesis off and on to be compared with the individual’s performance before implementation of the neuroprosthesis. Study designs for clinical trials of neuroprostheses can therefore be successful with only a small number of subjects, and FDA has considered this type of study design to be acceptable in the steps leading to the premarketing approval of neuroprostheses.
Increasing Clinicians’ Expertise in Conducting Clinical Trials
Designing, implementing, and conducting a large-scale clinical trial that has adequate controls and that can provide statistically significant outcomes data require health care professionals who are well trained in clinical trial methodology. This ensures that the therapies are administered in an appropriately controlled fashion, that surveys are properly conducted, and that the findings of the surveys and outcome measures are properly assessed. NINDS has recently recognized the need to provide intensive training in this area and is in the process of developing a course for clinical fellows and junior faculty in neurology or a neurosurgical subspecialty on the principles of clinical trial methodologies for neurology research.
Use of Multicenter Trials and Centralized Institutional Review Boards
In an effort to maximize the number of participants who can qualify for a trial, many large-scale clinical trials are designed by creating collaborative efforts between multiple research centers and trauma hospitals (Box 6-3). However, multicenter trials are costly and can be difficult to implement (NRC, 1993). Furthermore, a careful study design, careful study implemen-
|
BOX 6-2 Clinical trials of neuroprostheses can be designed so that a small number of patients can provide the information needed to provide proof of the efficacy of the prosthesis. The individual’s performance before use of the neuroprosthesis can be compared with the individual’s postoperative performance with the neuroprosthesis off and on. Studies are performed to assess the changes in performance over time to ensure the stable performance of the neuroprosthesis (a minimum of 12 months). A trial to examine a system for restoring hand grasp and release in people with C5-C6 tetraplegia assessed outcomes in each of three domains: impairment, disability, and handicap (Peckham et al., 2001). The hypothesis was that the person would have improvements in strength and range of motion of the hand (impairment measures); ability to grasp more objects in a standard pick-and-place test (disability measure); and ability to perform more activities of daily living (handicap measure). The protocol called for preoperative measurement of performance and postoperative measurement of performance with the neuroprosthesis off and on. Power analysis predicted that statistical significance would be achieved with less than 20 subjects, because the impact of the intervention was a significant restoration of capabilities when the device was on compared with the individual’s capabilities (nearly no function) in either the preoperative state or in the postoperative state with the device off. FDA allowed the application for premarketing approval to be submitted with less than 30 subjects. In a second clinical trial of a bladder neuroprosthesis, the individual’s performance was determined in the same three states described above: preoperatively and postoperatively with the prosthesis off and on (Creasey et al., 2001). A similar power analysis predicted that the study would achieve statistical significance with less than 20 subjects, and this was achieved for the primary outcome measure of the amount of residual urine in the bladder. Additional secondary measures were also evaluated. |
tation, and proper training of the participating physicians and nurses are required to ensure that the same standards of care and assessment are provided to each participant, regardless of where they receive the treatment being evaluated in the trial.
One large hurdle for multicenter trials has been the necessity of receiving the approval of the institutional review board (IRB) of each of the many research centers participating in the trial. The responsibility of the IRB is to ensure that human subjects are protected from all unnecessary harms associated with the study; however, for those responsible for conducting a multicenter trial, the task of obtaining IRB approval from multiple institutions is often time-consuming and cumbersome. As a solution to this problem, central IRBs began receiving accreditation in 2003. The
|
BOX 6-3 For more than 30 years, corticosteroids, including methylprednisolone, have been used to treat head injuries. Corticosteroids were given to patients to help control posttraumatic inflammatory changes, which are believed to contribute to neuronal degeneration. The use of this treatment was based on randomized clinical trials that included no more than a few hundred patients in each trial and a total of approximately 2,000 patients (Roberts et al., 2004). Meta-analysis of these trials suggested that the absolute risk of death in the corticosteroid-treated groups was about 1 to 2 percent lower than that for the controls (Alderson and Roberts, 1997). To confirm this benefit, researchers initiated the corticosteroid randomization after significant head injury (CRASH) trial. The CRASH trial included close to 20,000 patients who were recruited from more than 239 hospitals in 49 countries. The levels of coordination and integration of the data required in this trial are a model for future large multicenter trials. The CRASH trial analyzed the effects of early administration of a 48-hour infusion of methylprednisolone on the risk of death and disability after a head injury (Roberts et al., 2004). The CRASH trial found that the risk of death from all causes within 2 weeks of treatment was 3 percent higher in patients taking the corticosteroids than in those receiving a placebo. This study has two major limitations. First, the data were based on 2 weeks of follow-up data. The 6-month follow-up data have not yet been published. Second, the main cause of death was not noted; therefore, it is not known how many of the deaths were specifically due to the corticosteroid treatment. This study demonstrates the potential hazards of establishing treatments when a large randomized clinical trial has not been performed. As commented in an editorial published with the article, “[t]he key message of CRASH, however, is that applying treatments with unproven effectiveness is like flying blindly. In the future, we should avoid trusting in underpowered clinical trials with surrogate rather than clinical endpoints, and transferring evidence from one disease to another” (Sauerland and Maegele, 2004). |
private-industry sponsors of clinical trials prefer to use central IRBs for multicenter clinical trials, because a single process of review and approval can be used and the average time required to obtain approval is reduced (Loh and Meyer, 2004). However, the results of a survey of 125 major medical schools in the United States found that concerns about institutional liability and the loss of local representation in the review process are detracting from the use of central IRBs (Loh and Meyer, 2004). To increase academic and industry collaborations and reduce the administrative burden on local IRBs, the National Cancer Institute (NCI) developed a pilot program, the NCI Central Institutional Review Board (CIRB), which conducts a complete review of the trial but also allows local IRBs to focus on the implementation and specific issues related to the local conduct of
|
BOX 6-4
SOURCE: Adapted from NCI, 2004. |
the study (Box 6-4). This model system could be expanded for use in multicenter clinical trials in areas of research other than cancer, including spinal cord injury research.
Coordination of Care and Cure Efforts
It is important that ongoing efforts related to patient care and rehabilitation after a spinal cord injury be coordinated with efforts in developing therapeutic interventions for spinal cord injuries. A number of sites and systems can be used to conduct clinical trials of interventions for spinal cord injuries. The Model Spinal Cord Injury Systems of Care, funded by the National Institute on Disability and Rehabilitation Research (see Chapter 7), offers the resources of 16 hospitals and rehabilitation centers across the country with a known patient base and the staff and facilities that could be enlisted to conduct clinical trials. The resources of the Model Spinal Cord Injury Systems of Care have been used to conduct clinical trials of the drugs 4-aminopyridine (fampridine) and sildenafil (Viagra) (Northwest Regional Spinal Cord Injury System, 2000). In addition, most major hospital centers
and health maintenance organizations also have rehabilitation and treatment centers and unique patient registry resources. The U.S. Department of Veterans Affairs (VA) Spinal Cord Injury Service is another extensive resource with a large patient population and health care professionals with clinical expertise in rehabilitation medicine. VA, which has a number of clinics throughout the United States, has an already established infrastructure that could be used to help clinical trial administrators educate and recruit members of the community into clinical trials. Furthermore, VA has a Cooperative Studies Program to promote clinical trials throughout the VA health care system.
Standardization of Clinical Trial Outcome Measures
Currently, clinicians have more than 30 assessment tests and surveys that they can use to examine individuals with spinal cord injuries and assess their progress toward recovery (Table 6-4; see also Appendix D). Each of the individual measures in these tests and surveys focuses on a specific area of function or quality of life. However, a standard set of outcome measures is not available. This is particularly problematic in multicenter clinical trials, in which data from multiple centers must be compared to determine the efficacy of a treatment intervention. Furthermore, a single assessment scale that could provide a standardized rating of the severity of an injury and assess an individual’s progress toward recovery would be useful for researchers, clinicians, and individuals with spinal cord injuries.
One of the most frequently used scales is the American Spinal Injury Association Impairment Scale, which measures the degree of paralysis and loss of sensation (Ditunno et al., 1994). This five-point scale scores the effect of an injury on the basis of muscle strength and the severity of loss of sensory function and has been widely adopted to establish a standardized language for describing an individual’s spinal cord injury (Young, 2002) (see Chapter 2). However, the scale’s minimal resolution prevents sensitivity to small but significant changes in function (NINDS, 2003; Ellaway et al., 2004). Furthermore, the scale is focused on motor function and does not address bowel, bladder, and other functional limitations and does not assess quality of life and activities of daily living.
There is a need to develop a common set of integrated outcome measures specifically designed to assess a patients’ recovery and response to treatments and experimental therapies. The ISRT clinical initiative recommended a set of tests to ensure appropriate evaluation of the physiology of individuals with spinal cord injuries before and after treatments; however, this set has yet to be incorporated into a standard of care for all individuals with spinal cord injuries (Table 6-5). Five European spinal cord injury centers are collaborating to develop a standardized protocol for outcomes
TABLE 6-4 Tools to Assess Spinal Cord Injury in Humans
|
Functional recovery |
||
|
|
American Spinal Injury Association (ASIA) International Standards for Neurological Classification |
|
|
|
• |
Analyzes the effect that the injury has on both motor and sensory systems |
|
|
• |
Is based on the extent of injury and muscle strength |
|
|
• |
Uses an alphabetical score from A (the most severe) to E (the least severe) |
|
|
• |
Insensitive to small but significant changes in motor and sensory functions |
|
|
• |
May not be sensitive enough to detect even several spinal levels of regeneration in thoracic injuries |
|
|
• |
Does not specifically address functions that affect a patient’s quality of life |
|
|
• |
Does not assess pain, bowel, bladder, or sexual function |
|
|
Functional Independence Measure (FIM) |
|
|
|
• |
Is an 18-item, seven-level ordinal scale |
|
|
• |
Is designed to assess areas of dysfunction in activities that commonly occur |
|
|
• |
The scale has few cognitive, behavioral, and communication-related functional items |
|
|
• |
Is not specific for spinal cord injuries but is designed to assess neurological, musculoskeletal, and other disorders |
|
|
Functional Assessment Measure (FAM) |
|
|
|
• |
Was developed to augment the FIM |
|
|
• |
Specifically addresses functional areas that are relatively less emphasized in FIM, including cognitive, behavioral, communication, and community functioning measures |
|
|
• |
The scale has few cognitive, behavioral, and communication-related functional items |
|
|
• |
Is not specific for spinal cord injuries but is designed to assess neurological, musculoskeletal, and other disorders |
|
|
Spinal Cord Independence Measure (SCIM) |
|
|
|
• |
Is specifically designed to assess spinal cord injuries and to be sensitive to changes |
|
|
• |
Analyzes self-care, respiration, and sphincter management and mobility |
|
|
• |
Is more sensitive than FIM for spinal cord injuries |
|
|
Electrophysiology |
|
|
|
• |
Assesses MEPs or SSEP |
|
|
• |
Stimulates corresponding cortical areas of the brain and records the response in target nerves to see if connections are still functional |
|
|
• |
Is correlated to impairment of locomotor activity |
|
|
• |
Is noninvasive |
|
|
• |
Electrical activity may not coordinate with function |
|
|
• |
Hard to assess subtle but critical improvements to circuitry |
|
|
• |
Does not assess pain, bowel, bladder, or sexual function |
assessment (Curt et al., 2004). In addition, the International Collaboration on Repair Discoveries (ICORD) has recently published a set of guidelines for clinical trials (ICORD, 2004) and is in the process of developing clinical outcome measures for each type of intervention.
Given the heterogeneity between individuals in terms of the types and severities of their spinal cord injuries (see Table 1-2 and Chapter 2), not only is a common set of outcome measures required, but an integrated rating scale for the assigned set of outcome measures should also be designed. This will improve efforts to define clinical trial end points, monitor the progression of an individual’s treatment, and examine the effects of the treatment intervention on the multiple complications associated with a spinal cord injury.
TABLE 6-5 ISRT’s Set of Standard Tests to Assess Patient Outcomes
|
The heterogeneity of spinal cord injuries not only provides a reason for developing this scale but also presents challenges. These challenges can be addressed, however, by looking at similar rating scales that have been developed for other neurological disorders that have variable severities, including the Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS) and the Unified Parkinson’s Disease Rating Scale (UPDRS). ALSFRS is used to assess 10 behaviors, including swallowing, speaking, and movement, by using four to five defined functional status end points for each behavior. For example, in the handwriting evaluation, the choices are
-
normal;
-
slow or sloppy, all words are legible;
-
not all words are legible;
-
able to grip pen but unable to write; and
-
unable to grip pen.
UPDRS was developed in 1987 in response to the need for a rating scale that could monitor the longitudinal progression of Parkinson’s disease (Fahn et al., 1987). Like ALSFRS, this scale includes groups of questions devoted to motor function, activities of daily living, and behavior and mood. The scale is used to quantify an individual’s total disability and can be used to monitor the progression of the disease.
Currently, both the ISRT and the Christopher Reeve Paralysis Foundation are in the process of developing new assessment techniques for use in large-scale clinical trials of interventions for spinal cord injuries. Coordination and collaboration are needed to develop a standard set of assessment measures to ensure valid comparisons of data between these and other groups. This effort could benefit from a consensus conference, similar to the World Federation of Neurology Airlie House Therapeutic Trials in ALS workshop (Munsat, 1995), that could be sponsored by the New York State
Spinal Cord Injury Trust, to develop a set of outcome measures for spinal cord injury.
Ensuring Independent Evaluations
Investigator bias, either deliberate or unintended, can also significantly affect the interpretation of the results and analysis of the outcomes of clinical trials. Although independent peer review is designed to address these concerns, not every study, especially early-phase trials, enters peer review. Furthermore, reviewers are rarely given access to patients so that the reviewers can conduct their own evaluations, and the reviewers do not usually have access to the raw data from the study. Because of potential conflicts, it is important that evaluation of the findings from the trial be performed by unbiased coinvestigators or others researchers who were not part of the study. In response to concerns regarding patient safety, the policy of the National Institutes of Health requires that multisite clinical trials of interventions that may involve a risk to the participant establish a data safety monitoring board that is independent of IRBs and that is responsible for ensuring that the clinical trial is conducted with the highest regard for patient safety and ethical standards and to ensure the credibility of the clinical trial and the validity of the study results.
Increasing Industry Involvement
The level of investment in research and development on interventions for spinal cord injuries by the pharmaceutical and medical device industries is difficult to determine. A number of clinical trials of medications have focused on improving bowel, bladder, and sexual function (see Appendix G); and the Pharmaceutical Research and Manufacturers of America’s New Medications in Development database indicates that a number of medications for alleviating neuropathic pain are in phase I and phase II clinical trials (PhRMA, 2004a).
The potential financial incentives for industry to invest in the research and development of interventions to treat spinal cord injuries may be limited for a number of reasons, including the following:
-
Further research is needed on the basic mechanisms of neuronal injury and repair to target therapeutic approaches.
-
Only recently have the science and experimental methodology reached the stage at which the screening of large numbers of candidate drugs and compounds is possible.
-
Spinal cord injury is not a single outcome; rather, the types of
-
injuries vary, injury occurs at different locations on the spinal cord, and the severities of the injuries vary widely.
-
Because of the relatively small numbers of potential patients—an estimated 247,000 people in the United States have a spinal cord injury, and an estimated 11,000 new cases occur each year—and because of the heterogeneity of the secondary complications, the population of individuals with spinal cord injuries is small and is further fragmented, making the market for medications and other therapeutic interventions for spinal cord injuries even smaller.
Additionally, a variety of other issues confront the development of any new therapeutic product:
-
Financial costs. It is estimated that it costs an average of $800 million to go through the drug development and approval process from the identification of a drug target through FDA approval of an efficacious product (Tufts Center for the Study of Drug Development, 2001). The expense of launching a new drug is even more expensive, and the cost increased 55 percent from 1995 to 2002 (FDA, 2004b).
-
Time. The drug and device development process is also very time-consuming. For example, it takes on average 12 to 15 years before a drug is ready to market to the general patient population. As a result, from the time that a compound is discovered and a 20-year patent is filed, it takes on average 17 to 18 years for a pharmaceutical company to recoup its costs.
-
Competition. The therapy could be displaced by a newer and more effective treatment before the company realizes a profit.
One mechanism used to provide incentives to pharmaceutical companies is the Orphan Drug Act (P.L. 97-414). Signed into law in 1983 to stimulate the research, development, and approval of drugs for the treatment of rare diseases (defined as conditions that affect 200,000 people or less in the United States), the federal legislation provides two major incentive mechanisms: sponsors are granted 7 years of marketing exclusivity after approval of the orphan drug product, and sponsors receive tax incentives for clinical research on the product.
In 2004, 21 years since the legislation was enacted, only approximately 200 drugs and biological products have qualified for orphan drug status (FDA, 2004a). It is unclear whether the incentives provided by the Orphan Drug Act are sufficient to attract pharmaceutical industry investment in therapeutic interventions for spinal cord injuries. For diseases that affect more than 200,000 people, it is possible to obtain orphan drug status for interventions needed by certain patient subgroups for specific indications.
This has been the case for spinal cord injury; for example, Acorda was granted orphan drug status in developing the drug 4-aminopyridine (fampridine) to treat spasticity, since spasticity affects less than 200,000 individuals with chronic spinal cord injuries. In addition, Proneuron received orphan drug status for its autologous incubated macrophage therapy (Procord), which is designed to improve motor and sensory neurological outcomes in individuals with acute complete spinal cord injuries.
Some of the health outcomes of a spinal cord injury—such as spasticity, chronic pain, and pressure sores—also affect individuals with other diseases and conditions. The existence of these larger markets offers a potentially greater incentive for the private sector to invest in research and development. For example, medications to alleviate spasticity would benefit not only individuals with spinal cord injuries but also individuals with multiple sclerosis. This is especially compelling for interventions for the alleviation of pressure sores, a chronic problem affecting many elderly and bed-ridden patients.
To increase the amount of industry investment in spinal cord injury research, mechanisms need to be developed to provide incentives and overcome impediments. Public-private partnerships, joint postdoctoral internships between industry and academia, and other mechanisms that would facilitate collaborative efforts and move the science closer to clinical applications should be explored. As an initial step in this process, a conference or workshop is needed to bring together the relevant stakeholders to discuss current barriers and develop collaborative approaches and incentives. One topic could be modifying the standards used to obtain “orphan” status to be based on the percentage of the United States population affected by the disease or disorder rather than the total number of affected individuals. Discussions are needed among the range of stakeholders in the development of new therapeutics for spinal cord injuries, including federal and state health agencies; professional societies in neuroscience and clinical medicine; academic institutions; basic and clinical researchers; and biotechnology, medical device, and pharmaceutical companies.
Coordination and Expansion of Spinal Cord Injury Registries and Databases
Registries are online systems for storing and relating information about individuals (Box 6-5). A population-based spinal cord injuries registry would collect standardized information on the incidence, type, and causes of spinal cord injuries for a geographically defined population, including spinal cord injury mortality among cases not seen or admitted for hospital care. This information would allow greater insights into how spinal cord injuries might be prevented but would be of limited use in the development
|
BOX 6-5 The Center for International Blood and Marrow Transplant Research administers a database of clinical information on recipients of blood and bone marrow transplants that includes data on patients throughout the treatment process, beginning with the time of diagnosis and continuing through all subsequent phases: the time of administration of chemotherapy, the phase of decision making to perform a transplant, the time that the patient receives high-dose myeloablative therapy, the time that the patient is reinfused with stem cells, the immediate posttransplantation period, the complications that occur immediately posttransplantation, and eventually, the posttransplantation outcomes, such as disease remission. As is the case for spinal cord injury clinical trials, relatively few patients are eligible for clinical trials of bone marrow transplants. In addition to collecting data from an international network of transplant centers, the registry facilitates multicenter collaborations among the researchers and clinicians at those centers. They disseminate information to physicians, patients, researchers in other fields, and the general public. University researchers and pharmaceutical companies also use the data in the database as control groups, although they are charged for use of the data. In addition, the registry has a contract with the National Marrow Donor Program to oversee patient advocacy. The Consortium of Multiple Sclerosis Centers established The North American Research Consortium on Multiple Sclerosis (NARCOMS): Multiple Sclerosis Patient Registry in 1993 to speed the development of new therapies and health care services by facilitating research on multiple sclerosis and reducing the time and cost of research studies. The registry is a database consisting of functional, accessible information that investigators use to develop research strategies and survey issues related to multiple sclerosis. As of June 2004, the number of registry participants reached more than 24,000. Online enrollment has recently become available for this registry. The availability of the website has made it easier and faster for participants to enroll. The goal of the registry is to develop a computerized database with 35,000 participants who will be monitored over time by the use of semiannual updates. In 1973 the National Cancer Institute initiated the Surveillance, Epidemiology, and End Results (SEER) Program, which currently collects and publishes cancer incidence and survival data from 14 population-based cancer registries. Included in the SEER database is information on more than 3 million cancer cases, and approximately 170,000 new cases are added each year. Since the inception of the SEER Program quality control has been an integral part of the effort and the quality and completeness of the data is evaluated each year. A valuable innovation in the SEER registry is that it provides web-based access to registry data and analytic tools that can be used by both researchers and policy makers. |
of treatments. At the other end of the spectrum, a spinal cord injury registry could record specific outcome measures for individuals with spinal cord injuries and track those outcome measures over time, including data about the outcomes of specific treatments. This type of registry could be used to understand the effectiveness of specific treatments, including practices or programs for improving functional outcomes and quality of life. Spinal cord injury registries could also provide a mechanism for identifying and contacting potential participants regarding clinical trials or notifying individuals about new therapeutic interventions.
By knowing the prevalence of certain conditions and outcomes, it is possible to determine how many individuals in each study group will be needed for accurate determination of whether a treatment is really safe or effective or whether an apparent effect was due to chance. Most importantly, a spinal cord injury registry would increase the level of understanding of the natural course of injury and repair, which in turn would allow the design of meaningful clinical trials.
At present there is no nationwide coordination of spinal cord injury registries. However, several efforts that have the potential to fill this gap are under way or are being initiated. The Model Spinal Cord Injury Care Systems database has more than 25,000 initial hospitalization records for individuals with spinal cord injuries and is the largest available resource specifically focused on spinal cord injuries (Nobunaga et al., 1999). The Trauma Care Systems Planning and Development Act of 1990 (P.L. 101-590) stipulates that states receiving federal assistance for trauma care must establish a central data reporting and analysis system for trauma care data. Thus far, 38 states have general trauma registries that allow them to assess the incidence and prevalence of various traumatic injuries (including traumatic brain injury and spinal cord injury) and to develop prevention programs. The Department of Veterans Affairs has initiated efforts to centralize its registry of veterans with spinal cord injuries (VA, 2002). To facilitate multicenter clinical trials, the Christopher Reeve Clinical Trials Network is also developing an extensive patient database in coordination with five European rehabilitation centers that receive patients from acute-care hospitals.
For spinal cord injuries, there are issues regarding the logistics of obtaining informed consent from people whose personal health information will be used in the registry. Because a willingness to participate is often influenced by the individual’s condition, all conditions may not be accurately represented in the registry. For example, the authors of a prospective study of the feasibility of establishing a registry of stroke patients in Canada concluded that the registry was virtually useless because so few people agreed to participate and those few who did agree tended to be the least severely injured, which resulted in a registry with a highly biased sample of
the general population of stroke victims (Ingelfinger and Drazen, 2004; Tu et al., 2004). Although the experiences of researchers and clinicians who work with individuals with spinal cord injuries suggest that they are overall quite willing, and even eager, to participate in similar registries, the issue of sampling bias is too important to leave unexamined in any patient registry.
Important considerations in designing databases include:
-
scalability (it must be able to grow);
-
flexibility (it must be able to accommodate different types of data and be modifiable as needs evolve or new measures are incorporated); and
-
reliability (data entry practices must be consistent and verification of accuracy must be performed on an ongoing basis; often, the same patient is entered into the registry twice or the nature and the extent of data entry practices differ between sites).
Globally, data registries are at risk of significant bias because of privacy issues that have made the collection and use of health data more difficult for researchers (Ingelfinger and Drazen, 2004). Although such restrictions are generally imposed because of legitimate interests in protecting individuals from the misuse of their personal information, research efforts intended to help patients have also been restricted as an unintended consequence. The implementation of the Health Insurance Portability and Accountability Act Privacy Rule in 2003 imposed new restrictions on how patient health information can be shared among researchers. Although some of those requirements can be waived, most IRBs have been reluctant to do so. Other countries have imposed similar laws.
There are also concerns about the use of a legacy database—one that has already been created, often with a different application in mind. These databases are often out of date, the information is difficult to verify, and the structure of the database may be incompatible with new formats, potentially resulting in information exchanges that corrupt the new or the old system. This has been accomplished in other cases by establishing standard templates for health data, such as the Health Level Seven (HL7) project or Integrating the Healthcare Enterprise. Few data registries have reached their maximum potential, and spinal cord injury researchers would be well advised to seek input from successful pioneers in registry development.
Efforts should be made to develop standardized protocols for patient registry systems so that registries can be coordinated and used to assist in identifying candidates for participation in clinical trials and provide information on upcoming clinical trials to individuals with spinal cord injuries (see Chapter 7). This may require the establishment of a new set of databases that are flexible and capable of being integrated.
The coordination of all this information will provide an invaluable tool
for spinal cord injury research, will facilitate increased participation in clinical trials, and will provide data sets that can be used for studies investigating the long-term treatment outcomes from therapeutic interventions and the safety of those interventions.
ACCELERATING PROGRESS
Neither the scientific community nor the community of individuals with spinal cord injuries is content with the limited therapeutic options currently available for the treatment of spinal cord injuries. There is an obvious and urgent need to identify and test new interventions and to accelerate the pace of research, particularly in moving laboratory findings to clinical practice. A spinal cord injury involves serious and traumatic adverse changes to the human body, and an extensive research effort is needed to develop treatment approaches for the range of health outcomes that individuals with spinal cord injuries face.
Challenges arise in dealing with new experimental therapies that look promising but that are not yet in clinical trials with human subjects. Some individuals are willing to take chances on interventions that may endanger their safety but that may also offer the possibility of functional improvements. Efforts are needed to assist these individuals in understanding the current status of clinical trials. This includes identifying those trials open for the recruitment of participants and providing information on the potential health risks of experimental therapies.
RECOMMENDATIONS
Recommendation 6.1: Facilitate Clinical Trials
Mechanisms should be implemented that will facilitate the implementation of clinical trials while observing the established standards for the protection of human subjects in clinical research, including:
-
Utilize and coordinate existing facilities and resources in acute care, chronic care, and rehabilitation to support multicenter clinical trials.
-
The use of central institutional review board mechanisms in conjunction with local institutional review boards should be explored to facilitate coordinated multicenter studies.
-
Patient registries and databases should be coordinated and expanded to improve mechanisms to conduct clinical trials and facilitate patient recruitment by increasing awareness of ongoing clinical trials among potential participants.
-
A set of standardized clinical outcome measures should be devel-
-
oped. This may include a rating scale that is capable of integrating functional outcomes from different spheres of disability.
-
Clinical trial design should be a multidisciplinary effort and should incorporate, as appropriate, small “n” methodologies for early-phase clinical trials to ensure the rapid advancement of new therapies. Initial clinical trials that are promising should then be followed up with larger-scale clinical trials.
Recommendation 6.2: Increase Industry Involvement
The National Institutes of Health and the Food and Drug Administration (in collaboration with state and other federal agencies, professional societies, academic institutions, nonprofit organizations, and the pharmaceutical and medical device industries) should explore mechanisms that can be used to link federal, state, academic, and nonprofit efforts with those of industry with the goal of increasing the investment and involvement of the private sector in the development of therapeutic interventions for spinal cord injuries.
REFERENCES
Alderson P, Roberts I. 1997. Corticosteroids in acute traumatic brain injury: Systematic review of randomised controlled trials. British Medical Journal 314(7098): 1855-1859.
Berry C, Kennedy P. 2003. A psychometric analysis of the Needs Assessment Checklist (NAC). Spinal Cord 41(9): 490-501.
Blight A. 2004, February 24. Challenges of Conducting Clinical Trials on Spinal Cord Injury in Industry. Presentation at the Institute of Medicine Workshop on Translational and Clinical Research Workshop, Washington, DC. Institute of Medicine Committee on Spinal Cord Injury.
Bracken MB. 2000. Methylprednisolone and spinal cord injury. Journal of Neurosurgery—Spine 93(1 Suppl): 175-179.
Bracken MB, Holford TR. 2002. Neurological and functional status 1 year after acute spinal cord injury: Estimates of functional recovery in National Acute Spinal Cord Injury Study II from results modeled in National Acute Spinal Cord Injury Study III. Journal of Neurosurgery 96(3 Suppl): 259-266.
Bracken MB, Collins WF, Freeman DF, Shepard MJ, Wagner FW, Silten RM, Hellenbrand KG, Ransohoff J, Hunt WE, Perot PL. 1984. Efficacy of methylprednisolone in acute spinal cord injury. Journal of the American Medical Association 251(1): 45-52.
Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J. 1990. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the second National Acute Spinal Cord Injury Study. New England Journal of Medicine 322(20): 1405-1411.
Bracken MB, Shepard MJ, Holford TR, Leo-Summers L, Aldrich EF, Fazl M, Fehlings M, Herr DL, Hitchon PW, Marshall LF, Nockels RP, Pascale V, Perot PL Jr, Piepmeier J, Sonntag VKH, Wagner F, Wilberger JE, Winn HR, Young W. 1997. Administration of methylprednisolone for 24 or 48 hours or tirilazad mesylate for 48 hours in the treatment of acute spinal cord injury: Results of the third National Acute Spinal Cord Injury randomized controlled trial. Journal of the American Medical Association 277(20): 1597-1604.
Catz A, Itzkovich M, Agranov E, Ring H, Tamir A. 1997. SCIM—Spinal Cord Independence Measure: A new disability scale for patients with spinal cord lesions. Spinal Cord 35(12): 850-856.
Clifton GL, Knudson P, McDonald M. 2002. Waiver of consent in studies of acute brain injury. Journal of Neurotrauma 19(10): 1121-1126.
Creasey GH, Grill JH, Korsten M, Sang H, Betz R, Anderson R, Walter J, Implanted Neuroprosthesis Research Group. 2001. An implantable neuroprosthesis for restoring bladder and bowel control to patients with spinal cord injuries: A multicenter trial. Archives of Physical Medicine and Rehabilitation 82(11): 1512-1519.
Curt A, Schwab ME, Dietz V. 2004. Providing the clinical basis for new interventional therapies: Refined diagnosis and assessment of recovery after spinal cord injury. Spinal Cord 42(1): 1-6.
Dekkers W, Boer G. 2001. Sham neurosurgery in patients with Parkinson’s disease: Is it morally acceptable? Journal of Medical Ethics 27(3): 151-156.
DHHS (U.S. Department of Health and Human Services). 1996. Protection of human subjects, informed consent and waiver of informed consent requirements in certain emergency research: Final rules (21 CFR Part 50, 45 CFR Part 46). Federal Register 61(192): 51500-51533.
Dietrich WD. 2003. Confirming an experimental therapy prior to transfer to humans: What is the ideal? Journal of Rehabilitation Research & Development 40(4 Suppl 1): 63-69.
Ditunno JF, Young W, Donovan WH, Creasey G. 1994. The international standards booklet for neurological and functional classification of spinal cord injury. Paraplegia 32(2): 70-80.
Ditunno JF Jr, Cohen ME, Hauck WW, Jackson AB, Sipski ML. 2000. Recovery of upper-extremity strength in complete and incomplete tetraplegia: A multicenter study. Archives of Physical Medicine and Rehabilitation 81(4): 389-393.
Dobkin BH, Havton LA. 2004. Basic advances and new avenues in therapy of spinal cord injury. Annual Review of Medicine 55: 255-282.
Dumont RJ, Verma S, Okonkwo DO, Hurlbert RJ, Boulos PT, Ellegala DB, Dumont AS. 2001. Acute spinal cord injury. Part II. Contemporary pharmacotherapy. Clinical Neuropharmacology 24(5): 265-279.
Ellaway PH, Anand P, Bergstrom EMK, Catley M, Davey NJ, Frankel HL, Jamous A, Mathias C, Nicotra A, Savic G, Short D, Theodorou S. 2004. Towards improved clinical and physiological assessments of recovery in spinal cord injury: A clinical initiative. Spinal Cord 42(6): 325-337.
Fahn S, Marsden CD, Calne DB, Goldstein M, eds. 1987. Recent Developments in Parkinson’s Disease. Florham Park, NJ: Macmillan Health Care Information.
FDA (Food and Drug Administration). 2004a. Office of Orphan Product Development. [Online]. Available: http://www.fda.gov/orphan/index.htm [accessed December 21, 2004].
FDA. 2004b. Innovation, Stagnation: Challenge and Opportunity on the Critical Path to New Medical Products . Washington, DC: U.S. Department of Health and Human Services.
Hanigan WC, Anderson RJ. 1992. Commentary on NASCIS-2. Journal of Spinal Disorders 5(1): 125-131.
Harding A. 2004. More compounds failing phase I. The Scientist 18(13): 5.
Huang H, Chen L, Wang H, Xiu B, Li B, Wang R, Zhang J, Zhang F, Gu Z, Li Y, Song Y, Hao W, Pang S, Sun J. 2003. Influence of patients’ age on functional recovery after transplantation of olfactory ensheathing cells into injured spinal cord injury. Chinese Medical Journal 116(10): 1488-1491.
Hurlbert RJ. 2000. Methylprednisolone for acute spinal cord injury: An inappropriate standard of care. Journal of Neurosurgery: Spine 93(1 Suppl): 1-7.
ICORD (International Collaboration on Repair Discoveries). 2004. ICCP Workshops on Spinal Cord Injury: Report on the First Working Group. [Online]. Available: http://www.icord.org/ICCP/ICCP_SCI_Guidelines1.doc [accessed February 21, 2005].
Ingelfinger JR, Drazen JM. 2004. Registry research and medical privacy. New England Journal of Medicine 350(14): 1452-1453.
IOM (Institute of Medicine). 1999. A Report on the Sponsors of Cancer Treatment Clinical Trials and Their Approval and Monitoring Mechanisms. Washington, DC: National Academy Press.
IOM. 2001. Small Clinical Trials: Issues and Challenges. Washington, DC: National Academy Press.
Kleitman N. 2004. Keeping promises: Translating basic research into new spinal cord injury therapies. Journal of Spinal Cord Medicine 27(4): 311-318.
Lev M. 2004, August 27. Fetal-cell surgery raises hopes, fears. A procedure that involves aborted fetuses is drawing spinal-injury patients from the U.S., but a lack of clinical trials concerns researchers. Chicago Tribune. p. C4.
Loh ED, Meyer RE. 2004. Medical schools’ attitudes and perceptions regarding the use of central institutional review boards. Academic Medicine 79(7): 644-651.
Matthew G., 2001. Neurobiology: Molecules, Cells, and Systems. 2nd ed. Malden, MA: Blackwell Science.
Maynard FM, Reynolds GG, Fountain S, Wilmot C, Hamilton R. 1979. Neurological prognosis after traumatic quadriplegia. Three-year experience of California regional spinal cord injury care system. Journal of Neurosurgery 50(5): 611-616.
Meyers AR, Andresen EM, Hagglund KJ. 2000. A model of outcomes research: Spinal cord injury. Archives of Physical Medicine and Rehabilitation 81(12 Suppl 2): S81-90.
Munsat TL. 1995. World Federation of Neurology Research Group on Neuromuscular Diseases Subcommittee on Motor Neuron Disease. Airlie House guidelines—Therapeutic trials in amyotrophic lateral sclerosis. Journal of the Neurological Sciences 129(Suppl): 1-10.
NCI (National Cancer Institute). 2004. The Central Institutional Review Board Initiative. [Online]. Available: http://www.ncicirb.org/ [accessed August 22, 2004].
NINDS (National Institute of Neurological Disorders and Stroke). 2002. Facilities of Research in Spinal Cord Injury. [Online]. Available: http://grants2.nih.gov/grants/guide/notice-files/NOT-NS-02-011.html [accessed October 4, 2004].
NINDS. 2003. Translating Promising Strategies for Spinal Cord Injury Therapy February 3–4, 2003, Bethesda, Maryland. [Online]. Available: http://www.ninds.nih.gov/news_and_events/proceedings/sci_translation_workshop.htm?format=printable [accessed August 9, 2004].
Nobunaga AI, Go BK, Karunas RB. 1999. Recent demographic and injury trends in people served by the model spinal cord injury care systems. Archives of Physical Medicine and Rehabilitation 80(11): 1372-1382.
Northwest Regional Spinal Cord Injury System. 2000. SCI Forum—Fampridine (4-AP) Study at UW. [Online]. Available: http://depts.washington.edu/rehab/sci/fampridine.html [accessed November 11, 2004].
NRC (National Research Council). 1993. Clinical Trials and Statistics: Proceedings of a Symposium. Washington, DC: National Academy Press.
Peckham PH, Keith MW, Kilgore KL, Grill JH, Wuolle KS, Thrope GB, Gorman P, Hobby J, Mulcahey MJ, Carroll S, Hentz VR, Wiegner A, Implantable Neuroprosthesis Research Group. 2001. Efficacy of an implanted neuroprosthesis for restoring hand grasp in tetraplegia: A multicenter study. Archives of Physical Medicine and Rehabilitation 82(10): 1380-1388.
PhRMA (Pharmaceutical Research and Manufacturers of America). 2004a. New Drug Approvals in 2003. [Online]. Available: http://www.phrma.org/newmedicines/resources/2004-01-22.123.pdf [accessed November 22, 2004].
PhRMA. 2004b. Medicine in Development for Women. [Online]. Available: http://www.phrma.org/newmedicines/resources/2004-03-02.124.pdf [accessed November 11, 2004].
Pocock SJ, 1983. Clinical Trials: A Practical Approach. New York: Wiley, Inc.
Quest for new cures. 2003. The Pfizer Journal 7(3): 4-11.
Ramer MS, Harper GP, Bradbury EJ. 2000. Progress in spinal cord research: A refined strategy for the International Spinal Research Trust. Spinal Cord 38(8): 449-472.
Redmond DE Jr, Freeman T, American Society for Neural Transplantation and Repair. 2001. The American Society for Neural Transplantation and Repair considerations and guidelines for studies of human subjects. The Practice Committee of the Society. Approved by Council. Cell Transplantation 10(8): 661-664.
Roberts I, Yates D, Sandercock P, Farrell B, Wasserberg J, Lomas G, Cottingham R, Svoboda P, Brayley N, Mazairac G, Laloe V, Munoz-Sanchez A, Arango M, Hartzenberg B, Khamis H, Yutthakasemsunt S, Komolafe E, Olldashi F, Yadav Y, Murillo-Cabezas F, Shakur H, Edwards P, CRASH Trial Collaborators. 2004. Effect of intravenous corticosteroids on death within 14 days in 10008 adults with clinically significant head injury (MRC CRASH trial): Randomised placebo-controlled trial. Lancet 364(9442): 1321-1328.
Rosenfeld JV, Gillett GR. 2004. Ethics, stem cells and spinal cord repair. Medical Journal of Australia 180(12): 637-639.
Sauerland S, Maegele M. 2004. A crash landing in severe head injury. Lancet 364(9442): 1291-1292.
Smithline HA, Gerstle ML. 1998. Waiver of informed consent: A survey of emergency medicine patients. American Journal of Emergency Medicine 16(1): 90-91.
Sugarman J. 1999. Ethical considerations in leaping from bench to bedside. Science 285(5436): 2071-2072.
Sugarman J, McKenna WG. 2003. Ethical hurdles for translational research. Radiation Research 160(1): 1-4.
Tu JV, Willison DJ, Silver FL, Fang J, Richards JA, Laupacis A, Kapral MK, Investigators in the Registry of the Canadian Stroke Network. 2004. Impracticability of informed consent in the registry of the Canadian Stroke Network. New England Journal of Medicine 350(14): 1414-1421.
Tufts Center for the Study of Drug Development. 2001. Backgrounder: A Methodology for Counting Costs for Pharmaceutical R&D. [Online]. Available: http://csdd.tufts.edu/NewsEvents/RecentNews.asp?newsid=5 [accessed November 11, 2004].
VA (U.S. Department of Veterans Affairs). 2002. Spinal Cord Dysfunction Registry. [Online]. Available: http://www.sci-queri.research.med.va.gov/Registry.htm [accessed August 10, 2004].
World Medical Association. 2004. World Medical Association Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects. [Online]. Available: http://www.wma.net/e/policy/pdf/17c.pdf [accessed November 11, 2004].
Young W. 2002. Spinal Cord Injury Levels & Classification. [Online]. Available: http://www.sci-info-pages.com/levels.html [accessed September 30, 2003].































