
4
Identifying and Understanding Adverse Medical Device Events
“The hardest thing over the time that he was growing up—in the first year, mostly—was establishing my credibility with the doctors as a reporter to them…. [T]hat was the hardest thing, people not believing … that there was something going on and that I wasn’t just a hysterical mother.”
Nancy Harder, parent, 2004
Communication gaps between patients or parents and physicians are a longstanding concern in medicine and can cause considerable distress to parents. Poor communication can contribute to adverse events or other harms when physicians do not give credence to patient or family reports of problems, as recounted in the quote above from the mother of two children who have spina bifida and rely on cerebrospinal fluid shunts and other medical devices. Inadequate communication can also create problems when patients and family caregivers are inadequately prepared to fulfill their responsibilities for using or maintaining complex medical device. As care has shifted out of the hospital into the home, parents are bringing children home with ventilators, feeding tubes, monitors, and other complex or unfamiliar devices. This is stressful enough without the additional stress of poor training and education about the device use and problem identification. Communication gaps may reflect a physician’s lack of awareness of the problems that families and patients face in safely using medical equipment at home.
As emphasized in Chapter 1, the migration of care from hospital to home has brought many benefits, but it also presents risks as parents and families assume responsibilities for device operation, maintenance, and problem recognition once assigned to health care professionals. Surveillance programs, which have other limitations described in this chapter, have yet to adjust to the changed circumstances of much patient care.
The identification, reporting, and analysis of serious adverse device events and device failures and malfunctions are important elements of the
U.S. Food and Drug Administration’s (FDA’s) overall program of postmarket surveillance. A primary aim of the agency’s adverse event reporting program is to identify serious problems with a device (or its use) that become evident after a device is marketed when—depending on the device—it is used with many more patients, with different patient populations (e.g., children), in different ways (e.g., involving ad hoc modifications for pediatric use), for different purposes, in new and possibly less well-equipped settings, over longer periods, and, sometimes, by less experienced or skilled clinicians and care teams. Systematic clinical studies are often a superior tool for assessing these dimensions of device use, but such studies are not realistic for the entire array of devices that enter the market each year. Moreover, just as premarket studies may fail to detect rare events, so may postmarket clinical studies.
Although FDA is most interested in reports of serious unanticipated events, the adverse event reporting program also collects information that can be useful in understanding certain already recognized risks, for example, patient deaths by entrapment in the rails of hospital beds. In addition, reports of device failures and malfunctions—even when they have not caused harm—can help FDA and manufacturers to detect hazards that arise from aberrations in the manufacturing, distribution, modification, maintenance, storage, or reprocessing of a medical device. Adverse event reports can also lead to improvements in the design of a device. For example, in response to problem reports, manufacturers have redesigned cardiac pacemakers to make them substantially less susceptible to electromagnetic interference from modern necessities such as microwave ovens and cellular telephones (Niehaus and Tebbenjohanns, 2001).
For the most part, the public health goals and the limitations of postmarket surveillance policies and programs apply to both adults and children. Systems that support effective postmarket surveillance for patients generally are the foundation on which additions, adaptations, or emphases suited to children’s particular needs are then built. For example, the FDA guidance on assessment of pediatric medical devices cited in Chapter 2 makes sense only within an already existing structure for evaluating the safety and effectiveness of medical devices.
The first part of this chapter expands on Chapter 3’s description of the FDA program for adverse event reporting. It includes statistics on reports to FDA of adverse device events that involve children and presents examples of actual reports. This discussion is followed by a number of vignettes that illustrate the range of factors and devices that contribute to adverse medical device events with children and the complexities in identifying and understanding these events. Most of the vignettes depict events that result not from single faults or errors but rather from the interplay between weaknesses in some aspect of the design or manufacture of devices and the
circumstances of their use with children. Following the vignettes is a review of the sources of adverse events, the limitations of adverse event reporting, and FDA responses to these limitations. The chapter concludes with recommendations for the FDA.
ADVERSE DEVICE EVENT REPORTING AND FDA
As described in Chapter 3, FDA has authority for two programs of adverse event reporting that involve medical devices. The primary program receives mandatory reports of certain adverse device events from device manufacturers and user facilities and also accepts voluntary reports from health care professionals, consumers, and others. This program is a form of passive surveillance in that it awaits event reports. Active surveillance involves more direct effort by a sponsoring agency to obtain information, for example, through surveys. In addition, based on a sample of user facilities, FDA has created the pilot MedSun program, which includes some elements of active surveillance.
FDA provides Form 3500A (online at http://www.fda.gov/medwatch/getforms.htm) for manufacturers, user facilities, and importers to use for mandatory reporting of serious adverse events and problems involving devices, drugs, and biologics. (Vaccines have a separate reporting system.) The first page of the form asks for information about the
-
patient (including age, sex, and weight);
-
event or product problem, including an open-ended description of the problem;
-
product, including identifying information (e.g., for devices, the brand name, model, manufacturer, model and lot numbers) and other details (e.g., whether a device was an implant and if explanted, whether it is available for examination, and what concomitant medical products or therapies in use);
-
outcome (e.g., whether it involved a death or required some kind of intervention); and
-
initial reporter (e.g., contact information, whether a health professional).
The second page of the mandatory reporting form requests additional information from user facilities and importers (e.g., where the event occurred, when they became aware of it, who to contact for further information) and manufacturers (e.g., whether they evaluated the device, whether they took any remedial action.
For voluntary reports, FDA provides Form 3500, which has a first page that is almost identical to Form 3500A but has no second page. FDA also offers the option of online reporting for voluntary reporters. The voluntary
reporting form, the online option, and the instructions for reporting clearly require reading skills and knowledge above the levels possessed by many consumers. For example, the form uses terms like “relevant history,” “congenital anomaly,” “concomitant products,” “event abated,” and “labeled strength” (FDA, 2003o). The agency urges consumers who want to report an event to have their physician complete the form.
Both mandatory and voluntary reports involving devices are compiled in the Manufacturer and User Facility Device Experience database (MAUDE). After certain information is removed (e.g., patient age, facility name), the reports are made available in a searchable public database. FDA and manufacturers have access to the full reports to support their analyses.
Table 4.1 shows the number of adverse event reports received by FDA from late 1984 to 2004 by major category of reporter, requirement for reporting (mandatory or voluntary), and type of event as designated by the person reporting it. The great majority of reports in MAUDE are submitted by manufacturers. One of the most notable trends shown in the table is the shift of adverse event reports to the alternative summary reporting option after its introduction by FDA in 1995. In recent years, such summary reports have accounted for more than half of total reports, for example, nearly 98,000 of the almost 152,000 reports received in 2004. The sizeable increase in adverse event reports (primarily injuries and malfunctions) from 1992 through 1994 has been attributed, in part, to reports of problems with silicone breast implants, which account for almost one-third of all reports from manufacturers (GAO, 1997).
Mandatory user facility reports account for less than 3 percent of the reports in Table 4.1. This number is, however, somewhat deceptive because FDA attempts to eliminate duplicate reports from the statistics so that a facility report that goes to both FDA and the manufacturer (and then to FDA) is not counted twice. (Facilities are supposed to report to FDA directly only if an event involves a death or the manufacturer of a device is not known.) Voluntary reports from health care professionals and consumers also account for a small percentage of reports (about 3 percent each year).
Unlike some patient safety programs described later in this chapter, FDA does not require or encourage reports of close calls from user facilities. In contrast, manufacturers are required to report device-related malfunctions, including those that could cause a death or serious injury if they recurred. When close calls involve situations with the potential to recur and cause harm, reports of such events may provide valuable signals if manufacturer and FDA analysts are prepared to notice them.
FDA sometimes discovers deficiencies in manufacturer reporting of adverse events and product problems (or their systems related to such reporting) during quality systems inspections, through investigations of incidents, and in other ways. The agency typically responds with letters that
TABLE 4.1 Adverse Event Reports Submitted to FDA, Late 1984 Through December 2004
|
|
<1985 |
1985 |
1986 |
1987 |
|
Manufacturer reportsa |
||||
|
Death |
13 |
585 |
543 |
516 |
|
Injury |
109 |
9,483 |
11,738 |
9,589 |
|
Malfunction |
28 |
8,812 |
7,096 |
7,596 |
|
Other |
2 |
62 |
10 |
5 |
|
SUBTOTAL |
152 |
18,942 |
19,387 |
17,706 |
|
User facility reportsb |
||||
|
Death |
|
|||
|
Injury |
||||
|
Malfunction |
||||
|
Other |
||||
|
SUBTOTAL |
0 |
0 |
0 |
0 |
|
Distributor and importer reportsc |
||||
|
Death |
|
|||
|
Injury |
||||
|
Malfunction |
||||
|
Other |
||||
|
SUBTOTAL |
0 |
0 |
0 |
0 |
|
Voluntary reportsd |
||||
|
Death |
|
31 |
27 |
21 |
|
Injury |
|
345 |
482 |
288 |
|
Malfunction |
|
520 |
472 |
349 |
|
Other |
22,602 |
2,097 |
2,170 |
1,827 |
|
SUBTOTAL |
22,602 |
2,993 |
3,151 |
2,485 |
|
Summary reportse |
0 |
0 |
0 |
0 |
|
GRAND TOTAL |
22,754 |
21,935 |
22,538 |
20,191 |
|
1988 |
1989 |
1990 |
1991 |
1992 |
1993 |
1994 |
|
565 |
730 |
951 |
1,133 |
1,528 |
1,339 |
1,870 |
|
8,366 |
9,845 |
11,809 |
18,521 |
52,894 |
61,885 |
79,537 |
|
6,677 |
9,298 |
16,840 |
24,796 |
21,583 |
45,608 |
48,629 |
|
7 |
7 |
4 |
15 |
13 |
38 |
35 |
|
15,615 |
19,880 |
29,604 |
44,465 |
76,018 |
108,870 |
130,071 |
|
|
7 |
287 |
250 |
266 |
||
|
2 |
1,285 |
1,229 |
2,338 |
|||
|
6 |
1,083 |
988 |
989 |
|||
|
0 |
142 |
337 |
554 |
|||
|
0 |
0 |
0 |
15 |
2,797 |
2,804 |
4,147 |
|
|
11 |
18 |
49 |
|||
|
251 |
1,103 |
1,803 |
||||
|
33 |
139 |
274 |
||||
|
6 |
13 |
121 |
||||
|
0 |
0 |
0 |
0 |
301 |
1,273 |
2,247 |
|
32 |
19 |
319 |
32 |
4 |
5 |
61 |
|
194 |
364 |
140 |
54 |
77 |
280 |
1,292 |
|
294 |
255 |
252 |
85 |
95 |
167 |
1,508 |
|
1,716 |
1,664 |
1,894 |
3,610 |
4,439 |
3,013 |
2,015 |
|
2,236 |
2,302 |
2,605 |
3,781 |
4,615 |
3,465 |
4,876 |
|
0 |
0 |
0 |
0 |
0 |
0 |
0 |
|
17,851 |
22,182 |
32,209 |
48,261 |
83,731 |
116,412 |
141,341 |
|
|
1995 |
1996 |
1997 |
1998 |
|
Manufacturer reportsa |
||||
|
Death |
1,773 |
1,389 |
1,019 |
1,021 |
|
Injury |
51,752 |
38,236 |
31,122 |
18,554 |
|
Malfunction |
50,125 |
37,830 |
32,833 |
31,960 |
|
Other |
28 |
631 |
2,299 |
2,485 |
|
SUBTOTAL |
103,678 |
78,086 |
67,273 |
54,020 |
|
User facility reportsb |
||||
|
Death |
211 |
346 |
326 |
276 |
|
Injury |
2,315 |
3,173 |
3,892 |
2,556 |
|
Malfunction |
780 |
1,091 |
1,293 |
860 |
|
Other |
657 |
697 |
657 |
446 |
|
SUBTOTAL |
3,963 |
5,307 |
6,168 |
4,138 |
|
Distributor and importer reportsc |
||||
|
Death |
19 |
27 |
35 |
13 |
|
Injury |
1,661 |
3,606 |
1,364 |
189 |
|
Malfunction |
164 |
213 |
169 |
289 |
|
Other |
169 |
150 |
70 |
50 |
|
SUBTOTAL |
2,013 |
3,996 |
1,638 |
541 |
|
Voluntary reportsd |
||||
|
Death |
73 |
63 |
67 |
75 |
|
Injury |
1,559 |
864 |
835 |
963 |
|
Malfunction |
1,367 |
1,494 |
1,299 |
1,523 |
|
Other |
782 |
565 |
405 |
391 |
|
SUBTOTAL |
3,781 |
2,986 |
2,606 |
2,952 |
|
Summary reportse |
2,755 |
6,292 |
21,682 |
36,190 |
|
GRAND TOTAL |
116,190 |
96,667 |
99,367 |
97,841 |
|
NOTE: Represents most current data (March 31, 2005) for period through December 31, 2004. Yearly report counts are updated periodically to account for delayed data entry issues (e.g., backlog of reports not entered). aReceived since December 1984 (MDR Regulation, December 13, 1984). bReceived since 1992 (Safe Medical Devices Act of 1990). cReceived since 1992 (Safe Medical Devices Act of 1990). Distributors reported since December 19, 1998 (FDA Modernization Act of 1997). |
||||
outline the problems and needed corrections. Only rarely are criminal penalties sought.1 FDA staff could not cite cases in which user facilities had been penalized for failure to comply with their mandatory reporting obligations (personal communication, Thomas P. Gross, M.D., Director, Division of Postmarket Surveillance, CDRH, January 28, 2005).
Table 4.2 shows the number of reports submitted that identified adverse events as involving patients under age 21. (These data were supplied by FDA from their internal database. As noted above and in Chapter 3, the public database does not include information on age or birth date.) The table shows no entries for the summary reporting option because this option does not require information on patient age.
The numbers presented in Table 4.2 are undoubtedly an undercount of all reported events that involved children (leaving aside all actual events). The fields on the reporting form that request birth date or age information are sometimes not completed, perhaps because the information is not immediately available to the reporter. In FDA’s analysis of reports of patient entanglement in hospital bed side rails—an event associated with high rates of death (65 percent of reports) and injury (23 percent of reports)—age was not included for 36 of 111 of the reports in MAUDE (Todd et al., 1997a,b). Of the 75 cases for which age data were provided, 5 percent involved patients under age 17. Even if event reporting were more complete, it would be difficult to assess the extent of a problem without knowing the population at risk (the denominator problem as discussed elsewhere in this report and in Appendix D).
Box 4.1 presents several excerpts from reports to FDA of adverse events that involved children. The examples (which include the full narrative text of the reports) illustrate that reports vary greatly in the amount and usefulness of the information provided. Some offer a relatively clear picture of an event; others are incomplete. Reports nearly always focus on the immediate circumstances surrounding an event and thus are limited in the extent to which they point to contributing system factors, for example, understaffing.
These examples of reports make evident some of the challenges in investigating adverse event reports, especially when the investigator is organizationally removed from the event, as is usually the case for manufacturer or FDA staff. For example, although manufacturers (and FDA) can often follow up with reporters to collect additional information, manufacturers may not have access to the device for inspection, and important information
TABLE 4.2 FDA Adverse Event Reports Involving Individuals Under Age 21 (1999–2004)a
about the device (e.g., brand and model number and even manufacturer) may not have been documented. A later section of this chapter returns to these and other limitations of adverse event reporting programs.
When FDA receives an adverse event or device malfunction report from a manufacturer or other party, it (actually a contractor) checks the report, codes certain information if it has not been coded already by the reporter, and enters the report into the database.2 FDA has developed codes for both clinical outcomes (e.g., cerebral hemorrhage) and device outcomes (e.g.,
|
BOX 4.1 Device: Circumcision clamp FDA Device Classification: Clamp, circumcision Problem Description Circumcision using [circumcision] clamp. Clamp was loose, resulting in laceration of the glans penis with loss of tip. Infant was transferred to another hosp for urologic consultation and surgery to repair damaged penis. (MAUDE Report No. 257649) Device: Vacuum extractor FDA Device Classification: Extractor, vacuum, fetal Problem Description Infant boy was delivered at [time] on [date]. Delivery was complicated by a prolonged second stage. Infant suffered hemorrhage beneath scalp at birth. Infant was admitted to neonatal intensive care unit and was placed on ventilator at [time]. Infant expired at [time] on [date]. The cause of death was the hemorrhage. It is speculated that the hemorrhage resulted from the use of a vacuum extractor with a defective gauge. Gauge on the device registered in the green “safe” zone even though excess vacuum was being produced. This defect was confirmed by testing the device using a pressure transducer. There would have been no way the user of the device would have known that the gauge was defective and that a dangerous level of vacuum was being produced … Device manufacture date is 1/17/96. (MDR Access No. M763107) Device: Nasal dressing FDA Device Classification: Bandage, liquid Problem Description The nasal dressing was placed in the pt in 2004 subsequent to sinus surgery. Two days later the nasal dressing fell apart while trying to remove it. The hosp health professional tried to remove the dressing again in 2004 and was unable to do so. The next day the dressing was surgically removed. Manufacturer Response User was not able to provide lot number, therefore mfg data is unavailable. The device did not fail, but was apparently cut or teased apart in an attempt to remove it. Surgically removed sample showed the pvc pouch with foam inside was fully intact, three weeks following initial surgery; indicating that the core of the product did not come apart; but was wedged and had to be surgically removed. User selected a 3 cm adult size contributing to the difficulty of removal. More suitable choices, when dealing with a small child’s anatomy, include: the smaller removable dressing; model rr 200 which is 1/3 smaller than the device used. The dissolvable dressing; commonly used for pediatric cases because they can be trimmed to fit any size/shape anatomy, and do not require physical removal. (They dissolve away over time.) (MAUDE Report No. 1064611-2004-00002) |
|
Device: Reusable I.C. nebulizer with tubing FDA Device Classification: Nebulizer (direct patient interface) Problem Description Reporter feels 2 safety issues re: device are not being addressed by mfr. 1. Mouth piece has 8 mm × 17–18 mm rubber flap that disconnects easily. Reporter has found a child with this in its hand. 2. Same flexible rubber used for inside screw cap. Size is 12 cm which could easily be pulled off and block child’s airway. Response from mfr was that user should read instructions. Many of the users cannot or will not read instructions completely. This device is mainly for home use. (MAUDE Report No. 492408) Device: Pediatric peritoneal dialysis system FDA Device Classification: System, peritoneal, automatic delivery Problem Description Home patient’s (hp) foster mother reports a system error 2240 alarm in a drain during treatment on the homechoice machine. At the time of the alarm hp’s foster mother noticed the homechoice set pt line had disconnected from the hp’s transfer set. Hp’s healthcare professional (hcp) and foster mom both state that foster mom was not properly connecting hp’s transfer set to the homechoice set pt line and this is what caused them to disconnect. Hcp administered prophylactic antibiotics. Hp was admitted to the hospital on 12/14/99 for diarrhea and at the same time was monitored and subsequently diagnosed with peritonitis. Hp was discharged from the hospital on 12/21/99. Hp’s course of treatment is vancomycin 25 mg/l for 10 days and gentamycin 10 mg/l for 10 days. Manufacturer Response Hp’s foster mom just started taking care of this child about one week prior to this event and states she had no training on how to connect his transfer set to the homechoice set pt line. Since this occurrence the foster mom has received training from the hcp on sterile technique, operation of the machine and how to connect hp’s transfer set to the pt line. (MAUDE Report No. 1423500-1999-01553) Device: Infant heel warmer FDA Device Classification: Pack, hot or cold, disposable Problem Description In preparation of capillary blood draw, a liquid infant heel warmer was applied to pt’s foot causing a 2nd degree burn covering 33 percent of the foot. Manufacturer Response The suspect device was discarded by user facility. Lot info is not available. Health care provider could not verify that packet was “kneaded” for 30 to 60 seconds as indicated in instructions for use. Heel warmer was secured to pt by 2 “pampers.” There is no indication in the instructions for use to do this, however there is no contra indication either. (MAUDE Report No. 1216677-2004-00012) |
electrical failure, balloon rupture) (see FDA, 2001a,b). These outcome codes help agency staff to set priorities for the review of reports.
The first priority for FDA analysts is the review of reports of “Code Blue” events, the list of which includes pediatric deaths, multiple deaths, fires, explosions, or anaphylactic reactions. The contractor who first receives and codes adverse event reports notifies staff of the Center for Devices and Radiological Health (CDRH) within 24 hours of receiving a report of one of these events. Certain events—such as a cluster of injuries in a single facility—may prompt an emergency response. Otherwise, an investigation may lead to a public health notification, change in labeling, or other response based on the conclusion that a problem exists with a device or its use. An investigation may also lead to a determination that no action is warranted because the event was not related to a problem with a medical device or its use. The CDRH analysts who review reports are, in general, responsible for groups of products that are associated with a medical specialty or that have common design or material characteristics.
Less than 1 percent of reports involve high-profile events. In fiscal year 2003, the major problems identified through analysis of adverse events
included failures in aortic connector devices that resulted in hemorrhage and death, meningitis associated with cochlear implants, aneurysm-related deaths associated with endovascular grafts, hospital bed fires, toxic shock syndrome associated with a particular brand of tampon, off-label use of an adhesion barrier, and saline leakage in the access port of the lap band adjustable gastric band (FDA, 2004b). Some of these problems were identified through relatively short-term analyses of a few event reports, whereas others were the result of a retrospective analysis of up to 10 years of reports. Responses included FDA public health notifications or manufacturer withdrawals of products.
“International vigilance reports” are also a high priority for staff review. These reports are transmitted by agencies (“national competent authorities”) responsible for surveillance in other countries. They typically involve the recall of products that have significant potential for harm.
The next priority for review is reports of events that are not in the high-priority category but that are also not so familiar that they are either reported through the summary report option described in Chapter 3 or identified by an automated report screening process (see below). These intermediate-priority reports account for about a third of the total. Again, reviewers may determine that no follow-up action is needed or they may recommend follow up.
The lowest priority for review applies to summary reports and reports flagged by an automated screening process that searches for certain well-recognized device-event combinations (e.g., silicone breast implants and capsular contracture, which is a tightening of the scar tissue surrounding an implant). Summary reports now account for about half of all reported events. The automated screening procedure picks up or flags about 15 percent of individual reports, and about 10 percent of these reports are reviewed by staff each month but only as a check that the screening tool is performing as intended.
Although FDA staff generally do not look for trends or changes in the summary reports and the reports flagged by the automated screening process, manufacturers (under the quality systems regulations described in Chapter 3) are supposed to monitor their event reports for trends and changes in frequency or severity of adverse events. Such monitoring could prompt further investigation and action (e.g., a recall).
Except for the small group of high-profile events, no specific rules define when a single report or series of reports should prompt further investigation of MAUDE reports, follow-up inquiries to manufacturers or facilities, epidemiologic study, or review of the clinical literature. Reviews and assessments of reports and other information about device hazards and judgments about appropriate FDA responses have a considerable subjective component. Resource constraints limit the agency’s ability to
investigate reports that do not involve deaths and other high-profile events.
It is worth reiterating that in addition to adverse event reports, FDA may learn of potential problems with a marketed device or its use in other ways, including during inspections of facilities and as part of ongoing manufacturer efforts to refine and improve a product. Problems may also be detected during postmarket clinical studies sponsored by manufacturers, the National Institutes of Health (NIH), or others. Other avenues of problem identification include published case reports of unusual or unexpected problems, presentations at medical conferences, or informal conversations associated with such meetings. Such conversations were an early indicator of a possible link between cases of meningitis and cochlear implants (see Appendix F).
ANATOMY OF ADVERSE DEVICE EVENTS: ILLUSTRATIVE VIGNETTES
“I never really thought about reporting that [problem] in particular…. There are just millions and millions of things that can go wrong.”
Melisande Statz-Hill, parent, 2004
This mother of a young child cared for at home with multiple medical devices was making two points. First, it did not occur to her to report (even to the home health agency) a problem that seemed to involve error in the use of a device—in this case overtightening by a nurse of ties for a tracheostomy tube—rather than a malfunction of the device itself. The second point was that the opportunities for something to go wrong—even for a family with private-duty nursing support and a home health company that specialized in pediatric patients—seemed endless.
To illustrate the many kinds of adverse device events (and device malfunctions and close calls) and the challenges of analyzing such events, the committee developed several vignettes or synthetic case histories. They are intended to convey both the diversity of device events and the interplay of variables associated with events and their aftermath. These variables include the complexity of the device and its management, the setting of care and its characteristics, the characteristics of the patient (e.g., developmental stage), the circumstances of the family (e.g., understanding of how to operate a device correctly at home), the opportunities for (or impediments to) reporting the event, and the resources brought to bear on understanding the event. The examples do not attempt to represent proportionately the distribution of reported (much less actual) adverse events by type of device, problem, or reported consequences.
Each story below is simplified to highlight issues of interest. Some describe situations that are reasonably common and even accepted as “nor-
mal” (albeit unwanted). Others describe unusual situations that especially challenge those attempting to understand the event and prevent it from occurring again. Most of the stories point to the importance of considering human factors (human behavior and human systems and their interaction with devices) in the design of devices and the interconnection of devices and their accessories.
Although this report has tended to focus on more complex, high-risk medical devices (mainly Class III devices) that undergo clinical testing, several of the vignettes underscore that serious adverse events often involve less complex but very widely used devices such as catheters, accessory tubing, and syringes. Other vignettes describe problems associated with long-term use of an implanted or partly implanted device, that is, problems that cannot be expected to be evident in the relatively short-term clinical studies that are usually submitted as part of the FDA approval or clearance process for medical devices. Absent systematic long-term studies of medical device safety and effectiveness, such adverse events—as well as rare short-term events—may only slowly reveal themselves in usual clinical care. The importance of long-term studies of medical devices used with children is discussed further in Chapter 6.
Each vignette is synthesized from a variety of sources, including case reports in the medical literature, reports in FDA’s adverse event database (MAUDE), training materials for the MedSun program, experiences of committee members, presentations or discussions during public committee meetings, webpages for clinicians and patients and their families, news reports, and similar sources. No example depicts specific child and family circumstances exactly, although each story draws from real experiences. The fictional names, personal situations, and institutional details have been created to add a human dimension to the abstraction of adverse event or case reports and also to underscore points emphasized in this report.
|
Vignette A: Close call with aspirated syringe cap. This vignette involves a close call with a simple medical device, a syringe with a cap. Other children who have aspirated syringe caps have died. The example highlights the importance of careful communication with parents about the safe use of simple but potentially dangerous devices. The father of 9-month-old Julia brought her to the primary care clinic because she was clearly uncomfortable and seemed to be running a fever. The doctor diagnosed an infection and prescribed an oral antibiotic to be administered with a syringe. He gave Julia’s father illustrated instructions and also demonstrated |
|
how to use the syringe, which was not an oral syringe for medications but a needleless hypodermic syringe with a fenestrated cap that allowed medicine to be drawn into the syringe with the cap in place. That afternoon Julia’s father drew the medicine from the bottle through the fenestrated cap into the syringe, but he stopped to answer the phone. When he returned, he administered the medicine by placing the tip of the syringe into the child’s mouth, not noticing that the clear cap remained on the syringe. The baby immediately started gagging, and the father realized he had forgotten to remove the syringe cap. He quickly placed the child on his shoulder as he called his wife; a few seconds later he saw the cap in his daughter’s mouth and retrieved it. Although she was screaming, she was no longer gagging or gasping. Julia’s parents raced her to the clinic, where a doctor evaluated the child, finding her upset but okay. After calming Julia and her parents, this doctor substituted an oral syringe—with a very distinctive cap—for the parents to use for measuring and administering the antibiotics. Consistent with the clinic’s patient safety and quality improvement policies, the doctor reported the problem internally. The clinic patient safety officer found other reports in the medical literature of aspirated hypodermic syringe caps, some involving deaths. By the end of the year, the clinic had put in place a policy that only oral syringes should be used in the delivery of oral medicines with infants and small children, and staff were working on better education strategies for parents, including a “teach back” step during which the patient or caregiver demonstrates use of the device to the physician or nurse. The safety officer reported the event to FDA, even though it was not required. Nonetheless, the “lessons learned” were essentially confined to the clinic. Device involved: Hypodermic syringe. Proximate cause: Lay user error. Institutional/system factors: Communication shortfall—use of device demonstrated to parent but without “teach back” or “show me” step to assess the parent’s understanding; lack of warning about the cap hazard; clinic or physician choice of (less expensive) hypodermic rather than oral syringe that is designed for administration of liquid medications by mouth; failure of physician/health care team to remove cap of hypodermic syringe prior to giving it to parents to use. |
|
Design factors: Presence of cap obvious when syringe used for injection, but hazard less apparent with oral use; fenestrated cap design allowed cap to stay in place while medicine was drawn into syringe. Comment: Safety principles would suggest removing the opportunity for human error altogether, that is, redesigning the device because there will always be the potential for user (especially a lay user) to forget instructions or fail to appreciate risks and dangers. Further reading: Kurtzweil (1994); Family Practice News (2000); ISMP (2001); Schillinger (2004). |
|
Vignette B: Circumcision clamp injury. As described later in this chapter, injuries involving circumcision clamps have prompted an FDA safety alert. In this story, hospital personnel were unaware of the alert. As in this incident, inexperienced users of a device—even a “low-tech” circumcision clamp—contribute to adverse device events, but deficient hospital systems of training and credentialing for procedures—which would likely not be mentioned in an adverse event report—can play a role. The physician, a new pediatric resident, was preparing to circumcise newborn baby John. The basic instrument was a Mogen-type clamp. The resident had watched the procedure several times and had been supervised while performing a few. During the procedure, the baby suffered a slight laceration of the penis. Fortunately, the injury was minor and easily treated, but the baby was kept an extra day in the hospital. More serious injuries—including amputation—have been associated with clamp defects and procedural errors. The hospital investigation revealed that the physician had limited experience in performing the procedure and lacked training with the device model used, its assembly, and the safety measures specific to that device, including inspecting the clamp for size and alignment. Investigators found that components of the device were not properly aligned, and the device had been incorrectly repaired using incompatible replacement parts. They could not determine when the device had last been inspected for alignment. FDA and other warnings about problems with certain circumcision clamps were unknown to physicians within the hospital. The hospital considered the injury to be too minor to require a report to the manufacturer or FDA, but it did institute a new policy for routine inspection of clamps. |
|
Device involved: Circumcision clamp. Proximate cause: Use error: physician failed to determine that the device used met use specifications and was undamaged. An experienced user, or one adequately trained with the use and assembly of the device used, might have recognized a problem before harm occurred. Institutional/system factors: Inadequate training; inadequate procedures for disseminating manufacturer and FDA advisories and making appropriate changes in internal policies and practices; incorrect repair by hospital personnel; lack of policy to label clamps to indicate size; lack of policy for periodic inspection of device for wear or proper alignment. Design factors: Design prone to misalignment. Comment: The process of physician education and training is changing from the traditional “see-one, do-one, teach-one” approach to “see several, ask questions (e.g., about differences in patient anatomy and clinical situations, what risks to anticipate and prepare for) and then do several procedures under direct supervision to evaluate procedural and evaluative skills and judgment before eventual independence.” Further reading: ECRI (1999); FDA (2000g). |
|
Vignette C: Deprogramming of cochlear implant. Children’s play can affect device performance. In this case, the static electricity charges generated by playground equipment created enough energy to deprogram a cochlear implant, requiring surgical replacement. Prompt reporting and evaluation of such events can lead to device modifications that protect future children. Some years ago, during an afternoon visit to the home of her cousins, Jennifer went with the rest of the family to a nearby park, where the cousins enjoyed using the playground equipment. Jennifer had been born with severe hearing loss. When she was 18 months old, she received a cochlear implant. With intensive language development therapy, she did very well. After returning from the visit, Jennifer complained that she couldn’t hear. A few days later, after he had extensively questioned Jennifer and her mother, examined the implant site, and performed a diagnostic assessment of the device, the surgeon who had implanted the device confirmed that it had failed. Based on some past |
|
experiences and conversations with colleagues at conferences, he suspected that device had been damaged by static electricity from plastic playground equipment. He scheduled Jennifer for surgery to remove the device and implant a newer model. He also told Jennifer’s mother what he thought had happened and reassured her that it was nothing she could have been expected to foresee. The surgeon sent the explanted device to the manufacturer with a description of the circumstances and his conclusions. Such reports led to refinements in the device materials and electronics to shield the implant’s circuits from damage and protect the software programming from being changed by static electricity charges. The manufacturer changed the implant’s labeling to caution physicians, families, and, when appropriate, patients about the wide variety of activities that may lead to a static discharge. Device involved: Cochlear implant. Proximate cause: Exposure of device to electrostatic charge. Institutional/system factors: Possible underreporting and slow investigation of risks to device performance based on problem reports. Design factors: Lack of shielding and filtering to protect against static electricity. Comment: The device’s design did not anticipate certain environmental hazards, for example, the build-up of an electrostatic charge as a child uses a plastic playground slide or a tubular slide in an indoor play center. Even getting into a car with new tires can result in electrostatic energy when a child touches the door handle. Reports of implant deprogramming have led to design refinements, including changes in materials (e.g., plastic replacing metal in the external processor unit), which allowed for better isolation of static electrical energy. In addition, devices can now be reprogrammed by an external computer (taking less than 5 minutes), for the occasion when a child finds that plastic slide irresistible. |
|
Vignette D: Orthodontic headgear injury. In this vignette, a child’s orthodontic headgear became dislodged while he was sleeping, and one of its sharp and pointed metal arms embedded itself in the child’s lower eyelid. Orthodontic headgear is commonly prescribed by orthodontists to correct the alignment and position of the teeth. Safe use of devices by patients or families depends on their adequate education and understanding of safety issues, including how |
|
to handle a very sharp and pointed object when applying it, removing it, or otherwise living with it. Twelve-year-old William was very fortunate. He was wearing orthodontic headgear—sometimes called a facebow—that had been prescribed three months earlier to straighten his misaligned teeth. One morning he awoke with a sharp pain under his eye. His cry of pain brought his parents to his room, where they found a laceration just below their son’s right eye. The ridged, rod-like arm of the headgear had somehow become dislodged, and its sharp tip had sprung out to cut the boy’s face. His parents called the orthodontist, who recommended that they go to the emergency room; instead, the parents took the boy to his pediatrician who examined him and stitched the laceration. After consulting with the pediatrician, William’s mother took him to another orthodontist, who determined that a safer device would be suitable. No one involved understood that the incident could be reported to FDA or thought about reporting it to the manufacturer. The boy’s former orthodontist was relieved not to be involved in a lawsuit. He did not reconsider his practices for using facebows, perhaps because he thought that might indicate that his practice had been deficient. Devices involved: Orthodontic facebow. Proximate cause: Device design, including hazardous points, allowed dislodgement from user movement during sleep. Institutional/systemic factors: Poor communication about a hazard with rare but sometimes severe consequences; continued professional use of hazardous device design despite subsequent development and marketing of safer devices for most situations. Design factors: Importance of taking use environment (motion during sleep) into account; safer designs available for many patient situations. Comment: Fear of litigation may be a disincentive to reporting or acknowledging problems. Professionals who are primarily involved in office-based care may not be aware of reporting options or may find reporting burdensome. Further reading: Samuels and Jones (1994); Samuels et al. (1996); Blum-Hareuveni et al. (2004); WTTG (2004). |
|
Vignette E: Infection from flawed bronchoscopes. Infections have many possible causes, and linking a device to an infection can take |
|
time. In this case, good hospital infection control systems led to a fairly early identification of a problem that led to recall of a medical device. Not all hospitals were successfully notified of the recall. Jerry was a 2-year-old who thought his mother’s shiny round earrings looked good enough to eat—so he tried. He choked, and the earring went directly into his windpipe. His parents took him to the emergency room of a nearby hospital, where an X ray showed the earring was lodged about 5 inches below his vocal cords. To retrieve the earring, the doctor used a bronchoscope, a tool that allows the physician to see the inside of the airways, remove foreign objects, take samples of tissues or secretions, or clean out obstructed or infected areas. After an otherwise successful procedure, Jerry developed a bacterial infection that is common in patients with cystic fibrosis but uncommon in patients with normal lungs like Jerry. The bacterium was quickly identified and successfully treated with a short extension of Jerry’s hospital stay. Shortly afterward, hospital infection control and epidemiology staff identified an increased incidence of this kind of infection in patients not normally at risk. Their investigation identified the bronchoscopy procedure as a common factor among affected patients. This focused their attention on the facility’s bronchoscopes and the procedures for cleaning, disinfecting, and inspecting them between uses. After intensive scrutiny of the devices and the procedures, they suspected that the design of one of the devices played a role. In the meantime, through their professional contacts, the infection control staff learned that similar problems had recently been reported to the device manufacturer and FDA. Several weeks later, through the same informal communication channel, they learned that the manufacturer was recalling the device, and several days later, the risk management department received a letter to that effect. All involved were surprised to see subsequent news stories about problems at a prominent academic medical center whose physicians had not been promptly notified of the bronchoscope problem and recall because the notification letter had been misdirected. Although the risk management staff would still use their professional network, the department decided to subscribe to an online device recall tracking system to provide an extra margin of security against delayed or misdirected recall notifications. Devices involved: Bronchoscope; bronchoscope reprocessing units. |
|
Institutional/systemic factors: Lack of effective and timely procedures for problem notifications and recalls to facilities and professionals. Design factors: Design of a threaded bronchoscope port connector that could not be adequately cleaned or disinfected. Comment: Devices may not be initially suspected as sources of infection, and making a definitive link can involve considerable investigation and testing. Manufacturers and hospitals have room for improvement in managing device recalls. Further reading: FDA (2002k); Jurasek (2003); Kirschke et al. (2003); Srinivasan et al. (2003, 2004). |
|
Vignette F: Effect of growth on implanted defibrillator. Implants and other devices used with adults are often adapted for use with children by simply making the devices smaller. For implants, the use of a smaller device may be adequate initially, but the child’s growth may eventually require replacement with a larger implant. For some devices, this is expected, but for others, growth-related issues only become evident through long-term follow-up. This vignette describes the latter situation. Maxine was 3 years old when she was diagnosed with Long Q-T Syndrome, a rare condition that episodically and unexpectedly caused her heart to beat so fast that it would not pump blood effectively to her brain and other vital areas. She required electrical shocks to save her life. After several drugs to control her heart’s electrical activity proved ineffective, the child’s cardiologist suggested a surgically implanted cardiac defibrillator (ICD) that would monitor the heart’s rhythm through wires (leads) placed on the sensitive areas of electrical activity within the heart. If the device detected an unsafe rhythm, it would fire a shock to Maxine’s heart. ICDs have saved the lives of many adults and children. The device fired twice during the next 4 months, each time averting a full cardiac arrest. Maxine’s parents continued to take her to the cardiologist every 4 months for follow-up studies of the device’s functioning. They got good reports during each of these visits. Then one morning, Maxine suddenly fell to the carpet, clutching her chest and crying. As he was trained to do, the father immediately called 911. At the hospital, doctors determined that the device had misfired and had given the girl a shock. Based on a chest |
|
X ray, hospital staff concluded that one of the leads had fractured. Possibly it had been stretched during a growth spurt following the child’s most recent visit to her cardiologist. Maxine’s parents were surprised to learn that nontherapeutic firing of shocks was a known problem. Physicians said that ongoing monitoring of the device was the best way to check for lead fractures or changes in lead position, but periods of rapid growth could sometimes cause the kind of problem Maxine experienced. Devices involved: ICD and leads. Proximate cause of adverse event: Device malfunction, unintended shocks. Institutional/system factors: Limited counseling of family about potential for device performance problems and, hence, the need for lead monitoring and replacement as the child grows. Design problem: Possible lead failure due to patient growth; device not returned to manufacturer for analysis. Other comment: The ICD has been studied intensively with adults, but pediatric studies are scattered. Large groups of pediatric patients have not been studied prospectively. One study of 29 patients found 38 chronic complications, the most frequent being lead failure. Interestingly, the size of child at the time of the implant was not a factor in outcome, but growth of the child in weight, height, or surface area was directly correlated with lead failure. Unfortunately, researchers have not identified a clear threshold for predicting such failure and have called for a large prospective study, perhaps using a multi-center registry or network. Further reading: Silka et al. (1993); Alexander et al. (2004). |
|
Vignette G: Fatal data entry error. For operators of medical devices, understanding of and adherence to safe procedures for entering and checking patient and treatment data for medical devices is critical. Familiarity with the routines of data entry can lead operators to take shortcuts, forego cross-checking or rechecking of information, and assume incorrectly that a device is operating consistent with the treatment plan. In this vignette, data entry errors and failures to check dose levels led to the administration of lethal levels of radiation. Billy had been diagnosed with brain cancer at age 10. His long-term prognosis was not good, but his physicians thought radiation |
|
treatment could provide some additional years of reasonably good quality life with his family. At the very outset of the treatment, a hospital staff member incorrectly entered the radiation dose information, and no one ever again confirmed the prescribed dose with the settings on the radiation therapy unit. Four weeks after the final treatment, Billy experienced progressive skin eruptions at the area where the radiation beam was directed. A consulting dermatologist questioned the medical physicists at the treating facility about the radiation treatments. The boy’s physicians concluded that he had been exposed to a severe radiation overdose. A few months later, Billy died. Once attention had focused on the radiation dose, the hospital called in a consulting engineer to investigate. When he checked the settings in the electronic memory of the therapy unit, he discovered the dosage error. The facility’s staff began to fault the equipment’s software for not catching the error, but the engineer inquired whether the erroneous setting was a correct setting for any category of pediatric patient. It was. The system’s software would have not “queried” the dose for that reason. At the time, the institution did not have real-time dosimeters that would have detected the dose error. The hospital settled a lawsuit brought by Billy’s parents. Devices involved: Linear accelerator, radiation therapy simulator. Proximate cause: Staff error in entering data and failure to check the treatment settings against the prescribed dosage before each treatment. Institutional/system factors: Failure to follow institutional policy to double check data entry before each treatment; lack of real-time dosimeters; inappropriate staff reliance on software to detect error (dose was not outside the range for all patients); inadequate staffing of radiation oncology department. Device factors: Poor software design of data entry menu. Further reading: Adapted from FDA (2002n, Case Study 16). For a report on serious radiation device adverse event involving software, see Doyle (2000). |
|
Vignette H: Growth-related complication from gastrostomy tube (g-tube) design. A device designed for use with adults may not be suitable for a growing child. In this story, the doctors chose a |
|
specific gastrostomy tube because it could be placed with less invasive surgery and the design featured a disc that provided just enough tension to reduce leakage. As the child grew, the disc-tension design caused too much pressure, which caused the disc to embed in and then erode through the stomach wall. Robert, now 14, had survived a serious brain injury when he was 12. His parents visited him daily at the rehabilitation hospital where he received physical therapy and supportive care. The family was grateful for his gastrostomy tube, which allowed him to receive all of his nutrition through the tube inserted directly into his stomach. This tube had been placed 17 months ago in the radiology suite of the adult hospital where he was originally treated. It had seemed to work well, but then the area adjacent to the tube showed leakage and some redness. The rehabilitation hospital decided it was prudent to return Robert to the original hospital for evaluation. The work-up there found that the inner disc of the tube, which held it against the stomach wall, had actually burrowed into the inner lining of the stomach, going completely through the stomach wall. Surgeons had to remove that portion of the stomach where the disc was buried. They placed a different type of gastrostomy tube during the surgery, which required an additional 2 weeks of hospitalization. When the parents learned that the new gastrostomy tube would not cause the same problem, they asked why they were not told of the risk with the original device and whether the problem had been reported. They wanted other parents to be vigilant about this complication if it was possible. Devices involved: Percutaneous gastrostomy tube with triangular retention disc. Proximate cause: Tension from fixed distance between internal and external discs of the g-tube, which caused pressure on the tissues of the child’s growing abdominal wall; embedding of corners of triangular-shaped internal retention disc not noticed, possibly due to the child’s inability to communicate discomfort in a specific manner. Institutional/system factors: Lack of procedures to evaluate distress in patient with communication limitations; lack of adequate protocol for monitoring implanted devices in such patients; tube placed by radiologist who would not be expected to be involved in follow-up management of the device; no apparent hand-off of device management to other physician involved in child’s care. |
|
Design factors: Lack of device mechanism to measure the disc tension or allow adjustment for a child’s growth; failure of device labeling to mention growth considerations; disc shape possibly contributed to embedding. Other comment: Authors of a case report on this problem recommend checking the tension of the device by regularly spinning the tube around the retention disc. They also suggest scheduled replacement of the tube, given the predictable growth of a child’s abdominal wall. Some pediatric gastroenterologists favor tubes with circular retention discs but would still recommend replacement of these with a device that did not provide such tension as soon as the ostomy track was mature. In children’s hospitals, placement by interventional radiologist is probably not standard practice (personal communication, Norberto Rodriguez-Baez, M.D., Division of Gastroenterology and Nutrition, Department of Pediatrics, University of Texas Southwestern Medical Center, March 24, 2005). Further reading: Cahill et al. (2004). |
|
Vignette I: Parent’s mistake with home infusion pump. As in this story, treatments once confined to hospitals are also taking place in the home and school. Parents and others now provide care and cope with medical devices—and problems—that formerly were the domain of health professionals. Training for parents on how to operate a device may be limited and include neither directions on how to assess or troubleshoot problems nor evaluation of a caregiver’s capacity to deal with mistakes or malfunctions. Katie was 2 years old when she was diagnosed with a resistant bacterial infection of her femur. She was sent home with an infusion pump that would deliver several weeks of intravenous (IV) antibiotics through a central venous catheter. While in the hospital, Katie had gone through four different standard IV lines (catheters). Few usable vein sites were left, so she received the central line. Katie’s mother attended classes at the hospital on how to manage the line and change the dressings around the skin entry site. She was confident that she had the necessary skills and information to participate safely in her child’s care. A nurse was to come out to the house for at least one of the three doses per day. One evening, Katie’s mom was feeling particularly frazzled and distracted because both siblings were fussing and her husband was |
|
out of town. She forgot to prime the tubing—that is, fill it with fluid to remove air—before putting the tubing into the cassette unit of the automatic pump and then attaching that set-up to her child’s IV. After she turned the pump on, it sounded an alarm within seconds, and the pump display clearly read “air in line.” Katie’s mom turned off the pump, removed the tubing from the pump’s cassette, and desperately tried to remember what to do. Meanwhile, the air in the IV line migrated through the tubing into Katie’s veins. The child stopped breathing. As she had been trained to do, Katie’s mom gave her rescue breaths, which revived the child. The mother then saw that she had not clamped the tubing with the air in it and immediately realized what had happened. She called 911 and an ambulance took Katie to the emergency room, where she was treated and released. Subsequently, Katie’s mom took the girl to the hospital’s emergency room for all of her antibiotic doses until the infection resolved. Devices involved: Portable IV infusion pump. Proximate cause: Failure to prime tubing (remove air from tubing) prior to connecting tubing to patient’s IV; failure to clamp tubing. Institutional/system factors: Inadequate parent education on safe device operation and troubleshooting; possible poor selection of patient for family-delivered home therapy (multiple children, only one adult routinely at home). Device factors: Lack of warning messages other than “air in line” on the device display; no message to “disconnect tubing from child” or “clamp tubing close to the child.” Further reading: Laskey et al. (2002). |
|
Vignette J: Injury from pediatric use of orthopedic device. This story illustrates how adverse outcomes can occur when physicians lack sufficient information to guide the use of complex, high-risk devices with children. In this example, a child with severe facial abnormalities underwent craniofacial reconstruction using a complex rigid external distraction osteogenesis device. Howard was born with his upper jaw and cheek area so under-developed that his lower jaw protruded and his teeth were misaligned. His eyes looked like they were sinking into his cheeks, and his underdeveloped midface and crowded airway created speech problems. When Howard was almost 8 years old, his parents con |
|
sulted a craniofacial group that offered “stretching” of the child’s midface using a process similar to that used earlier to widen his palate. The process, which is common in orthodontal work, is called distraction osteogenesis. It takes advantage of the fact that bones are constantly remodeling, especially in the growing child or in healing bone. During the procedure, the surgeon strategically makes cuts in the bone, after which traction is applied to maintain a specified distance between the bone segments. Active bone-building cells then construct new bone to fill in the gap and thus elongate the bone. The procedure had been well studied in the elongation of leg bones in adults, but now the surgeons were applying the theory to the complex set of bones of a child’s skull and face. During surgery, the base of the device system, called a halo, was screwed into Howard’s skull to anchor various rods, bars, and brackets attached to his facial bones. Shortly after Howard’s discharge from the hospital, the area around two of the screws in his skull had become very red and swollen and was draining yellowish material. A CT scan showed that the screws had gone too far into the skull and were touching the brain’s surface. Surgeons removed and replaced the screws, but after two more days, doctors found the new screws had actually gone partially into the brain. Fortunately, Howard suffered no brain damage. No one could explain how the problem happened; the boy had only been lying in bed or on the couch at home. After removing the old hardware, doctors tried a new device still under testing and development. This device allowed for more facial and mouth movement and also for more precision in setting the force and angle of force that the tension bars applied to the halo element that is attached to the skull. This device worked well and within 2 years, Howard had good functional and cosmetic results. Still, his parents wondered about how the child’s growing bones would develop because the device was too new to have long-term results. Howard’s physicians read about a very similar case in a journal case report, which mentioned studies of the halo device but noted the lack of data about important questions in pediatric use of the device. Device involved: Modified halo rigid external distraction system. Proximate cause: Excess force applied to skull bones by traction apparatus; direction of force and lesser thickness and density of child’s skull also likely factors. |
|
Institutional/system factors: Lack of agreed-upon guidelines for the use of procedures that have not been evaluated with children; lack of commitment to systematic long-term evaluation of complex orthopedic devices. Design factors: Manufacturers and clinical investigators have not systematically studied or modeled the complex force vectors required in the application of this type of device to developing bones in a child’s face and skull. Other comment: Absent systematic clinical studies, physicians using complex devices may lack adequate instructions about safe and effective methods of application and appropriate patient follow-up and monitoring. They likewise may lack sufficient information about the prospect of long-term benefits and short-term and long-term harms to guide clinical and family decisions about the use of a device with children. Further reading: Dormans et al. (1995); Le et al. (2001). |
|
Vignette K: Difficult-to-detect implant problem. This story provides another example of how an ill child’s physical dimensions can affect device performance. With attentive parents who consulted and followed the device manufacturer’s instructions, the child received the careful evaluation needed to reveal a difficult-to-detect problem. Delay could have been fatal; catheter malfunctions with this device have been linked to deaths of both children and adults. The parents of 4-year-old Sarah found themselves confused by their daughter’s increasing muscle tightness, high fever, and itching. Sarah had cerebral palsy, but they had not seen her muscles twisted up like this since she’d had an intrathecal Baclofen pump placed in her abdomen. The hockey puck-sized pump delivered the muscle-relaxing Baclofen drug through a small catheter to the spinal fluid. Before that, Sarah’s muscle spasticity meant that she could not walk easily or play like other children. Then the pattern changed, and Sarah showed signs of lethargy; her muscles seemed too “floppy” rather than too tight. To her parents, she seemed to have switched from showing symptoms of an underdose to showing symptoms of an overdose, at least according to the information on a wallet-sized card supplied by the pump manufacturer. The card, which Sarah’s parents consulted as had been stressed by the child’s physicians, instructed patients to |
|
check immediately with their neurologist. The neurologist found that the pump’s battery and reservoir of medication were adequate but that the girl’s symptoms were consistent with Baclofen withdrawal. He ordered a study to check, in particular, the integrity of the tubing. The study showed no leaks, but the doctor recommended a surgical evaluation, given that Sarah was getting more and more ill. Surgical exposure of the device revealed that the tubing was cracked in the segment closest to the pump, an area of the tubing that is relatively inflexible and that passed over the bony prominence of Sarah’s pelvic bone. Like many children with cerebral palsy, Sarah was seriously underweight, so she had little padding to prevent tubing wear at the site it passed over her bones. The surgeons replaced the tubing. Approximately 6 months later, Sarah again showed symptoms of an overdose. During another surgery, her doctors found the tubing cracked in the same area. (For technical reasons, it was virtually impossible to discover or verify this problem without surgical exploration.) This time, however, Sarah’s neurologist and surgeon had read a report in a professional journal that concluded that the infusion pump needed to be placed differently in children such as Sarah, to avoid compression at a spot where the tubing was relatively inflexible. (The compression led first to an underdose and then to a fracture and subsequent overdose.) This seemed reasonable, so the surgeon adjusted his surgical procedure. He later learned that the manufacturer had adjusted its implantation directions. Device involved: Intrathecal Baclofen pump. Proximate cause: “Stiff” portion of catheter tubing in friction against active child’s protuberant bone, causing “nick” or “crack” in tubing. Institutional/system factors: Possible slow detection of problem due to underreporting. Design factor: Device large relative to size of very young child with cerebral palsy (average 4-year-old girl with cerebral palsy weighs 24 pounds compared to normal weight of 35 pounds); stiff tubing stressed by initial implantation strategy. Other comment: Extensive manufacturer website addressed many aspects of device operation and troubleshooting. The information and warnings on the manufacturer’s information card helped parents to recognize problem and seek assessment that identified the life-threatening device malfunction. Further reading: Coffey et al. (2002); Dickerman and Schneider (2002); Dickerman et al. (2003); Gooch et al. (2003). |
|
Vignette L: Losing track of an implant. This case illustrates a relatively “forgotten” device, one that was important during a child’s early years but not in adolescence, when responsibility for device management shifted to the adolescent. This shift has risks as well as benefits related to adolescent inexperience and failure to appreciate or recognize the need for vigilance in device maintenance. Without ongoing involvement in device use or maintenance, parents may lose track of what is happening, especially if the device is not seen as critical to life and health. Elizabeth, a 16-year-old with cystic fibrosis, had a “port-o-cath” central venous catheter placed when she was 6 years old. This catheter had provided intravenous access for countless admissions and treatments for pneumonia, each requiring long courses of antibiotics. Once Elizabeth entered her teenage years, the surgically implanted device (which consisted of a self-sealing medication reservoir that was attached to a catheter that ended in the superior vena cava) was rarely used. Elizabeth requested that she take over the device’s weekly care, which involved flushing the catheter. The catheter was imbedded near her breasts and privacy had become paramount to Elizabeth. After following the routine successfully for an extended period, Elizabeth forgot to flush the catheter for several weeks. Then, when she tried it one morning, she had to exert great force and still was unsuccessful. Later that day at dance practice, she experienced sudden and sharp chest pain. The school called an ambulance, which took her to the emergency room of the hospital where she had been treated before. When the girl’s parents arrived at the emergency room, they told the staff that her lung disease was stable. Neither Elizabeth nor her parents thought to mention the central line, and the staff saw nothing in the girl’s medical record that alerted them to the line’s presence. (Mention of the device was buried in progress notes, and the record included no device equivalent of a medications list.) Because Elizabeth was having trouble breathing, the emergency personnel obtained a chest X ray, which showed that the catheter had separated from the reservoir and had migrated through the vein into the right side of the heart, with part of it headed out into the small vessels of the lungs. Elizabeth then told the physicians and her parents that the device had been neglected for some weeks and that she had been unable to successfully flush it that morning. She underwent surgery to remove the fractured catheter. |
|
Device involved: “Port-o-cath” central venous catheter. Proximate cause adverse event: Old central line tubing with thrombus formation that made it difficult to flush; likely fracture of line during forceful attempt to clear it. Institutional/system factors: Lack of clear documentation in medical record at the hospital/emergency room citing device presence; incomplete medical history from patient and family; school personnel unaware of device and unable to advise emergency medical responders; patient’s physicians lost track of device, failed to assess continued need for it, and failed to monitor patient adherence to and awareness of device maintenance requirements. Design problem: Thrombus and fibrin clot formation common in catheters; lifespan of catheter system not known or publicized. Other comment: Notwithstanding the importance of respecting an adolescent’s developing maturity and the value of a wellness model of care, these patient-centered strategies risk a loss of vigilance with respect to device risks, safe maintenance, and adult oversight. The adolescent years challenge parents in many ways, especially when the adolescent has a serious but stable medical condition. In these situations, monitoring of a teen’s health maintenance practices can be a source of tension, and teens may be reluctant to admit that they have not followed instructions and need assistance. Further reading: Fratino et al. (2001). |
|
Vignette M: Insufficiently evaluated procedure. In this vignette, an interventional radiologist attempted to remove a clot from the end of a central venous catheter using a procedure that had been reported in the literature but not systematically evaluated. During the procedure, the tip of the original patient’s central line sheared off and immediately drifted into the end of a vein within the lungs. The child suffered no immediate harm but required surgery to replace the catheter. After Hannah was diagnosed with acute lymphoblastic leukemia, doctors placed a central venous catheter in her chest to deliver the required chemotherapy. The catheter was used frequently for both delivery of medication (its primary purpose) and blood drawing for ongoing laboratory studies. For a 3-year-old facing six months of chemotherapy, easy access for blood draws reduced her pain and stress. |
|
After 3 months, Hannah’s catheter became clogged at the catheter tip. The catheter still allowed the free flow for chemotherapy but not for blood draws. The clog resulted from a natural process of fibrin formation that occurs on the end of every catheter that dwells within the lumen of a vein for an extended period. The interventional radiologist offered to perform a relatively new procedure that involved going into a vessel in Hannah’s leg with a second catheter that had a loop at its end to grab and strip the fibrin debris off the end of the other catheter. The goal was to salvage the original catheter. This workaround had been reported in the pediatric interventional radiology literature and seemed reasonable given that the only alternative was to replace the catheter, a surgical procedure requiring general anesthesia. During the procedure, the clot was stripped, but with it, the tip of the original central line sheared off and floated away, coming to rest deeply within the vessels of Hannah’s lungs. It could not be retrieved. Hannah experienced no symptoms, but required another central line to be placed surgically. The radiologist was completely surprised by the event, having never heard of or imagined such a possibility. She discussed the event with colleagues and reported it to hospital risk management, which did not evaluate whether it was reportable under FDA rules and did not otherwise investigate or act further. Hannah’s parents really did not understand what happened; they were focused entirely on their child’s health and did not consider action against the hospital. The girl’s oncology team wondered whether the expected benefit of trying the new procedure really justified the risk, which was not well defined because the procedure had not been systematically evaluated with children. The radiologist hoped to see future studies that assessed the risk of fracture or shearing of central venous lines when tension is applied to them during a stripping procedure. Devices involved: Normal central venous catheter; modified central venous catheter. Proximate cause: Use of device beyond its specifications. Institutional factors: Lack of expertise and training in procedure; lack of opportunity for learning and early problem identification by others due to failure to report adverse event. Design factor: Catheter prone to clotting. Other comment: “Workarounds” (as described in Chapter 2) are common to fix or modify—rather than surgically remove and re |
|
place—a device that is implanted. This is justified if the risk incurred is low compared to replacement. When a procedure and the requisite skills have not been evaluated with significant numbers of patients, the risk can be difficult to assess. Further reading: Knelson et al. (1995); Haskal et al. (1996); Bessound et al. (2003). |
|
Vignette N: Fatal error in connecting tubing. Correct assembly of devices is critical for safety. In this vignette, one tube was confused for another because their end-fittings looked similar. The result was fatal when a child’s oxygen tubing, under pressure, was inadvertently connected to his IV pump. Although training nurses to assemble devices is important, a device design that would prohibit such a deadly hook-up would be more effective at avoiding this human error. Nurse Johnson independently cared for four children on a busy pediatric ward. One patient, 9-year-old Andrew, had been hospitalized after a severe asthma attack, requiring oxygen therapy and aerosolized treatments as well as IV antibiotics. He was absorbed in his video game when Johnson arrived to connect the tubing for his afternoon antibiotic dose. A respiratory therapist had just finished Andrew’s breathing treatment, delivered using the oxygen source from the wall. She placed that oxygen tubing across the bed rail, but forgot to turn the oxygen flow off. Johnson stepped in beside Andrew’s bed and hung the bag and tubing of IV antibiotics she had carried in the room with her. Andrew complained that the nurse was in the way of his video game and she stepped aside. Then, when she reached over the video-game control wires, she mistakenly took hold of the oxygen tubing, the end of which was very similar to the tubing for the IV bag. She next fitted the wrong tubing to the pump, which was already connected to Andrew’s IV in his arm. This sent air into the IV set, and then into Andrew, creating a fatal air embolism. Andrew gasped and died instantly. When the resuscitation effort failed, shocked hospital staff set in motion the institutional procedures for notifying a family of a child’s unexpected death. Although the resuscitation effort had disrupted the physical scene around the child’s bed, the improperly connected tubing remained in place, and interviews with nurses |
|
and therapists provided a clear picture of the circumstances. Also, later review of a chest X ray taken during the resuscitation (to check emergency placement of a tracheal tube) found air disseminated throughout many of the major blood vessels. This indicated that the suspected air embolism was indeed the mechanism of death. Devices involved: IV set and tubing; nebulizer and tubing; video wires. Proximate cause: Use error: failure to connect tubing correctly. Institutional/system factors: Inadequate professional training and institutional reinforcement about fundamental importance of tracing tubing and electrical connections from origin point to patient. Design factor: Tubing connectors that allow incorrect connections; failure to prominently label tubing that is not protected from incorrect connections. Other comment: Mandatory standards for tubing connector design are not in place. Further reading: ISMP (2003); ECRI (2004b). |
|
Vignette O: Fatal incubator malfunction. The total dependency of infants requires that pediatric devices have redundant safety mechanisms. In this case, an infant incubator overheated, resulting in the death of a baby. The noisy, busy environment of the neonatal intensive care unit contributed because nurses did not hear the device’s alarm. Brittany, a 2-week-old newborn in the intensive care nursery of a hospital, was resting inside of the protected world of her isolette (incubator). Within this world, all vital environmental variables—oxygen, humidity, heat, and light—are fixed to support a fragile baby’s existence. The nursery was understaffed and, as usual, it was noisy with many alarms, infant cries, telephones, and beeping monitors. Brittany’s isolette alarm went off, but none of the nurses noticed. The alarm was, in effect, cognitively “subtracted out” by similar incoming auditory stimuli. Eventually, a nurse noticed that the isolette display showed an abnormal blanked-out reading. When found, Brittany had suffered irreversible, fatal hyperthermia (excessive body temperature). The isolette had malfunctioned and had gotten so hot that the mattress tray had melted. The facility and the manufacturer initially could not determine the source of the problem, and a literature search turned up no |
|
similar events. Consultant engineering specialists eventually determined that a brief power interruption had disrupted computer control of the device, which produced an abnormal display and allowed the fatal overheating. The power interruption had been caused by loosened wires within a replacement power plug on the device’s power cord. The replacement had occurred before the unit was shipped to the hospital. The investigators also found that the high temperature back-up thermometer had been improperly serviced by the manufacturer. In another critical failure, the hospital engineering staff had not conducted the usual incoming inspection of the device after a communication error left them unaware that the device had arrived. The hospital risk manager reported the incident to FDA and the manufacturer. The manufacturer, after filing two reports that the investigation was continuing, filed a third report that described the wiring problem. Some time after Brittany’s physician had told her family the shocking news of the baby’s death and offered support for the family in their grief, the physician and other hospital personnel—consistent with institutional policies and procedures and with help from the family services unit—met with the family again. They again expressed their deep regrets, but they also described what they had learned, explained what they were doing to prevent such a tragedy from occurring again, and answered the family’s questions. Later, the hospital and the family agreed on a financial settlement, which was followed by a settlement with the manufacturer. Devices involved: Incubator, back-up thermometer, detachable power cord, skin temperature probe. Proximate cause: Manufacturing error in installing power cord; improperly adjusted back-up thermostat. Institutional factors: Failure to conduct initial product inspection; communication failures; environmental noise; understaffing. Further reading: Adapted from FDA (2002n, Case Study 8). |
SOURCES OF ADVERSE DEVICE EVENTS
The stories above illustrate many sources of adverse device events and close calls. Box 4.2 presents one categorization of event sources and provides brief additional examples of each.
The critical source of an adverse device event may be as profound as a shortfall in basic scientific understanding of a disease or physiological process, for example, the long-term effects of incubator-supplied oxygen on
|
BOX 4.2 Science/evidence base or engineering concepts
Device design (including design of accessory devices and software)
Manufacturing process
Labeling
User facility administrative and patient safety systems
|
children’s eyes or the interaction between a child’s body and a material used in a device. The source may also be as ordinary as a typing or data entry error for a programmable device, an error which, although mundane, can have tragic consequences for an individual child and family. One goal of human factors engineering and other safety strategies is to design devices and the systems in which they are used in ways that either limit the oppor-
User facility device inspection and maintenance
Environmental conditions at site of use
Operator/user training and supervision
Device operation by individuals or teams
Tampering, sabotage, or counterfeiting
SOURCE: A starting point for the categories used here was the classification scheme devised by ECRI for its Medical Device Safety Alert database (see http://www.mdsr.ecri.org/); see also Bruley, 1994; FDA, 2002n; ECRI, 2005. |
tunity for certain types of use errors or block them from having harmful consequences.
Compared to drugs, use errors tend to be much more variable in nature for devices, reflecting the greater physical diversity of medical devices and their means of human use. Some adverse drug events actually stem from errors in the use or design of medical devices, for example, infusion pumps.
A device problem may sometimes be quite obvious. For example, a medical device may visibly warp or crack. Certain kinds of use errors, such as faulty connection of tubing, may also be quickly evident. In this respect, the proximate cause of many device adverse events may be clearer than with many events involving drugs, including those involving interactions with other drugs.
In other cases, extended investigation may be required to determine that a bad outcome or adverse event is related to a medical device. Infections, which can have many possible causes, are a case in point. A device-related infection (e.g., one associated with a design flaw or deficient sterilization practices) may only be suspected when an unusual pattern or trend in infections is noticed and other explanations appear inadequate. Linking the infection to a medical device can require combination of laboratory and epidemiological studies, as was the case with the earlier story (vignette N) about infected bronchoscopes (see also the investigation of meningitis in patients with cochlear implants discussed in Chapter 6).
Sophisticated devices with many different components present particular challenges for adverse event investigation. A recent article on the possible hazards of telemedicine used a number of adverse event reports in MAUDE to illustrate how incidents involving complex, software-controlled technologies (which often integrate components from several manufacturers) can be extremely difficult to understand, recreate, and diagnose (Johnson, 2003; see also Johnson, submitted for publication).
As noted throughout this report, although adverse device events sometimes have single causes, they often have multiple contributing factors. In addition, events often involve multiple devices (and multiple people interacting with the devices), each of which may need to be evaluated as a possible cause or contributor.
A few studies of adverse events and medical errors have looked at pediatric populations. For example, an analysis of voluntary, anonymous reports to a network of neonatal intensive care units reported 1,230 events in a 27-month period (Suresh et al., 2004). Nearly half of the errors resulted from failures to follow policies or protections and approximately a quarter each from inattention and communication problems. During the last 10 months of the study period, 2 percent of the events resulted in serious harm and 25 percent in minor harm. Another study reported that a complication related to medical care was found with 0.8 percent of all hospital discharges of children in 1996 (McCormick et al., 2000). A more recent analysis of a large database of inpatient admissions from 1988 to 1997 found rates of hospital-reported medical errors between 1.81 and 2.96 per 100 discharges (Slonim et al., 2003). Rates were significantly higher for technology-dependent or special needs children, a finding consistent with studies of adults.
LIMITATIONS OF ADVERSE EVENT REPORTING PROGRAMS
Problems with the passive or spontaneous reporting of adverse events or health problems are hardly unique to medical devices or FDA programs (see, e.g., O’Neil et al., 1993; Cullen et al., 1995; IOM, 2000c, 2004b; Wald and Shojania, 2001b; Samore et al., 2004). Other public health agencies, medical product manufacturers, health care facilities, and patient safety organizations experience similar problems. Notwithstanding the limitations, event reporting remains an important component of existing and evolving programs to protect patients and improve public health.
FDA’s program for the reporting and analysis of adverse device events has been the subject of at least three reports by the Government Accountability Office (GAO, formerly the General Accounting Office). The first two, issued in 1986 and 1989, reported significant weaknesses (GAO, 1986, 1989). A 1997 report credited FDA with improvements (some in response to legislative changes) but stated that “FDA does not systematically act to ensure that the reported problems receive prompt attention and appropriate resolution” and thus does not function satisfactorily as an early warning system for problem medical devices (GAO, 1997, p. 2). The report also cited the agency’s slow review of reports of device malfunctions that did not result in harm but that might nonetheless have served as early warnings of problems before they caused harm. In 2003, FDA itself characterized its program of postmarket surveillance as “not working well” (FDA, 2003n, unpaged).
In addition to criticizing FDA, the 1997 GAO report also criticized the quality of user facility reporting. It cited delayed reports, failure to submit semiannual summary reports, and lack of critical information (e.g., type of device, outcome of event). It proposed that feedback to reporting facilities of information about the outcome of a report might improve knowledge of device problems and encourage better reporting. The report acknowledged the agency’s response that providing such feedback would require substantial resources. As described below, the pilot MedSun program responds to some of the GAO’s criticisms by providing more feedback and other interaction with personnel at participating facilities.
A recent estimate of underreporting of medical device-associated adverse events came from an analysis of data from National Electronic Injury Surveillance System (NEISS), which has information on consumer product-related injuries based on emergency department records from a probability sample of hospitals (Hefflin et al., 2004). The analysis found the number of reports was “four times greater than the annual number of adverse event reports received by medical device-regulating surveillance systems” (Hefflin et al., 2004, p. 246). This analysis may provide some sense of the magnitude of serious, device-related problems experienced by patients outside the hospital. NEISS does not capture data on injuries treated in other areas of a hospital, which would include injuries associated with device errors or
malfunctions that were, for example, experienced and managed in the intensive care unit.
Underreporting is also suggested when a public health notification or recall brings attention to a problem and FDA then sees a sudden increase in reports of the problem. One example reported in the literature involved a vacuum extractor used to assist vaginal delivery of a newborn (see, e.g., Ross et al., 2000).
The limitations of passive or spontaneous reporting of adverse device events apply generally to both adults and children. It is possible, however, that the events affecting children could be subject to higher or lower rates of underreporting or poor-quality reporting. The committee is not aware of any relevant comparative studies.
Problems with Reporting of Adverse Events
“The ‘grapevine’ system of reporting appears to be relied upon by physicians and other health professionals. In many subspecialties, including cardiology and neonatology, there is a small network of pediatric experts. It is common for these physicians to share stories of mishaps or near misses in an effort to prevent others from making the same mistakes. This dependence by many individual physicians on this kind of information sharing is clearly not sufficient…. [However,] there is no quick, simple system to allow reporting of medical device usage (both successful and unsuccessful). Current electronic databases [of adverse reports] are often difficult to locate and can be cumbersome or time consuming to use.”
American Academy of Pediatrics (AAP) et al., 2004b
Important as informal professional communication networks are, they are not adequate to the task of systematically identifying and communicating the array of problems that can arise with the design, production, distribution, and use of medical devices. Such networks also are not universal and may not reach those clinicians or other users most in need of information about problems with medical devices. Furthermore, although informal communications may lead to alterations in professional practice, they may or may not reach hospital risk managers, device manufacturers, FDA, or others in a position to respond more comprehensively to device problems once they are identified. In some respects, informal communication might be viewed as a “workaround,” a way of compensating for some of the inadequacies of formal surveillance programs.
Contributors to Underreporting
Several of the vignettes presented earlier in this chapter described failures to report adverse events and also suggested some of the reasons for
underreporting. In addition to ignorance of reporting requirements or opportunities, reasons include workload pressures, liability considerations, misunderstanding of privacy regulations, concerns about competitors, and lack of adequate institutional procedures and other support for reliable reporting.
As users of devices, clinicians may not be aware that devices are “reportable” products and that FDA has an adverse device event reporting program. Clinicians also may be accustomed to “working around” certain kinds of device problems without recognizing them as reportable events (AAP et al., 2004b; Bright, 2004).
At the institutional level, patient safety programs have tended to focus more on medication errors than device errors. Institutions have been slower to develop structures and procedures to support the reporting, understanding, and prevention of adverse device events, including education of clinicians about their role in identifying adverse device events and device malfunctions or failures. Institutions may likewise lack reliable mechanisms for learning about and implementing device recalls and public health notifications that advise changes in practices involving a device.
Some institutions have been intimidated or confused by the patient privacy provisions of the Health Insurance Portability and Accountability Act of 1996 (HIPAA, P.L. 104–191). On its MedWatch website, FDA has clear messages that “the Privacy Rule specifically permits covered entities (such as pharmacists, physicians, or hospitals) to report adverse events and other information related to the quality, effectiveness, and safety of FDA-regulated products both to the manufacturers and directly to FDA” (FDA, 2003b, unpaged). In addition, the Privacy Rule permits (but does not require) covered entities to disclose—without getting a patient’s authorization—protected health information to parties (for example, manufacturers) that are conducting postmarket surveillance required by FDA (45 CFR 164.512(b)(1)(iii)(D)). (State laws can be more conservative than federal law, but most are consistent with respect to public health exceptions.)
It is the committee’s sense that confusion about HIPAA and the legal discretion of user facilities and professionals to disclose patient information related to adverse events to FDA or manufacturers remains a problem. The actual instructions for Form 3500A for reporting adverse events do not mention HIPAA, although MedWatch has a notice about HIPAA that is displayed on the page that includes links to forms and information about reporting safety problems. As discussed in Chapter 6, HIPAA also is a concern for manufacturers and investigators collecting information for required postmarket studies.
When health care professionals and institutions do report adverse device events, they do not always include essential information about the event, the device (e.g., specific model), and any attached accessories. Front-
line clinicians may know little or nothing about the history of a device (e.g., whether it has been refurbished), and they may not be aware of the difference between a device brand and a specific device model. If not documented at or near the time of an event, these kinds of details may be difficult to reconstruct or collect later. Details about generic, seemingly innocuous products such as many kinds of tubing may be practically unavailable.
In addition, concerns about liability related to possible errors in the use of a device may affect whether professionals and user facilities report a problem, how they characterize the nature and source of the problem (e.g., use error versus design problem), and whether they provide a manufacturer with a device for evaluation.3 This concern exists despite the confidentiality protections offered by FDA’s statute and regulations for patients, physicians, and other initial reporters of events, and (in most situations) user facilities (21 USC 360i(b)(2); 21 CFR 803.9; 21 CFR 20.63(f)). Identifying information about these reporters is not included in the public MAUDE database, and such information in the internal FDA database is not releasable in responses to requests under the Freedom of Information Act. Internal facility records documenting an adverse event and its investigation are not so protected.
In contrast to user facilities, the names of manufacturers and devices are included in the public MAUDE database. Such information is a necessary means of identifying problems with specific devices (including use errors) and disseminating that information to clinicians, user facilities, and patients. Anonymous reporting, which may be constructive for some purposes, is not appropriate in this case. Notwithstanding regulatory requirements and public health arguments, manufacturers may understandably be concerned that their reports will attract attention from lawyers who specialize in medical product and malpractice litigation (see, e.g., Quinley, 2001).
Moreover, competing companies have access to the public MAUDE reports. One long-time observer of medical device regulation suggested that competitive pressure “is a powerful deterrent [to reporting events], leading companies to file no more than the least amount necessary under a law—
|
3 |
MedSun training materials distinguish between accident (adverse event) investigations and forensic investigations. “The goals of an accident investigation are to determine what happened, why it happened, and which corrective actions and preventive measures can be taken. The goal is not to assign blame…. Forensic investigations are performed in relation to litigation, arbitration, and contract issues…. [The goal] is to provide a clearly stated, reasonable biomedical engineering or medical opinion on the cause of the accident at deposition or trial…. Some investigators see the assignment of blame as one fundamental goal of a forensic investigation. In this regard, however, it is important to remember that in the end, legal liability is determined by juries and courts” (FDA, 2002n, p. 3-2). |
especially since these reports can be read selectively” (Dickinson, 2004c). Both liability and competitive considerations create incentives for manufacturers to interpret an adverse event as resulting from user error rather than to cite device design, labeling information, and similar factors that might have caused or contributed to the event.
Overreporting
Although the discussions of problems with adverse event reporting systems tend to focus on underreporting and incomplete reporting, what might be called overreporting is also a problem. That is, user facilities, health care professionals, and consumers may report events that are not related to a medical device (or its use) but rather to the patient’s underlying medical problem or some other circumstance. They may also report trivial events. FDA has provided guidance to user facilities and others about what not to report, but it is obvious from a scan of recently submitted reports that the advice is not always followed. In 1993, FDA concluded that of approximately 2,834 reports it received from user facilities, only 664 should have been submitted (cited in GAO, 1997, p. 17).
Inappropriate reporting adds to the burden on FDA staff and on the manufacturers who initially receive most adverse event reports that are submitted to FDA. Manufacturer complaint files include many reports that are screened out as not reportable.
Shift of Care from Hospital to Home
Several of the vignettes presented above involve a particular patient safety challenge—the shift of complex care from the hospital to home. This shift reflects, in part, the progress in biomedical science and engineering that allows more adults and children, first, to survive severe medical problems and, second, to live at home with supportive technologies. Little information is available about medical device safety or adverse events in the home (see, e.g., CHCPR, no date; Lantos and Kohrman, 1992; AAP, 2000b; Tucker, 2004; Bruno and Ahrens, 2005).
Pediatric home health care is, essentially, a stepchild in patient safety programs. The movement of care and devices out of the hospital has not been matched by programs to encourage the awareness, documentation, investigation, and reporting of adverse device events. In the committee’s experience (including its discussions with parents), many if not most patients and families are likely to be unaware that FDA has a role in device safety and provides for written or online submission of problem reports from consumers. The committee found virtually nothing about the reporting of adverse events by home health agencies, which may not be even
aware of events and may, in any case, focus exclusively on resolving problems (e.g., clarifying operating instructions for families, troubleshooting problems, swapping out malfunctioning devices) without regard to their legal responsibilities for event reporting.
Recognizing that medical device safety in the home is a neglected area, CDRH has created a home health care committee. Among other resources, the committee has created a checklist that provides useful, basic guidance in relatively nontechnical language for families or patients using medical devices at home (FDA, 2003l). The major checklist headings advise
-
As a homecare medical device user, you should know how your device works.
-
Take care of your device and operate it according to the manufacturer’s directions.
-
Always have a back-up plan and supplies.
-
Educate your family and caregivers about your devices.
-
Keep children and pets away from your medical device.
-
Contact your doctor and home health care team often to review your health condition.
-
Report any serious injuries, deaths, or close calls.
A number of organizations have endorsed the FDA checklist, and it may be freely copied and distributed by professional societies, patient advocacy groups, and others. The committee encourages its wider distribution, especially by groups such as the American Academy of Pediatrics and the National Association of Children’s Hospitals and Related Institutions that are involved with technology-dependent children.
Identifying, Documenting, and Investigating Possible Adverse Events
The identification, reporting, and investigation of adverse events, device problems, and close calls is an important task that is most reliably performed in the context of clearly defined organizational structures and procedures maintained by manufacturers, health care providers, FDA, and other relevant organizations (e.g., engineering consulting groups). Individual patients and families at home and even office-based physicians and nurses are, in general, not well prepared to investigate such events, even if they recognize them as possibly related to a problem with a device or its use. Within health care institutions, well-functioning quality management and patient safety programs should ideally provide a culture that supports the recognition, reporting, and investigation of device safety problems as well as procedures that make it easy to do so. (It is not clear that this observation applies to home health agencies that are not well integrated into the culture
of a larger health care organization that has strong patient safety programs and norms.)
Figure 4.1 depicts in a simplified form the basic steps to be followed in a hospital or other facility once an obvious or possible adverse event (or close call) is recognized. The figure highlights the importance of documenting the circumstances of an event and maintaining the scene until a preliminary investigation can be conducted. In some cases, investigations are
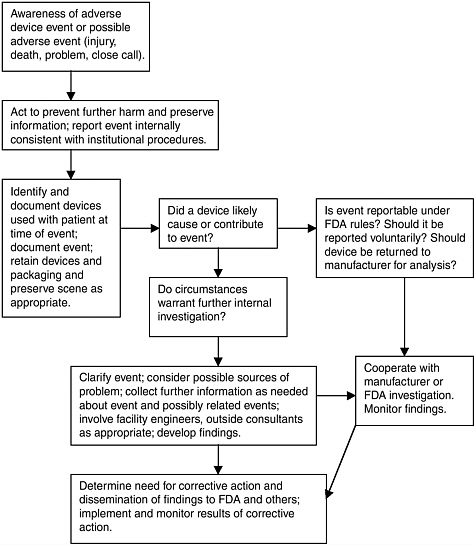
FIGURE 4.1 Identifying and investigating an adverse device event in a health care facility (adapted from MedSun training materials [FDA, 2002n]).
hindered because a device or its key accessories are not available for examination or the device has been cleaned or changed in some way (e.g., its electronic memory erased) that eliminates or compromises relevant information. An important feature of the MedSun program is training in procedures to follow after an event is recognized (e.g., properly impounding and storing devices involved in an incident and otherwise securing and protecting the integrity of relevant materials, records, and information).
Even when a problem with a device is obvious, for example, when a device has fractured, the contributing factors may not be obvious. A fracture could result from a design flaw, a short-term manufacturing lapse, use of the device outside its specifications or directions for use, or other causes or combination of causes. It may be impossible later to identify the source of an adverse event or close call if the setting and circumstances surrounding the problem have not been carefully and accurately documented and if the device and related packaging and accessory devices are no longer available for analysis. Documentation is a particular concern with events that occur in home settings where family members rather than professionals are primarily responsible for the day-to-day operation of devices.
Manufacturer examination of retrieved devices is often useful in determining whether and why the device failed or malfunctioned or whether a design feature contributed to a user error.4 Implant retrieval, however, faces a number of obstacles, including confusion about the ownership of a device, provider fears of liability, provider emphasis on fixing problems, costs of retrieving and returning devices, and lack of formal procedures for obtaining patient consent. A 2000 NIH document stressed the importance of device retrieval for device research and recommended that the information card for patients with implants provide an opportunity for patients to consent to implant retrieval (NIH, 2000).
To the extent that documentation of the circumstances of an adverse device event depends on information from the patient, some details may not be available when the patient is an infant or very young child. (Limits on communication with infants and young children may also contribute to adverse events, for example, when children cannot provide important information about what they are experiencing or when fear interferes with their ability to cooperate with a procedure.) Participants may also provide incon-
|
4 |
To cite one recent case, as part of its program to analyze products returned from physicians, one company identified a small number of implanted defibrillators that had a battery shorting problem that could lead to rapid battery depletion (Medtronic, 2005). Based on additional testing, the company estimated that perhaps between 0.2 percent and 1.5 percent of the devices in question could develop the problem. Using its registry information for the devices, the company provided physicians with a list of possibly affected patients. |
sistent accounts of an event, and those recording or investigating these accounts may introduce inaccuracies or biases that complicate the search for causation. As noted above, liability and competitive concerns also have the potential to bias reports.
The initial focus of adverse event investigations is on identifying the immediate circumstances and causes of an event, for example, a use error or manufacturing flaw. Human factors analysis and root-cause analysis go beyond the immediate or proximate source of an adverse event to identify underlying and potentially preventable device, use, environmental, and other organizational or systems factors that contributed to the event (see, e.g., Sawyer, 1996; Murff et al., 2001;Wald and Shojania, 2001b). As described below, human factors analysis can also be used prospectively to identify and avoid device design features that promote use errors.
Determining Whether a Problem Exists
Although it is important for those immediately involved in an adverse event or other problem involving a medical device to take the steps identified above and to assist in a health care organization’s investigation of the event, the organization itself may not have the critical expertise needed to evaluate an event or problem. Often, that expertise resides with the manufacturer of a device. In some cases, health care organizations or manufacturers may call on outside engineers or other consultants for assistance and confidential evaluations of a problem or potential problem with a device.
As noted above, the nature of an adverse event, malfunction, or other problem associated with a device is sometimes such that those involved can feel reasonably confident that a device problem exists, for example, that the packaging is faulty or that the device has arrived with an element incorrectly assembled. The primary questions then focus on the extent of the problem (e.g., certain lot numbers or all lot numbers) and its origin (e.g., a single random manufacturing aberration, an aberration affecting several product lots, a problem with an accessory device, or a design characteristic that becomes a problem only in unusual circumstances).
In other cases, it may be much more difficult to link an unwanted outcome (e.g., an infection or a surgical injury) to a problem with the device. The outcome may be one that can occur as a result of the patient’s medical condition or that is a known risk of treatment for the condition.
When adverse events are analyzed, one question is whether the type of event is occurring uniquely or more frequently among patients who are treated with a particular medical device. As discussed further in Appendix D, answering this question requires data about both the frequency of the event in question (numerator data) and the population with and without the device who are potentially at risk of the event (denominator data).
Ideally, additional information would also be available about other patient characteristics (e.g., age, gender, severity of illness) that might affect the likelihood of the event. Adverse event databases suffer deficiencies in all these areas.
Further, because underreporting, incomplete reporting, and biased reporting are such severe problems with passive adverse event reporting systems, it is expected that reported instances of a particular device-associated event will typically be only a fraction of all instances of the event. Underreporting and the lack of comparative population information necessary to construct event rates are important reasons for the interest in surveillance based on large automated population databases such as those of big HMOs
For some devices, it may be possible to make estimates for some missing variables using information from the manufacturer, for example, device tracking registries or registries created as a condition of marketing approval. Other registries such as those created by professional groups or academic medical centers may likewise be useful in making estimates. FDA analysts may also seek data on adverse events from public health databases, as in the investigation of meningitis cases among recipients of cochlear implants cited above and discussed in Chapter 6.
In addition, data from premarket clinical studies may be evaluated. A case in point involves reports of subacute thromboses and possible hypersensitivity reactions following introduction of a drug-eluting stent. In October 2003, FDA issued a public health advisory on the topic. A month later it issued another notice stating that the agency’s review of pre-approval clinical trial data indicated that the rate of subacute thromboses was the expected rate for such stents and that many of the hypersensitivity reactions may be related to the drug therapy associated with the stenting procedure (Alonge, 2004).
Improved epidemiologic research capacity would help the agency tap alternative information sources. FDA funding restricts the time that analysts can spend both reviewing reports of serious adverse events and investigating those reports that are suggestive of a problem with a device or its use.
Responding to Problems
As described in several vignettes above, the most immediate response to an adverse event may be rescue interventions undertaken by health care professionals (or parents at home). In some cases, the event may prompt no investigation. In other cases, an affected institution may identify a problem with a device and take action to change or restrict the use of the device, perhaps without reporting the problem to either the manufacturer or FDA. Such isolated responses deprive other patients and physicians of potentially valuable safety information, although professional communication net-
works may disseminate information about the problem and the response, which, in turn, may lead to action in some other institutions.
Reports to manufacturers and FDA allow wider communication of problems. As reported by GAO, an FDA analysis found that approximately 25 percent of all classified device recalls were linked to adverse event reports, but about half of recalls involving Class I devices were associated with such reports (GAO, 1997).
In some cases, a single incident report or even a close call may be sufficient to prompt a response. For example, in 1985, an infant was electrocuted when a sibling connected the electrode lead wires for the child’s apnea monitor to a power source (cited in GAO, 1997). FDA issued a safety alert and asked all manufacturers of home apnea monitors to evaluate their devices to determine whether changes were needed. The agency subsequently changed the criteria for clearing new devices to require that devices be designed to protect against this risk. Eventually, it issued warnings to hospitals about the continued use of devices with unprotected leads that were sold before the change in clearance criteria was applied to hospital monitors (FDA, 1993a).
Chapter 3 identified several short-term responses available to FDA and a manufacturer once a problem with a device is identified. These responses range from developing warnings or advice for practitioners to recalling a product. Table 4.3 summarizes such responses to device problems for the years 1998 to 2004.
TABLE 4.3 Medical Device Class I Recalls and Safety Alerts, Public Health Advisories, and Notices, 1998–2004
|
|
1998 |
1999 |
2000 |
2001 |
2002 |
2003 |
2004 |
|
Class I Recalls |
5 |
7 |
16 |
9 |
11 |
14 |
28 |
|
Web Notifications |
0 |
0 |
0 |
0 |
3 |
3 |
3 |
|
Public Health Advisories, Notifications, Notices, Letters, Safety Alerts, and HHS News Items |
17 |
12 |
4 |
6 |
6 |
4 |
2 |
|
Talk Papers |
1 |
0 |
0 |
0 |
0 |
1 |
0 |
|
FDA Press Release |
2 |
1 |
0 |
0 |
0 |
1 |
11 |
|
NOTES: The reference to Safety Alerts, Public Health Advisories, and Notices in the title is taken from the FDA title for this information in the main FDA safety page (FDA, 2005d). Labels for these notifications have changed over time. Updates for safety notices are counted as original notices. Most press releases cover recalls that are also reported in the first line of the table. SOURCE: FDA Medical Product Safety Information (FDA, 2005d) (cited May 11, 2005), FDA CDRH Public Health Notifications (FDA, 2005h) (cited May 11, 2005), and ECRI Health Devices Alerts Database. FDA lists Class I, II, and III recalls in its monthly Enforcement Reports (see FDA, 2005b). |
|||||||
As noted earlier, adverse event reports can direct attention to problems with the design of a device and, thus, provide opportunities for device refinement or innovation. Not surprisingly perhaps, manufacturers’ responses to adverse event reports as found in MAUDE usually provide no indication that they might be considering a report as a resource for evaluating device design.
The committee did not investigate FDA procedures for issuing and following up on safety alerts, but it does have some concerns about whether FDA has adequate resources to analyze adverse event reports and to develop responses for problems that are not seen as high-priority but that nonetheless may pose real risks to children in particular. The problems with orthodontic headgear are a case in point. In a November 2004 story on a child blinded by a mishap with orthodontic headgear, a Washington, D.C., television station criticized FDA for not having taken any action to protect children although FDA staff acknowledged that they were aware of the problem (WTTG, 2004). A search of FDA databases yielded three reports of headgear-related eye injuries (in 1990, 1997, and 2002) as well as additional reports related to other problems, including device breakage. Reports of blinding injuries have also appeared in the literature for at least 30 years (see, e.g., Samuels et al., 1996; Blum-Hareuveni et al., 2004). The American Association of Orthodontists presented guidelines and cautions on the use of these devices in 1975, but apparently has not revisited the issue since then (AAO, 1975). A committee inquiry to FDA found an investigation of headgear injuries had been initiated, and in spring, 2005, an article on headgear safety appeared in FDA and You, an online publication for health educators and middle and high school students (FDA, 2005c). It is not clear whether further dissemination may occur to get this information before those facing decisions about the use of orthodontic headgear (personal communication, Thomas P. Gross, M.D., Director, Division of Postmarket Surveillance, CDRH, June 3, 2005).
One likely reason that the headgear-related risks have not attracted more attention is that the devices have been used by millions of children with few reports of injury. These products are also not as attention getting as more “high-tech” devices such as implants. Furthermore, headgear injuries and other incidents typically occur at home and, if not severe, they may be treated by office-based practitioners who are not required to report adverse device events and are likely unaware of voluntary reporting options. Emergency room physicians who treat serious injuries associated with these and other medical devices may likewise be unaware that they can voluntarily report events to FDA. Nonetheless, the long and continuing history of headgear incidents and the rare but devastating injury raise questions about whether lack of resources has limited FDA’s ability to investigate and respond.
FDA has been more attentive to the problems with circumcision clamps as described in Box 4.1 and Vignette A. A few years ago, CDRH analysts noticed a large enough number of lacerations and other events related to the clamps (105 reports of injuries between July 1996 and January 2000) that it undertook an investigation that led to a safety alert in 2000 (FDA, 2000g). Subsequently, reports of injuries dropped, but the agency was concerned that incidents were still occurring and being reported, so it reiterated its warning in an item in its Patient Safety News series in 2002 (FDA, 2002a). The FDA warnings on circumcisions clamps followed—and cited—several earlier notices by ECRI, a private nonprofit technology assessment and health care research organization (see ECRI, 1993, 1995, 1997, 1999, and discussion at end of this chapter).
Public health notifications are not sufficient responses to some device problems. Recalls of a product may be necessary. Whether a recall is undertaken at a manufacturer’s own initiative, as a result of an FDA request, or after an FDA order (which is rare), the recall will not reach its goal of protecting patients if information about the recall does not get to those who need it. The same observation, of course, holds true for dissemination of information about labeling changes or new advice about the appropriate use of a device.
FDA recognizes weaknesses in the recall process. As summarized in one overview, recalls can go unnoticed for various reasons, for example, “recall information doesn’t get into the right hands, registered letters are sent to old addresses, hospitals don’t see notices because they are swamped with so many other responsibilities, or perhaps there are mixed signals on the urgency of the problem” (Rados, 2003, unpaged).
Vignette E cited problems with misrouted information about the recall of a bronchoscope. It noted that hospitals can subscribe to information services that automatically alert them to device recalls and other relevant safety information. FDA’s MedWatch program also allows people to sign up for automatic e-mail safety alerts that cover devices, drugs, biologics, and dietary supplements. An alert system limited to devices may, however, make it easier for those concerned specifically about device safety to focus on device problems. In addition to helping professionals evaluate problems and gain insights from peers, listserves and similar tools can also help disseminate safety information.
FDA INITIATIVES TO IMPROVE ADVERSE EVENT REPORTING AND RELATED ACTIVITIES
FDA clearly recognizes limitations of adverse event reporting programs in general as well as particular concerns with its own program. Sometimes with congressional direction, it has undertaken or planned a number of
initiatives to respond to certain limitations and concerns. The MedSun pilot program, the human factors initiative, and other activities described below are examples. For the most part, their focus is general, although some include attention to pediatric issues.
MedSun
Compared to the agency’s primary spontaneous or passive event reporting program, the pilot MedSun program was created as a less inclusive but more intensive effort to better identify and understand problems with the safe use of medical devices. The program provides for more attention to close calls, more education of participating facility representatives to improve the level and quality of event reporting and analysis, and more feedback and interaction to determine the nature of device problems and close calls as a basis for preventing future problems and improving patient safety within health care facilities. These educational and feedback features offer incentives for institutional participation that are absent in the traditional program. MedSun now has a waiting list of interested facilities (FDA, 2004a). With the possible exception of certain special studies, the program will not collect denominator data that would allow the calculation and comparison of problem rates.
As shown in Table 4.4, by June of 2005, MedSun had recruited over 350 hospitals, of which 22 were acute-care general children’s hospitals and 2 were acute-care pediatric specialty hospitals. These participating facilities
TABLE 4.4 Cumulative Number of Facilities Recruited Into MedSun
|
|
2002 |
2003 |
June 2005 |
|
Nonpediatric hospitals with 100+ beds |
57 |
167 |
307 |
|
Children’s hospitals of any size |
2 |
8 |
22 |
|
Children’s rehabilitation hospitals of any size |
1 |
2 |
2 |
|
Nursing homes of any size |
8 |
19 |
21 |
|
TOTAL |
68 |
196 |
352 |
|
NOTE: Nonpediatric hospitals with fewer than 100 beds and other types of facilities such as home health and outpatient clinics are not included. Not all sites that have been recruited have received program orientation. SOURCE: Personal communication, Thomas P. Gross, M.D., Director, Division of Postmarket Surveillance, CDRH, June 10, 2005. Data provided by CODA, Inc. |
|||
constitute about 6 percent of all hospitals and 12 percent of children’s hospitals. (Nationally, the United States has approximately 4,900 short-term, nonfederal community hospitals [AHA, 2004] and approximately 160 short-term [nonpsychiatric] children’s hospitals [NACHRI, 2003].) The modest overrepresentation of children’s hospitals in the MedSun implicitly acknowledges the particular value society places on learning about and protecting the health of its youngest members, even though children are, overall, a generally healthy population.
MedSun also includes 21 nursing homes. It does not include any psychiatric hospitals or federal hospitals. It also does not include independent outpatient centers or independent home health organizations, although some participating health systems include such entities. Participating facilities must agree to participate for at least 12 months, but FDA hopes they will agree to renew annually.
An outside contractor (CODA) manages the pilot program. Its responsibilities include providing assistance to participants on mandatory and voluntary adverse event reporting and receiving and processing mandatory event reports before forwarding them to the manufacturer or FDA or both. The contractor also is involved (with assistance from another contractor) in analyzing and providing feedback on event reports.
MedSun requires participating facilities to designate two staff contacts, one from their risk management or quality improvement area and one from their biomedical or clinical engineering staff. The program provides three hours of training in reporting adverse events or close calls. In addition, the contractor and FDA have organized two conferences for MedSun participants, and they have made or plan to make slide sets on special topics (e.g., pediatrics) available for educational and promotional use in participating facilities. MedSun participants can also request FDA analyses of MedSun or MAUDE reports on device problems.
Another benefit for participating institutions is a newsletter that summarizes adverse event reports received by FDA, presents analyses of issues by FDA staff, and provides other information of interest. From 1992 to 2003, FDA distributed a somewhat similar newsletter for subscribers from all user facilities. Acknowledging resource constraints, the newsletter announced its discontinuation, claiming that it “had finally served its purpose” of providing training, education, and feedback on adverse event reporting (FDA, 2003a). This claim is not convincing given the picture presented in this chapter.
As part of an active surveillance element of the MedSun program, participants agree to respond to periodic rapid response surveys that focus on specific product problems or concerns. For example, 29 hospitals have participated in a 6-month study to identify cases of thrombosis or hypersensitivity reaction that occur within 30 days of implantation of a drug-eluting
stent (MedSun, 2004, p. 2). FDA asked cardiac catheterization laboratories in study facilities to complete surveys at the beginning and end of the study and to complete another questionnaire to report any events involving drug-eluting stents. Results have yet to be reported publicly. FDA has planned similar surveys involving other devices.
The committee is not aware of special surveys that have involved only the children’s hospitals in the pilot MedSun program. It has learned that both Child Health Corporation of America (CHCA) and the National Association of Children’s Hospitals and Related Institutions (NACHRI) have been working with MedSun to assist the project in gaining substantial input from pediatric hospitals (personal communication, Cheri Throop, R.N., Chief Quality Adviser, CHCA, April 22, 2005). One of the recommendations at the end of this chapter encourages FDA and MedSun participating children’s hospitals to serve as a resource for the broader involvement of children’s hospitals in device safety.
One recent addition to MedSun is the Medical Device Engineering Network (M-DEN), which provides an interactive query and comment option for participants to share questions, experiences, and advice. The discussions may involve problems or concerns that would not normally be reported to FDA (Crowley et al., no date). Several teleconferences already have been organized on topics of interest to biomedical engineers.
MedSun has only recently approached its recruitment goals, and the program has yet to train representatives from many of the recently recruited facilities. Data collection began in February 2002 (FDA, 2004a). Because the pilot program is still in its early stages, it is not ready for systematic evaluation of its performance in meetings its goals. The committee understands that relatively few reports are being submitted each year, on average, by the MedSun facilities, but no public information is yet available. At some point, a comparison of the reports submitted by participating facilities with those received through the passive reporting system will be needed. The recommendations at the end of this chapter include suggestions for the evaluation plan.
Exploring Computer-Based Surveillance and Improved Device Coding
FDA has been interested in the potential applicability to adverse device events of some strategies being used or tested with adverse drug events. For example, the agency supported a study to compare possible device surveillance strategies and evaluate whether computer-based surveillance could reliably detect device adverse events and hazards, which were defined as “a state of increased risk related to device use” (Samore et al., 2004, p. 333). The study, which was conducted at a large medical center that had experience using computer-based strategies for detecting adverse drug events,
concluded that any of the surveillance strategies it investigated detected only a minority of the adverse events that were identified by the strategies collectively and that each strategy had significant limitations. An update of the first published report on the study is currently being prepared (personal communication, Thomas P. Gross, M.D., Director, Division of Postmarket Surveillance, CDRH, February 4, 2005).
A particular focus of investigators was a “computer-flag” strategy that was embedded in the hospital’s computerized patient record system. It involved nurse review of flagged records using a protocol devised in consultation with FDA. The flags were based on “detection rules” for seven categories of events (e.g., complications and hazards related to various types of catheters), and one goal was to identify hazards before they caused harm. This approach yielded more information on adverse events than the hospital’s voluntary adverse event reporting system, but it missed some important events. Disappointingly, the positive predictive value of the flags was low, that is, the great majority of flagged events did not, upon investigation, involve a device-related problem or hazard. Thus, its utility for detecting potential problems before they could cause harm was limited.
Nonetheless, the computer-flag strategy—in combination with other tools—helped investigators better understand the clinical environment. “It appeared that the typical health care worker response to a device problem was to fix it or retrieve a new device that worked and then move on, an appropriate solution at the individual patient level but not an effective systems approach” (Samore et al., 2004, p. 333). This observation was reiterated in committee discussions with clinicians.
To consider patient perspectives, investigators conducted a postdischarge patient survey. It found that people focused on simple, common devices that caused discomfort rather than what are normally defined as serious adverse events. Events reported in the survey had no overlap with other reports.
Another strategy—retrospective review of medical records for ICD-9-CM codes5 that indicate a likely device problem (e.g., code 996.01, mechanical complication due to heart valve prosthesis)—was useful in identifying problems that occurred prior to hospitalization. Still, with ICD-9-CM codes, device problems can only be described by broad category rather than by individual device. Thus, this strategy is not well suited to identify a
|
5 |
The International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) is used in assigning codes to diagnoses and procedures associated with hospital utilization. It is based on the World Health Organization’s International Classification of Diseases, Ninth Revision (ICD-9). In the United States, the National Center for Health Statistics and the Centers for Medicare and Medicaid Services have responsibility for maintaining the system. |
particular device model or brand for analysis of problems in its design, manufacture, or use.
As discussed further in Chapter 6, whether for written or electronic records, a fundamental problem is the lack of an accepted, feasible coding system for devices that is equivalent to that for drugs and allows recording of sufficient device-specific information in the medical record. Chapter 6 includes a recommendation on the development of common standards and approaches for capturing use and outcomes data for implants and other medical devices. Building on extensive experience with device coding and its limitations, FDA recently held a conference to explore new strategies for improving device coding.
Human Factors Analysis
FDA’s work in the area of human factors analysis supports steps to prevent device problems and to analyze problems once they occur. As described in Chapter 3, the field of human factors engineering focuses on how people use technologies and how human characteristics (e.g., cognitive capacities, expectations, and physical limitations) interact with characteristics of products and work environments. FDA has been interested in the application of human factors analysis to medical device safety since the 1970s, and its human factors program has worked with manufacturers on the incorporation of human factors engineering principles in the design of medical devices (see, e.g., Carstensen, 1996; Gross, 1996; Sawyer, 1996; FDA, 2000b).
Human factors analysis can also contribute to improvements in the evaluation and characterization of adverse events. One recent activity involved the development of a model for this purpose based on literature review and interviews with device users, primarily nurses (Kaye et al., 2003). The preliminary model defined several broad contributors to device problems related to unmet user needs (e.g., device does not indicate when it is operating improperly), user perceptions (e.g., monitor display is not easy to see), user cognition (e.g., device operates differently from most similar devices), and user actions (e.g., device can be improperly connected to other devices). These categories, which emerged from interviews, include many situations that give rise to the kinds of workarounds described in this chapter and Chapter 2.
Additional Activities
Another FDA initiative is the use of Systematic Technical Assessment of Medical Products (STAMP) teams to examine adverse events, including deaths and serious injuries, associated with selected devices (FDA, 2004v).
Teams, which include experts outside FDA, have examined surgical staplers and clips and laparoscopic trocars (devices that penetrate the abdomen and pelvis to allow insertion of laparoscopes and surgical instruments). The first such examination focused on shunts used with hydrocephalus (FDA, 1999f).
The agency also participates in the Quality Interagency Coordination (QuIC) Task Force, a department-wide patient safety initiative. One study in this context has audited Medicare patient records to identify certain events (other than infections) related to central venous catheters. In initial findings, the devices were associated with a 2 percent rate of events such as misplacement of the catheter or pneumothorax (air or gas in the space surrounding the lungs, often called a collapsed lung) (Gross, 2004).
OTHER REPORTING AND ANALYSIS OF ADVERSE EVENTS
Beyond the activities of health care facilities, manufacturers, and FDA as described above, it is worth noting that a number of other public and private programs include the reporting and analysis of adverse events as part of broader health care quality and patient safety programs. Notable among other federal agencies is the program overseen by the National Center for Patient Safety of the U.S. Department of Veterans Affairs (http://www.patient.safety.gov). Examples of one state and two private programs are briefly described below. Most patient safety programs do not emphasize adverse device events as such.
State Reporting Program: New York
A number of states, including New York, Pennsylvania, and Oregon, have created some form of adverse event or patient safety reporting program. New York State has required since 1985 that hospitals report certain types of adverse events. In 1998, it required a more comprehensive, Internet-based system—the New York Patient Occurrence and Tracking System, or NYPORTS (Hevesi, 2003; see also New York State Department of Health, 2004). Under the program, covered facilities are to report the most serious incidents or “occurrences” within 24 hours.
Of 54 defined types of incidents, the program has classified 19 as “most serious,” including unexpected patient deaths and equipment malfunctions that result in patient harm. Facilities are to investigate and report on the causes of the incidents within 30 days using a standard investigation and reporting format, which should document a root-cause analysis of the occurrence. (Hospitals can report information electronically, but the state’s diagnostic and treatment clinics cannot.) The state Department of Health uses the data for a variety of quality improvement and patient safety purposes.
The New York program is more comprehensive than FDA’s medical product reporting in that it covers problems not related to medical products, for example, surgery on the wrong patient or wrong part of the body. In contrast to MedWatch, which focuses on manufacturer analysis of problems, New York puts more demands on facilities to investigate and report incidents in a systematic fashion that supports problem identification and quality improvement activities. Information is shared with facilities, but individual reports are protected from public disclosure. The New York program thus has some features in common with FDA’s pilot MedSun program.
One problem that the New York program shares with FDA is underreporting of events. A recently released audit of the program documented underreporting of serious events and late or missing investigation reports of serious occurrences (Hevesi, 2003). Of the nearly 5,800 reports that the audit said should have been reported within 24 hours, 84 percent were not.
Private Reporting Programs
A variety of other private organizations concerned with health care quality and patient safety include some attention to safety problems with medical devices. For example, the Institute for Safe Medication Practices has described hazards linked to the design or use of medical devices used to administer medications. Patient safety and quality improvement initiatives sponsored by consumer groups such as the National Consumers League and professional societies such as the American College of Cardiology (ACC) and the American Thoracic Society may likewise cover device issues. For example, the ACC was a joint sponsor of a conference that considered shortcomings in postmarket surveillance of cardiovascular devices (O’Shea et al., 2004). (Other sponsors included FDA, the Agency for Healthcare Research and Quality, and the trade group AdvaMed.)
Joint Commission
The Joint Commission on the Accreditation of Healthcare Organizations is best known for its long history of accrediting and setting detailed standards for hospitals and other health care organizations. Many of these standards involve the safe use of medical equipment.
As part of its standards, the Joint Commission has also identified a set of sentinel events that are subject to reporting and review. Sentinel events, which are incidents that “signal the need for immediate investigation and response,” are defined as unexpected occurrences that involve “death or serious physical or psychological injury, or the risk thereof” and do not result from the patient’s medical condition (JCAHO, 2005a, unpaged).
Thus, they include both serious adverse events and close calls. Accredited health care organizations are expected to have internal policies and procedures for analyzing and responding to sentinel events, including the application of a root-cause analysis.
Certain sentinel events are reviewable by the Joint Commission when voluntarily reported by hospitals or when otherwise identified (e.g., through a newspaper story or a patient report). One type of reviewable event is a perinatal death that is not related to a congenital condition in an infant with a birth weight greater than 2,500 grams. A root-cause analysis of the 84 such events reported between 1995 and 2004 found that the most frequent contributing factor was communication problems (JCAHO, 2004).
Between 1995 and the end of 2004, the Joint Commission reviewed nearly 3,000 sentinel events (JCAHO, 2005b). The organization continues to be concerned about a low level of voluntary reporting, which limits the utility of the effort as a source of information about the nature and causes of events. One early review of the Joint Commission program suggested that if it had had the same yield as New York’s event reporting program, it would have received as many as 21 times the reports it actually did during the period reviewed (Wald and Shojania, 2001a).
ECRI
The most comprehensive private program of adverse event reporting and analysis related to medical devices is maintained by ECRI, a private nonprofit health services research and technology assessment organization. Among other activities related to patient safety, ECRI gathers and investigates reports of incidents involving medical devices from health care providers, patients, and manufacturers around the world. It provides investigative and consulting services to health care providers, governmental health agencies, and other organizations. Each year the organization receives more than 1,000 high-quality reports of medical device adverse events and publishes scores of original hazard reports on specific device models as well as problems generic to classes of medical devices. In 1973, ECRI’s problem reporting program served as a model for the newly emerging FDA Device Experience Network.
ECRI’s monthly journal, Health Devices, includes independent medical device evaluations (e.g., recent evaluations of infusion pumps [ECRI, 2004c]) and reports on device safety. Another publication, Health Devices Alerts, provides weekly reports on medical device hazard and recall information, product safety alerts, reported problems and recommended responses, and published research on medical devices. In addition to the reports on circumcision clamps cited earlier, a number of ECRI reports
have dealt with safety issues related to devices used with children, including incubators, cribs, ventilators, and automated external defibrillators.
Under contract to FDA, the organization developed the education and training materials for recognition, investigation, and root-cause analysis of medical device adverse events for the pilot MedSun program. ECRI also assists in the analysis of the MedSun problem reports. In addition, ECRI has a contract with FDA to help harmonize FDA’s medical device product codes with the Global Medical Device Nomenclature (GMDN). It recently drafted a white paper for FDA on the automatic identification of medical devices.
CONCLUSIONS AND RECOMMENDATIONS
One theme of this report is that an effective regulatory program for evaluating and monitoring the safety of medical devices in general is a necessary foundation for efforts to safeguard children in particular. Thus, steps to improve FDA’s programs for the reporting of adverse device events overall should benefit children as well as adults. To promote more focused attention to pediatric issues, Chapter 7 includes a recommendation (7.1) that FDA identify a focal point of responsibility for pediatric issues within the Center for Devices and Radiological Health to evaluate the adequacy of the Center’s use of pediatric expertise and its attention to pediatric issues in all aspects of its work to promote medical device safety.
Another theme of this report is that medical device safety is a shared responsibility. The recommendations below start with FDA but extend to include manufacturers, health professionals, user facilities, and patients and families. Chapter 7 extends this discussion.
Within FDA, adverse event reporting and improvement should be understood in the entire context of the agency’s activities to protect patients and promote medical device safety from the early stages of device development through the end of a device’s useful life. These activities include guidance for developers of devices, premarket evaluations, systematic postmarket clinical studies of selected devices, public health notifications and additional information for users of devices, quality system inspections of manufacturers, and other strategies. The FDA program itself should be seen as part of a more expansive system of public and private programs and actions to safeguard patients and improve health outcomes.
FDA Adverse Event Reporting
As part of a larger system of postmarket surveillance and device safety regulation, a passive or spontaneous program of reporting has a role to play in detecting unexpected device problems (including problems with the use
of a device) and increasing understanding of certain already recognized problems. Adverse event reports may provide the first signal that a problem exists with a device or its use or both. Adverse event reporting is particularly important for medical devices in pediatric use because events involving children are often unusual, are sometimes extreme, and identify problems in a patient population that often has not been studied before a device is marketed.
Certainly, this report and many other analyses of spontaneous event reporting programs across diverse realms make clear that such programs have significant limitations. These limitations include underreporting, poor-quality reports, delayed reports, reports of problems not associated with a device (i.e., false-positive reports), and lack of information needed to compute and compare rates of events. Efforts to investigate a worrisome event report may be frustrated by distance in time and place from the event, with consequent loss of critical information about the circumstances surrounding the event and unavailability of the suspect device or devices for analysis. The adequacy of FDA resources for event analysis is also a concern.
Initiatives to increase the spontaneous reporting of adverse events present a dilemma. On the one hand, there is general agreement that serious events are underreported; on the other hand, there is concern that increased reporting would likely bring an increase in reporting not only of serious events but also of “noise,” that is, reports that are of no real interest, that are so poorly prepared as to be useless, or that do not even involve device problems or adverse events. Such reports waste the resources of all involved.
Nonetheless, the committee believes it is important for FDA to sustain and improve its adverse event reporting program and demonstrate its value. One objective should be to improve links between the reporting program and various FDA databases, including the databases for device recalls, enforcement, and public health notifications. For example, someone reporting or considering reporting a device problem through the online MedWatch option should be able to link easily and clearly to public health or recall notifications related to the device in question.
FDA should also consider how to encourage reporters to identify when an event involves a child. In some cases, a facility reporter or a manufacturer will know that an incident involved a child without having the child’s exact age. It would be desirable to give such reporters an explicit opportunity to mark whether an event involved a child (age unknown). (The committee recognizes that changing the adverse event reporting form is a major, complicated undertaking, but encourages that this change be evaluated the next time that FDA or Congress considers revisions.)
Recommendation 4.1: FDA should collaborate with industry, health care professionals and organizations, and parent and patient advocates to
-
focus more attention on adverse device events, including events involving children;
-
promote linkages between adverse event reporting systems, various FDA databases, and other safety programs;
-
update product labeling, patient information, and other communications to promptly reflect safety-related findings from analyses of adverse event reports; and
-
issue yearly reports on results from adverse event analyses, including findings involving children.
Recommendation 4.2: FDA should continue educational and communication programs to promote recognition and useful reporting of serious adverse device events and device problems by hospitals and other user facilities. Such encouragement should continue whether or not requirements for mandatory reporting by user facilities are eventually eliminated with the effective implementation of the MedSun program. Reporting by user facilities of events possibly related to devices should continue to include deaths, serious injuries, and device malfunctions.
In addition, as suggested earlier in this chapter, FDA should continue its efforts to educate providers about HIPAA and the legality and value of providing information to support postmarket surveillance. Such information includes not only adverse event reports but also data for required postmarket studies as discussed in Chapter 6.
The legislation creating pilot MedSun program provided that mandatory reporting requirements for user facilities should end when the program is fully implemented. Before that happens, the evaluation of the program should consider MedSun’s performance both as an active surveillance system (e.g., responding to FDA inquiries, conducting special studies) and as a spontaneous reporting system for detecting serious unexpected device events. Given that many manufacturer investigations and reports of adverse events start with reports from user facilities, one question is whether it is prudent to eliminate mandatory reporting for these facilities, even if the limitations of such reporting are recognized and facilities are not sanctioned for failure to report. It would be unfortunate if user facilities felt even less responsibility to report serious events and deaths in the future.
The careful evaluation of the pilot MedSun program will be critical. Although a formal evaluation is premature given that the pilot program is not fully implemented, FDA should be putting in place the data collection resources it will need for the evaluation. The evaluation should include an assessment of the extent to which reporting by non-MedSun facilities generates signals of significant device problems that are not reported by MedSun
facilities (because they did not experience them or because they either did not detect them or did not report them). It is important for FDA to audit participant performance, including the periods when initial participants rotate out of the program and new facilities replace them.
Recommendation 4.3: FDA’s plan for evaluating MedSun’s performance as a replacement for and improvement on mandatory user facility reporting should include, among other elements:
-
assessment of ongoing program and participant facility success in educating facility personnel about identifying, evaluating, and reporting adverse device events and improving the quality, timeliness, and usefulness of event reports;
-
determination of the extent to which the sample of MedSun participating hospitals—including children’s hospitals—represents the relevant range of facility characteristics and experiences, including representation of both academic medical centers and community hospitals and sufficient representation of facilities with device-oriented specialties and procedures;
-
comparison with the mandatory user facility reporting system, including the extent to which either program produced reports for FDA or manufacturers of emerging hazards, important close calls, or other significant events (including those involving children) that were missed or delayed by the other; and
-
evaluation of the active surveillance components of the program in reducing harm to patients, promoting constructive communication between facilities and FDA, and improving timely knowledge of the nature and extent of selected device problems, including errors in the use and design of devices.
Prior to formal evaluation of MedSun, the committee encourages FDA efforts to extend to other institutions the lessons the agency and participants learn as they implement the program. For example, after their value has been assessed and revisions considered, the training materials developed for MedSun participants could be made more widely available. It is reasonable to provide MedSun participants with incentives to participate, but the written materials used in the program are only a small part of these incentives. Likewise, FDA should consider whether the information in the newsletter provided for MedSun participants could be used, at least in part, as a communication tool to inform interested parties in other facilities and encourage timely, complete, and appropriate reporting of adverse device events and other device problems to manufacturers and FDA. Despite the agency’s claim
that its discontinued user facility newsletter had served its purpose, the problems of facility underreporting and poor-quality reporting remain significant.
The committee commends FDA for the oversampling of children’s hospitals in the MedSun program. The MedSun participating children’s hospitals should be considered not only as a particular resource for investigation of safety questions related to children but also as a resource or base for a broader set of device safety activities involving children’s hospitals, CHCA, and NACHRI.
Recommendation 4.4: Within the pilot MedSun program, FDA and participating children’s hospitals should serve as a resource for the broader involvement of children’s hospitals in patient safety programs to identify, evaluate, respond to, or prevent problems with the use and design of medical devices. In addition, FDA should promote efforts to link or otherwise employ event reporting, device recall, safety notification, and other databases within and outside FDA to better assess and report on device safety issues involving children.
Information generated by MedSun could also prove more broadly useful. For example, it could be shared with academic clinicians and engineers to stimulate studies to identify device redesign or other strategies to prevent identified problems.
This chapter has noted the lack of a practical, precise coding scheme for medical devices that allows identification of specific models and brands of implants and other devices. Chapter 6 includes a recommendation (6.2) for the development and adoption of common device coding and other standards and approaches for capturing and linking use and outcomes data for medical devices.
Manufacturers
Sophisticated manufacturers recognize good adverse event reporting as a resource to help them learn about and correct problems with existing devices and identify areas for design refinement or product innovation. If adverse event reporting programs for devices are to improve device safety, manufacturers must receive timely and useful event reports, maintain sound procedures for evaluating these reports, and respond to identified problems on a timely basis. FDA regulations provide detailed direction on manufacturer responsibilities, and site inspections include a review of manufacturer compliance.
In addition to designing and redesigning devices to protect against unsafe use, promoting the safe use of devices is another important responsibility of device manufacturers. For some complex, high-risk implants and other devices, safe and effective use may require professionals to develop
new procedural and assessment skills. Some manufacturers have established mechanisms to develop and evaluate the competency of professionals to use such devices, and expectations for training and competency may be reflected in the labeling of the device. Training associated with such devices should cover the identification and reporting of adverse events.
Recommendation 4.5: When FDA mandates or agrees to device labeling that requires professionals to be trained in the safe and appropriate use of a medical device, the training should include information on the identification of adverse events, voluntary adverse event reporting under MedWatch, and user facility and manufacturer medical device reporting (MDR) requirements.
In addition, for complex devices that involve monitoring or operation by patients or families, manufacturers should provide directions about when and where to seek help, advice on reporting problems, and instructions, warnings, and troubleshooting guidelines that are understandable to nonprofessionals. Some manufacturers already have strong patient education and assistance programs. For certain home-use devices, FDA should consider requirements that manufacturers of certain devices (e.g., the orthodontic headgear mentioned earlier) affix labels stating that injuries related to the device can be reported to FDA.
FDA inspections of manufacturers should continue to include, as part of quality systems inspections, attention to complaint handling and event investigation and reporting. As discussed in Chapters 3 and 7, such inspections are occurring substantially less frequently than required by law.
Health Professionals and Professional Organizations
For many if not most medical devices, health care professionals who care for children occupy the critical intersection between device manufacturers and children and their families. They are well positioned to understand devices, evaluate their successes and failures with individual children, receive early warning of problems through professional networks, and determine what kinds of education health care workers—and patients or families—need to use devices safely. Significant complications with devices are often first reported at professional meetings without prior reporting to FDA, manufacturers, or other patient safety programs. Child health professionals are thus an essential but underdeveloped resource for identifying and reporting adverse device events. The challenge is how to better employ this resource to protect patients.
One difficulty is that pediatricians and other child health professionals are bombarded with advice, guidance, directives, and educational materials of all sorts. The likelihood that these incoming messages will change behav-
ior (or even be read) should certainly not be assumed, especially if financial and other pressures work in opposing directions. Nonetheless, incremental opportunities exist to improve recognition by child health professionals that medical device problems are reportable events, that reporting events has the potential to stimulate product and process improvements to benefit children, and that reevaluation rather than acceptance of certain common problems may be warranted.
For example, following direct mailings, continuing medical education, and other efforts to increase reporting by professionals to the Vaccine Adverse Event Reporting System (VAERS), the proportion of all vaccine adverse event reports that were attributed to health care professionals increased from 11 percent in 1991 to 35 percent in 2001 (Zhou et al., 2003). The committee recognizes that medical device reporting involves a vastly larger array and diversity of products, but FDA can collaborate with professional societies to set priorities for educational efforts. The agency can work with pediatric and other professional societies and journals, residency programs, and other resources to add messages about recognizing and reporting adverse device events to the messages that are already being disseminated about reducing health care errors and improving the quality of care for children.
In discussions with professional groups such as the American Academy of Pediatrics (AAP) and others, the committee found a general willingness of the groups to become more involved in efforts to promote the safe use of medical devices with children (AAP et al., 2004b; ACC, 2004; ATS, 2004a). These efforts encompass both the reporting of adverse events and the expanded use of registries as well as other means of developing better information about the short- and long-term outcomes of medical device use.
Recommendation 4.6: Medical, surgical, and other organizations or societies that include health professionals who care for children should
-
establish working groups to evaluate problems as well as benefits in the pediatric use of devices of particular importance to their practice;
-
collaborate with existing public and private patient safety initiatives to add or expand attention to safe and appropriate use of medical devices with children;
-
establish standards for professional education and competency in the use of these devices; and
-
include as professional competencies the identification and appropriate reporting of device problems and the successful communication with patients and families about how to prevent, recognize, and respond to device problems.
Information from adverse event and case reports as well as systematic clinical studies and registry-based research will help provide a stronger evidence base for pediatric practice guidelines and standards of competency. These guidelines and standards should, in turn, reduce the unsafe and unnecessary use of devices.
Hospital and Other Device Safety Programs
Hospital and other patient safety programs that now focus almost exclusively on errors or problems involving other medical products and services can extend their reach. For example, for devices used with children, possible targets for such programs include certain types of common workarounds that have not been assessed adequately to determine the extent to which they constitute reactions to device problems, pose risks of their own, or warrant reconsideration of the way devices are used or designed.
One objective of the MedSun program is to encourage more coherence in user facility device safety programs. Children’s and other hospitals generally lack the kind of obvious focal point for medical device safety that pharmacists provide for drugs. Clinical engineering units, risk management departments, an array of clinical units, quality assurance programs, materials management divisions, purchasing departments, and other units share a fragmented and incomplete accountability for device safety.
Recommendation 4.7: Children’s hospitals and other user facilities should establish a focal point of responsibility for medical device safety. Tasks include reviewing and monitoring the adequacy of institutional programs in areas such as tracking of safety alerts and recalls, responding to safety alerts and recalls, training in adverse event evaluation and reporting, and factoring safety data or evaluations into device purchase decisions.
FDA should also charge its home health committee with investigating the role of home health agencies and vendors that supply home medical equipment in reporting adverse events and examining what might be done to support these providers. It is important that these organizations focus on identifying and resolving problems, but it is also important that serious problems be reported to manufacturers and FDA. A better understanding of problems with devices used in the home may promote refinements in the design of such devices, changes in the selection and monitoring of devices for home use, and improved information and training for patients and families.
Resources for Patients and Families
Given the continued movement of complex care into the home, FDA should seek more creative ways to publicize its device safety activities and resources to patients and families, particularly families caring at home for children who rely on complex, life-sustaining medical devices. The CDRH checklist on medical devices for home use is a good model that should be more widely disseminated, including by professional and provider groups such as AAP and NACHRI.
Adverse event reporting will hardly be a first priority for families who confront a problem with a device. Troubleshooting and getting assistance from health care professionals, manufacturers, and home care agencies or vendors will take precedence, especially if the problem involves a life-supporting device. Nonetheless, some families may appreciate the opportunity to report their experiences with device problems further, for example, by sharing what happened and what was learned with other families through various kinds of family and patient support groups.
Some families may learn that they can report problems to FDA. The agency sensibly advises consumers who wish to report adverse events to seek the assistance of their physicians, who can provide clinical and technical details that may be important in understanding and describing the nature of the problem. Some patients and families, however, may wish to report directly to FDA without involving a physician. They may, for example, worry about alienating a physician they depend on by complaining about a device that the physician prescribed. They may also feel that a physician has ignored or dismissed their observations about a problem because their description of an event or problem was not technically sophisticated. Such dismissal risks overlooking real problems observed by those who are with the patient for extended periods.
Although FDA may have qualms about the quality and utility of the information received from patients and families, it still should offer consumers easier opportunities to report. As discussed earlier, the consumer who wishes to report a problem through the agency’s MedWatch program now faces instructions that are not written with the layperson in mind.
FDA should enlist its home health committee and others in advising on the creation of a simpler event reporting form in lay language for consumer reporting of events. (Again, the committee recognizes that changes in Form 3500 or 3500A involve a lengthy process.) The agency’s online reporting option could also be modified to provide additional explanations and assistance aimed specifically at lay users. Ideally, the online reporting option would also be designed to provide some feedback to reporters, for example, by directing consumers to additional resources such as advice on discussing concerns with a manufacturer or vendor and instructions about returning a
device (with problem documentation) to a manufacturer or vendor. It could provide links to reports on safety problems with a device that is the subject of a consumer’s report. The committee recognizes that it is not feasible to provide individualized feedback for each consumer report, but information technologies have the potential to allow more than a computerized thank you or acknowledgment of a report.
Recommendation 4.8: FDA should continue to improve and expand its medical device safety resources for patients and families and its focus on devices used in the home and community by
-
working with patient, family, and consumer organizations, providers, and industry to make it easier for patients or their families to report device problems to manufacturers or FDA and to learn about resources to support the safe use of medical devices;
-
making online reporting and information resources more accessible by using language and directions appropriate for lay users; and
-
enlisting hospitals, home care agencies and vendors, and other professional and provider groups to promote patient and family understanding of how to use devices safely, when and how to seek help, and when and how to report problems.
The recommendations above cover many areas for improvement in the agency’s adverse event reporting program for medical devices. At the same time, Congress and FDA deserve credit for past and continuing efforts to improve the program. These efforts include, for example, creating an online reporting option, developing computerized aids to screen reports and identify problems, creating a more active surveillance initiative in the pilot MedSun program, and using adverse event reports to inform the agency’s human factors research program.
In addition to continuing efforts to improve the existing program and fully implement the MedSun program, FDA is investigating additional forward-looking or prospective strategies based on automated patient information systems that would not only improve the detection and investigation of adverse events but also identify device hazards or hazardous practices before they cause harm to patients. The agency recognizes that this strategy requires improved means of identifying medical devices for purposes of analyzing and responding to adverse event reports. In particular, codes for use in the medical record should allow identification of both the manufacturer and model of a device rather than, as now, a general category of device (e.g., apnea monitor).
The next chapter of this report shifts attention to a different dimension of postmarket surveillance of medical devices. It examines the monitoring by FDA of postmarket studies required by FDA.






































































