
1
Introduction
In 1956, John and Mary Holter were shattered when their son, Casey, was born with spina bifida and hydrocephalus (water on the brain). Doctors told the Holters that the opening of the spine on their son’s back could be repaired but that they had not yet found a successful way to manage the buildup of spinal fluid in Casey’s brain, a process that would eventually kill him.
Mr. Holter, a self-described “mechanic” who worked in a lock company’s research lab, decided to tackle the fluid problem. He developed a small, one-way valve that he thought would allow the brain fluid to drain and save his son’s life. He prevailed upon the child’s neurosurgeon, Eugene Spitz, to use the untested device, which was made of silicone, a new material. Casey lived an unexpected 5 years before dying from other causes.
The Holter valve made drainage or shunting of cerebrospinal fluid a practical reality. Today, after years of technical modification and adjustment in clinical procedures, about 30,000 shunt procedures are done in the United States each year. The life expectancy of children with hydrocephalus is now measured in decades, not years, and hydrocephalus is no longer the end of a family’s dreams.
(Baru et al., 2001; Basse, 2003)
Through the determined creativity of single individuals such as John Holter and the contributions of organized teams of medical and engineering researchers, advances in biomedical science and engineering—combined with achievements in public health—have brought innumerable benefits to millions of children and their families and communities. Notably, in addition to improved sanitation and nutrition, the development of vaccines to prevent common childhood diseases and antibiotics to treat infections have saved the lives of countless children. Although medical devices such as syringes, intravenous infusion equipment, and infant-sized catheters have played supporting roles in immunizations and antibiotic therapy, devices have figured most prominently in other areas of pediatric health care.
For example, mechanical ventilators and other respiratory support devices in combination with medications and other therapies rescue thou-
sands of fragile newborns each year, including babies born prematurely with lungs that are insufficiently developed to sustain independent breathing. Some infants rely on these devices only briefly, but others with chronic health conditions use these life-sustaining devices for years. As shown below in the photographs of older and newer ventilatory support equipment (Figures 1.1 and 1.2), reductions in the size of equipment and other advances now allow many children who rely on these devices to live at home with their families, attend school, and participate in community life.
To cite another example, children who once would have died from congenital heart conditions today survive with the aid of implanted devices such as mechanical heart valves, pacemakers, devices that close holes in the heart, and artificial tubes used to bypass malformed heart valves. The surgical procedures associated with these and other treatments typically require additional sophisticated equipment, including cardiopulmonary bypass systems (heart–lung machines that oxygenate and circulate the blood while the heart is stopped for surgery), devices that provide anesthesia, and equipment that monitors breathing, oxygen levels, and other critical physiologic variables and warns clinicians of impending trouble. Box 1.1 lists examples of life-saving and life-sustaining devices that benefit children.

FIGURE 1.1 Child in iron lung, World Health Organization, c. 1938 (Used with permission of IUPUI University Library Special Collections and Archives).

FIGURE 1.2 Zoe and Dad on a hike (Courtesy of Lynne Andersson and Stephen Andersson).
This report examines the regulation and surveillance of a wide range of medical devices used with children. In some cases, these devices are intended solely or primarily for use with children. Examples include newborn incubators, inhaled nitric oxide delivery units for certain newborns with respiratory problems, and “bililights” or “biliblankets” that provide phototherapy for infants with jaundice. The design and evaluation of these devices will necessarily focus on the children expected to use the devices.
Some devices now widely used with adults were first aimed at pediatric conditions. For example, the first external pacemakers were developed for children who were born with cardiac anomalies that required surgical correction and subsequent pacing of the heart rhythm. Initially, the devices
|
BOX 1.1 Mechanical ventilators: Negative-pressure ventilators or “iron lungs,” which were developed in the late 1920s by Drinker and Shaw, supported children with respiratory failure from poliomyelitis into the 1950s. Subsequently, the positive-pressure ventilator became widely used to support virtually all infants and children in need of assisted breathing related to acute or chronic respiratory failure. Although it is difficult to attribute improved outcome to any single factor when many aspects of care have changed, the management of respiratory distress using mechanical ventilation in premature infants is one area where the increased survival is well documented (Gregory et al., 1971; Farrell and Avery, 1975). Balloon atrial septostomy (Rashkind procedure): This nonsurgical, catheter-based procedure provides a way for oxygenated blood to get to the systemic tissues in infants with transposition of the aorta and the pulmonary artery (a congenital heart defect that starves the blood of oxygen). It stabilizes the newborn until definitive surgery can be performed. The development of the device and associated surgical techniques led to a dramatic increase in survival of affected infants. One-month survival rates rose from approximately 10 percent in the early 1960s to more than 90 percent after the procedure became widely used (Rashkind and Miller, 1968; Liebman et al., 1969; Rashkind et al., 1969; Rashkind, 1971, 1983). Cerebrospinal fluid shunt for hydrocephalus: For infants and others suffering from inadequate draining of cerebrospinal fluid from the brain, the hydrocephalus shunt has reduced mortality from approximately 50 percent to approximately 10 percent (Gilmore, 1990; Staal et al., 1997). The device consists of a silicone tube that is passed through the brain into the cerebrospinal fluid-containing ventricles and connected under the scalp to a one-way valve. This valve drains through a longer silicone tube into the abdominal cavity, the veins that drain into the heart or, rarely, other body cavities. Modern valves can have their drainage pressure characteristics adjusted noninvasively after implantation. Although still plagued with problems of infection and blockage requiring surgical repair, cerebrospinal fluid shunts allow more than half of children with hydrocephalus to avoid brain damage and developmental delay caused by prolonged increased spinal fluid pressure on the brain (Casey et al., 1997). Pulse oximetry: In years past, the standard technique for assessing the body’s oxygenation was to obtain a sample of arterial blood by needle puncture or through an indwelling catheter. This presented many complications for small infants with conditions that left them at risk for a life-threatening lack of oxygen. It required removal of blood, which might ultimately necessitate a transfusion if there were multiple samples, and it could be technically difficult to obtain the sample from a small baby. The development of the pulse oximeter, a device that passes a light wave through an extremity (e.g., a finger or toe) to “see” the color of the blood in the capillaries and, hence, assess the amount of oxygen, was a tremendous advance. It could measure oxygenation continuously, which was particularly valuable during operations and in the care of critically ill infants. |
plugged into wall sockets, which meant they severely limited the children’s mobility during recovery and were fatally useless during a power interruption. Although simple by today’s standards, the external battery-powered pacemaker was a revolutionary development, and it was soon followed by the implantable pacemaker. This innovation helped establish what is now a large and very successful medical electronics industry (see, e.g., Rhees and Jeffrey, 2000).
In many cases, medical devices used to help children have been initially developed for, tested with, and most frequently employed to treat adults, who constitute a much larger market for medical devices and medical treatments than children, who are, overall, a healthier segment of the population. As discussed further in Chapter 2, implants and other devices may require modification for use with infants and children to account for differences in overall body size as well as in the dimensions of body structures such as blood vessels. Furthermore, because children are not small adults, the design, use, or monitoring of certain devices may need to take into account not only size but also children’s physical, cognitive, and emotional growth and development.
Sometimes it will be quite obvious that a device originally developed for adults is not, in that form, suitable for some children, for example, when a particular type of implanted device is clearly too large for infants and toddlers. Other times problems with pediatric use of a device—such as more rapid or intense inflammatory reactions to implant materials than seen with adults—will not be evident during initial research and early clinical use.
It is important to recognize that while children may experience the benefits of a successful medical treatment for longer periods than older adults, they likewise may suffer the negative consequences of treatments or adverse events for many more years. As a case in point, disturbances in cardiorespiratory function at young ages may have lifelong neurological consequences, which is an argument for early intervention as well as for long-term monitoring of the consequences of device use and other interventions that may create such disturbances. Furthermore, if medical treatments interfere with growth and development, affected children may never completely “catch up.” For example, some devices for treatment of scoliosis are used in conjunction with a spinal fusion process that limits further growth of the spine and surrounding structures, including the rib cage. In young children, such a limitation in thoracic growth can even prove fatal if the lungs are unable to grow and develop sufficiently.
Although medical devices and associated surgical and medical procedures can often correct medical problems and allow children to live normal and active lives, not all problems can be fully corrected, and some children saved in infancy are still expected to die in adolescence or early adulthood.
Moreover, for many children with severe medical conditions, decisions about treatment are not limited to a one-time surgery or other episode of treatment. Rather, the balancing of potential benefits and harms is ongoing as new options for treatment are proposed or the shortcomings of past interventions are revealed.
Beyond treatment decisions, parents weigh risks in structuring their child’s daily life and determining the degree to which a tightly controlled, medically monitored environment should be moderated to allow a more normal childhood experience. For example, parents weigh the risk of damage to an implant against the opportunity for a child to play sports with his or her peers.
Often parents and clinicians must make decisions in the absence of good evidence about the relative safety and effectiveness of medical devices and procedures. Many complex medical devices used with children have not been systematically evaluated in pediatric populations. Even for adults, who are the typical subjects of clinical trials involving devices, uncommon problems may not be evident in clinical trials used to support applications for marketing approval. The clinical studies undertaken in support of a product’s approval for marketing are usually conducted for relatively short periods in carefully controlled populations that do not fully represent the population of expected users (e.g., patients with multiple health conditions). For that reason, once medical products enter the market, government health agencies, clinicians, manufacturers, and others sometimes continue to study them for longer periods and with broader populations. In certain circumstances, the U.S. Food and Drug Administration (FDA) can require such studies. Policymakers have also created requirements for manufacturers, health care facilities, and others to report problems—adverse events—that are caused by or associated with legally marketed drugs, devices, and other medical products.
Until 1976, federal officials had limited authority to regulate the safety or effectiveness of medical devices. In that year, Congress added the Medical Device Amendments (P.L. 94–295) to the Federal Food, Drug, and Cosmetic Act (P.L. 75–717). By this step, Congress acknowledged the increasing sophistication and importance of medical device technology while also recognizing that both the benefits and the risks of this advanced technology warranted more systematic attention by FDA, manufacturers, and others.
In the past decade, with enactment of the FDA Modernization Act of 1997 (P.L. 105–115) and the Medical Device User Fee and Modernization Act of 2002 (P.L. 107–250), Congress streamlined certain regulatory procedures for medical devices. The 2002 legislation also included several provisions related to pediatric uses of devices, one of which called for this
report on postmarket surveillance of medical devices used with pediatric populations.
Postmarket surveillance of medical devices used with children is a little-investigated topic. Again, this is partly because the market for most medical products is concentrated among adults, especially older adults, so attention tends to follow clinical and market realities. Moreover, assessments and discussions of medical product regulation and patient safety tend to focus more on pharmaceuticals than on medical devices. Thus, pharmacoepidemiology is a relatively well-established area of epidemiologic inquiry while medical devices epidemiology is not. In the device arena, no readily identified organizations exist in parallel to the International Society for Pharmacoepidemiology, the International Society for Pharmacovigilance, or the Academic Programs in Pharmacoepidemiology. (These organizations devote some attention to device epidemiology, but it is not a major focus.) In addition, discussions of medical product regulation—for drugs and vaccines as well as medical devices—have tended to concentrate on the regulatory requirements and processes related to the approval or clearance of products for marketing rather than on the subsequent evaluation or surveillance of their performance.
It is appropriate to put a high priority on keeping unsafe and ineffective medical devices and other products from entering the market in the first place. It is also reasonable to ask whether the system of postmarket surveillance established under the Federal Food, Drug, and Cosmetic Act provides adequate safeguards for the use of medical devices once the devices have been approved for marketing. This report considers that question as it relates to children specifically.
As the committee that developed this report reviewed the literature, consulted knowledgeable individuals, and considered the questions before it, several themes emerged. They are:
-
Children and their families benefit from safe, effective medical devices. Timely access to such devices prevents premature deaths and significantly improves the quality of life for children and their families.
-
Systematic attention to children’s needs and characteristics is important in medical device design, use, and evaluation because children differ from adults in important ways. For devices developed primarily for use with adults but with expected or possible pediatric uses, such attention can encourage the early identification of potential pediatric benefits and harms and the early consideration of modifications in the design or use of a medical device that will minimize risks and safeguard child patients. In addition, long-term studies may be necessary to understand developmental effects and the long-term balance of benefits and harms of pediatric use of long-term implants and certain other devices.
-
An effective regulatory program for evaluating and monitoring the safety of medical devices in general is a necessary foundation for efforts to safeguard children in particular. This basic foundation of device regulation then requires the addition of pediatric expertise, guidelines, and other resources.
-
The regulation of medical devices reasonably differs from the regulation of drugs. Medical devices are more variable than drugs in their mode of operation, range of function, dependence on user skills, and potential for harm. Many are simple and very low risk. For some complex devices, the use of conventional randomized, controlled trials to provide evidence of safety and effectiveness may not be feasible or ethical. In addition, much device innovation and development is characterized by ongoing product modification and improvement with a relatively rapid rate of product turnover, which may complicate some aspects of long-term monitoring and evaluation of safety and effectiveness.
-
A careful assessment of medical device regulations weighs potential positive and negative outcomes, including whether the potential negative effects of a regulation are acceptable. Just as the balance of potential benefits and harms should be considered when medical devices are reviewed, so should policymakers weigh the potential outcomes of regulations and consider possible unintended and unwanted consequences, for example, the discouraging of beneficial innovations and refinements in medical devices.
-
The shift of medical device use from institutions to homes, schools, and the community complicates postmarket surveillance. The migration of care out of the hospital into patient homes has brought many benefits, but it also presents risks as families and patients have taken on many roles in the operation, maintenance, and troubleshooting of complex medical devices that were formerly performed by health care professionals. In addition, for complex devices now used in the home, the opportunity and responsibility for identifying and reporting possible device-related adverse events has shifted, in considerable measure, to patients, families, office-based physicians, home care nurses, home health agencies, and even school personnel. Of these groups, only home health agencies have legal obligations for reporting adverse events. Surveillance programs have yet to adjust to these realities.
-
Medical device safety is a shared responsibility. No matter how successful, government regulation of medical devices is not sufficient in itself to safeguard children or adults who use medical devices. Clinicians, health care providers, engineers, manufacturers, researchers and research funding agencies, patients and families, consumer groups, and others have central roles to play. Regulations backed by both incentives and sanctions do, however, contribute as part of a larger system of ethical, financial, professional, and other influences that support safe, effective health care.
ORIGIN OF STUDY TASKS AND OVERVIEW OF REPORT
In 2002, Congress passed the Medical Device User Fee and Modernization Act. The main focus of this legislation was the establishment of a system of user fees, similar to that already adopted for drugs, to support the more expeditious review by FDA of applications by manufacturers for approval to market devices. The legislation also included three provisions related to medical devices with possible pediatric applications. One called for pediatric expertise, when appropriate, in the review of applications for FDA approval to market a medical device (Section 210). A second provision directed the Secretary of the Department of Health and Human Services to provide guidance on the kind of information needed to assure the safety and effectiveness of medical devices intended for use with children and to protect children involved in clinical studies of such devices (Section 213).
A third section called for a study by the Institute of Medicine (IOM) to assess whether “the system under the Federal Food, Drug, and Cosmetic Act for the postmarket surveillance of medical devices provides adequate safeguards regarding the use of devices in pediatric populations” (Section 212). The study was also to examine
-
the FDA’s monitoring and use of adverse reaction reports, registries, clinical studies, and other postmarket surveillance activities;
-
the adequacy of FDA’s monitoring of commitments for further clinical studies made by manufacturers at the time of approval of specific medical devices;
-
the adequacy of postmarket surveillance studies to evaluate how children’s active lifestyles may affect failure rates and longevity for implanted devices; and
-
the length of postmarket surveillance studies of implanted devices, including whether studies continue long enough to evaluate the impact of children’s growth and development given the expected length of time that a child will have an implant.
Together, these legislative provisions constituted the statement of task and charge to the IOM, which is the health policy arm of the National Academy of Sciences. The IOM appointed a 13-member committee of experts to prepare this report.
Given its origins, one audience for this report is legislative and administrative policymakers and those who advise them. The report does not, however, assume technical knowledge of medical device regulation. It likewise does not assume expertise in pediatric medicine or medical device design or use. It is meant to be credible to technical experts but understandable to a broader audience, including consumer and patient advocacy groups. Parents and the general public are not primary audiences, although
the committee has sought examples that would be meaningful and understandable to general readers.
This report often focuses on complex devices that are implanted in the body for extended periods (e.g., pacemakers) or that can be expected to have serious health consequences if they fail (e.g., mechanical ventilators). Even relatively simple devices can, however, cause injury and even death due either to errors in their use or to shortcomings in their design, manufacture, distribution, maintenance, or instructions for use. For example, in 2002, FDA announced the Class I (high priority) recall of a bassinet with a drop-leaf work surface that could and had collapsed when used for the unintended purpose of holding an infant (FDA, 2002j). That same year, FDA reported the Class I recall of a foam-tipped oral swab following a report that the tip had dislodged and blocked a patient’s airway (FDA, 2002g). Regulatory and other strategies to protect children must also include means to detect and prevent harm from less complex and less intrinsically risky medical devices.
The remainder of this chapter defines a number of the concepts and terms included in the legislative charge for this IOM study. It also provides a brief overview of the history of medical device regulation and its context. Chapter 2 examines the rationale for particular attention to the special characteristics of children when medical devices are designed, evaluated, regulated, and used. The regulatory framework for postmarket surveillance is reviewed in Chapter 3.
Chapter 4 examines the nature, reporting, and analysis of adverse device events, including advantages and limitations of adverse event reporting as a surveillance tool. It includes a number of illustrative vignettes that depict the diversity of adverse device events involving children. Chapter 5 reviews FDA’s monitoring and reporting of manufacturers’ fulfillment of commitments for postmarket studies required by FDA, and Chapter 6 considers the adequacy of postmarket surveillance studies to assess the effects of children’s activity levels and growth and development on device performance. Beyond the recommendations presented in the preceding three chapters, Chapter 7 offers a more comprehensive view of FDA postmarket surveillance and its place in a broader system of shared responsibilities for improving the quality of health care and protecting adult and child patients from harm.
This report also includes several appendixes. Appendix A describes the committee’s information collection strategies. Appendix B provides an illustrative list of medical devices used exclusively or frequently with children. Appendix C reviews the nature of medical device innovation. Appendix D focuses on methodological issues for postmarket surveillance. Appendixes E and F present case studies of surveillance issues related to two complex devices—cerebrospinal fluid shunts and cochlear implants. The final appendixes provide a glossary and committee biographies.
SELECTED CONCEPTS AND DEFINITIONS
Discussions of medical device regulation are replete with legal and technical terminology that is unfamiliar to most audiences but reflects the complexity of medical products and their regulation. This section defines a number of terms found in the study’s statement of task as well as some related terms. Later chapters include more definitions. Chapter 2, for example, discusses the concepts of children’s active lifestyles and growth and development as they relate to certain device therapies, especially those involving implanted devices.
Pediatric Population, Children
Pediatrics refers to the health care of children. As part of guidance on premarket assessment of pediatric medical devices, FDA’s Center for Devices and Radiological Health (CDRH) stated that it considered pediatric use “to be any use of a medical device in a pediatric population … in which there is a primary pediatric indication. General indications, where considerable pediatric application is anticipated, are also included in this definition” (FDA, 2004p, p. 5). This report generally refers to devices with pediatric uses rather than to pediatric devices.
The term pediatric population may refer to all children or to a subgroup of children who share certain characteristics (e.g., a diagnosis or the use of a medical device). In broad usage, children are individuals, including infants and adolescents, who are not considered to be adults. From a legal perspective, nearly all states consider individuals aged 18 or above to be adults, although states may allow younger individuals to make decisions about medical treatment and other matters as adults under certain circumstances (English and Kenney, 2003; Campbell, 2004). In recent guidance on premarket assessment of devices used with children, CDRH has expansively defined children as those under the aged of 21, arguing that this upper age limit may be useful for some devices and clinical studies (FDA, 2004p). In a narrower usage, a child is an individual who is between the periods of infancy and adolescence. As discussed further in Chapter 2, definitions of these periods vary, even within FDA (see Chapter 2).
Medical Device, Implanted Device, Combination Product
Definitions
The key statutory distinction between a drug and a device is that a device does not work primarily through chemical means and does not
depend on metabolic action. As defined in the U.S. Code (21 USC 321(h)), a medical device is:
an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including any component, part, or accessory which is:
-
recognized in the official National Formulary, or the United States Pharmacopeia, or any supplement to them;
-
intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals; or
-
intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes.
Consistent with scientific and technological developments, a more recent definition adopted by the European Union (EU) explicitly excludes products that work by immunological mechanisms. The EU definition also includes software necessary for a device to operate properly.1 The EU definition goes beyond diseases to recognize explicitly that devices are used in connection with injuries and disabilities as well as for contraception. In addition, the definition acknowledges the importance of products such as implants and prostheses that replace or modify the anatomy.
FDA regulations use two slightly different definitions of implants or implanted devices. The regulations related to investigational devices (see Chapter 3) define an implant as “a device that is placed into a surgically or naturally formed cavity of the human body if it is intended to remain there for a period of 30 days or more” (21 CFR 812.3(d)). The regulations on medical device tracking (see Chapter 3) refer to implanted devices as those “intended to be placed into a surgically or naturally formed cavity of the human body for more than 1 year to continuously assist, restore, or replace
the function of an organ system or structure of the human body throughout the useful life of the device” (21 CFR 821.3(f)).2
An increasing number of medical products combine elements such as a drug and device or a biological product and device.3 Examples of such products include insulin pumps (computer-controlled, wearable devices that deliver periodic doses of insulin as programmed by the patient) and drug-eluting stents (devices that hold open blood vessels and are coated with slow-release drugs to reduce new blockage of the vessels). As defined by FDA, a combination product consists of two or more regulated components that are physically combined or linked, packaged together as a single unit, or packaged separately but required to be used together (21 CFR 3.2(e)). The product’s primary mode of action determines which unit of FDA has primary jurisdiction over the product (21 CFR 3.4; FDA, 2004e). Thus, because the primary action of the drug-eluting stent is to open the blood vessel, it is primarily regulated by CDRH. In contrast, a drug-eluting disk that delivers chemotherapy agents for brain tumors is primarily regulated by the Center for Drug Evaluation and Research.
Although this report often will refer to a medical device, “a” medical device may actually be composed of several device subsystems or individual devices that work together as a system to achieve the desired effect. For example, a left ventricular assist device that helps the heart beat recently received special FDA approval (a humanitarian device exemption or HDE as described in Chapter 3) for use with children. The device is described as consisting of four major subsystems—a pump, an external controller, a clinical data acquisition system, and a patient home support system—as well as assorted accessories, including batteries, a battery charger, and a shower kit to protect the device during showering (H030003, FDA, 2004b). The pump subsystem itself consists of the pump housing within which three components move blood through the pump, a titanium inlet/inflow cannula
(tube), a Dacron outflow graft, an implanted probe to measure blood flow, and a cable to connect the implanted pump to the external battery pack and controls. Each component must function safely and effectively in concert with the other components.
Device Classes, Approval, Clearance
Since 1976, the legal classification of medical devices has provided for three classes of devices based in large measure on the risk they pose. Class I devices, which include such mundane items as tongue depressors, lice combs, toothbrushes, and bedpans, are the least regulated. They do not require review by FDA before they can be legally marketed. Class III devices, which include implants and other high-risk devices, are the most regulated. For these devices, manufacturers must submit clinical evidence of safety and effectiveness and secure approval by FDA prior to marketing. Manufacturers of Class II devices face an intermediate level of regulation, including a clearance process that usually does not require the submission of clinical data. These three classes of devices and their regulation are discussed further in Chapter 3.
In addition to these three broad classes, FDA uses a more detailed nomenclature system—involving some 1,700 categories of medical devices—to support its regulatory activities. FDA is participating in efforts to “harmonize” device nomenclature internationally. Much of that work now focuses on the possible merger of two systems, the Universal Medical Device Nomenclature System (UMDNS) developed by ECRI (formerly the Emergency Care Research Institute) in the United States and the Global Medical Device Nomenclature (GMDN), which is owned by the European Standards Organization (Lumpkin, 2004). To illustrate this kind of detailed device classification scheme, Appendix B lists a subset of terms from the UMDNS for devices that have uses with children. As discussed in Chapters 4 and 6, this level of detail, while useful for some purposes, does not reach the level of detail about a specific device (e.g., lot number, model, brand) necessary for some safety and surveillance purposes.
Between the detail of the UMDNS and GMDN and the generality of the three broad FDA risk-related device classifications are other ways of categorizing devices that are useful for certain objectives.4 For example, for purposes of establishing scientific panels to review devices, FDA regulations
group devices into 16 categories based mostly on medical specialty (Box 1.2) (21 CFR 862–892).
Safeguard, Safety, Risk, Harm, Hazard, Benefit, Effectiveness, Efficacy
A safeguard protects someone or something from harm. FDA’s statute and regulations do not define safe or safety as such but do describe criteria for determining that a medical product is safe. Specifically, “there is reasonable assurance that a device is safe when it can be determined, based upon valid scientific evidence, that the probable benefits to health from use of the device for its intended uses and conditions of use, when accompanied by adequate directions and warnings against unsafe use, outweigh any probable risks” (21 CFR 860.7(d)(1)). There should also be adequate demonstration “of the absence of unreasonable risk of illness or injury associated with the use of the device for its intended uses and conditions” (21 CFR 860.7(d)(1)).
Thus, from a device regulation perspective, safety is relative. A product with great expected benefits and significant (but not unreasonably high) risks may be judged to be safe, while a product with lesser expected benefits and the same level of risk may be judged unsafe.
Safety is also relative in other respects. What is safe for an adult may not be safe for a child. What is safe for an adolescent may not be safe for an infant.
Few if any medical products are completely free from risk. Risk refers to a potential harm or the potential of an action or event to cause harm. Specific risks can be characterized along several dimensions, including the probability of a given harm as well as its likely severity and duration. A harm is a hurtful outcome of an event or action. A hazard is a potential
|
BOX 1.2 Anesthesiology Cardiovascular Clinical Chemistry and Clinical Toxicology Dental Ear, Nose, and Throat Gastroenterology and Urology General and Plastic Surgery Hematology and Pathology Immunology and Microbiology Neurological Obstetrical and Gynecological Ophthalmic Orthopedic Physical Medicine General Hospital and Personal Use Radiology SOURCE: 21 CFR 862–892. |
source of harm. Harms may be immediate or long term and may be common and anticipated or rare and unexpected.
A benefit is a positive or valued outcome of an action or event. A potential benefit is a positive but uncertain outcome, for example, the desired result of an experimental intervention.
Just as safety is not explicitly defined, FDA’s statute and regulations do not define effectiveness explicitly but instead set forth criteria for determining effectiveness. Specifically, “[t]here is reasonable assurance that a device is effective when it can be determined, based upon valid scientific evidence, that in a significant portion of the target population, the use of the device for its intended uses and conditions of use, when accompanied by adequate directions for use and warnings against unsafe use, will provide clinically significant results” (21 CFR 860.7(e)(1)). As is true for safety, a device that is effective for an adult may not be effective for a child.
Effectiveness is sometimes differentiated from efficacy, with the former term used to describe the achievement of desired results in actual practice and the latter to the achievement of such results in controlled studies. In 1998 guidance on clinical evidence of effectiveness for drugs and biological products, FDA states that as used in that document, “the term efficacy refers to the findings in an adequate and well-controlled clinical trial or the intent of conducting such a trial and the term effectiveness refers to the regulatory determination that is made on the basis of the clinical efficacy and other data” (FDA, 1998f, p. 1).
Adverse Event, Close Call or Near Miss, Device Failure or Malfunction, Error
An adverse event is an instance of harm during patient care or research that is not the result of the individual’s disease or medical condition. Thus, a death due to cancer while a patient is receiving chemotherapy through an infusion pump is an adverse outcome but not an adverse event. A death due to the incorrect setting of an infusion pump for chemotherapy is both an adverse outcome and an adverse event. The charge to the committee refers to adverse reactions, but FDA refers to adverse events. This report follows FDA usage.5
Adverse events are sometimes defined to include events that have the potential to cause harm, such as close calls or near misses that could have resulted in harm but did not. To cite an example, when a device malfunc-
tions but a caregiver notices it and responds quickly enough to avert injury, that event is a close call or near miss.
Adverse events and close calls may result from a device failure or malfunction. As defined in federal regulations, a device failure “means a device does not perform or function as intended, and includes any deviation from the device’s performance specifications or intended use” (21 CFR 822.3(c)). Elsewhere, the regulations define device malfunction in similar terms as “the failure of a device to meet its performance specifications or otherwise perform as intended” (21 CFR 803.3(n)). Devices may fail or malfunction in myriad ways—breaking outright, leaking, catching fire, clogging, crimping, warping, experiencing software “bugs,” and otherwise deviating from intended performance.
Adverse events may also occur when devices are improperly used, operated, assembled, monitored, stored, maintained, or selected by health care workers, families, or patients. These events are kinds of health care errors, which another IOM report has defined as “the failure of a planned action to be completed as intended (i.e., error of execution) or the use of a wrong plan to achieve an aim (i.e., error of planning)” (IOM, 2000c, p. 28). Because errors in the use of medical devices may reflect flaws in the design of the devices and because reference to user errors may contribute to a counterproductive culture of blame within health care organizations, FDA tends now to refer to use error rather than user error.
The reporting of adverse events and device malfunctions is discussed further in Chapters 3 and 4. The focus of these discussions is on adverse events detected in normal patient care rather than in research. The reporting of adverse events in research is governed by separate policies.
Premarket and Postmarket
As with many government regulatory agencies, FDA has developed a specialized vocabulary to describe its responsibilities, activities, organizational units, and regulated entities. Much of this terminology has its basis in statutory language and distinctions. In referring to premarket and postmarket rather than premarketing and postmarketing activities, this report follows the statutory language that provided for this study and the usual (but not invariable) practice of FDA in describing activities that occur either prior to or following the entry of a medical product into the market.
Premarket regulatory processes include evaluations, decisions, and other activities that occur before the marketing of a medical product consistent with legal requirements (see Chapter 3). Thus, the development and evaluation of information about a device’s safety and effectiveness and the approval or clearance of a product for marketing are premarket activities.
Likewise, the authorization to test an unapproved device with humans is a premarket activity.
Postmarket evaluations, activities, and decisions occur after regulatory approval, clearance, or registration of a medical product for marketing. As discussed further in Chapter 3, the major device-related postmarket responsibilities of FDA involve its programs for adverse event reporting and focused surveillance or follow up of selected products, sometimes including required clinical studies.
Surveillance and Surveillance Tools
From a broad public health perspective, surveillance may be defined as the “ongoing, systematic collection, analysis, interpretation, and dissemination of data regarding a health-related event for use in public health action to reduce morbidity and mortality and to improve health” (CDC, 2001, p. 2). Most but not all surveillance involves unwanted events such as deaths and injuries or hazardous or potentially hazardous situations such as exposure to unsafe medical products, communicable diseases, toxic substances, or unsafe workplaces (see, e.g., Halperin et al., 1992; Tilson, 1992; Friis and Sellers, 2004).
Broadly, then, postmarket surveillance of medical devices refers to programs that seek to protect public health by systematically collecting, analyzing, and communicating information about events involving or potentially involving legally marketed medical devices. More narrowly, Section 522 of the Federal Food, Drug, and Cosmetic Act uses the term Postmarket Surveillance to describe one type of surveillance, specifically, activities that FDA may—after a device is approved or cleared for marketing—require manufacturers to undertake to gather safety and, sometimes, effectiveness information for a small group of Class II and Class III devices (21 USC 360(l)). As described by FDA and discussed further in Chapter 3, the primary objective of Section 522 Postmarket Surveillance “is to study the performance of the device after marketing as it is to be used in the general population for which it is intended … [with a focus on] morbidity or mortality … [and on] device failure and its attendant impact on the patient” (FDA, 1998b, unpaged). Unless otherwise noted, this report uses the term postmarket surveillance in its broad sense (and indicates the narrower usage by referring to Section 522 Postmarket Surveillance).
In addition to studies ordered after a device has been approved or cleared for marketing, surveillance studies may be undertaken to investigate important unanswered questions that exist at the time a device is considered for approval. As a condition of approving a device, FDA can require that manufacturers collect more information about the safety or effective-
ness of a device. This report uses the term postmarket studies or postmarket study commitments to refer collectively to condition-of-approval studies and Section 522 studies that are ordered after a device enters the market.
Some postmarket studies continue to follow individuals who participated in the clinical trials or other studies that were used to support an application for FDA approval or clearance of a medical device.6 Other postmarket studies involve registries of new patients. A registry is a system for collecting information about a class of individuals or patients who have in common a disease, injury, condition, medical procedure or product, or similar characteristic. The term registry is sometimes used narrowly to refer to the database itself and sometimes more broadly to refer to analyses and studies based on registry information. For the latter, this report generally refers to registry studies or registry-based studies.
A major tool of FDA postmarket surveillance is an adverse event reporting system for collecting and analyzing information about product failures or harms related to or potentially related to medical products. For medical devices, the emphasis is on the reporting of device failures and malfunctions and device-related deaths or serious injuries. As discussed in Chapters 3 and 4, the primary FDA program of adverse event reporting for medical products, MedWatch, relies on passive surveillance; that is, it awaits reports that manufacturers, health care facilities, health care professionals, and others decide to submit. In addition, FDA has created the Medical Device Safety Network (MedSun, formerly known as the Medical Product Surveillance Network), a pilot program that involves selected hospitals and nursing homes). This program includes some elements of active surveillance, for example, a request that all or some participating institutions collect information on a specific problem or event.
In addition to surveillance undertaken or directed by FDA, manufacturers for implanted devices such as pacemakers and defibrillators conduct active surveillance of these products. Health care providers, some state governments, accrediting groups, and various other private organizations also have surveillance programs for identifying patient safety problems, although medical devices usually do not figure prominently in these programs.
Certain activities to build additional knowledge about the safety or effectiveness of a marketed medical device—for example, some clinical tri-
|
6 |
A guidance document on clinical trials involving medical devices cites this definition of clinical trial: “a prospective study comparing the effect and value of intervention(s) against a control in human subjects” (Friedman et al., 1985; cited in FDA, 1996c). Many clinical studies used to support FDA approval of medical devices do not have prospective control groups. Chapter 6 and Appendix D discuss research strategies and issues. |
als sponsored by the National Institutes of Health (NIH)—are not normally described as surveillance. Similarly, manufacturer studies to support the approval of new indications for the use of a device (e.g., use for a different medical condition) are not usually viewed as surveillance, although they may generate important information about device safety or effectiveness that is relevant to previously approved indications.
If postmarket surveillance identifies safety problems involving a device or its use, manufacturers may recall the product, modify its design or manufacturing process, or change information about how or for whom it should be used. Manufacturers and regulators may advise clinicians, health care organizations, and sometimes patients or consumers to cease using the product, limit its use to certain patient groups or clinical purposes, or change processes for using the product (e.g., by adjusting equipment settings in different ways).
EVOLUTION OF MEDICAL DEVICE REGULATION
Regulation of medical devices has tended to lag behind regulation of pharmaceuticals.7 When Congress passed the original Pure Food and Drugs Act (P.L. 59–384) in 1906, it banned interstate and foreign commerce in misbranded and adulterated drugs, food, and drinks—but not medical devices. The legislation provided for the seizure of prohibited products and for fines and imprisonment for those engaging in prohibited commerce. The legislation was the culmination of years of advocacy and agitation for federal action to protect consumers from impure, unsafe, and mislabeled foods and medicines. (Table 1.1 provides a time line of significant events in the evolution of medical product regulation in the United States.)
Although the 1906 Act did not cover devices, the U.S. Postal Service (under its general authority to act against mail fraud) could pursue cases of mail order fraud involving devices. For example, a 1929 FDA report mentioned cooperation with the Postal Service in cases involving “a cap device alleged to grow hair, and a supposedly electronic belt and insoles for the treatment of rheumatism and kidney ailments” (as quoted by Hutt, 1989, p. 101).
FDA first received authority to regulate medical devices in the 1938 Federal Food, Drug, and Cosmetic Act. Among many other important provisions, that legislation authorized factory inspections, directed that drug and device labels provide adequate directions for safe use, extended controls to cosmetics, and eliminated the requirement that fraud be proved in
TABLE 1.1 Selective Time Line of Key Dates in Development of the Food and Drug Administration’s Regulatory Authority over Medical Products, Especially Devices
|
1848 |
Drug Importation Act. Intended to stop the import of adulterated drugs. |
|
1902 |
Biologics Control Act. Intended to protect the purity and safety of serums, vaccines, and similar products used to prevent or treat diseases in humans. |
|
1906 |
Pure Food and Drugs Act. Provided first major federal regulation of drugs. Did not apply to medical devices. (Fraudulent medical devices covered under postal fraud regulations.) |
|
1938 |
Federal Food, Drug, and Cosmetic Act. Required premarket review of new drugs for safety. Gave FDA authority over adulterated or misbranded therapeutic devices. |
|
1940 |
Food and Drug Administration moved from the Department of Agriculture to predecessor of the Department of Health and Human Services. |
|
1941 |
Insulin Amendment. Required FDA to test and certify purity and potency of insulin. |
|
1944 |
Public Health Service Act. Covered a broad spectrum of health concerns, including regulation of biologics. |
|
1954 |
Voluntary program of drug reaction reporting. Created through collaboration of FDA with American Society of Hospital Pharmacists, the American Association of Medical Record Librarians, and (later) American Medical Association. |
|
1962 |
Drug Amendments (Kefauver-Harris). Expanded FDA responsibilities to ensure drug safety and effectiveness. Effectiveness must be proved by “substantial evidence.” |
|
1966 |
Fair Packaging and Labeling Act. Required all products in interstate commerce to be honestly and informatively labeled, including medical devices. |
|
1973 |
FDA guidelines for voluntary reporting of adverse events. |
|
1976 |
Medical Device Amendments. Redefined “device.” Required manufacturers to give FDA notification of new devices introduced to market. Gave FDA authority to describe good manufacturing practices, to approve and ban certain devices before marketing, and to require notification, replacement, or refund by makers of defective products. |
|
1984 |
Medical Device Reporting (MDR) regulations. Required device manufacturers and importers to report device-related deaths, serious injuries, and malfunctions to FDA. |
|
1990 |
Safe Medical Devices Act. Required facilities using medical devices to report incidents related to a death, serious illness, or serious injury to FDA or manufacturers. Provided for mandatory Postmarket Surveillance by manufacturers for implanted or life-supporting devices that might cause death or serious harm. Gave FDA authority to impose civil penalties and, under certain circumstances, recall devices. |
|
1992 |
Global Harmonization Task Force established to promote international harmonization in regulation of medical devices. |
|
1992 |
Medical Device Amendments. Required semi-annual reports from user facilities. Expanded requirements for registration, certification, documentation, reporting, and surveillance of medical devices. |
|
1993 |
MedWatch created. Allowed consumers and health care professionals to report adverse events. |
|
1997 |
Food and Drug Administration Modernization Act. Accelerated FDA review of devices, regulated advertising of devices for unapproved uses. |
|
2002 |
Medical Device User Fee and Modernization Act. Provided for user fees for premarket reviews of medical devices. Included provisions for this and other pediatric studies or analyses. |
|
2004 |
Medical Devices Technical Corrections Act. Required a report on barriers to the availability of devices intended for children and expanded provision for electronic labeling. |
|
SOURCES: Hutt, 1989; Merrill, 1994; Higgs, 1995; FDA, 1999d, no date; Flannery, 2002; Whitmore, 2004. |
|
cases of false product claims. It also required that new drugs—but not new devices—be shown to be safe before they were marketed.
As described in one review of the history of device regulation, “[p]aradoxically, just after the FDA was given adequate statutory authority to police the safety and labeling of devices, a flood of fraudulent devices began to appear on the market” (Hutt, 1989, p. 105). The agency devoted considerable resources to such devices in the 1940s and 1950s.
Congress passed another broad piece of major legislation with the Drug Amendments of 1962 (P.L. 87–78, sometimes called the Kefauver-Harris Amendments for its sponsors). The legislation significantly expanded regulatory requirements for drugs but not devices. Notably, it required manufacturers to show evidence of effectiveness as well as safety before marketing, to report adverse events for marketed drugs, and to include information about risks as well as benefits in medical advertisements. Before becoming involved in clinical studies of investigational drugs, research subjects had to give their informed consent.
Proposals for the regulatory reforms of the early 1960s had originally covered medical devices, but those provisions were set aside in favor of a focus on drugs. At the time it was understood that “Congress would return to the matter of device legislation within a matter of months,” but despite many calls for action, Congress waited to do so until 1976 (Hutt, 1989, p. 106). In the interim, FDA capitalized on similar statutory definitions for drugs and devices to classify certain innovative products as drugs rather than devices, and the courts acquiesced to this interpretation of the 1938 legislation. For example, the Supreme Court sustained FDA’s categorization of a laboratory screening device (an antibiotic sensitivity disk) as a “drug,” which made it subject to premarket regulations. The Court reasoned that “the word ‘drug’ is a term of art for the purposes of the Act, encompassing far more than the strict medical definition of that word. If Congress had intended to limit the statutory definition to the medical one, it could have so stated explicitly” (United States v. Bacto-Unidisk, 1969, unpaged).
Acknowledging concerns about the safety of increasingly sophisticated and complex medical devices, the Secretary of the Department of Health, Education and Welfare (now the Department of Health and Human Services) established a committee to consider the regulation of medical devices. In 1970, the committee, which was chaired by Dr. Theodore Cooper (director of what was then the National Heart and Lung Institute), recommended that regulation of devices be tailored to characteristics of devices rather than essentially copying provisions established for drugs. For example, the committee recommended that device regulation be keyed to the variability in the risks presented by different kinds of devices (Study Group on Medical Devices [Cooper Committee], 1970). The committee also undertook a literature review that identified (as reported to a congressional committee) more than 700 deaths and 10,000 injuries linked to medical devices, including 512 deaths or injuries attributed to heart valves, 89 deaths and 186 injuries linked to heart pacemakers, and 10 deaths and 8,000 injuries attributed to intrauterine devices (U.S. Congress, 1973, as cited in OTA, 1984 and Hutt, 1989).
Responding to the recommendations of the Cooper Committee, FDA conducted an inventory of medical devices then on the market. It also began work to classify medical devices based on the level of risk and appropriate regulation. The organizational unit responsible for devices became the Bureau of Medical Devices and Diagnostic Products, matching the Bureau of Drugs in organizational standing. That unit, which was merged with a unit responsible for radiological health in 1982, was renamed the Center for Devices and Radiological Health in 1984, the name that remains today.
The Medical Device Amendments of 1976 (P.L. 94–295) extended FDA authority to regulate devices. As recommended by the Cooper Committee, the legislation distinguished the specifics of device regulation from those of drug regulation in several respects, for example, creating the three-tier classification described earlier, which links regulatory requirements to risk. The 1976 legislation also gave FDA authority to create a system for reporting adverse events associated with devices. Going beyond the earlier voluntary program for reporting adverse device-related events, FDA issued regulations in 1984 that required manufacturers and importers of devices to report information indicating that a device might have caused or contributed to a death or serious injury. They were also to report malfunctions with the potential to cause death or serious injury.
The Safe Medical Devices Act of 1990 added requirements that hospitals and other “user” facilities report to FDA and manufacturers any events indicating that a device had caused or contributed to a death. It also required user facilities to report to manufacturers events suggesting that a device had caused or contributed to serious patient harm. The legislation established new requirements that manufacturers track certain kinds of
high-risk medical devices, and it gave FDA the authority to order recalls of devices under certain circumstances. It further provided that FDA direct manufacturers to conduct additional information collection activities for certain implants and other devices with the potential to cause serious harm. Although the Safe Medical Devices Act increased the scope of device regulation, it also gave FDA the authority to approve—through a Humanitarian Device Exemption (HDE)—certain medical devices for small user populations without requiring substantial clinical evidence of effectiveness. Since HDE regulations went into effect at the end of 1996, 30 HDEs have been approved, several of which provide for pediatric use.
Reflecting a growing sentiment that regulations—or the way they were administered by FDA—were interfering with the timely introduction of important new medical products, the 1997 FDA Modernization Act reversed some provisions of the 1990 legislation. It eliminated certain requirements for adverse event reporting and ended provisions for mandatory postmarket surveillance studies in favor of FDA discretion to order studies or information collection for certain kinds of devices. The legislation further focused FDA resources on higher risk devices and authorized the creation of a new adverse event reporting system based on a sample of hospitals and other user facilities.
The Medical Device User Fee and Modernization Act of 2002, which called for this IOM study, provided for a system of user fees for FDA premarket reviews. Among other provisions, the legislation directed FDA to prepare a report on the effects of and compliance with surveillance requirements. The legislation also authorized additional appropriations for postmarket surveillance activities, but Congress did not appropriate these funds.
Other FDA regulations set forth requirements for the protection of human participants involved in research on medical devices and other medical products. As briefly described in Chapter 6, these human research protection regulations include special protections for children (IOM, 2004a).
In addition to special provisions related to research involving children, Congress and FDA have directed attention to children in some other areas, mostly to expand the availability and testing of pharmaceuticals for pediatric use. Some provisions have, however, dealt either with medical devices specifically or with all products regulated by FDA. For example, the Best Pharmaceuticals for Children Act of 2002 directed FDA to create an Office of Pediatric Therapeutics to be responsible for coordinating and facilitating FDA activities that affect children and the practice of pediatrics. As discussed further in Chapter 2, the Medical Device User Fee Amendments of 2002 directed the agency to develop guidance on the assessment of medical devices used with children and to make pediatric expertise available when issues involving children arise or may arise, for example, in the review of
medical device applications or studies of medical devices. In the Medical Devices Technical Corrections Act of 2004, legislators directed FDA to report on “barriers to the availability of devices intended for the treatment or diagnosis of diseases and conditions that affect children” (P.L. 108–214, Section 3). That report was issued by FDA in fall 2004 (see Chapter 7 for a summary).
MEDICAL DEVICE REGULATION IN CONTEXT
As suggested above, the extension of FDA’s regulatory authority over devices was a response to the increasing complexity and sophistication of medical devices. This increase has many causes, including the stimulus to scientific and engineering innovation provided by World War II, the post-war acceleration of public and private investments in biomedical and bioengineering research, the growth of academic medical centers, and the expansion of public and private health plans that pay for medical treatments.
Today’s medical devices constitute an extremely varied category of medical products—some as simple and low risk as an infant cap, others as complex and high risk as a cardiac pacemaker. The medical device industry likewise is quite variable, as described in Appendix C. It includes small firms with a single product (or variants on a core product) and large companies with diverse product portfolios and substantial resources to devote to research and development, marketing, and government relations. Compared to the pharmaceutical industry, the device industry includes a larger proportion of small firms, and patents are less important as a source of competitive advantage. In this environment, few incentives may be present to develop, modify, and test medical devices to meet the special needs and characteristics of children, given that children constitute a relatively small market for most complex medical devices. (For further discussion, see FDA, 2004y.)
Differences between drugs and devices extend to clinical research. The classic models for medical product research were developed with drugs, not medical devices, in mind. The conventional distinctions between Phase I, II, and III clinical trials for drugs do not readily fit device trials. Instead, discussions of device trials sometimes differentiate between feasibility or pilot studies and pivotal studies.8 As described above, clinical testing is not required for Class I and most Class II devices.
Compared to the drug industry, the product development cycle in the device industry tends to involve a more continuous process of refinement and innovation. Instead of single molecules (and a relatively small array of formulation options such as pills or liquids), devices typically involve a number of components or features that change as a result of minor or major alterations in design, materials, manufacturing processes, or other characteristics.9
As illustrated in Figure 1.3, the movement from product concept to approved product typically takes many years as is also true for drugs. For both drugs and devices, the development process sometimes ends with the abandonment of a product that fails to prove itself technically, clinically, or competitively. To the extent that drugs and devices differ, the differences may warrant adaptations in regulatory and evaluation strategies. Still, the public should expect medical devices to be safe and effective and should expect FDA to fulfill its responsibilities in this regard.
In recent years, CDRH has applied a model of the medical device product cycle to guide the conceptualization and evaluation of its procedures for device evaluation and regulation (Feigal, 2002). As shown in Figure 1.4, the cycle begins with a concept followed by initial development and testing, a phase that includes consultation with FDA about the evaluative methods and information needed to support approval or clearance of the device. The cycle continues through FDA approval or clearance (if the evidence warrants) and then moves on to product marketing and commercial use. Typically, problems with a device or continued innovation and improvement lead to its eventual departure from the market, although not necessarily from clinical use or FDA surveillance. As noted above, some
|
|
randomly assigned to receive the experimental drug or a standard treatment or a placebo. (Information collection activities conducted after marketing approval are sometimes referred to as Phase IV studies or trials. Such studies are often much less rigorous and may be intended more to achieve market awareness than to build scientific knowledge.) This phase classification of clinical trials is not routinely applied to device studies. More commonly, the earliest device investigations using human subjects are termed pilot or feasibility studies, and subsequent studies are referred to as pivotal studies. Pilot or feasibility studies with a small number of human subjects provide an initial clinical assessment of device safety, an opportunity to modify the prototype device to improve performance, and, sometimes, a period of important learning about the technical process and skills required to use the device safely and effectively. Pilot studies also provide experience and information useful in designing so-called pivotal studies, which recruit larger numbers of research participants and often involve multiple study sites and centers. |
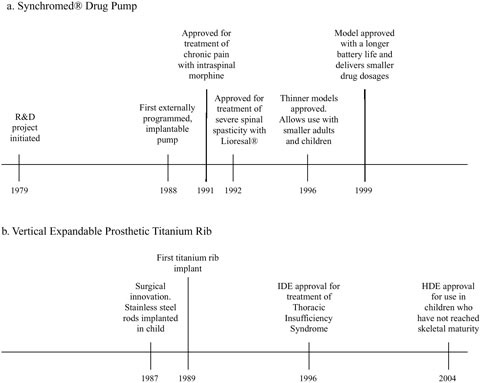
FIGURE 1.3 Time lines of key dates in development of the Synchromed® drug pump and the Vertical Expandable Prosthetic Titanium Rib (VEPTR) (Doctor’s Guide, 1996; P860004-S042, FDA, 1999; H030009, FDA, 2004a; Sansom, 2004; personal communication, Paul Citron, Committee Member, November 16, 2004).
implants may remain in a patient indefinitely, and other devices may survive in hospitals, nursing homes, or home use long after better devices or other therapies have become available.
One theme of this report is that government regulation of medical devices cannot by itself safeguard children or adults who use medical devices. The availability of safe and effective medical devices and their safe and effective use depends on the knowledge, skill, creativity, and integrity of many individuals and organizations on the frontlines of clinical care and device development and production.
At the same time that attention is paid to these individuals and organizations in their own right, policymakers need to view these actors as parts of a health care system and market for medical services and products that are characterized by a strikingly complex set of structures, policies, processes, resources, ethical values, and incentives that interact in ways that are often difficult to anticipate. Thus, those creating or modifying regulatory
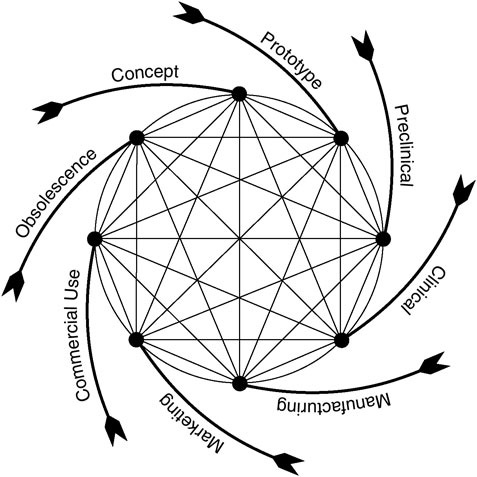
FIGURE 1.4 Total product life cycle for medical devices (Feigal, 2002).
and other public policies should consider how such interactions may support or compromise the achievement of social and policy goals. That requires looking beyond FDA policies to, for example, the research priorities of NIH, the coverage and reimbursement policies of Medicare and other health plans, and the patient safety initiatives of public agencies such as the Agency for Healthcare Research and Quality and private agencies such as the Joint Commission on the Accreditation of Healthcare Organizations. It also requires an understanding of the processes of device innovation and the characteristics of the device industry, and an appreciation of the potential for policies to have unintended and unwanted effects.
The broad goal of applying a systems perspective to health care is to improve the quality of care by understanding features of organizational and social systems that help individuals and groups perform correctly and con-
sistently to achieve desired results. In the context of medical device safety and postmarket surveillance, a systems perspective also means looking beyond individual errors with medical devices to identify the human factors and system characteristics that contribute to such errors.
Another dimension of improving the quality of care involves strengthening the evidence base for clinical practice and the translation of that science base into guidelines and other tools, processes, or systems that successfully influence practice. Many common medical practices—for example, a range of unlabeled uses of drugs and medical devices with children—have not been subjected to systematic clinical investigation to document their safety and effectiveness in practice.
In response to the particular shortfalls in the knowledge base for pediatric care and in the availability of medical products evaluated for use with children, Congress, FDA, NIH, and others have sought to create a mix of incentives and requirements to expand pediatric research and reduce barriers to the development of drugs, devices, vaccines, and other medical services that improve children’s health and well-being. The next chapter describes why children’s needs and characteristics warrant special attention and pose challenges to those developing, evaluating, and monitoring medical devices.





























