
C
The Dynamics of Pediatric Device Innovation: Putting Evidence in Context
Annetine C. Gelijns, Ph.D., Brigid Killelea, M.D., Michael Vitale, M.D., Vipul Mankad, Alan Moskowitz, M.D.*
INTRODUCTION
There are several compelling reasons to study the dynamics of device innovation and evaluation in pediatrics. First, this innovation process has been much less studied than its counterparts in pharmaceuticals or biotechnology, although devices address many of the same critical clinical needs as drugs. Yet, there are important differences between pharmaceutical and device innovation, such as the structure of the industry, the role and importance of academic medical centers, and the degree of ongoing, incremental innovation that is the hallmark of the device evolution process. In device innovation, not only does the device undergo incremental change, but there is also substantial change in the ways in which device recipients are managed, including implantation/insertion procedures, their hospital care, and outpatient management. Such changes occur both in the pre- and postmarket settings, which raise challenges for the evaluative enterprise.
A second reason to study the device innovation process is that there are important unmet needs for novel or improved pediatric devices. These needs differ in many ways from those of the adult population. For example, children need smaller devices; a growing number of congenital heart defects require heart valves and occlusion devices sized appropri-
ately for infants (AAP et al., 2004). Physiologic differences also play a role; children have higher heart rates, and this may reflect itself in more rapid calcification of prosthetic valves, which affect the materials used in these products. Children also have different inspiratory flow rates and different abilities to coordinate manual tasks. For example, devices for inhaled administration of drugs need to accommodate timing the activation of an inhaler with the child’s intake of air. Lifestyle differences also play an important role for designing devices for children. For example, cochlear implants have been known to deprogram in response to contact with plastic playground slides (see Appendix F). Since children’s life spans are longer, devices that are left in place for long periods of time, such as cranio-facial prostheses, may show increased rates of polymer plate absorption. On the other hand, devices may need to be replaced more frequently as they wear out; growing children may require larger devices as they age (AAP et al., 2004).
A third reason to study this topic is that the evaluation of pediatric devices offers unique challenges. Similar to devices for use in adults, devices for use with children are subject to U.S. Food and Drug Administration (FDA) regulatory requirements. When FDA clearance or approval of a device requires clinical data, trials may provide important information on safety and efficacy. However, even the most rigorous trials have inherent limitations in terms of measuring long-term outcomes (especially in younger patients) and the ultimate generalizability of their findings. Given the small patient populations for many pediatric devices, premarket studies are often single-arm studies or registries as part of a Humanitarian Device Exemption (HDE) approval process, which may lead to increased uncertainty in terms of the knowledge gained. The incremental nature of the device innovation process and the inherent limitations of premarket studies argue for ongoing monitoring and evaluation of the outcomes in widespread clinical use. Ongoing evaluation is costly, however, and of particular concern in the realm of pediatric medical devices, where the markets are small and the economic incentives to innovate weak.
This paper explores these issues in pediatric devices, and, among the broad spectrum of devices that range from tongue blades to imaging machines, focuses specifically on the more technologically sophisticated end of the spectrum. It first reviews the main players in device innovation and the policy environment in which they operate. It then analyzes the dynamics of pediatric device innovation and evaluation, and the challenges inherent in designing and conducting premarket and postmarket clinical trials. The paper concludes with some observations about possible analytical, institutional, and economic solutions to improving the knowledge base about devices, while at the same time, fostering much needed innovation in this area.
THE PLAYERS IN MEDICAL DEVICE INNOVATION
In today’s knowledge-based economy, medical device innovations arise within a complex network of public and private sector institutions, including universities, national laboratories, and industrial firms (Gelijns and Thier, 2002). These institutions operate in an environment increasingly affected by governmental rules and incentives, which, in turn, shape the interactions among these institutions.
The Medical Device Industry and Its Markets
The Second World War brought about a fundamental transformation of the medical device industry. Wartime research stimulated many broad advances in science and engineering, such as microwaves, radar, ultrasound, and new materials. In the late 1940s, the transistor was invented—ushering in the era of low-power electronics and microelectronics (Gelijns and Rosenberg, 1998). These would prove to be of great benefit to the development of medical devices and had much to do with the post-war growth of the industry. Over the past few decades, the medical device industry has expanded dramatically. By 1992 there were roughly 1,700 different types (not including model variations) of medical devices, developed by approximately 10,000 to 11,000 device firms, either domestic or foreign, operating in the United States (FDA, 1992). By 2001, the number of firms had increased to about 16,000. These firms introduce over 7,500 new and modified products into health care annually; the vast majority of these, however, fall into low-tech medical equipment categories (Feigal, 2003).
The medical device industry is a highly fragmented industry consisting of start-ups and giant corporations. In 2001, 67 percent of publicly traded medical device firms had fewer than 20 employees, and only 6 percent had more than 500 (U.S. Department of Commerce as referenced in AdvaMed, 2004). The role played by firms of different size on the medical device industry is not entirely clear, although it is often asserted that small firms play a disproportionately large role in making the initial innovations (Lewin Group, 2000). These companies are research focused, specializing in the “front end” of R&D. Perhaps not surprisingly, a study by the Wilkerson Group concluded that “nearly all significant new and innovative products and procedures were pioneered by start-up companies.” Indeed, in their survey they cite 29 major advances in therapy, all of which are attributed to start-ups (Wilkerson Group, 1995).
By comparison, large firms are crucial in determining the eventual commercial success of new devices (NAE, 2003). Their organizational assets include the following. First, they have an ability to navigate the com-
plex regulatory requirements surrounding the introduction of new products into health care. Firms build skills in managing clinical trials and serving the needs of regulatory agencies over time through multiple product filings. Second, large companies often have considerable skills in manufacturing and marketing. First-mover advantages are not always of primary importance to eventual success in the marketplace when new technologies possess certain significant commonalities with earlier ones (e.g., magnetic resonance imaging [MRI] with computer tomography [CT] scanning). Although the large multinational firms often entered the MRI field late, they quickly traversed the ground already covered by pioneering small firms, and asserted their skills in marketing and servicing, as well as their already established reputations in the CT field, to assume dominant positions in MRI (Gelijns and Rosenberg, 1998; Trajtenberg, 1990). Third, these companies understand the purchasing patterns of multiple stakeholders in a complex hospital environment. Hospitals prefer to contract with a limited number of suppliers, and so device companies need to offer a full product line of compatible products to meet buyers’ needs. As such, large companies have a clear advantage over small companies who may manufacture only one device.
The global medical devices market has more than doubled since 1991, when the worldwide market was about $70 billion (The Lewin Group), to $169 billion in 2000 (AdvaMed, 2004). The United States market accounted for 43 percent of the 2001 total.1 Although it has been declining in recent years, the United States has long run a positive balance of trade in medical device categories (in 2002 this surplus averaged about $3.3 billion; U.S. International Trade Commission). The market can be subdivided into various broad segments (Wilkerson, 1995). These include specialty devices and implantable products, medical and surgical supplies, imaging systems and other equipment (such as patient telemetry monitoring), in vitro diagnostics, and, the more recently expanding, health information systems segment. According to current estimates, the demand for medical devices is expected to grow, with the domestic demand for implantable devices expected to exceed $24 billion by 2007, up from $8.7 billion in 1997 (Freedonia Group, 2003). But within these apparently large markets, companies capture relatively few sales from any single product. Even “blockbuster” products in this industry rarely exceed $100 million. The medical device industry is very dynamic, characterized by short product life cycles.
Patents provide less protection than in pharmaceuticals, and competitors are able to invent around existing patents. Consequently, research activity is intense, with 11.5 percent of sales invested in R&D for all device firms (Figure C.1), and probably up to 18 percent of sales reinvested in R&D by the more innovative firms, a figure comparable with that of pharmaceutical companies (AdvaMed, 2004).
There is a paucity of data on the universe of medical device firms manufacturing pediatric devices and the size of these markets. Children represent about 30 percent of the population and less than 12 percent of personal health care spending (NACHRI, 2001). Looking at the total number of hospitalizations in 2000, there were 36,417,565 for adults and 3,501,901 for children other than normal newborns—about 10 percent of the adult population (NACHRI, 2001). Of course, the spectrum of hospitalizations in children differs from adults, and may not involve therapeutic devices in the same proportion as in adult hospitalizations. Indeed, pediatric medical device markets are typically very small. For example, the market for left ventricular assist devices (LVADs) for children over 6 years of age is expected to be around 150–200 devices annually.2
Universities and Their Academic Health Centers
The private sector relies heavily upon an infrastructure of institutions and research activities within the academic realm (Gelijns and Rosenberg, 1999). Research universities are key players in the medical device innovation system, with advances in physics, materials sciences, optics, analytical methods, and computer science often being generated in departments of physics, chemistry, computer sciences, or engineering schools. Moreover, in recent decades bioengineering research has emerged as a separate discipline, with 70 universities and colleges offering bioengineering degrees in 1998 (NAE, 2003).
Within universities, academic medical centers (AMCs) play a particularly important role in the development of medical devices. Around the country, academic medical centers may have a slightly different organizational structure, but they are generally comprised of a medical school, its teaching hospital, a potential network of affiliated hospitals, and a nursing school. An academic medical center may also involve a school of dentistry, a school for allied health professionals, and a school of public health. These complex, multifunctional organizations have a three-pronged mission: (1) they train clinicians and biomedical researchers and, thereby, shape the distribution of medical skills and specialties; (2) they provide advanced
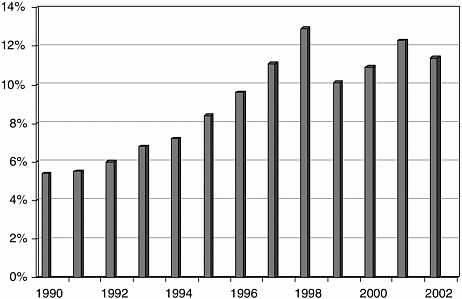
FIGURE C.1 Medical device R&D spending as a percent of sales.
SOURCE: S&Ps Compustat. Data from publicly traded companies. From AdvaMed (2004).
specialty and tertiary care, and as such are early adopters of the “latest” in technology. With a large patient population, a wide referral base, and a research-oriented attending staff, academic medical centers also attract patients with rare diseases, making them an ideal place to develop and test new devices and orphan drugs; and (3) they conduct a whole range of biomedical research activities, ranging from laboratory-based fundamental research to population-based clinical studies. Such evaluative studies may yield clinical data that may provide support for FDA approval, coverage by payers, and postmarket surveillance of pediatric devices. There are around 250 children’s hospitals in the United States, which account for a substantial percentage of all National Institutes of Health (NIH)-funded pediatric research (NACHRI, 2001). Almost all of these centers participate in clinical trials and health-related research, and about one-third of these centers have child health research centers where interdisciplinary research is conducted (NACHRI, 2001).
In the United States, AMCs, and basic biomedical research in particular, have been major beneficiaries of post-war science policy. The research budget of NIH was nearing $30 billion in 2003, and nearly 80 percent of its extramural monies are spent in AMCs, providing strong support for their research enterprise.3 At the same time that biomedical research spending
|
3 |
Award data available at: http://grants.nih.gov/grants/award/award.htm. Accessed 3/9/2005. |
increased significantly in the post-war years, health insurance coverage of the American people expanded, which has paid AMCs handsomely for their patient care and educational activities. There is a close interdependence, as we will explore, between AMCs and device firms throughout the different stages of the innovation process. These close university–industry interactions were also fostered by public policy, such as the creation of the Bayh Dole Act in 1980 (P.L. 96–517), which provided incentives to patent and license the findings of publicly funded research.
THE POLICY ENVIRONMENT
The government plays a multifaceted role in the device innovation process. It supports medical R&D, regulates the marketing approval of new devices and provides postmarket oversight. It also pays for clinical interventions through Medicare, Medicaid, and numerous other benefit programs.
Federal Support for Research and Development
Federal support for R&D in medical devices flows through multiple institutional and disciplinary channels. Although the majority of medical device-related R&D funds are spent in AMCs, federal agencies also fund basic and applied research in academic science departments and engineering schools, federal laboratories, and industry proper.
Compared to other countries that are members of the Organisation for Economic Co-operation and Development, the United States spends the largest amount of its overall research budget in the life sciences (over 50 percent). The largest single medical research funding agency is the NIH, and most of its nearly $30 billion budget is spent on extramural research in AMCs, particularly in non-human bench research. In 2003, NIH funding of children’s hospitals and pediatric departments was approximately $961 million.
Only a small portion of the NIH budget is used to create opportunities for the development of devices. In 2000, NIH created the National Institute for Biomedical Imaging and Bioengineering (NIBIB) “to improve health by promoting fundamental discoveries, design and development, and translation and assessment of technological capabilities” (NIBIB, 2005). NIBIB’s fiscal year 2004 budget was over $288 million (DHHS, 2004).
The pre-existing NIH institutes also have device R&D programs, including programs specifically for children. A case in point is the National Heart, Lung, and Blood Institute (NHLBI). The NHLBI created the artificial heart program to support the development of a family of devices to assist the failing heart and to rehabilitate patients with heart failure. In 2003, the NHLBI requested proposals to develop novel circulatory assist
devices for infants and children. This followed the recommendations of a NHLBI task force to invest more funds in the area of pediatric cardiovascular disease (NHLBI, 2002). They also recommended establishing a clinical trials network to implement multi-center, randomized studies for assessing new and emerging therapies. Part of the network’s charge is to create registries for the longitudinal follow-up of children, which would address late risk of specific heart defects and treatments.
In 2000, Congress passed the Children’s Health Act of 2000 (P.L. 106–310), which created the Pediatric Research Initiative to be housed within the Office of the Director of NIH. The objectives were to increase support for pediatric research, strengthen collaborative efforts among institutes and centers, and speed development of pediatric clinical drug trials. No new funds were appropriated by Congress, but the Office of the Director allocated $5 million for the initiative from its discretionary account for one year only (FY2002). (According to an analysis by Gitterman and colleagues, between 1998 and 2005 when the NIH budget doubled, pediatric spending by NIH increased by an average annual rate of 12.8 percent compared to an overall NIH average growth rate of 14.7 percent. The proportion of the total NIH budget devoted to the pediatric portfolio declined from 12.3 percent to 11.3 percent.)
Regulation
The FDA is charged with the formidable task of approving medical products as they enter the market and regulating devices in the postmarket period. Broadly speaking, the Medical Device Amendments of 1976 expanded the FDA’s responsibility in this field, with the intention to ensure that new devices brought to market were both safe and effective. It divided medical devices into three classes, with high-risk devices (i.e., devices that are life-supporting or sustaining, that are of substantial importance in preventing impairment of health, or that have a potential for causing risk of injury or illness) being grouped in Class III. These Class III devices, about 10 percent of all devices, must demonstrate safety and efficacy in clinical trials before the FDA grants marketing approval.4
FDA regulation of medical devices has been undergoing significant change. The introduction of the Safe Medical Devices Act of 1990, for example, established new requirements for premarket, as well as postmarket studies. As far as premarket studies are concerned, device manufacturers are now required to conduct more rigorous studies with appropriate, and where possible, randomized controls. In the area of postmarket surveillance, a number of separate mechanisms exist for collecting data. Device manufacturers as well as health care providers must submit adverse event reports to FDA if a device may have caused or contributed to a death or serious injury. In the case of high-risk devices, companies must keep track of patients, and, in certain cases, the FDA requires post-approval clinical studies to detect possible risks associated with device use, as well as to provide information on effectiveness. The changes incorporated in the Safe Medical Devices Act should encourage higher quality device evaluations and provide more useful information about safety and efficacy. At the same time, some of these changes also have important consequences for the timely introduction of new medical devices to market, and for the degree of financial risk and costs associated with the innovation process.5
For some Class III devices, especially those targeted to a narrow market such as is often the case in pediatrics, gathering sufficient evidence for premarket approval may be a lengthy and/or risky process because of the small number of affected patients. Therefore, for diseases that affect fewer than 4,000 people per year in the United States, the FDA may grant a Humanitarian Device Exemption. An approved HDE allows for marketing of these devices without the rigorous clinical and non-clinical PMA effectiveness requirements, provided that there is no alternative device, the device would otherwise not be available, and the manufacturer shows that reasonable safety and probable benefit to the patient outweighs the risk of injury or illness. An approved HDE authorizes limited marketing of a Humanitarian Use Device (HUD) (FDA, 2001). There have been 130 HDE applications since the inception of the HDE program, but only 30 have been approved. Manufacturers are not allowed to make profits on these devices; they may
|
|
To support marketing approval decision for a Class II device, or in some instances a 510(k) submission (about 10 percent of submissions), a sponsor must conduct clinical studies. If a device poses a significant risk (Class III), the sponsor must submit a request for an Investigational Device Exemption (IDE) to the FDA. Following clinical studies, a device may be approved for marketing through a so-called premarket approval decision (PMA). |
only recover the costs of production. Currently, there are six active HDEs for pediatrics, including the LVAD, a heart valve, bladder stimulator, and fetal bladder stent, gastric stimulator, and cultured skin. Table C.1 list some representative pediatric devices approved for marketing or as part of an HDE.
TABLE C.1 Some Recently Approved Devices Tested for Use in Childrena
Reimbursement
Decisions about medical coverage and reimbursement for use of pediatric devices are made primarily by individual state Medicaid programs and by commercial health insurance companies. These decisions influence not only the quality of health care for children, but also the innovation process, marketing, and availability of devices for children. Absence of or insufficient reimbursement for a pediatric procedure or the costs of devices used during the procedure is often a significant barrier in development of a new pediatric device.
Within the Medicaid program, each state may develop its own benefit package (covering services that are deemed “reasonable and necessary”) and set reimbursement rates within the broad framework of the 1965 Social Security Legislation and federal regulations. In doing so, some states use coverage decisions made by the Medicare program, while others have their Medical Directors make state-specific policies for coverage or use guidelines developed by commercial insurance companies in their regions. Until recently, few commercial payers (with some notable exceptions such as the Blue Cross and Blue Shield process) provided clear descriptions in their contracts of how coverage decisions were made, and there was no systematic process for gathering input from clinicians and consumers. In recent years, however, commercial payers are increasingly basing their coverage decisions on both clinical and economic evidence.
Following a coverage decision, billing codes are a necessary, critical pathway for reimbursement of services and procedures and the use of pediatric or other devices. The coding system, which was primarily designed for general use, often doesn’t contain specific codes for pediatric devices, which may have different development and production costs than adult devices. This increases the risk of inadequate reimbursement for pediatric devices, which is a disincentive for innovation. Until recently, the coding revision and coverage decision process did not have input from Medicaid programs. The newly created system of public consultations to inform this process will also need to include pediatric providers, advocates for children with device needs, and device manufacturers to ensure that information that could affect pricing is taken into consideration.
Finally, if a code exists or is created for devices used with children, decisions by payers to set a reimbursement level vary and are subject to political pressures and negotiations. A recent Centers for Medicare and Medicaid Services (CMS) decision expanded the coverage of an implantable cardioverter–defibrillator (ICD) coupled with a requirement for development of additional evidence through large-scale, prospective, observational studies or registries (McClellan, 2005). Further characterization of the subgroups for which the device would be beneficial and of the real-world
clinical outcomes will add further information to allow the CMS to use public funds more effectively. A challenging task would be to link reimbursement decisions to postmarket surveillance of pediatric devices since it will require coordination of multiple state-operated Medicaid programs and commercial payers.
THE DYNAMICS OF DEVICE INNOVATION
The device innovation cycle can be seen to consist of various, partly overlapping, stages. These stages include research in the physical sciences and engineering, human physiology and pathophysiology; the development of novel product ideas, device prototypes, and manufacturing methods; the evaluation of devices in animals and humans; the modification of existing products; and the discovery of new indications for use. Generally, the development of a new device prototype is driven by the conjoining of new technological capabilities with the perception of unmet clinical needs.
Because the markets for medical devices are often fragmented and relatively small, the medical device industry has historically not invested heavily in basic research. In fact, the medical device industry often has been heavily dependent on scientific and technological capabilities that have been developed in other sectors of the economy. Medical device innovation exploits research and new technological capabilities and components that have been developed by universities, the military, the electronics industry, and a range of firms manufacturing essential, specialized materials, such as high-quality glass for fiber optics or inert materials for prosthetic devices. In addition to these developing technological capabilities, new insights into human physiology and pathophysiology have become increasingly central to the development of new devices. Understanding the electrophysiology of the heart, for example, has been critical for designing a pacemaker or implantable defibrillator, as is circulatory physiology for designing artificial hearts and circulatory assist devices. Similarly, renal physiology has been crucial in elucidating the pharmacodynamics and pharmacokinetics of hemodialysis, which, in turn, has contributed to the development of improved dialysis machines. These insights often have been generated in academic medical centers.
Development of Device Prototypes
The development of a new device prototype can occur either in industry or in a clinical setting, most often an academic medical center. Clinicians and academic researchers not only identify the need for a new device or for improvements in existing devices, but they may also be the innovators and builders of the original prototype because of their role as sophisticated
users. The importance of academic faculty in the development of device prototypes has been documented for a whole range of devices, such as the automated clinical chemical analyzer, renal dialysis machines, intrauterine devices, catheters, laparoscopes, fiber-optic endoscopes, and MRI machines (von Hippel and Finkelstein, 1979; Shaw, 1987; Gelijns, 1991; Gelijns and Rosenberg, 1995, 1998).
In the pediatrics realm, the Vertical Expandable Prosthetic Titanium Rib (VEPTR) is a recent device created by a practicing orthopedic surgeon. Dr. Robert Campbell recognized the need to insert a rib spacer to avert respiratory failure and early death in children with thoracic insufficiency syndrome from spinal and chest wall deformities (FDA, 2004a; Sansom, 2004). The device, which is curved, is inserted between ribs and allows the lungs to expand to a larger thoracic volume. As the child grows at roughly 6-month intervals, the device can be expanded through a simple outpatient surgery. The titanium rib has been implanted in children as young as 6 months of age, and is ideally left in place until the respiratory system is close to maturity. This surgically implantable device, which has been used under an HDE status, gained FDA approval in 2004 after it was used in approximately 250 children.
If initially developed in academia, innovators often discover that they are unable to advance the project because critical, enabling technologies are missing or are too technologically specialized to develop within the laboratory or elsewhere in the university. It is at this point that a partnership is often formed between the academic researcher and an industrial firm, which has the applicable technological expertise and interest in the proposed application.
Most devices are evaluated in animals and undergo bench testing before they are used in clinical testing. The reliability of the pediatric LVAD, for example, is generally tested in mock loop systems, and pre-clinical testing of the device, depending on the age of the recipient, can be done in dwarf pigs or calves.
Premarket Clinical Evaluation
Following bench and animal testing, some Class II and all Class III devices enter the stage of human evaluation. The clinical data generated by such testing form the basis for obtaining FDA premarket and payer coverage approval, and thereby lead to widespread market access. A small percentage of devices (i.e., Class III devices and a small subset of 510(k) devices) require rigorous safety and efficacy evaluation. As a result, a modest but growing number of devices undergo randomized, controlled trials or other prospective comparative studies. In recent years, clinical trial spending on devices by industrial firms has been growing substantially.
Clinical Trials Organizational Infrastructure
One can divide up the clinical trials infrastructure into organizations that coordinate the activities of clinical trials and those that actually provide the patient care, that is, clinical sites that conduct clinical trials. Device manufacturers, academic medical centers, and contract research organizations (CROs) all are engaged in clinical trials coordination, and each offers different strengths to the process. Device manufacturers have the greatest familiarity with the engineering and scientific principles of the devices, but few firms are large enough to have the personnel to design, conduct, and analyze trials. Moreover, they have financial interest in the outcome of studies, which might influence the credibility of the results. Academic medical center faculty has also been involved in the design, conduct, and analysis of clinical trials. However, during the last decade, contract research organizations have captured part of this market (Moskowitz, 2003). CROs are private, for-profit organizations that are engaged in the management of clinical trials, including protocol design, patient recruitment, data collection, data management, monitoring, and analysis. CRO usage among the medical device industry is not common; only 13 percent of medical device firms employ CROs (whereas 90 percent of drug firms use CROs), and few CROs have extensive experience conducting implantable device trials (Centerwatch, 2005).
Academic medical centers, community-based hospitals, and practices can be venues to enroll and treat patients, and collect data on their outcomes. Academic medical centers have traditionally been involved with the testing of prototype devices and have served as the source of patients for more involved clinical trials. In particular, they are the venue of care for more complex patients, which is common among patients who need implantable devices and invasive procedures.
Clinical Trial Design and Its Challenges
There is a spectrum of research designs employed in testing pediatric devices, ranging from randomized comparative trials to single-arm studies with historical controls. The first stage of clinical testing is usually a single-arm, prospective study in a diseased population to assess the feasibility of use in humans. These studies are generally small in size, and they provide only preliminary evidence about short-term safety and efficacy. The major value of these studies is that they offer prima facie evidence for designing larger-scale, pivotal trials. Optimally, these pivotal trials would be randomized or otherwise appropriately controlled studies and would incorporate a spectrum of endpoints. Over the past 20 years, there has been a transition from intermediate, physiological endpoints to more clinically relevant pa-
tient outcomes, such as survival, functional capacity, and quality of life. The advantage of the trend toward more clinically relevant outcome measures is that clinical and economic decision making is enhanced; the drawback is that it generally takes longer periods of observation to achieve these measures of outcome.
In comparison to pharmaceutical trials, device trials must contend with unique ethical, logistical, and methodological challenges. A major constraint in designing pediatric trials can be found in patient recruitment. In general, pediatric diseases are low prevalence, affecting the ability of researchers to recruit a sufficient number of participants in a reasonable period of time. Given the small market potential for many pediatric devices, there is already little economic incentive to pursue the development of new pediatric devices. This is compounded by the increased development costs associated with having to conduct comparative trials with large sample sizes, which would require a greater number of clinical centers or a longer enrollment period to capture the needed trial population. When randomized or other prospectively controlled studies are not feasible within a reasonable time frame, the FDA allows single-arm studies that use objective performance criteria derived from prior studies. Alternatively, they may allow comparative studies with a greater chance of a random variation error or, in other words, accept a higher p-value to establish statistical significance.
Another challenge, in comparison to pharmaceutical trials, is choosing the right time to take a device into the pivotal trial stage. A pharmaceutical compound generally does not undergo substantial change as it progresses through the various phases of clinical trials, except that development may be discontinued if the profile is undesirable, and a modified compound may enter pre-clinical and phase I trials. Devices, however, undergo extensive modifications and refinements during the clinical evaluation stages. These modifications are not only in the device itself, but also in the clinical management of a patient with a device. A case in point is the REMATCH trial, which evaluated the efficacy and safety of a left ventricular assist device for long-term therapy of end-stage heart failure patients. During this trial, various changes were implemented, such as a modification of the driveline, the introduction of a locking screw ring to prevent detachment of the blood-transport conduits to and from the pump, and a clinical protocol to better prevent and manage driveline infections with antimicrobial agents and laminar flow operating rooms. Such modifications in the device or clinical management can be accommodated in the design of clinical trials. In the REMATCH trial, for example, there was no change in the predetermined sample size (Rose et al., 2001). If, however, the device design or clinical management change offers a substantial change in the measures of outcome, additional patients may need to be recruited to accommodate specific subgroup analyses.
Once the optimal time to begin a pivotal clinical trial is established, decisions concerning which venue and which clinicians to engage in testing a particular device can have a major effect on how the results of the trial will be interpreted and whether the device achieves broader usage. In contrast to pharmaceuticals, the efficacy of a surgically implanted device can be linked to the skill of the implanting surgeon. If there is substantial variation in skill among trial investigators, the results of the trial may be difficult to interpret. A positive average outcome for the experimental therapy may only be positive because of a few exceptionally well-skilled clinical sites, and a negative average outcome may only be negative because of a few, poorly skilled clinical sites. Clinical researchers must be on the guard for such outcomes. Trials typically offer a separate randomization scheme for each clinical site so that each site is balanced with respect to the number of experimental and control patients that they treat. Moreover, examining the effect of study site on the primary outcome is a routinely performed analytical step. One strategy to assure uniformity of skill is to engage in a pilot trial or run-in period, which will not be counted in the final analysis. Another is to limit participation to individuals that have a particular skill level. Conducting a trial in a highly specialized center with unique surgical expertise may result in a successful trial, but may not be generalizable to widespread usage or provide useful information on the economic value of using the device in less specialized centers.
Pediatric patients are considered a vulnerable population in the context of conducting clinical research. Research that presents more than a minimal risk without the chance of direct benefit to the child is unlikely to be approved by an Institutional Review Board (IRB). Those trials that are approved involve special considerations regarding the informed consent process. Only the parent or legal guardian has legal standing for signing a statement of informed consent for a minor (the age cut-off varies by state). However, older children, who have the capacity to understand the activities involved in trial participation, need to give their assent for the participation.
Blinding, an important technique for controlling observational bias when evaluating the safety and efficacy of a new clinical intervention, is an issue in invasive or implantable device trials. Obviously, it is impossible for the clinician that implants a device to be blinded. Patient blinding is usually not possible when the comparative therapy is not a device. Thus, randomization is more of a problem in device trials because of the lack of blinding. This is true in particular when there is a life-threatening illness. Here both patient (their family) and physician will have expectations that the device is their best hope and would be devastated to learn, up front, that they would not receive the preferred therapy. This could deter some patients and physicians from entering into a device trial, while others might enroll but seek treatment outside the protocol if they didn’t receive the therapy they wanted.
This might lead to a loss-to-follow-up or out-of-protocol crossover, which could ruin a small-scale trial. The ethical dilemma here is heightened when there are no alternative therapies and assignment to a control arm means essentially no therapy (Moskowitz et al., 1997).
Measuring survival in trials that compare devices and medical therapies poses methodological challenges. When device therapy involves a high upfront operative risk, but subsequently a reduced mortality compared to the control group, the survival curves are likely to cross. Analyzing the differences between such curves depends on the analytical method chosen and the time frame of the analysis. Most analytical methods (e.g., log-rank, Wilcoxon test) average risk over the follow-up period. So, extending or reducing the follow-up time has the potential to reverse the ordering of relative efficacy because less or more weight, respectively, will be given to the mortality in the peri-operative period (Rose et al., 1999).
Measuring the effects of treatment on children’s quality of life is a more complex task than for adults, largely because children are developing (Rosenbaum and Saigal, 1996). Any assessment of functional status must be performed in a developmental context. Key aspects of quality of life (such as physical, emotional, and social function) develop rapidly as the child ages, which means that a group of questionnaires must be developed that are specific for an age group. Similarly, age-adjusted normative values are needed to put the measured values in the context of the general population. For younger children at an earlier stage of intellectual development, investigators must rely on parents or caretakers to act as proxies for direct patient-based responses. Despite these challenges, there has been considerable progress in the field of quality of life assessment for children, with new survey instruments, both generic and disease-specific, being developed and validated in a range of pediatric conditions (Drotar, 1998; Eiser and Morse, 2001; Koot and Wallander, 2001).
Assessment of the economic value of devices is challenging in pediatric populations for many of the same reasons that it is difficult to assess clinical outcomes. The small patient populations that frequently result in single-arm studies or registries hamper not only the identification of treatment effects, but also leave the assessment of cost-effectiveness without a comparator. The short-term nature of many randomized trials makes it difficult to accurately assess the long-term economic impact of treating a particular patient population for the individual payer. Economic analyses typically use outcomes such as costs per life year saved or quality-adjusted life-year (QALY) saved, which require survival projections. In the case of children with long life expectancies, these projections are more difficult to make and, consequently, involve greater uncertainty. Moreover, the tendency to conduct pediatric device trials in highly specialized treatment centers makes it difficult to infer the economic value of the use of devices in less special-
ized centers. In recent years, there has been an increase in economic evaluations for pediatric populations. Ungar and Santos (2003) created a pediatric economic database and documented a 7-fold increase in publications between 1980 to 1984 and 1995 to 1999. Currently, the database contains over 1,000 citations of full economic evaluations from January 1980 to the end of December 2003. However, searching the database for device-related economic evaluations, we found only 30 citations analyzing 11 different device categories (e.g., cochlear implants, amplatzer catherization techniques for occlusion of atrial septal defects, and laparoscopic spelenectomy). Most of these evaluations were conducted in the last 5 years, indicating an emerging trend.
In short, rigorous trials can provide important evidence about the efficacy, safety and—more recently—the economic impact of new pediatric devices. Regulatory decisions then have to combine this empirical evidence with qualitative judgments about the acceptability of the trade offs between benefits and risks associated with new technologies, and payers have to combine the empirical evidence with qualitative judgments about the acceptability of the trade-offs between benefits and costs. Such trade-offs depend upon the disease context and available alternatives, the preferences for the outcomes at stake, and the amount of uncertainty in achieving them. Given that premarket trials are based on a sampling process, there will always be uncertainties. Attempting to eradicate uncertainty is impossible, and attempts to bring the level of uncertainty down to minute levels would be costly in terms of the time and expense of the premarket development process, as well as the indirect expense of holding off a promising therapy from patients. Diminishing these uncertainties will require widespread clinical use and analyzing the outcomes in the postmarket setting.
Adoption of a New Device, Feedback, and Continued Innovation
The adoption of a new device in widespread clinical practice does not signal the end of the development process. In fact, widespread use is typically a prerequisite for garnering insights about the technology that provide important feedback to the R&D sector, either in industry or academia, about necessary improvements to optimize ease of use and the associated outcomes. These second-generation devices then re-enter the cycle of preclinical and clinical evaluative studies.
Ongoing innovation, however, does not only take place in R&D laboratories, but also in clinical practice itself. A common phenomenon is that, with further experience, improved strategies of managing patients with a device may emerge, including changes in the operative intervention, post-operative management, and outpatient care. In addition, the selection of
patients tends to change and expand. An interesting example can be found in laparoscopic surgery. Consider the transition from open surgical procedures to minimally invasive surgical approaches. These laparoscopic procedures tend to minimize post-operative pain and recovery time, and may reduce the treatment cost per patient. As a result, the target population for these procedures has expanded. Often this includes less sick patients for whom the risks of the procedure are now acceptable or sicker patients who initially were too risky to be candidates. This potential to expand the target population suggests that elasticity of demand for medical services is greater than commonly supposed.
In addition, totally new indications for use may emerge from the application and mastery of seemingly routine practices. Most medical devices achieve new indications by transfer from one organ system to another, although these transfers often require design modifications. The first endoscopes, for example, were used for cystoscopy early in this century. In the 1960s, after the development and introduction of fiber-optics, gastrointestinal endoscopy and gynecological laparoscopy became well established. A further extension of endoscopy depended on the eventual joining of television cameras to the scope, which facilitated their use in procedures such as arthroscopy. Widespread use is often a precondition for identification of these new uses, and clinical practice itself is a central source of medical innovation. “Learning by doing” in clinical practice, which may suggest modifications in the technology and the design of confirmatory trials, is widespread and confers broad health and economic benefits. A study of the top 20 blockbuster drugs from 1993 found that secondary indications exceeded 40 percent of revenues, and that a similar pattern held for medical devices (Gelijns et al., 1998)
Postmarket Evaluation
There are various reasons, as suggested above, why it is important to collect outcomes data in the postmarket setting. Premarket trials have limited time frames and seldom measure long-term effectiveness or safety. In pediatric clinical trials, the long life expectancy of children offers ample opportunity for late consequences of a disease, or treatment, to develop, which may be unknown at the time of treatment. The delayed consequences of radiation treatment of the face for teenage acne, for example, were only seen decades after treatment as an increased incidence of thyroid cancer. Trials also intentionally limit patient heterogeneity and, therefore, may not be generalizable to all potential recipients, who will receive the device in the postmarket setting. Moreover, premarket trials are often conducted in specialized centers, and as the device disseminates to other participants, the outcome parameters may change.
The iterative nature of medical device development argues for continued monitoring of devices as they are used in general clinical practice. There are various ways in which postmarket data can be collected. Mandatory and voluntary reporting of device-related deaths and serious adverse events to the FDA by manufacturers and clinicians has been plagued by underreporting. As part of the voluntary reporting system, the FDA has been implementing the MedSun system since 2002, an Internet-based pilot reporting system, comprised of over 180 hospitals and nursing homes.
The FDA could also mandate more postmarket studies or registries as part of the PMA approval. Clinical trials in the postmarket setting may differ substantially from premarket studies in their target populations, endpoints, and comparison groups. While FDA-related trials in the premarket setting may utilize a placebo control group (although not often with devices), postmarket studies are more concerned with comparisons between alternative treatment options. Postmarket studies are more apt to use a general practice setting than “centers of excellence” and to expand the target population beyond those seen in premarket studies. Moreover, they are more likely to include a broader range of outcomes, including functional status, quality of life, and economic endpoints facilitating much more accurate and meaningful estimates of the cost-effectiveness of new treatment options.
Specialty societies or regional authorities might also independently initiate such registries. For example, consider ECMO or extracorporeal life support (ECLS), which is a modified form of cardiopulmonary bypass, used in children and adults (Zapol et al., 1979). As the use of ECMO gained in popularity in 1984, the Neonatal ECMO Registry was established and began collecting data on in-hospital outcomes from clinical centers. To date, ECLS has been employed in more than 26,000 neonatal and pediatric patients with an overall survival rate of 68 percent (Lequier, 2004). In the United Kingdom, clinical trials are now being conducted to assess the long-term outcomes of ECMO. Another case in point is the New York State Cardiac Advisory Committee, which required all hospitals in New York State to collect data on the in-hospital outcomes for pediatric patients undergoing surgery to correct congenital cardiac defects. The risk-adjusted outcome rates for the specific hospitals can be used in quality improvement programs. Generally speaking, either hospitals or the device manufacturers conduct these postmarket observational studies. A more recent model of postmarket data collection involves LVADs. As a condition of marketing and reimbursement approval of the HeartMate™ LVAD for destination therapy (long-term implantation) in patients with advanced heart failure, the manufacturer must collect long-term data on patient outcomes and device performance for all patients receiving the implant. Three major government agencies (FDA, CMS, and NIH) have put for-
ward funds to support the registry, which would be coordinated by an independent organization. The participating hospitals would provide inkind support for data collection efforts, while industrial firms would provide additional financial support. The expectation is that industrial firms would assume the responsibility for the registry over time. Models like this might provide interesting formats for the pediatric device world where private sector funds are limited.
CONCLUDING OBSERVATIONS
Incentives for innovation in the area of pediatric devices are far less compelling than for the adult population. Generally speaking, this area is characterized by small markets, limited reimbursement, and formidable challenges to conducting premarket trials. The perception is that these obstacles have resulted in unmet needs for diagnosing and treating diseases in children. Many diseases could benefit from improved or novel devices (such as pediatric LVADs for cardiomyopathies). Although, in some areas, the demand has been partially met by clinicians modifying adult devices for use in children (such as the use of adult biliary stents for pediatric intravascular placement); this process is affected by the risk of little oversight of good manufacturing practices. Federal agencies have called for more data collection to better assess these unmet needs.
Although the extent of the problem is not defined in detail, expert opinion indicates that a strong case can be made for stimulating clinical innovation (including device innovation) in pediatrics (FDA, 2004b; see also AAP et al., 2004). Such innovation could lessen the substantial emotional and economic toll imposed by childhood diseases. However, as this paper argues, there is an equally strong case to be made for a more rigorous knowledge base about the effectiveness, safety, and economic impact of device modalities for children. Premarket clinical trials are limited in their ability to provide insights about long-term effectiveness and safety, resulting in uncertainty about the ultimate value of these devices. Moreover, this uncertainty is exacerbated by the fact that devices keep evolving long after they have moved out of the clinical development phase into widespread clinical practice itself. Observations by users about the limitations of devices are fed back into the R&D process and may lead to subsequent modifications. In addition, physicians modify the clinical management strategies for their device patients (e.g., implantation techniques, infection prevention techniques, or means to diminish bleeding), and change the selection criteria for patients eligible for device therapies. This evolution, which is largely based on tacit know-how and usually not subjected to formal experimental testing, further heightens the uncertainty level about the clini-
cal and economic impact of devices, and, hence, our understanding about best practices in caring for patients.
Yet, there is a tension between stimulating innovation and increasing the requirements for collecting more data on the clinical and economic impact of device therapies, which is exacerbated by the small size of many of the markets and affected patient populations. Attempting to eradicate all clinical uncertainty in the premarket phase is impossible, and attempts to bring the level of uncertainty down to minute levels would be costly in terms of the time and expense of the premarket development process, not to mention the indirect expense of holding off a promising therapy from patients. Diminishing these uncertainties requires widespread clinical use and ongoing outcomes assessment in the postmarket setting. However, even if we emphasize postmarket data collection, we need ensure that this in itself does not impose an additional disincentive to pediatric device innovation.
What then are some of the options for achieving this? Facilitating the process of premarket and postmarket trials requires solutions that lie within the analytical, institutional and financial realms. First of all there are various ways of decreasing the sample size for premarket clinical trials. This is important because, in general, pediatric disease populations are small enough that enrollment times would be lengthy and threaten the usefulness and validity of the results of the trial as well as drive up the related costs. Techniques such as Bayesian analysis, which utilize prior probabilities in the hypothesis testing, relaxing the tolerance to random variation error (i.e., utilizing more relaxed confidence intervals), or utilizing non-concurrent control groups and objective performance criteria, each can reduce the required sample size compared to a classic randomized controlled trial. Another consideration is to eliminate requirements for extensive premarket studies in children when the device in question has been used widely in adults, the pathophysiology of the disease is similar in adults and children, and the adaptation of the devices requires no major technological changes (FDA, 2004b).
Another important set of initiatives can be found in the institutional realm. We need to strengthen the institutional infrastructure for conducting clinical trials for devices in pediatrics by creating networks of pediatric hospitals. A case in point is childhood cancer, which constitutes a fairly small population. Nearly 95 percent of children with cancer under age 15 are treated at institutions that are affiliated with the Children’s Oncology Group, resulting in more than 70 percent of cancer patients in this age group being enrolled in one or more clinical trial (Tejeda et al., 1996; Bleyer et al., 1997). Although the group focuses on evaluation of multi-modal cancer therapy (i.e., effective use of combinations of surgery, radiation, and chemotherapy), this network, with appropriate public or private funding,
may be used for evaluation of surgical, radiological, and drug delivery devices. These U.S. hospital networks should also become involved in data collection in the postmarket setting, which requires better integration of data collection for these studies with the data collection systems used in everyday practice of medicine. These institutions could be induced to participate through better reimbursement rates for the care they provide or through direct subsidy for data collection efforts by research agencies. The NIH, for example, sponsors the pediatric heart disease clinical research network to study new interventions for congenital heart disease.
This brings us to the financial realm. There is an increased need for public–private partnerships for funding premarket trials of innovative pediatric devices (as per the philosophy of the NIH Roadmap) and postmarket studies. The products of this research are subject to the classic public good argument, where health benefits would accrue to the public-at-large, but investors would be unable to derive a sufficient return on their investment. Alternatively, one could also argue for increasing the financial incentives to the private sector, which would be able to raise private funds for developing and evaluating novel pediatric devices. Among suggested incentives are decreased or capped liability or better reimbursement for pediatric device therapies.
The need to obtain better knowledge about the clinical and economic impact of devices, especially in the postmarket setting is a general one. The pediatric case, however, offers unique challenges in that the incentives to innovate are weak as a result of small populations, which also complicates the clinical trial process. Therefore, the case is especially strong for the creation of novel partnerships between the public and private world that would experiment with new analytical, institutional, and economic models.
REFERENCES
AdvaMed. 2004. The Medical Technology Industry at a Glance. Available at: http://www.advamed.org/newsroom/chartbook.pdf. Accessed 11/15/04.
American Academy of Pediatrics (AAP), Ambulatory Pediatric Association (APA), American Pediatric Society (APS), et al. 2004 (August 20). Letter to Food and Drug Administration re: Response to FDA’s Request for Comments on the Availability of Appropriately Designed and Adequately Studied Medical Devices.
Bartlett RH, Andrews AF, Toomasian JN, et al. Extracorporeal membrane oxygenation for newborn respiratory failure: forty-five cases. Surgery 1982;92:425–433.
Bleyer WA, Tejeda H, Murphy SB, et al. National cancer clinical trials: children have equal access; adolescents do not. J Adolesc Health 1997;21:366–373.
Centerwatch, Clinical Trials Listing Services. Available at: http://www.centerwatch.com/. Accessed 3/10/05.
Department of Health and Human Services (DHHS). Statement by R Pettigrew on FY 2005 NIBIB Budget Request. Available at: http://www.hhs.gov/budget/testify/b2004040ln.html.
Drotar, D. (ed). 1998. Measuring Health-related Quality of Life in Children and Adolescents: Implications of Research and Practice. Mahwah, NJ: Lawrence Erlbaum Associates.
Eiser C, Morse R. Quality of life measures in chronic diseases in childhood. Health Technol Assess 2001;5(4).
Feigal DW, Gardner SN, McClellan M. Ensuring Safe and Effective Medical Devices. N Engl J Med 2003;348(3):191–192.
FDA (Food and Drug Administration), Office of Device Evaluation. 1992. Annual Report Fiscal Year 1992. Rockville, MD.
FDA, Center for Devices and Radiological Health. 2001. Humanitarian Device Exemptions: Questions and Answers; Final Guidance for Industry. Available at: http://www.fda.gov/cdrh/ode/guidance/1381.html. Accessed 11/12/04.
FDA. 2004a. New Humanitarian Device Approval: Vertical Expandable Prosthetic Titanium Rib (VEPTR)–H030009. Available at: http://www.fda.gov/cdrh/mda/docs/h030009.html. Accessed 10/20/05.
FDA. 2004b. Report to Congress: Barriers to the Availability of Medical Devices Intended for the Treatment or Diagnosis of Diseases and Conditions That Affect Children. Washington, DC: FDA. Available at: http://www.fda.gov/cdrh/pediatricdevices/rtc100104.html. Accessed 07/08/05.
Freedonia Group. 2003. US Implantable Medical Device Demand to Exceed $24 Billion in 2007. Available at: http://www.the-infoshop.com/press/fd161115_en.shtml. Accessed 3/8/05.
Gelijns AC. 1991. Innovation in Clinical Practice: The Dynamics of Medical Technology Development. Washington, DC: National Academy Press.
Gelijns AC, Rosenberg N. The dynamics of technological change in medicine. Health Affairs 1994 Summer;13(3):28–46.
Gelijns AC, Rosenberg, N. 1999. Diagnostic Devices: An Analysis of Comparative Advantages. In Mowery D, Nelson R (eds). The Sources of Industrial Leadership in Seven Industries. Boston, MA: Cambridge University Press. Pp. 312–358.
Gelijns AC, Thier SO. Medical innovation and institutional interdependence: Rethinking university-industry connections. JAMA 2002;287(1):72–77.
Gelijns AC, Rosenberg N, Moskowitz AJ. Capturing the unexpected benefits of medical research. N Engl J Med 1998;339(10):693–698.
Koot HM, Wallander JL. 2001. Quality of Life in Child and Adolescent Illness. East Sussex: Brunner-Routhledge, New York:Taylor and Francis, Inc.
Lequier L. Extracorporeal life support in pediatric and neonatal critical care: A review. J Intensive Care Med 2004;19(5):243–258.
Lewin Group. 2000. Outlook for Medical Technology Innovation: Will Patients Get the Care They Need? Report 1, The State of the Industry. The Health Industry Manufacturers Association.
McClellan MB, Tunis SR. Medicare coverage of ICDs. N Engl J Med 2005;352(3):222–224.
Moskowitz AJ. 2003. Report of the Panel on the Medical Devices and Equipment Industry. In: The Impact of Academic Research on Industrial Performance. Washington, DC: National Academy Press. Pp. 77–114.
Moskowitz AJ, Reemtsma K, Rose EA, Gelijns AC. 1997. Clinical Outcomes in Surgery. In: Sabiston DC, Lyerly HK (eds.). Textbook of Surgery: The Biological Basis of Modern Surgical Practice. 15th ed. Philadelphia, PA: WB Saunders. Pp. 36–54.
National Academy of Engineering (NAE). 2003. The Impact of Academic Research on Industrial Performance. Washington DC: National Academy Press.
National Association of Children’s Hospitals and Related Institutions (NACHRI). 2001. All Children Need Hospitals. Available at: http://www.childrenshospitals.net Accessed 11/12/04.
National Heart, Lung, and Blood Institute. 2002. Report of the Task Force on Research in Pediatric Cardiovascular Disease. Available at: http://www.nhlbi.nih.gov/resources/docs/pediatric_cvd.pdf. Accessed 3/9/05.
National Institute of Biomedical Imaging and Bioengineering (NIBIB). 2005. Untitled. Available at: http://www.nibib1.nih.gov. Accessed 6/17/05.
New York State Cardiac Advisory website. Available at: http://www.health.state.ny.us/nysdoh/heart/pediatric/message.htm. Accessed 3/9/05.
Rose EA, Moskowitz AJ, Packer MP, et al. The REMATCH trial: rationale, design, and end points. Ann Thorac Surg 1999;67:723–730.
Rose EA, Gelijns AC, Moskowitz AJ, et al. Randomized Evaluation of Mechanical Assistance for the Treatment of Congestive Heart Failure (REMATCH) Study Group. Long-term mechanical left ventricular assistance for end-stage heart failure. N Engl J Med 2001; 345(20):1435–1443.
Rosenbaum PL, Saigal S. Measuring Health-Related Quality of Life in Pediatric Populations: Conceptual Issues. In Spilker B. (ed.). Quality of Life and Pharmacoeconomics in Clinical Trials. 2nd ed. 1996. Philadelphia, PA: Lippincott-Raven Publishers. Pp. 785–791.
Sansom W. Titanium rib becomes 1st new FDA-approved spine deformity treatment in 40 years. HSC News, The University of Texas Health Science Center at San Antonio. 2004;37(37):unpaged. Available at: http://www/uthscsa.edu/hscnews. Accessed 10/20/05.
Shaw BF. 1987. The Role of the Interaction between the Manufacturer and the User in the Technological Innovation Process. Ph.D. dissertation. University of Sussex, Sussex, United Kingdom.
Tejeda HA, Green SB, Trimble EL, et al. Representation of African-Americans, Hispanics, and whites in National Cancer Institute Cancer Treatment Trials. J Natl Cancer Inst 1996;88:812–816.
Trajtenberg M. The use of multivariate regression analysis in contrast-detail studies of CT scanners. Medical Physics 1984;11(4):456–464.
Tufts Center for the Study of Drug Development (TCSDD). Cost issues increasingly drive U.S. formulary and clinical guidelines. Impact Report 2004; 6(5).
Ungar WJ, Santos MT. The Pediatric Economic Database Evaluation (PEDE) project: establishing a database to study trends in pediatric economic evaluation. Med Care 2003; 41(10):1142–1152.
Von Hippel EA, Finkelstein SN. Analysis of innovation in automated clinical chemistry analyzers. Sci Public Policy 1979;6(1):24–37.
Wilkerson Group. 1995 (June). Forces Reshaping the Performance and Contribution of the U.S. Medical Device Industry. The Health Industry Manufacturers Association, Washington, DC.
Zapol WM, Snider MT, Hill JD, et al. Extracorporeal membrane oxygenation in severe respiratory failure. JAMA 1979;242:2193–2196.

























