
E
The Regulatory History of Cerebrospinal Fluid Shunts for Hydrocephalus
Stephen J. Haines, M.D.,* Jeffrey P. Blount M.D.**
STATEMENT OF PROBLEM
Cerebrospinal fluid (CSF) shunts are implantable devices inserted by neurosurgeons to treat hydrocephalus. Shunt insertion and revision are the operations most commonly performed by pediatric neurosurgeons (1). There is little question that these devices have saved the lives of thousands of children and reduced morbidity in tens of thousands more, yet CSF shunts demonstrate a substantially higher degree of failure or adverse outcomes than most approved devices in current use (2). The U.S. Food and Drug Administration (FDA) has regulatory authority over these and all medical devices. Historically the FDA has taken a limited regulatory approach toward CSF shunts. The purpose of this paper is to provide background about hydrocephalus and its surgical treatment and to examine the effect that such an approach may have had on the development, safety, and effectiveness of CSF shunts.
BACKGROUND AND DEFINITIONS
Anatomy and Physiology
Cerebrospinal fluid is continuously made (predominantly although not exclusively) within normal hollow cavities of the human brain called ven
tricles. The largest of the ventricles are the two lateral ventricles, which occur in parallel and have a shape that is complex but that can be broadly described as the appearance of a medially angulated letter C with a tail extending from their back (Figure E.1). The tail is the occipital horn while the top part of the letter C is the frontal horn and the inferior and more lateral part is the temporal horn. Smaller singular third and fourth ventricles are midline structures in direct communication with the lateral ventricles. At the base of the fourth ventricle in the brain stem (laterally out the foramen of Luschka and medially out the foramen of Magendie), the CSF escapes the middle of the brain and freely flows into the subarachnoid space that extends around the outside of the brain and down into the spine to surround the spinal cord and nerve roots.
Cerebrospinal fluid is generated by small fronds of pink-orange tissue within the ventricles called choroid plexus (Figure E.2). Blood flows into and out of the choroid plexus via the choroidal vessels, and the CSF is continuously generated from within the choroid plexus in an energy-dependent process. The rate of production of CSF is about 0.2–0.3 cc/
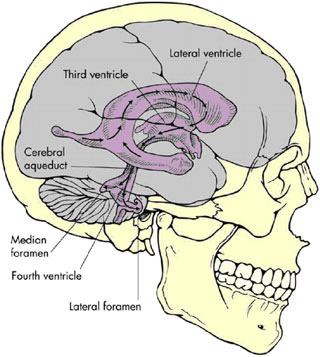
FIGURE E.1 Ventricles of the human brain (3). (Source: Brian J, Warner D. Atlas of anesthesia: Scientific principles of anesthesia. Miller R, Schwinn DA, eds., 1997. Used with permission of Current Medicine, Inc., via ImagesMD.com.)
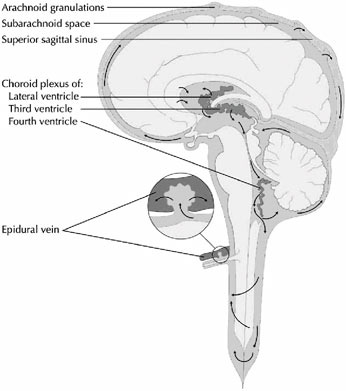
FIGURE E.2 The cerebrospinal fluid system (4). (Source: Digre K. Idiopathic Intracranial Hypertension Headache. Current Pain and Headache Reports. 6(3):217–225. Used with permission of Current Science, Inc., via ImagesMD.com.)
minute or 35 cc/hr. The total capacity of normal ventricles in adults or older children (above age 2) is about 35 cc, and another 120 cc of CSF surrounds the spinal cord and nerve roots. Thus, the total amount of CSF in the adult or large child is typically about 150–160 cc. Yet the rate of daily production of CSF is about 3 times that amount.
This imbalance is corrected by the resorption of CSF back into the bloodstream, which occurs primarily along the superior sagittal sinus and to a lesser degree from the epidural veins. Structures called arachnoid granulations (Figure E.2) extend extensively from the venous sinuses and serve to reabsorb CSF back into the bloodstream. As part of the plasma volume, it in turn is filtered by the kidneys. Thus, there is a complex, one-way, and tightly regulated circulation of CSF from the choroid plexus within the ventricles, through some narrow interventricular passageways (foramina or aqueducts), over the surface of the brain, and into the bloodstream. A variety of pathologic processes can disturb this delicate circulatory pathway, resulting in a relative or absolute imbalance between the amount of
fluid produced and reabsorbed. Some of these pathologic processes are congenital (e.g., congenital obliteration or stenosis of aqueducts or obliteration of the resorbtive capacity of the arachnoid granulations), while others are acquired (e.g., infections, intracranial hemorrhage, or residual hemorrhage from trauma or tumors). The resulting imbalance leads to a relative accumulation of cerebrospinal fluid within the ventricles of the brain that is called hydrocephalus.
Hydrocephalus
Hydrocephalus (sometimes referred to as “water on the brain”) is a condition in which an excess of cerebrospinal fluid accumulates in the brain. In most cases, this is associated with an increase in the CSF pressure, which can be measured by placing a fluid filled catheter in the ventricle and connecting it to a manometer or strain gauge. CSF pressure is most commonly measured in centimeters of water rather than millimeters of mercury because manometric readings utilize the CSF itself for measurement and CSF has the same density as water. As the normal CSF pressure changes with age, the definition of “high” pressure varies with age as well. The normal pressure in an adult is thought to be less than 20 cm of water. However, a pressure of 15 cm of water in a normal adult may be abnormally high in an infant.
There are less common circumstances when the measured CSF pressure in the ventricle may be in the normal range. For example, ventricular enlargement associated with excess CSF volume causes brain dysfunction (“normal pressure hydrocephalus”). This condition is typically observed in elderly adults. Their scans do not show loss of brain surface volume, and they may be successfully treated with CSF shunts. A full discussion is beyond the scope of this paper, however.
Hydrocephalus must be distinguished from ventricular enlargement caused by loss of brain volume (sometimes called “hydrocephalus ex vacuo”), which may result from an acute or chronic injury to the brain. In the latter situation, the ventricular cavities are enlarged, CSF pressure is normal, but, unlike those with normal pressure hydrocephalus, MRI or PT scans show loss of brain volume. It is the generalized loss of brain substance (which causes the ventricular enlargement) that reflects the underlying brain disorder that is responsible for their brain dysfunction. CSF shunts do not help this condition.
Classification Systems
Hydrocephalus may be classified by the location of the primary CSF space enlargement. “External” hydrocephalus occurs when the subarach-
noid space surrounding the brain is enlarged. The ventricles may be modestly to moderately enlarged or rounded, but the intracranial pressure is normal. This condition may also be known as benign macrocrania of infancy or benign extraventricular hydrocephalus (BEH). Ventricular shunts are not used to treat external hydrocephalus. The more common and more serious “internal hydrocephalus” is notable for ventricular enlargement and elevation of intracranial pressure and is usually treated by implanting a ventricular shunt.
A further classification of internal hydrocephalus is that of “obstructive” versus “communicating.” In the former, the CSF formed inside the ventricles cannot flow through its normal pathways (i.e., is obstructed) from reaching the absorbtive arachnoid villi along the sagittal sinus. Common sites of obstruction are the cerebral aqueduct (which connects the third and fourth ventricles) and the outlets of the fourth ventricle. Less commonly obstruction may occur at the foramen of Monroe or within the ventricles. Any diffuse injury (e.g., infection, hemorrhage, or trauma) in the brain has the possibility of eliciting scarring and inflammation that may contribute to the obstruction.
Another classification is based on etiology, but this is generally used in conjunction with the above classification scheme. This classification system defines broad classes of insult to the brain that resulted in the hydrocephalus. Examples include congenital, post-infectious, post-hemorrhagic, and post-traumatic7 hydrocephalus.
Diagnosis of Hydrocephalus
Hydrocephalus is typically suspected because of age-dependent signs of increasing intracranial pressure. In infants, this manifests as head growth that exceeds the normal rate, bulging of the normally flat anterior fontanelle (soft spot) of the skull, and behavioral signs, including irritability and unexplained vomiting. In its later stages, there can be continuous downward gaze of the eyes (“sun setting”) and lethargy.
Older children and adults will complain of headache, nausea, and vomiting. Because the head cannot grow to accommodate significant increases in CSF volume after the age of 2 or 3 years, older children and adults are at increased risk for significant elevations of intracranial pressure (ICP). As such, hydrocephalus may cause double vision, papilledema (swelling of the optic nerve that can be seen by eye examination with an ophthalmoscope), confusion, and lethargy. Left untreated, this can progress to coma and death.
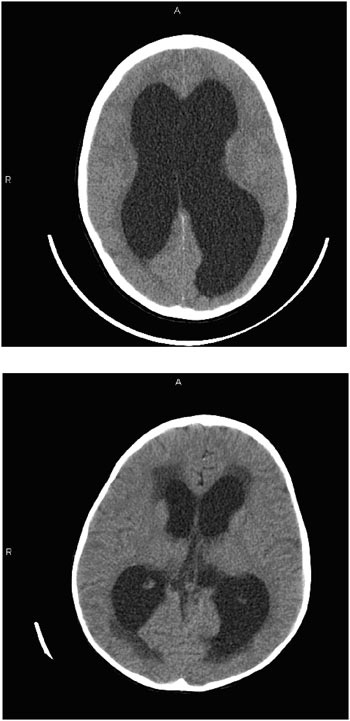
To diagnose hydrocephalus, doctors examine an image of the brain, using either CT or magnetic resonance imaging. CT provides excellent definition of the ventricles and is sufficiently rapid that sedation is rarely
necessary even in young children. As such, CT imaging has traditionally occupied the cornerstone of radiographic assessment of the child with hydrocephalus. Figure E.3 compares the CT scans of two children. The first scan reveals distention of the ventricles in a child with hydrocephalus. The second scan, from a different child, reveals transependymal flow at the tips of the lateral ventricles. This darker color within the brain substance at the tips of the ventricles is thought to result from fluid within the substance of the brain. Whether the fluid is egressing through the tissue from the distended ventricles or failing to gain access to the ventricles from the tissue remains an issue of controversy.
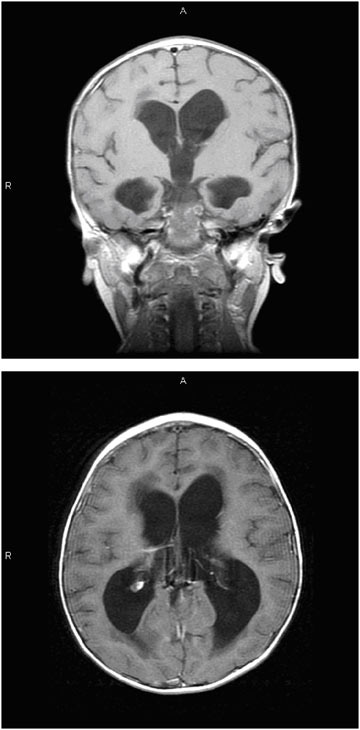
MRI provides images in multiple planes (axial, coronal, and sagittal) and provides better tissue definition. As such, MRI may provide important additional information that may elucidate etiology and facilitate classification and treatment. Figure E.4 is a sequence of MRI images of a child with hydrocephalus secondary to an intracranial tumor (which is not evident in these images). In this sequence, the left image shows the rounded, full appearing ventricles in the coronal plane. The image on the right shows ventricular distention and transependymal flow, which is evident as the darkened areas in the tissue at the tips of the ventricles in an image taken in the axial plane.
Prognosis
The contemporary prognosis for hydrocephalus is largely dependent upon the degree to which it is recognized and successfully treated and followed. The natural history of untreated hydrocephalus is ominous. While some patients are able to arrive at an equilibrium in which excess CSF volume is compensated by head growth and brain volume reduction to result in normal intracranial pressure, most are not. Even those that reach a compensated equilibrium often suffer cognitive and physical impairments related to the effects of adjusting to chronically increased pressure on brain development. Prior to the development of effective shunt systems, the natural history of most infants with hydrocephalus was that of progressive neurologic decline, macrocephaly (potentially grossly dysmorphic), and early death.
TREATMENT AND COMPLICATIONS
Treatment
The treatment of hydrocephalus is surgical. Although some medications have been shown to reduce the rate of CSF formation (acetazolamide, digoxin, and furosemide are the most common), they rarely are sufficient to relieve the symptoms of significant hydrocephalus. It is unusual to significantly delay
surgical treatment once the diagnosis is established. Occasionally these medications can be used to temporize until surgical therapy is undertaken, but there is no significant role for medical therapy in the long-term, contemporary treatment of hydrocephalus.
Historically, three conceptual surgical approaches have been undertaken to treat hydrocephalus: reduction of the rate of formation of CSF by ablation of the choroid plexus, establishment of alternative pathways for the spinal fluid to reach the arachnoid granulations, and shunting of the fluid to a body cavity where it can be absorbed (5).
Prior to the development of valve-regulated CSF shunts, attempts were made to decrease the formation of CSF by surgical removal of the choroid plexus (choroid plexectomy). This intervention was developed because the choroid plexus was the site where CSF was known to be generated. Later, it became evident that CSF also egresses directly from the brain tissue itself. While occasionally successful in establishing a “compensated” state, choroid plexectomy was rarely curative and has been relegated to be of only historic interest with an important exception. Choroid plexectomy may be highly useful in treating hydranencephaly—intrauterine destruction of the brain caused from infections or infarcts, which results in loss of the cortex and replacement with CSF.
A number of surgical interventions have been proposed to bypass the site of obstruction to CSF flow surgically. The first historical efforts attempted wholly intracranial diversion from the trapped ventricles. One such procedure (popularized by Torkildsen during the early 1950s) involves placement of a valveless tube from the ventricle to the subarachnoid space (usually the cisterna magna at the base of the skull). Despite initial claims of success, ventriculocisternal diversion did not provide long-term control for hydrocephalus and is no longer used.
Another approach involved the surgical creation of an alternative pathway past an obstruction to the subarachnoid space and arachnoid granulations. Anatomically, the most inviting location to pursue this was through the floor of the third ventricle. Hydrocephalus causes distention of the third ventricle with thinning of the floor so that an opening can be made to the subarachnoid space (third ventriculostomy) without disturbing any functional neurological tissue. When done as a major operation involving widely opening the skull (craniotomy), it had significant risk of death and disability. In the 1950s and 1960s, emerging capabilities in stereotactic neurosurgery allowed the procedure to be performed with less morbidity than with open approaches. However, the puncture was made without direct visualization or x-ray guidance and severe complications occurred.
In the late twentieth century, advances in endoscopy led to a resurgence of interest in third ventriculostomy. Contemporary endoscopes allow excellent visualization of the floor of the third ventricle. Endoscopic third ven-
triculostomy is an important procedure in contemporary management of hydrocephalus, but it is an effective intervention only for a subset of patients with hydrocephalus. Long-term results of and patient selection for endoscopic third ventriculostomy remain topics of considerable interest and continued investigation (6).
Valve-Regulated CSF Shunts
The first reliable valve-regulated CSF shunt was developed in the 1950s by John Holter after his son was born with spina bifida and hydrocephalus. Holter was a machinist and, faced with a condition for which no reliable treatment was available, designed and produced the Holter valve in conjunction with neurosurgeon Eugene Spitz. This valve was rapidly adopted and represented a major breakthrough in the treatment of hydrocephalus. Other differential pressure valves were subsequently developed, and major and minor modifications of devices and techniques followed (2).
Like all shunts the initial shunt consisted of a ventricular catheter, a valve, and a distal catheter. The ventricular catheter is the portion of the shunt that penetrates the skull and the brain tissue. The valve prevents overdrainage of cerebrospinal fluid. In the absence of a valve, fluid can rapidly drain down the tube and cause acute drops in the intracranial pressure. Acutely this causes severe headaches, vomiting, and lethargy and may contribute to the collapse of the ventricles. Subsequently, ventricular collapse may cause the surface of the brain to pull away from the inner surface of the skull. This can result in the disruption of draining veins and the accumulation of potentially life-threatening subdural hematomas. The distal catheter is the portion of the shunt that drains the CSF from the valve to its ultimate body cavity for absorption. The most popular location for the distal drainage catheter in early shunts was the right atrium of the heart. The catheter was usually inserted through the jugular vein, requiring sacrifice of the vein or one of its major tributaries. In some cases, it was even placed directly in the atrium by an operation through the chest.
With time and experience, a number of complications related to the placement of CSF shunt catheters within the vascular system were identified. When such catheters stopped working, the revision operation could be quite challenging (7). In 1968, a study concluded that the relative position of the catheter in the superior vena cava was critical to its continued function: the rate of malfunction rose rapidly as the catheter rose from the level of the sixth to the fourth thoracic vertebra as visualized on a chest x-ray. Routine revisions were recommended to prevent such malfunctions (8). Although this observation identified that shunt failure was common in infants in the first 36 months after placement, it took the authors 11 years to accumulate and publish that experience. Another method for predicting
the need for elective lengthening of the atrial catheter was reported in 1976 (9). Other sites for the distal catheter were explored, before and after the introduction of the Holter valve, including the mastoid (10), sagittal sinus (11), gall bladder (12), and pleural (13) and peritoneal (14) cavities. Because of the ease of revision and less serious cardiovascular and infectious complications of the ventriculo-peritoneal shunt, it became the most popular site for catheter implantation to date, with atrial and pleural shunts a distant second and third in frequency of utilization (15).
Complications Drive the Development of the Modern CSF Shunt
The most common problems with valve-regulated shunts are obstruction and infection (16), (17), (18). The youngest patients seem at highest risk of shunt failure (19), (20). Obstruction may occur in any part of the system (ventricular catheter, valve, or distal catheter) (2), but the most common site is the ventricular catheter. Figure E.5 shows an endoscopic view of a ventricular catheter partially covered in fibrinous debris. Strands of debris can be seen to be gradually occluding one of the holes of this ventricular catheter. Gradual occlusion of the ventricular catheter by similar tissue is thought to be the most common cause of VP shunt malfunction. Other causes of malfunction include discontinuity of the system (disconnec-
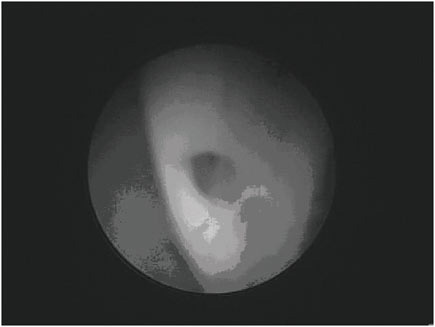
FIGURE E.5 Endoscopic view of a ventricular catheter partially covered in fibrinous debris. (Courtesy of Jeffrey P. Blount, M.D.)
tion or fracture of tubing), migration of the system (often, but not always, associated with disconnection or fracture), perforation of an organ at the site of the distal catheter, or development of a cyst around the end of the distal catheter (particularly related to shunts with distal catheters in the peritoneum).
To decrease the number of shunt obstructions, a number of technical innovations focused on the ventricular catheter. It is hypothesized that ingrowth of choroid plexus tissue into the small proximal holes of the ventricular catheter was the mechanism by which ventricular catheter obstruction occurs. Consequently, several catheters were developed that attempted to prevent ingrowth of choroid plexus. The most popular was the flanged catheter that featured soft silastic flanges at the most distal end of the ventricular catheter. In theory, the flanges were to keep choroid away from the holes in the ventricular catheter. Unfortunately, in clinical practice they were found to be optimal sites for choroid plexus adherence. This adherence made necessary shunt revisions difficult. The flanges increased the surface area of the device, which, in turn, increased the frequency of intraventricular hemorrhage from avulsion of the choroid plexus.
The number and size of the holes in the proximal part of the ventricular catheter have also been modified in an ongoing effort to reduce the rate of ventricular catheter failure. No significant improvement in overall ventricular catheter longevity has been realized despite differences in size, number, and positioning of holes in the proximal catheter.
Another recent technical innovation involving the ventricular catheter is the impregnation of the catheter with antimicrobial agents. This is discussed further in the section on shunt infection.
Valve design has similarly undergone extensive, continuous technical revisions and innovations. The earliest prototype of the Holter valve featured two rubber condoms with slits at either end of a piece of tubing. Other early valves featured simple differential pressure mechanisms that allowed flow when pressure in the proximal part of the shunt reached a given value. These valves prevented continuous drainage but did little to prevent siphoning or over-drainage. To improve valve function, the valve was moved to the distal end of the shunt. The distal slit valve included a tiny slit in the distal end of the peritoneal catheter, which provided sufficient resistance to CSF outflow to function meaningfully as a valve. The Raimondi distal spring valve (a peritoneal valve) was less successful. This valve featured a spring-like device in the distal end of the peritoneal catheter that functioned to resist CSF outflow. Unfortunately, the spring provided sufficient rigidity that hollow viscus perforation was a frequent and serious problem. Concern over this event led a small group of pediatric neurosurgeons to take the unprecedented step of writing to the FDA in 1978 to petition for the removal of the device. As a result of this action and
word-of-mouth dissemination of the knowledge of bowel perforation complications, the distal spring valve fell quickly out of use.
Advances in the understanding of CSF physiology fostered technologic advances in valve design in the 1980s and early 1990s. Prior to this, all shunt valves functioned through differential pressure. The next sequence of valve design added flow control to differential pressure. Several valves were marketed that incorporated these concepts (e.g., Orbis-Sigma 1, Delta valve).
Another recent innovation in shunt valve design was the introduction of valves that can be externally adjusted to change the pressure setting of the valve, allowing the performance characteristics of the valve to be changed without requiring surgical replacement (21). These valves were used to treat some forms of slit ventricle syndrome, to allow for changes in normal intracranial pressure with growth from infancy to adolescence, and to prevent subdural hemorrhage consequent to shunt over-drainage. Overall effectiveness for each indication remains to be definitively demonstrated.
The shunt design, marketing, and pediatric neurosurgery communities hoped that such advancements, based on improved understanding of CSF physiology, would predictably result in real and demonstrable improvements in shunt longevity (22). Unfortunately, as documented in the multi-centered prospective shunt design trial, these aspirations were not realized, and rates of shunt failure remained relatively unchanged (23), (24).
Infection
Infectious complications have plagued shunts since they first were implanted (25), (26), (27), (28). Infection is an important problem for two main reasons. First, shunt infections are associated with significant morbidity, mortality, and expense. Second, shunt infections are largely iatrogenic events that have proven markedly resistant to extensive efforts aimed at reducing or eliminating them (26), (28), (30), (31). Shunt infection may result in ventriculitis, which is similar to meningitis except that it is centered within the ventricles of the brain rather than diffusely throughout the subarachnoid space. The infection itself may injure the brain and result in developmental delay, direct neurologic deficits, or seizures. The patient’s immune response to the infection may be similarly damaging to the brain and worsen the injury and resulting deficit. The infection may cause obstruction of the shunt with secondary elevation of intracranial pressure and risk for cerebral herniation. If undetected, shunt infections may progress to severe neurologic injury or death (32), (33).
Treatment is invasive and expensive. Infection occurs at a rate of 7–10 percent of operations (34). Surgical shunt removal with implantation of a temporary drain and intravenous antibiotics are the cornerstone of care
(35). Once the infection is cleared, a second operation is needed to insert a new shunt. A recent estimate suggested that treatment of an individual shunt infection costs in excess of $50,000 (36). Overall risks of shunt failure and infection correlate with clinical experience of the institution where implantation is performed (37). In-hospital mortality varies by surgeon and hospital experience, ranging from 0.1 percent to 0.8 percent of hospital admissions (38).
The most common microorganisms to cause shunt infections are Staphylococci from the skin. Other cutaneous bacteria are frequently implicated, and enteral organisms (from the gut) may rarely be implicated. The preponderance of cutaneous organisms is strongly suggestive that the critical event in the pathophysiology of shunt infections is inoculation of the shunt with skin bacteria at the time of shunt insertion. A wide variety of interventions have been proposed to reduce the risk of shunt infection. The only intervention that has been proven to reduce the rate of infection is the provision of intravenous antibiotics prior to the initiation of shunt surgery (39). Clinical studies also indicate that a dedicated program toward reducing shunt infection often results in reductions of shunt infection rate. In poorly controlled, retrospective reviews, individual centers have reported utilization of protocols that resulted in lower shunt infection rates. However, no single technique or procedure has significantly reduced infection rates across time and multiple clinical centers (40), (41), (42). The number of properly controlled, prospective trials is very limited (43).
The manifestation of the shunt infection is dependent upon the anatomic location of the shunt. Early CSF shunt systems employed the atrium for the distal catheter placement. As such, blood and heart valve infections were frequent. Many of these infections were not easy to diagnose. In fact, catheters can be colonized by staphylococcus aureus without causing overt infectious signs in the shunted child. However, chronic exposure to the organism could lead to immune-complex glomerulonephritis (an inflammatory kidney disease) that might result in kidney failure (44). This condition is called “shunt nephritis” (45), (46). Shunt infection investigators were intrigued by shunt nephritis through the 1970s, but as the peritoneal shunt became more popular, other issues in shunt infection dominated publications (Figure E.6).
One of the principal advantages in utilizing the peritoneum for distal catheter placement is the relatively minor damage imparted by a shunt infection (47), (48), (49). The peritoneum has a well-recognized capability to seal-off/wall-off localized infections. Continued CSF accumulation within the loculated peritoneum can result in a localized collection of infected CSF called a pseudocyst (50). The severity of infection-related complications is significantly less for peritoneal shunts, and the recognition of this and the
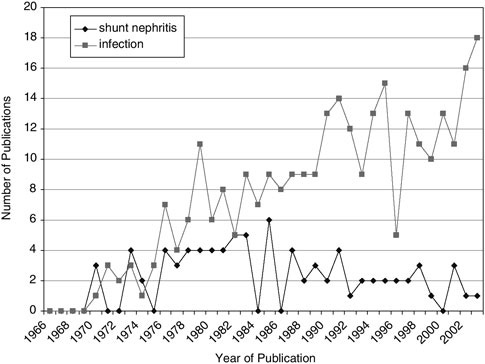
FIGURE E.6 Shunt infection publications by year.
gradual increase in the number of peritoneal shunts represents a successful hallmark in the history of CSF shunts.
The focus on infection and the inability to eliminate it as an important complication have led recently to the introduction of shunt components, such as ventricular and distal catheters, impregnated with antibiotics (51). Several prototypes are currently available and preliminary reports suggest their potential effectiveness (52).
Other Complications
A host of other complications has been described. All have important implications for shunted patients and the health care system. Because the shunt is an implant, its repair requires an operation. Virtually all infections require that the infected shunt be removed and replaced. Obstruction, disconnections, fractures, and migrations all require surgical correction. Organ perforation may require shunt repair or replacement in addition to surgical intervention. Hemorrhage can sometimes cause additional brain
damage. The spread of a brain tumor outside of the central nervous system through a shunt can seriously complicate the treatment of the tumor. Heart, lung, and kidney failure, which can result from shunt infection or malfunction, can be life-threatening.
Reports of Complications
To examine the identification and attempts to devise ways of treating and preventing complications, we performed a search of the MEDLINE and OLDMEDLINE databases with the following strategy:
|
1 |
Exp HYDROCEPHALUS/co and exp HYDROCEPHALUS/su [Complications, Surgery] |
560 |
|
2 |
exp Cerebrospinal Fluid Shunts/ae, mo [Adverse Effects, Mortality] |
1,713 |
|
3 |
1 or 2 |
2,184 |
The 2,184 articles identified were then screened by title and abstract (where necessary) to identify those that dealt directly with complications of CSF shunts. A category of “general” was identified for comprehensive reviews of CSF shunts that might contain discussions of complications. This category of publication was used primarily in the early days of shunt experience. Only 13 of the 128 articles in this category were published after 1976.
The articles discussing complications were grouped according to type of complication. The large category of “malfunction” includes general studies of malfunction and specific studies of occlusion, disconnection (including fracture of the catheter), migration (of a portion of the system away from its site of implantation), and perforation of an organ by the distal catheter of the shunt system.
The large category of infection includes general studies of shunt malfunction and specific studies of shunt nephritis, infected abdominal cysts, or peritonitis.
Other identifiable categories of complication include uninfected abdominal cysts, intracranial hemorrhage caused by the shunt, tumor metastases through the shunt, pneumocephalus (symptomatic air in the head related to the shunt), slit ventricles (ventricles that become smaller than normal after shunting and are associated with symptoms), cardiopulmonary (including heart failure, pulmonary embolism, lung dysfunction, or catheters that perforate the heart or become loose in the vascular system), and death. Articles describing other complications or studying multiple types of complications were classified as “miscellaneous.” Table E.1 shows the distribution of the 1,272 articles. (The entire reference list is available upon request.)
TABLE E.1 Articles on Shunt Complications by Type of Complication
|
Complication |
|
Articles |
|
|
|
|
# |
% |
|
Malfunction |
|
179 |
14.1 |
|
|
Occlusion |
59 |
4.6 |
|
Disconnection |
16 |
1.3 |
|
|
Migration |
29 |
2.3 |
|
|
Organ perforation |
75 |
5.9 |
|
|
Infection |
|
388 |
30.5 |
|
|
Infection |
293 |
23.0 |
|
Abdominal: cyst or peritonitis |
7 |
0.6 |
|
|
Shunt nephritis |
88 |
6.9 |
|
|
Abdominal Cyst |
(no infection) |
43 |
3.4 |
|
Hemorrhage |
|
55 |
4.3 |
|
Abdominal |
metastasis |
54 |
4.3 |
|
Pneumocephalus |
|
28 |
2.3 |
|
Slit ventricles |
33 |
2.6 |
|
|
Cardio-pulmonary |
68 |
5.3 |
|
|
Mortality |
12 |
0.9 |
|
|
Miscellaneous |
412 |
32.4 |
|
|
Total |
1,272 |
|
The relative frequency of publication of types of complication by year is shown in Figure E.7. The early dominance of cardio-pulmonary complication reports is replaced by dominance of infection reports after 1976 and is explained by the evolving predominance of ventriculo-peritoneal shunts.
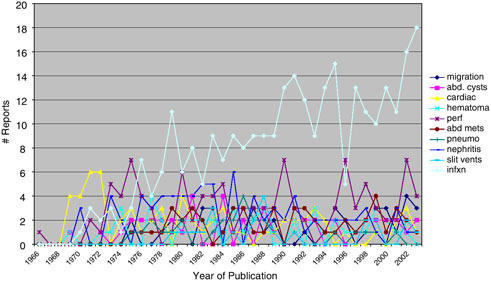
FIGURE E.7 Literature reports of shunt complications.
A cursory review indicates that CSF shunts have many different serious complications, including death, which may result from unrecognized or untreated malfunction and infection as well as a host of less common situations.
Issues of Growth and Active Lifestyle
Three specific issues related to growth and lifestyle has been overtly addressed by shunt investigators. The first is the need to revise atrial catheters placed in young children, discussed above. The second is the development of a strategy to avoid revision of peritoneal catheters because of growth. The third is the fracture of catheters under the skin as they cross from the neck to the chest.
Early in the experience with peritoneal catheters, some were concerned that excessive catheter length might be problematic (perhaps wrapping around the bowel and leading to bowel obstruction or causing discomfort because of amount of foreign material) and used catheters just long enough to enter the peritoneal cavity at the time of placement. Children could outgrow these catheters, requiring surgical revision to lengthen the peritoneal end. A catheter that would expand with growth was reported in 1975, but did not achieve widespread acceptance (53). Another theory suggested that a specific, optimal length of catheter could be determined for each patient, avoiding a presumed problem related to excessive catheter length (54). This approach likewise failed to achieve currency. A number of reports have suggested ways to add length to existing catheters (55), (56), (57). Over time, it became common practice to insert catheters long enough to allow for growth through maturity and to avoid elective operations that lengthened the tubing. Recently, this practice has been validated in the published literature (58). Because of the content and nature of the FDA reporting process, these problems are not identified in the formal postmarket surveillance process.
Finally, the issue of fractures of the distal catheter in the neck has been investigated (59), (60), (61). Although it has not been possible to definitively prove that these fractures are related to activity, this is hypothesized to be an important factor because of the consistent location of the fractures.
THE PERIOD OF REGULATION
Regulation Prior to Marketing
In 1976, amendments to the Federal Food, Drug, and Cosmetic Act gave the FDA its first real authority to regulate medical devices. Devices were placed in three classes. Class I devices “present minimal potential for harm to the user” and are the subject of the least regulatory control. Class II
devices “are those for which general controls alone are insufficient to assure safety and effectiveness, and existing methods are available to provide such assurances.” These devices are subject to special controls, such as special labeling or performance standards. For a Class II device that is used in supporting or sustaining human life, FDA must identify special controls that will provide adequate assurance of safety and effectiveness. Class III devices “are usually those that support or sustain human life, are of substantial importance in preventing impairment of human health, or which present a potential, unreasonable risk of illness or injury” Class III devices are subject to the most stringent regulation (62).
Although CSF shunts are necessary to sustain life in many patients with hydrocephalus and their failure can lead to death unless properly detected and treated, CSF shunt systems have been classified as Class II in the Code of Federal Regulations (21 CFR 882.5550) (63). In 1999, the Systemic Technology Assessment of Medical Products (STAMP) Conference (sponsored by the FDA) investigated the topic of CFS shunt technology. The introduction to the subsequent report indicates that the decision to classify shunts as Class II medical devices was “based upon the belief that standards could be written to assure the safety and effectiveness of marketed shunts, and that clinical experience had proven shunts to be reasonably safe and effective” (64).
The rationale expressed in the initial classification is similar to the rationale described above and, in addition, states that the panel responsible for the classification believed that “the characteristics of central nervous system fluid shunts and their components are reasonably well established.” It further stated that “the complications associated with these devices have been extensively reported in the literature,” and it acknowledged a mortality rate in patients treated with shunts of 5–35 percent. The panel noted that the device was primarily used in children. The Commissioner of FDA further commented that he doubted “that requirement [sic] premarket approval of these devices will improve the complication rate associated with their use” (43 FR 55714-55716, 1978).
Class II devices do not generally require “premarket approval” (PMA), but premarketing notification (510[k] notification) must be submitted to and cleared by FDA. This gives FDA the opportunity to assure that general controls and specific controls for the device are met. In FDA terminology, these devices receive clearance rather than approval. Applicants for clearance usually must show that the devices meet technical standards for biocompatibility, function, and reliability through bench testing. They must also demonstrate the device to be “substantially equivalent” to a device that was either marketed before May 28, 1976, or has been shown (through the notification or clearance process) to be substantially equivalent to such a device. Current, relevant technical standards are embodied in the Interna-
tional Organization for Standardization (ISO 7197:1997) and American Society for Testing and Materials (ASTM F647-94[2000]) documents.
The FDA maintains a public, searchable database of releasable decisions on 510(k) applications cleared after January 1999 (65). Table E.2 includes 26 codes that the FDA may use to describe its determinations about whether a device meets the criterion of substantial equivalence. The actual, online database, however, only includes decisions for devices that are found to be substantially equivalent. Those familiar with FDA confidentiality, trade secret, and privacy regulations might expect this, given the prominent use of “releasable” in the description of the database. Less expert users of the database might, however, expect to find negative decisions,
TABLE E.2 Codes for Determination of Substantial Equivalence by FDA for 510(k) Notifications
|
Code |
Description |
|
Determination of Substantial Equivalence listed in 510(k) database |
|
|
AN |
Approved Evaluation of Automatic Class III Designation |
|
ST |
Substantially Equivalent – Subject to Tracking Regulations |
|
PT |
Substantially Equivalent – Subject to Tracking and Postmarket Surveillance |
|
SF |
Substantially Equivalent – Waiting on Future Policies |
|
SS |
Substantially Equivalent – Special Labeling |
|
SE |
Substantially Equivalent |
|
SA |
Substantially Equivalent – Awaiting Device Approval |
|
SW |
Substantially Equivalent – Awaiting Drug Approval |
|
CS |
Substantially Equivalent – CLIA Submission |
|
SK |
Substantially Equivalent – Kit |
|
KD |
Substantially Equivalent – Kit with Drugs |
|
SI |
Substantially Equivalent – Market after Inspection |
|
SP |
Substantially Equivalent – Postmarket Surveillance |
|
PR |
Substantially Equivalent – Proposed Recision |
|
SU |
Substantially Equivalent – with Limitations |
|
SN |
Substantially Equivalent – for Some Indications |
|
SD |
Substantially Equivalent – with Drug |
|
Determination of Non-Substantial Equivalence listed in FDA decision codes |
|
|
FB |
Subject to 515(b) – Requires PMA |
|
NE |
Not Substantially Equivalent |
|
SC |
Not Substantially Equivalent – Cannot Market |
|
SL |
Not Substantially Equivalent – Improper Label |
|
RE |
Rescind Substantial Equivalence |
|
UD |
Unable to Determine Equivalence |
|
UO |
Unable to Determine Equivalence – Outstanding Drug Issue |
|
UR |
Not Substantially Equivalent – Unreliable Data |
|
OD |
Unable to Determine Equivalence – Outstanding Device Issue |
|
|
|
especially because the documentation for the database defines nine codes for various kinds of negative decisions.
A search of the 510(k) database through January 2005 (65) produced 166 records for code JXG (shunts, central nervous system), 8 records for HCA (catheter, ventricular), and 1 record for NHC (catheter, ventricular [containing antibiotic or antimicrobial agents]). HCD (cannula, ventricular) and GYK (instrument, shunt system implantation) were not included in further searches as these devices are not permanently implanted shunts. All 175 devices were cleared as substantially equivalent (SE). According to this database, the FDA has never required postmarket surveillance, tracking, special labeling, or placed limitations beyond those proposed by the manufacturer for CSF shunts.
The search also identified a valve system that is electromagnetically adjustable to 17 different pressure settings (67). All previously marketed shunts operated at a single, predetermined pressure. These pressure settings are susceptible to alteration by strong magnetic fields, such as magnetic resonance imaging scanners. Although not required by the FDA, this submission included a randomized clinical trial, comparing the programmable shunt to its non-programmable predecessor (21). To date, the FDA has not required a premarket approval application for any CSF shunt.
Based in part on the descriptions provided above, it is clear that the CSF shunt is considered to be a mature device from a regulatory point of view. Despite its use to “support or sustain human life” and its “substantial importance in preventing impairment of human health,” FDA does not require the intense scrutiny applied to Class III life-saving devices introduced into clinical practice after 1976. One must ask if this is a reasonable conclusion or if this conclusion is based on short-term effectiveness without sufficient consideration of long-term effectiveness and complications.
Postmarketing Surveillance
In 1999, the FDA convened the previously mentioned STAMP conference, “Shunt Technology: Challenges and Emerging Directions.” The conference reviewed “families of closely related medical devices having broad use and most often, several years of marketing experience, as well as, a significant potential for adverse events.” In presentations, patient advocates, industry representatives, and physicians familiar with cerebrospinal fluid shunts and the associated complications stressed continuing difficulty with problems of shunt function and infection. The report states that “over 60% of shunt patients manifest some type of shunt complication over their lifetime such as shunt obstruction, over-drainage, infection, device migration, disconnection and fracture.” The report summary mentions the possible development of patient information cards and online databases that
would allow more comprehensive collection of information on shunt complications using standard definitions, but makes no specific recommendations (64).
It is clear that the approved CSF shunt devices continue to have important problems with both function and infection. The existing system for monitoring these devices for problems should be examined for effectiveness both in identifying problems and in prompting corrective action.
The 1976 Medical Device Amendments granted FDA authority to issue regulations requiring adverse event reporting for marketed medical devices. FDA issued these medical device reporting regulations in 1984. A medical device reportable event as defined by the statute means:
(1) An event about which user facilities become aware of information that reasonably suggests that a device has or may have caused or contributed to a death or serious injury; or (2) An event about which manufacturers or importers have received or become aware of information that reasonably suggests that one of their marketed devices:
(i) May have caused or contributed to a death or serious injury; or
(ii) Has malfunctioned and that the device or a similar device marketed by the manufacturer or importer would be likely to cause or contribute to a death or serious injury if the malfunction were to recur. (21 CFR 803.3)
Subsequent adverse event reports were collected in the Medical Device Reports (MDR) database. To strengthen the reporting system, Congress enacted the Safe Medical Devices Act of 1990 (SMDA), which established requirements for reporting by device manufacturers, distributors, and user facilities. The FDA established a new database, Manufacturer and User Facility Device Experience (MAUDE), and began the transition from the MDR database in 1992. Since August 1996, MAUDE has been the exclusive adverse event report database.
In 1990, Congress also enacted separate regulatory authority for postmarket surveillance. The FDA website provides the following statement regarding the purpose of postmarket surveillance (68).
The primary objective of postmarket surveillance is to study the performance of the device after marketing as it is to be used in the general population for which it is intended. Generally, the primary variables to be studied are morbidity or mortality. The major interest lies in device failure and its attendant impact on the patient.
Postmarket surveillance is considered a warning system for the early detection of potential problems within a reasonable time of their first marketing. The intent of the regulation is to:
-
Identify problems
-
Provide safety warnings
-
Provide information not available from the medical device reporting regulation
-
Provide actual use of safety and effectiveness information.
ECRI (formerly the Emergency Care Research Institute), a not-for-profit health services research agency, maintains a version of the MDR and MAUDE databases with additional search capabilities. Both versions of the databases were used for this report. Searches were performed in December 2004. The strategy summarized in Table E.3 was used to search the MDR database.
This search produced 1,762 reports relevant to CSF shunts submitted to the MDR database from 1978 through 1996. Because of misclassifications leading to the inclusion of non-shunt devices and overlapping product codes that include CSF devices that are not implanted shunts (e.g., external ventricular drains, CSF shunt programmers, and devices for passing shunt catheters subcutaneously), the records were reviewed by the author (SJH) to identify misclassifications and reports not related to implanted CSF shunts. The final result was 1,555 reports regarding implanted CSF shunts.
The MAUDE database was searched in June 2004 with the strategy ((((“CEREBROSPINAL FLUID” OR CSF OR VP OR V-P OR VA OR VENTR* OR HYDROCEPH* OR CNS OR CENTRAL) AND SHUNT) OR (“NEURO VALVE” OR PUDENZ OR “SHUNT KIT” OR “POSTERIOR FOSSA” OR OSV OR DELTA)))). This produced 2,985 records of reports submitted from 1992 through 2004. These were reviewed for accuracy and a number of misclassifications by FDA product code were identified. Thirty-four were clearly shunts misclassified as another device (Table E.4). Others were non-shunt devices misclassified with a shunt code. Cleaning of the list resulted in 2,298 records of reports to the MAUDE database regarding problems with CSF shunts. An amended search in December 2004 increased the number of reports to 2,472.
Classification of Complications
In Part 2, we presented a classification of complications based on review of the literature. Both the MDR and MAUDE reports list reporter-classified outcomes as: death, serious injury, malfunction, or other. Because the MDR database could only be obtained in a text file, the “EFFECT TYPE” was identified by text searching the 1,762 reports that were identified in line S9 of the search strategy in Table E.3. All effect types other than “DEATH,” “SERIOUS INJURY,” or “MALFUNCTION” are grouped as “OTHER.” The MAUDE database was available in spreadsheet format allowing a more detailed search. The responses “OTHER,” “NO
TABLE E.3 Search Strategy (MDR Reports) for Adverse Events for CSF Shunts
|
Step |
Items |
Description (use 4 digit years) |
|
S1 |
56,618 |
SF=(AI OR ABS) |
|
S2 |
1,508 |
SO=(HEALTH()DEVICES?) |
|
S3 |
657 |
PC=(16-244?) |
|
S4 |
486 |
S3 NOT S1 |
|
S5 |
826 |
PC=(10-704? OR 16-151? OR 10-769? OR 15-588? OR 16-244? OR 17-090? 04 17-734?) |
|
S6 |
589 |
S5 NOT S1 |
|
S7 |
1,709 |
FP=(JXG? OR GYK? OR HCD? OR HCA?) |
|
S8 |
1,709 |
S7 NOT S1 |
|
S9 |
1,762 |
S7 OR S6 |
|
S10 |
111 |
S9 AND (OCCLU? OR BLOCK? OR OBSTRUCT*) |
|
S11 |
77 |
S9 AND (DISCONNECT? OR FRACTUR?) |
|
S12 |
6 |
S9 AND MIGRAT? |
|
S13 |
7 |
S9 AND (PERFORAT? OR EXTRU? OR PENETRAT?) |
|
S14 |
49 |
S9 AND (INFECT? OR VENTRICULITIS? OR MENINGITIS?) |
|
S15 |
1 |
S9 AND (PERITONITIS? OR PSEUDOCYST? OR PSEUDO (1N) CYST?) |
|
S16 |
0 |
S9 AND CYST (3N) ABDOM? |
|
S17 |
0 |
S9 AND CYST AND PERITON? |
|
S18 |
0 |
S9 AND (NEPHRITIS OR GLOMERULONEPHRITIS) |
|
S19 |
49 |
S9 AND (HEMORRHAG? OR HAEMORRHAG? OR HEMATOMA? OR HAEMATOMA? OR BLEED?) |
|
S20 |
0 |
S9 AND METASTAS? |
|
S21 |
1 |
S9 AND TUMOR? AND (ABDOM? OR PERITON?) |
|
S22 |
22 |
S9 AND (AIR OR PNEUMOCEPHALUS) |
|
S23 |
424 |
S9 AND (SLIT OR INTRACRANIAL()HYPOTENSION OR LOW()PRESSURE) OR OVERDRAIN? OR OVER()DRAIN?) |
|
S24 |
30 |
S9 AND (HEART CARDIAC OR ATRI? OR SUPERIOR()VENA()CAVA OR SUPERIOR()VENACAVA OR ENDOCARDITIS OR VEGETATION) |
|
S25 |
15 |
S9 AND ET=DEATH |
|
S26 |
9 |
S9 AND DIED |
|
S27 |
16 |
S25 OR S26 |
|
S28 |
388 |
S23 NOT (S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S19 OR S21 OR S22) |
|
S29 |
295 |
(S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S19 OR S21 OR S22)NOT S28 |
|
S30 |
0 |
S29 AND (HAIR OR FAIR) |
|
S31 |
24 |
S11 AND (HEART OR CARDIAC OR ATRI? OR SUPERIOR()VENA()CAVA OR SUPERIOR()VENACAVA OR ENDOCARDITIS OR VEGETATION) |
|
S32 |
11 |
S11 AND ET=DEATH |
|
S33 |
7 |
S11 AND DIED |
|
S34 |
12 |
12 S32 OR S33 |
|
S35 |
0 |
S11 AND (BRAIN()DEATH OR BRAINDEATH OR BRAIN()DEAD OR BRAINDEAD) |
|
S36 |
442 |
S11 AND (SLIT? OR INTRACRANIAL()HYPOTENSION OR LOW()PRESSURE OR OVERDRAIN? OR OVER()DRAIN?) |
|
S37 |
36 |
S11 AND S19 OR S20 OR S21 OR S22 OR S23 |
TABLE E.4 CSF Shunt Reports in the MAUDE Database Misclassified as a Non-Shunt Device
|
Listed FDA Product Code |
Actual Product |
Report # |
|
<blank> |
Shunt catheter |
36 |
|
|
Shunt catheter |
5,787 |
|
VP shunt |
9,189 |
|
|
VP shunt |
9,200 |
|
|
VP shunt |
9,201 |
|
|
VP shunt |
19,227 |
|
|
VP shunt |
20,112 |
|
|
KPM (peritoneo-venous shunt) |
VP shunt |
941 |
|
|
VP shunt |
16,815 |
|
FIQ (cannula, A-V shunt) |
Shunt catheter |
2,336 |
|
CAR (monitor, spinal fluid pressure, electrically powered) |
Shunt catheter |
5,860 |
|
LID (not currently listed, “ventriculo-amniotic” in database) |
VP shunt |
9,511 |
|
VP shunt |
15,964 |
|
|
VP shunt |
16,748 |
|
|
LXL (not currently listed, “valve-shunt-fluid” in database) |
VP shunt |
25,331 |
|
|
VP shunt |
28,084 |
|
|
VP shunt |
507,413 |
|
MAJ (catheter, percutaneous, intraspinal, short-term) |
VP shunt |
65,062 |
|
HCA (catheter, ventricular) |
VP shunt |
105,654 |
|
|
VP shunt |
174,723 |
|
VP shunt |
206,462 |
|
|
VP shunt |
210,976 |
|
|
VP shunt |
228,231 |
|
|
VP shunt |
238,155 |
|
|
VP shunt |
394,625 |
|
|
VP shunt |
437,978 |
|
|
VP shunt |
446,166 |
|
|
GBW (catheter, peritoneal; General and Plastic Surgery) |
VP shunt |
206,467 |
|
|
VP shunt |
220,542 |
|
VP shunt |
241,343 |
|
|
VP shunt |
241,350 |
|
|
VP shunt |
385,607 |
|
|
GWB (antisera, fluorescent, all types, streptococcus pneumoniae) (probable typographical error) |
VP shunt |
501,635 |
|
JQG (radiometric, F259, iron-binding capacity) |
VP shunt |
501,622 |
|
|
VP shunt |
501,934 |
ANSWER PROVIDED,” and blank fields were grouped as “OTHER.” The shunt reports classify as follows (Table E.5).
In the MDR database, the refined text search for “MORTALITY” (Table E.3, line S34 and 35) produced 12 reports suggesting shunt-related death. Of the 12 reports identified, 2 were of non-shunt devices misclassified as shunts leaving 10 shunt-related deaths.
TABLE E.5 Outcome as Reported for CSF Shunt Events in MDR and MAUDE Databases
|
Outcome |
MDR |
MAUDE |
||
|
Number |
Percent |
Reported number |
Percent |
|
|
Death |
15 |
0.9 |
8 |
0.3 |
|
Serious Injury |
810 |
46.0 |
1,364 |
55.2 |
|
Malfunction |
870 |
49.4 |
572 |
23.1 |
|
Other |
67 |
3.8 |
528 |
21.4 |
|
Total |
1,762 |
|
2,472 |
|
In the MAUDE database, eight official reports listed outcome as “DEATH.” The report narratives were examined in detail by the author. Four reports were found to state that the patient had died. In one instance, the reporter determined that the death was unrelated to the shunt. One report was the result of the filing of a malpractice suit alleging death related to the shunt. The other two patients died within 2 weeks of shunt operation and, therefore, would ordinarily be considered to have device-related mortality. Thus, of the eight official “DEATH” outcomes, six appeared to be definitely shunt related, one (the malpractice allegation) was possibly shunt related, and one was unrelated.
The report narratives were then text-searched for “DEATH” or “MORTALITY.” Where death was mentioned but no details were available, the death was considered shunt related. When all reports indicating patient death, either in the outcome section of the official report or as a result of the text search, were adjudicated by the author, there were 11 deaths related to shunt complications. This includes any death within 30 days of shunt operation and excludes one patient who died 5 months postoperatively from an intracerebral hemorrhage.
This analysis of the reporting of shunt-related death suggests that both the outcome section of the reports and the automated text search of the entire narrative file result in overlapping and incomplete ascertainment of specific problems.
Because there were simply too many reports for individual review, and accepting the likely inaccuracies suggested by the detailed review of reporting of “DEATH,” the computerized text searching of the MDR and MAUDE databases using the strategies described above was used to classify the reported complications using the classification scheme presented in Part 2. The MAUDE search done for Table E.6 utilized the strategy listed above, but used a different text searching program that was part of the preparation of data for the disproportionality analysis reported below, resulting in minor differences in identified mortality reports. The definition of “MALFUNCTION” in the “EFFECT TYPE” reporting analyzed above is differ-
TABLE E.6 Classification of Complications in the MDR and MAUDE Databases and Published Literature
|
Complication |
|
MDR |
MAUDE |
Literature |
|||
|
# |
% |
# |
% |
# |
% |
||
|
Malfunction |
|
201 |
12.9 |
526 |
21.3 |
179 |
14.1 |
|
|
Occlusion |
111 |
7.1 |
380 |
15.4 |
59 |
4.6 |
|
Disconnection |
77 |
5.0 |
125 |
5.1 |
16 |
1.3 |
|
|
Migration |
6 |
0.4 |
11 |
0.4 |
29 |
2.3 |
|
|
Organ perforation |
7 |
0.5 |
10 |
0.4 |
75 |
5.9 |
|
|
Infection |
|
36 |
2.3 |
75 |
3.0 |
388 |
30.5 |
|
|
infection |
36 |
2.3 |
72 |
2.9 |
293 |
23.0 |
|
Abdominal: cyst or peritonitis |
0 |
0 |
3 |
0.1 |
7 |
0.6 |
|
|
Shunt nephritis |
0 |
0 |
0 |
0 |
88 |
6.9 |
|
|
Abdominal Cyst |
(no infection) |
0 |
0 |
0 |
0 |
43 |
3.4 |
|
Hemorrhage |
|
29 |
1.9 |
54 |
2.2 |
55 |
4.3 |
|
Abdominal |
metastasis |
0 |
0 |
4 |
0.1 |
54 |
4.2 |
|
Pneumocephalus |
|
9 |
0.6 |
6 |
0.2 |
28 |
2.2 |
|
Slit ventricles |
442 |
28.4 |
109 |
4.4 |
33 |
2.6 |
|
|
Cardio-pulmonary |
24 |
1.5 |
6 |
0.2 |
68 |
5.3 |
|
|
Mortality |
12 |
0.8 |
5 |
0.2 |
12 |
0.9 |
|
|
Miscellaneous |
802 |
51.6 |
1,687 |
68.2 |
412 |
32.4 |
|
|
Total |
1,555 |
|
2,472 |
|
1,272 |
|
|
ent from the definition of “MALFUNCTION” for the purposes of complication classification (some malfunctions would be called “SERIOUS INJURIES” while some would not), thus explaining the differences in the totals for “MALFUNCTION” in the two tables. This classification follows:
ECRI also maintains a Health Devices Alerts Data Base, which collects data, reports, and alerts “from a wide variety of national and international patient safety organizations, ECRI product evaluations, member hospital reports, and accident and forensic investigations, in addition to FDA Enforcement Report data and manufacturer notices.” When searched with the following strategy ((((“CEREBROSPINAL FLUID” OR CSF OR VP OR V-P OR VA OR VENTR* OR HYDROCEPH* OR CNS OR CENTRAL) AND SHUNT) OR (“NEURO VALVE” OR PUDENZ OR “SHUNT KIT” OR “POSTERIOR FOSSA” OR OSV OR DELTA)))), there were 4 of 13 identified action items related to CSF shunts. A fifth was discovered serendipitously. Of 345 abstracts in published literature, 202 referred to CSF shunts. No alerts were identified.
ECRI defines action items as “reports of medical device problems, hazards, and recalls that have been verified by ECRI with the device manufacturer/distributor. Each Action Item includes ECRI’s specific recommendations and instructions to help those who have the affected product take
the actions needed to prevent harm” (69). The nine unrelated action items involved devices such as knee prostheses, airways, and pacemakers that use some of the terms in the search but are not CSF shunt devices. The five action items involved risk of fracture of a right angle connector leading to a recall of unimplanted connectors (from a batch of 4,600 units), debris in metal connectors leading to a recall of 156 units, abnormally high operating pressure of valve leading to recall of all unimplanted units (number not specified), mislabeled closing pressure on valves leading to recall of unimplanted valves (from a total of 60,760 distributed), and the distribution of ventricular catheters without holes in them. The two most recent actions were in 1992 and 2004.
Data Mining Techniques
We applied disproportionality analysis techniques (see Appendix D) to the shunt subset of the MAUDE database. Shunt complications in the database were identified by searching the text of the narrative descriptions of adverse events in the database (the details are given in Appendix D of the main report). This analysis identified only two associations between specific manufacturers and adverse event reports. Table E.7 shows the results of the analysis, identifying an excess of catheter disconnections associated with products of Company A and an excess of catheter migration associated with products of Company B.
We could not identify any action such as a recall, safety alert, or publication possibly related to the migration events identified for Company B. In the case of company A, concern was expressed in the literature regarding fracture (included in our definition of disconnection) in papers published in 1992 and 1995 (60), (70), (71). These papers deal with predominantly pediatric populations. A change in the formulation of the catheter was made by the company in response to concerns about an excessive number of catheter fractures. As the public data in the MDR and MAUDE databases does not include the age of the patient, we could not determine if children were overrepresented in the adverse event reports. No author has
TABLE E.7 Disproportionality Analysis of CSF Shunt Events in MAUDE Database
|
N |
E |
RR |
EBGM |
Manufacturer |
Event |
|
91 |
47.2 |
1.93 |
1.85 |
COMPANY A |
Disconnection |
|
6 |
0.982 |
6.11 |
1.70 |
COMPANY B |
Migration |
|
NOTE: N = number of events reported, E = number of events expected in the absence of association, RR = raw relative risk estimate (N/E), EBGM (Empirical Bayes Geometric Mean) is an adjusted relative risk estimate. For details see Appendix C. |
|||||
addressed the question of whether or not children are particularly susceptible to such fractures because of their activity levels.
Summary
Complications of CSF shunts have been the subject of clinical investigation since the first effective valve-regulated shunt was introduced in the 1950s. Important modifications in shunt procedures and design have been documented in the literature since that time. Since 1976, the FDA has collected reports from manufacturers and user facilities regarding death, serious injury, and malfunction related to CSF shunts. Analysis of these reports compared to the reports in the literature suggests that infections of CSF shunts have received much more attention in the literature than in the reports and that the problem of slit ventricles received greater attention in the MDR reporting system than in either the MAUDE system or the literature. The application of data mining techniques to the databases identified only one problem disproportionately associated with one manufacturer. There is suggestive evidence that this problem was also identified in the literature.
CONCLUSIONS
The tone for regulation of CSF shunts was set when the decision was made to classify them as Category II devices. This classification does not require premarket approval, which routinely requires submission of data from clinical studies. Viewed from today’s perspective, this classification may seem unusually lax for a device that treats life-threatening conditions and may have malfunctions associated with death or disability.
The contrast with the cochlear implant (see Appendix F) is instructive. The first version of that device was approved through the premarket approval process in 1984 (P830069). Multiple design changes and adaptations have been approved since that time. The device has a low failure rate (between 0.8 percent and 0.3 percent per year), and the implantation infection rate is relatively low (1.6 and 4.1 percent in two recent reports) (72), (73). Yet 15 reports of meningitis created sufficient concern to result in studies that led to recall of one brand of implant. A subsequent study identified 26 cases of meningitis in 4,264 children with cochlear implants. This represents a substantial increase over the expected rate of meningitis, but is far below the rate of 7 to 9 percent for CSF infections as seen with CSF shunts (74). CSF shunts treat life-threatening disease, and their failure is associated with life-threatening complications. Cochlear implants treat a serious, but not life-threatening condition and rarely are associated with life-threatening complications. Although the focus on meningitis associated
with cochlear implants did not arise through the FDA device reporting system, FDA attention to the problem did generate significant change in available devices and management protocols to decrease the patient risk. No similar effort has been directed toward CSF shunt-related infection.
As of June 2003, there were 171 CSF shunt-related premarket notifications in the FDA database, resulting in decisions affirming “substantial equivalence.” These decisions have allowed manufacturers to introduce many variations and modifications of the CSF shunt and its parts. For an important medical device with a relatively small market, the 510(k) notification approach may have been very effective and appropriate.
CSF shunts have been associated with very few manufacturing problems. The FDA has never recalled a shunt product. ECRI has issued only five health device alerts involving probably only several hundred actually distributed devices. The record suggests that the FDA’s mechanisms for assuring sound manufacturing processes, biocompatibility, sterility, and basic function have worked well to minimize mechanical malfunction, faulty modifications in design, and errors in the manufacturing process.
There is good evidence of rational progress in the clinical history of CSF shunting. Both technical (surgical) and technologic (device) advances are real and persistent across the history of this undertaking. Technical successes include the evolution toward peritoneal distal catheters, cessation of implantation of short catheters (that require elective lengthening), and the realization that compulsive sterile technique is associated with decreased infection rates. Technical disappointments include endoscopic placement of ventricular catheters (75) and lack of clear superiority of frontal versus occipital trajectories (76). Technologic successes include the development of silastic, differential pressure valves, and pliant peritoneal catheters. Technologic disappointments have included flanged catheters, variations in ventricular catheter hole size, distal spring valves, polyethylene shunts, and flow-regulated valves (insofar as no expected decrease in obstruction rate was seen). Programmable valves and antimicrobial impregnation are recent developments for which no final conclusion can currently be reached. All of these developments have occurred in a relaxed federal regulatory environment.
However, shunts continue to have clinically significant, sometimes life-threatening complications. As noted at the STAMP conference, over 60 percent of shunted patients will experience a complication, most of which require surgical revision of the device. The rates of infection and malfunction have been stable for the past two decades, and there is no convincing evidence that they vary by brand or type of device. In the setting of premarket notification rather than premarket approval, the burden of identifying adverse events or complications for the purposes of assuring that safety and effectiveness are maintained falls predominantly outside the premarket
review system, with a small role for the formal postmarket surveillance system.
That postmarket surveillance system does not appear to have resulted in any important change in the safety or effectiveness of CSF shunts. Despite more than 4,000 reports of device problems, the majority of which resulted in patient injury or, rarely, death, there have been only four recalls identified. All were manufacturer initiated recalls related to manufacturing or labeling issues. With the exception of infection, which has received far greater attention in the published literature than in reports to the FDA, the device problems reported to the FDA have reflected the type and distribution of the problems reported in the medical literature. As a result, one could conclude that the time and effort involved in reporting and reviewing those reports, at least as implemented, thus far, have not resulted in any important change in the safety, effectiveness, or clinical use of CSF shunts.
The current structure of the adverse event reporting system makes it difficult to search it for the details necessary to analyze specific complications. Frequently, the data necessary to classify the adverse event report in a clinically useful manner is simply not present. Additionally, variation in the understanding of types of events and errors in product classification makes accurate analysis nearly impossible. However, it is clearly beyond reasonable expectations or the resources of the FDA to impose reporting rules that require adherence to carefully specified definitions, more detailed information, and rigorous error checking.
The application of data mining techniques did identify one manufacturing problem (excessive catheter fracture) that was contemporaneously identified in the medical literature.
These data raise several questions. One could argue that postmarket surveillance, in its present form, offers no benefit and could ask if it should be abandoned for these devices. Alternatively, the question remains whether or not a strengthened monitoring effort that would allow the identification of device-specific rates of infection and malfunction could result in further progressive improvement in both malfunction and infection rates. Is it a problem that the system monitors for serious device complications but does not feed this information into a system designed to identify and implement solutions to the problems? Has the system been lulled into inaction by accepting as inevitable the relatively high rates of complications associated with CSF shunts?
For the present, the burden of identifying problems associated with CSF shunts and searching for solutions to those problems rests with the informal system of clinical observation and research. While real gains have been made and the life of a hydrocephalic child is markedly different than a generation ago, there is clearly much more to learn. Recent flow-controlled valves more closely emulate best available concepts of CSF physiology, but
have failed to reduce rates of obstruction. This failure has heightened collective awareness concerning our incomplete understanding of CSF physiology and the pathophysiology of hydrocephalus. The question of whether or not a higher level of FDA regulation and more intense focus on the CSF shunt would have resulted in a shunt with lower risk of infection and malfunction cannot be answered from this retrospective look at the problem. It is, however, an important question worthy of further exploration.
REFERENCES
1. Bondurant CP, Jimenez DF. Epidemiology of cerebrospinal fluid shunting. Pediatr Neurosurg 1995;23(5):254–258.
2. Drake JM, Kestle JR, Tuli S. CSF shunts 50 years on—past, present and future. Child’s Nerv Syst 2000;16(10-11):800–804.
3. Brian J, Warner D. 1997. Atlas of anesthesia: Scientific principles of anesthesia. Miller R, Schwinn DA, eds.
4. Digre K. Idiopathic intracranial hypertension headache. Current Pain and Headache Reports. 6(3):217–225.
5. Aschoff A, Kremer P, Hashemi B, Kunze S. The scientific history of hydrocephalus and its treatment. Neurosurg Rev 1999;22(2-3):67–93.
6. Iantosca MR, Hader WJ, Drake JM. Results of endoscopic third ventriculostomy. Neurosurg Clin N Am 2004;15(1):67–75.
7. Engelman RM, Ransohoff J, Cortes LE, Spencer FC. Complications of ventriculoatrial shunting for hydrocephalus requiring cardiac operation. Annals of Thoracic Surgery 1969;8(5):464–469.
8. Becker DP, Nulsen FE. Control of hydrocephalus by valve-regulated venous shunt: avoidance of complications in prolonged shunt maintenance. J Neurosurg 1968 March;28(3): 215–226.
9. Muke R, Glashoff M. The longitudinal increase of the internal jugular vein and the upper v. cava as measured between the mastoid to the heart: parameter for timing the reoperation after ventriculocardiostomy (author’s transl). Monatsschr Kinderheilkd 1976 April;124(4):157–161.
10. Svien HJ, Dodge HW Jr., Lake CF. Ventriculomastoid shunt in the management of obstruction to the aqueduct of sylvius in the adult; report of case. Mayo Clinic Proceedings 1952 May;2127(11):215–218.
11. Sharkey PC. Ventriculosagittal-sinus shunt. Journal of Neurosurgery 1965 April 22;362–367.
12. Smith GW, Moretz WH, Pritchard WL. Ventriculo-biliary shunt; a new treatment for hydrocephalus. 1958:Surgical Forum 9:701–705.
13. Ransohoff J. Ventriculo-pleural anastomosis in treatment of midline obstructional neoplasms. Journal of Neurosurgery. 1954 May;11(3):295–298.
14. Jackson IJ, Snodgrass SR. Peritoneal shunts in the treatment of hydrocephalus and increased intracranial pressure: a 4-year survey of 62 patients. Journal of Neurosurgery 1955 May; 12(3):216–222.
15. Cochrane DD, Kestle JR. The influence of surgical operative experience on the duration of first ventriculoperitoneal shunt function and infection. Pediatr Neurosurg 2003 June;38(6):295–301.
16. Griebel R, Khan M, Tan L. CSF shunt complications: an analysis of contributory factors. Child’s Nerv Syst 1985;1(2):77–80.
17. Guertin SR. Cerebrospinal fluid shunts. Evaluation, complications, and crisis management. Pediatr Clin North Am 1987;34(1):203–217.
18. McGirt MJ, Leveque JC, Wellons JC3, Villavicencio AT, Hopkins JS, Fuchs HE, et al. Cerebrospinal fluid shunt survival and etiology of failures: a seven-year institutional experience. Pediatr Neurosurg 2002;36(5):248–255.
19. Piatt JH Jr., Carlson CV. A search for determinants of cerebrospinal fluid shunt survival: retrospective analysis of a 14-year institutional experience. Pediatr Neurosurg 1993;19(5):233–241.
20. Tuli S, Drake JM. Multiple shunt failures: an analysis of relevant features. Child’s Nerv Syst 1999;15(2-3):79.
21. Pollack IF, Albright AL, Adelson PD. A randomized, controlled study of a programmable shunt valve versus a conventional valve for patients with hydrocephalus. Hakim-Medos Investigator Group. Neurosurgery 1999 December;45(6):1399–1408.
22. Drake JM, Kestle J. Rationale and methodology of the multicenter pediatric cerebrospinal fluid shunt design trial. Pediatric Hydrocephalus Treatment Evaluation Group. Child’s Nerv Syst 1996;12(8):434–447.
23. Drake JM, Kestle J. Determining the best cerebrospinal fluid shunt valve design: the pediatric valve design trial. Neurosurgery 1996;38(3):604–607.
24. Drake JM, Kestle JR, Milner R, Cinalli G, Boop F, Piatt J Jr., et al. Randomized trial of cerebrospinal fluid shunt valve design in pediatric hydrocephalus. Neurosurgery 1998;43(2):294–303.
25. Borgbjerg BM, Gjerris F, Albeck MJ, Borgesen SE. Risk of infection after cerebrospinal fluid shunt: an analysis of 884 first-time shunts. Acta Neurochir (Wien) 1995;136 (1-2):1–7.
26. Drake JM. Shunt infections. J Neurosurg 1996;855:985–986.
27. Walters BC. Cerebrospinal fluid shunt infection. Neurosurg Clin N Am 1992;3(2):387–401.
28. Corlett K. Septicaemia and meningitis in hydrocephalus. Report of a case complicating ventriculo-atrial shunt with a Spitz-Holter valve. Northwest Medicine 1960 November;59:549–551.
29. Kulkarni AV, Rabin D, Lamberti-Pasculli M, Drake JM. Repeat cerebrospinal fluid shunt infection in children. Pediatr Neurosurg 2001;35(2):66–71.
30. Bryant MJ, McEniery J, Walker DG, Campbell R, Lister B, Sargent P, et al. Preliminary study of shunt related death in paediatric patients. J Clin Neurosci 2004;11(6):614–615.
31. McGirt MJ, Zaas A, Fuchs HE, George TM, Kaye K, Sexton DJ. Risk factors for pediatric ventriculoperitoneal shunt infection and predictors of infectious pathogens. Clin Infect Dis 2003;36(7):858–862.
32. Enger PO, Svendsen F, Wester K. CSF shunt infections in children: experiences from a population-based study. Acta Neurochir (Wien) 2003;145(4):243–248.
33. Tuli S, Tuli J, Drake J, Spears J. Predictors of death in pediatric patients requiring cerebrospinal fluid shunts. J Neurosurg Spine 2004;100(5):442–446.
34. Cochrane DD, Kestle J. Ventricular shunting for hydrocephalus in children: patients, procedures, surgeons and institutions in English Canada, 1989-2001. European Journal of Pediatric Surgery 2002 December;12 Suppl 1:S6–S11.
35. Schreffler RT, Schreffler AJ, Wittler RR. Treatment of cerebrospinal fluid shunt infections: a decision analysis. Pediatr Infect Dis J 2002;21(7):632–636.
36. Darouiche RO. Treatment of infections associated with surgical implants. N Engl J Med 2004;350(14):1422–1429.
37. Smith ER, Butler WE, Barker FG. In-hospital mortality rates after ventriculoperitoneal shunt procedures in the United States, 1998 to 2000: relation to hospital and surgeon volume of care. J Neurosurg Spine 2004 February;100(2):90–97.
38. Smith ER, Butler WE, Barker FG. In-hospital mortality rates after ventriculoperitoneal shunt procedures in the United States, 1998 to 2000: relation to hospital and surgeon volume of care. [see comment]. Journal of Neurosurgery Spine 2004 February;100(2): 90–97.
39. Blum J, Schwarz M, Voth D. Antibiotic single-dose prophylaxis of shunt infections. Neurosurg Rev 1989;12(3):239–244.
40. Faillace WJ. A no-touch technique protocol to diminish cerebrospinal fluid shunt infection. Surg Neurol 1995;43(4):344–350.
41. Fitzgerald R, Connolly B. An operative technique to reduce valve colonisation. Z Kinderchir 1984;39 Suppl 2:107–108.
42. Gardner P, Leipzig TJ, Sadigh M. Infections of mechanical cerebrospinal fluid shunts. Curr Clin Top Infect Dis 1988;9:185–214.
43. Kulkarni AV, Drake JM, Lamberti-Pasculli M. Cerebrospinal fluid shunt infection: a prospective study of risk factors. J Neurosurg 2001;94(2):195–201.
44. Arze RS, Rashid H, Morley R, Ward MK, Kerr DN. Shunt nephritis: report of two cases and review of the literature. Clin Nephrol 1983;19(1):48–53.
45. Futrakul P, Surapathana LO, Campbell RA. Review of shunt nephritis. J Med Assoc Thai 1970 April;53(4):265–274.
46. Meadow R. Shunt nephritis: renal disease associated with infected ventriculo-atrial shunts. Dev Med Child Neurol 1973 February;15(1):83-84.
47. Bryant MS, Bremer AM, Tepas JJ3, Mollitt DL, Nquyen TQ, Talbert JL. Abdominal complications of ventriculoperitoneal shunts. Case reports and review of the literature. Am Surg 1988;54(1):50–55.
48. Ersahin Y, Mutluer S, Tekeli G. Abdominal cerebrospinal fluid pseudocysts. Child’s Nerv Syst 1996;12(12):755–758.
49. Rainov N, Schobess A, Heidecke V, Burkert W. Abdominal CSF pseudocysts in patients with ventriculo-peritoneal shunts. Report of fourteen cases and review of the literature. Acta Neurochir (Wien) 1994;1271-1272:73–78.
50. Gaskill SJ, Marlin AE. Pseudocysts of the abdomen associated with ventriculoperitoneal shunts: a report of twelve cases and a review of the literature. Pediatr Neurosci 1989;15(1):23–26.
51. Govender ST, Nathoo N, van Dellen JR. Evaluation of an antibiotic-impregnated shunt system for the treatment of hydrocephalus. [see comment]. Journal of Neurosurgery 2003 November;99(5):831–839.
52. Govender ST, Nathoo N, van Dellen JR. Evaluation of an antibiotic-impregnated shunt system for the treatment of hydrocephalus. J Neurosurg 2003;99(5):831-839.
53. Hammock MK, Milhorat TH, McClenathan JE. Expanding ventricular shunts for hydrocephalus in infancy and childhood. Dev Med Child Neurol Suppl 1975;(35):89–93.
54. Bridges N, Rifkinson-Mann S, Handler M, Epstein F. Planning ventriculoperitoneal shunts in infants and small children. Pediatr Neurosci 1988;14(4):191-195.
55. Goldenberg TM, Pritz MB. A simple method for distal catheter lengthening of ventriculoperitoneal shunts. Technical note. J Neurosurg 1992 November;77(5):810–811.
56. Mittal P, Chand M, Chhabra DK. A simplified technique for lengthening peritoneal catheters. Br J Neurosurg 1992;6(5):475–476.
57. Ninomiya H, Yoshimine T, Maruno M, Kato A, Hayakawa T. A peritoneal marker for optimal timing of ventriculo-peritoneal shunt revision in early childhood. Neurol Res 1998 September;20(6):526–528.
58. Couldwell WT, LeMay DR, McComb JG. Experience with use of extended length peritoneal shunt catheters. Journal of Neurosurgery 1996 September;85(3):425-427.
59. Langmoen IA, Lundar T, Vatne K, Hovind KH. Occurrence and management of fractured peripheral catheters in CSF shunts. Child’s Nerv Syst 1992 June;8(4):222–225.
60. Cuka GM, Hellbusch LC. Fractures of the peritoneal catheter of cerebrospinal fluid shunts. Pediatr Neurosurg 1995;22(2):101–103.
61. Ramsey DB, Chadduck WM. Premature fractures of platinum-cured Silastic shunts. Child’s Nerv Syst 1992 October;8(7):406–410.
62. CDRH (Center for Devices and Radiological Health). Device Classes. Web Page. Online 2002 November 21; Available from: URL: http://www.fda.gov/cdrh/devadvice/3132.html#class_3.
63. Pena C, Bowsher K, Samuels-Reid J. FDA-approved neurologic devices intended for use in infants, children, and adolescents. Neurology 2004;63(7):1163–1167.
64. CDRH. Shunt Technology: Challenges and Emerging Directions. Online 1999 January 8; Available from: URL: http://www.fda.gov/cdrh/stamp/shuntconf.pdf.
65. CDRH. Releasable 510(k) Database. Web Page. Online 2005 May 5; Available from: URL: http://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfPMN/pmn.cfm.
66. CDRH. File Layout for Releasable 510(k)s. Web Page. Online 2005 June 6; Available from: URL: http://www.fda.gov/cdrh/pmndoc.html.
67. CDRH. 510(k) Summary for HP Valve System (K974739). Online 1997 December 18; Available from: URL: http://www.fda.gov/cdrh/pdf/k974739.pdf.
68. CDRH. Device Advice: Postmarket Surveillance Studies. Web Page. Online 1 A.D. June 8; Available from: URL: http://www.fda.gov/cdrh/devadvice/352.html.
69. ECRI. Health Device Alerts. Online 2003 December 1; Available from: URL: http://www.ecri.org/demo/hdimagingdemo/hdimgfaq.html.
70. Ramsey DB, Chadduck WM. Premature fractures of platinum-cured Silastic shunts. Child’s Nerv Syst 1992 October;8(7):406–410.
71. Langmoen IA, Lundar T, Vatne K, Hovind KH. Occurrence and management of fractured peripheral catheters in CSF shunts. Child’s Nerv Syst 1992 June;8(4):222–225.
72. Cunningham CD, III, Slattery WH, III, Luxford WM. Postoperative infection in cochlear implant patients. Otolaryngol Head Neck Surg 2004 July;131(1):109–114.
73. Yu KC, Hegarty JL, Gantz BJ, Lalwani AK. Conservative management of infections in cochlear implant recipients. Otolaryngol Head Neck Surg 2001 July;125(1):66–70.
74. Reefhuis J, Honein MA, Whitney CG, Chamany S, Mann EA, Biernath KR, et al. Risk of bacterial meningitis in children with cochlear implants. N Engl J Med 2003 July 31;349(5):435–445.
75. Kestle JR, Drake JM, Cochrane DD, Milner R, Walker ML, Abbott R 3rd, et al. Lack of benefit of endoscopic ventriculoperitoneal shunt insertion: a multicenter randomized trial. J Neurosurg 2003;98(2):284–290.
76. Albright AL, Haines SJ, Taylor FH. Function of parietal and frontal shunts in childhood hydrocephalus. J Neurosurg 1988;69(6):883–886.



































