
2
Medical Devices for Infants, Children, and Adolescents
“‘We have to show we can make this work with the wrong equipment, and then convince someone to make us the right equipment,’ Lock explained. He told me that the first device he tried to create for children was an instrument to open a stenosis, or closure, of two portals to the heart: the aortic and the mitral valves. If Lock could dilate these valves using a tiny catheter, a child with the condition could avoid open-heart surgery. He went to … a prominent medical-device manufacturer. The company suggested that he use a catheter designed to open a small artery in the abdomen of an adult. ‘They told me there wasn’t a market,’ he recalled. So, for three years, Lock used the abdominal catheter to open the aortic and mitral valves of adults. This was relatively successful, and … [the company], convinced that there was an adult market, agreed to make an aortic-and-mitral-valve catheter—for adults. ‘As an act of charity only, they made a few pediatric-shaped catheters,’ Lock said. ‘It’s unlikely that we would ever have got the pediatric catheters built if there hadn’t been an adult market—which we had to invent.’”
Jerome Groopman, 2005, p. 361
This saga of medical device innovation illustrates both the special needs of children and the challenges of getting to the market a device that meets those needs. As discussed in this chapter, some medical devices may require no adaptation to be safely used with children, and some may be made smaller or otherwise successfully adapted for pediatric use. Other devices are not suitable for some or most children because, for example, they cannot be made small enough or they will interfere with or be compromised by children’s growth.
Many pediatricians and other children’s advocates are dismayed by the lack of satisfactory medical devices for children with certain serious medical
problems, and they have also expressed concern about the limited testing of device safety and effectiveness with children of different ages (AAP et al., 2004b; FDA, 2004y). These views echo those long expressed about pharmaceuticals for children (AAP, 1977, 1995). Doubtful that government or industry will invest significant resources to develop specialized devices and drugs for children, some children’s hospitals have begun their own programs to do so. For example, Children’s Hospital Boston has announced a Pediatric Product Development Initiative that is focusing on the initial stage of development for pediatric devices that have the potential to attract commercial investors once a promising prototype is produced (Kong, 2004).
Notwithstanding the desirability of medical devices that meet children’s needs, the reality is that children are not the primary patient population for most complex medical devices. Most children living in developed countries are basically healthy. Compared to adults, especially older adults, they are far less likely to die, be hospitalized, or suffer serious illness. In the United States in 2001, people under age 20—who constitute nearly 30 percent of the population—accounted for about 2 percent of deaths and about 11 percent of hospital discharges (excluding newborns) (Arias et al., 2003; Kozak et al., 2004). In 2003, over 80 percent of children had health status reported as “excellent” or “very good” compared to about 65 percent of people aged 18 to 64 and about 39 percent of people aged 65 or over (Schiller et al., 2004).
Children are, however, overrepresented or uniquely represented among certain causes of suffering and death, notably those associated with prematurity and congenital anomalies (a diverse group of malformations, deformations, and chromosomal abnormalities present at birth). In addition, although individuals under age 20 account for about 10 percent of fatal injuries (intentional and unintentional injuries, 2001 data), they account for over one-third of nonfatal injuries (2002 data) (NCIPC, 2001, 2002).
Children, particularly newborns, are also overrepresented or uniquely represented among patients treated with certain medical devices. These devices include infant incubators, devices for closing the patent ductus arteriosus (a congenital blood vessel defect), home apnea monitors, and devices for screening hearing in newborns.
The committee found current data on medical device use by age group to be virtually nonexistent, but children clearly form a small proportion overall of patients in need of or treated with devices for serious medical conditions. One consequence is that manufacturers and others assessing the market for new, refined, or modified devices have weak incentives to focus on children’s special needs and characteristics.
This chapter provides a specific pediatric context for the consideration of postmarket surveillance of medical devices used with children. It reviews definitions of pediatric subgroups (infant, child, adolescent) as well as two terms in the committee’s statement of task—growth and development and
active lifestyle. It also discusses ways in which the design and use of devices have been modified (or cannot yet be successfully modified) to accommodate children’s special characteristics. The concluding section describes how problems with pediatric use of medical devices may be identified: a priori based on a combination of expert understanding of children’s developmental characteristics and detailed knowledge of the operating characteristics of a particular device; during the clinical testing of a device with children to demonstrate safety and effectiveness to the U.S. Food and Drug Administration (FDA); and as experience with a device accumulates following its entry into the market.
DEFINITIONS
Infant, Child, Adolescent
As described in Chapter 1, this report generally uses the term children broadly to include all pediatric age groups. Definitions vary for the subgroups infant, child, and adolescent and tend to reflect the concerns and purposes of those developing them. The definitions serve primarily as general guides that encourage attention by clinicians, researchers, policymakers, and others to developmental differences—physical, cognitive, and psychological—within young populations and between younger and older populations.
In guidance documents on pediatric drug testing and assessment of pediatric medical devices, the FDA has defined infants as those younger than 2 years of age (FDA, 1994, 2004p; ICH, 2000).2 The discussion of drug testing notes that this is a period of rapid development in the central nervous system, immune system, renal and hepatic pathways of drug clearance, and body size. While this development is underway, products that are reasonably safe for adults and even older children may be riskier, even lethal, for babies. (An example is verapamil, an antiarrhythmic medication that should be avoided in the young infant.)
The guidance for medical devices does not single out infancy as a period of particular vulnerability, although instances of vulnerability can be cited. For example, studies have repeatedly found infection rates for implanted cerebrospinal fluid shunts to be higher in premature neonates than in other children (see, e.g., McGirt et al., 2003). With respect to certain medical procedures, however, infancy may be a preferred time for interven-
tion because of the infant brain’s particular plasticity, that is, its ability to mold or shape itself to accommodate to damage in one area with new growth and connections to other areas to achieve the same function (Johnston et al., 2001; Luciana, 2003). Studies of long-term outcomes are still important to determine whether such accommodation compromises other neurodevelopmental processes as the child matures.
Although the wording is slightly different in documents on drugs and devices, a child is regarded as an individual between the ages of 2 and 11 (FDA, 1994, unpaged; ICH, 2000; FDA, 2004p). For certain conditions and medical products, differences within this age group in cognitive and emotional development may be significant, particularly with respect to the use of medical devices that normally require user set-up steps, user programming, or other patient-initiated control. What is well beyond the capabilities of a 3-year-old may be quite manageable for a 10-year-old. Physiologic differences within the 2-to-11 age group also may be important. For example, younger children whose bones heal more quickly may be safely treated for femoral fractures using a body cast, whereas children aged 6 to 12, who heal more slowly, may benefit from surgical treatment that requires a shorter period of incapacitation (see discussion of titanium nails below).
For drugs and devices, FDA documents differ on the age range for adolescents. For drugs, the range is ages 12 to 15 (“up to age 16”) (FDA, 1994, unpaged). For medical devices, however, the upper end of the range is age 20 (or “up to the age of 21”) (FDA, 2004p, p. 4). In specifying this broader range, the guidance on medical devices states: “Given the scope of medical devices and the impact that a device could have on a growing adolescent, as well as the effect growth could have on the device, we believe that including the upper age limit identified above may be useful for some devices and device clinical trials” (FDA, 2004p, p. 4). No specific instances are cited. The guidance also notes that “the descriptions are somewhat arbitrary and that, in fact, the subject’s weight, body size, physiological development, neurological development, and neuromuscular coordination may often be more appropriate indicators than chronological age” (FDA, 2004p, pp. 4–5).3
Growth and Development
From infancy through adolescence, children mature physically, cognitively, and emotionally. Pediatrics as a medical specialty arose out of recognition that the care of children should be informed by knowledge of how children’s growth and development may affect and be affected by clinical care. One question for this report, as examined in Chapter 6, was whether postmarket surveillance studies continue long enough to evaluate the effects of growth and development on the longevity of implanted devices.
Growth in size is one of the most obvious aspects of the human passage from birth to adulthood. As discussed further below, some implants have limited or suboptimal pediatric applications because they cannot grow as the child grows or they interfere with growth. Many implants (e.g., artificial blood vessels [synthetic conduits]) can be replaced with larger sizes as a child grows, but repetitive surgery for this purpose presents risks, including difficulties related to dissection through accumulated scar tissue. Children’s growth and development may dictate a more frequent monitoring schedule than is necessary with adults.
Growth can also shift the location of an implanted device, for example, when an implant fixed to bone migrates to an undesired place as the bone grows. One goal for innovation in biotechnology is to devise materials and processes that allow implants to grow with a child or work in ways that do not interfere with growth.
In addition to physical growth and size, other developmental differences or changes may be relevant to medical product use. For example, as children develop, they not only get larger but also gain in strength, dexterity, and gross and fine motor skills (Pena et al., 2004). These gains may be necessary before they can independently operate certain devices such as patient-operated drug infusion or electrical stimulation systems.
Metabolic, hormonal, and other developmental differences may be relevant for some implanted and other devices (e.g., cardiopulmonary bypass machines) that come into contact with a child’s tissues. Questions have, for example, been raised about the long-term consequences of extensive infant exposure to a chemical used in making plastic for the tubing employed in an array of devices for neonatal intensive care (Rais-Bahrami et al., 2004).
To cite yet another kind of developmental consideration, clinicians and researchers have identified tactile, visual, and auditory overstimulation as a concern for ill infants who depend on ventilators and other medical devices to assist their breathing, nutrition, and other functions (ATS, 2003). Guidelines for clinicians have suggested modifications in the setting and use of treatments to minimize such stress.
Cognitive and psychosocial development and social environments are relevant to the safe pediatric use of certain medical devices, particularly
complex devices used outside the hospital. For chronically ill children reliant on medical devices, psychological and intellectual development (assuming their condition permits it) includes learning how to manage the devices independently and safely. For example, older children who have a tracheostomy can learn to manage a device that permits them to talk with the tracheostomy tube in place.
Sometimes psychosocial development brings risks. For example, older adolescents may be less receptive than children and younger adolescents to parental monitoring of adherence to practices necessary for safe and effective device use. In a similar vein, a clinician in a large pediatric diabetes center has written that “[t]eens are probably the least reliable group to start on the [insulin infusion] pump” because they easily learn to use it but “are typically preoccupied with many other things, and the pump quickly goes down on the priority list” (Ahern, 2001, unpaged).
Risk-taking behavior by adolescents is, generally, a long-standing public health concern (see, e.g., Rolison and Scherman, 2002; Kelley et al., 2004; Steinberg, 2004). Some recent research suggests that the areas of the brain that limit such behavior may not fully mature until a person reaches the mid-twenties (Giedd, 2004).
Finally, one consequence of children’s developmental characteristics is that children often depend on their parents or other adults to provide their medical history to clinicians and to answer and ask questions about a medical problem or its care. Many survey-based measures of pediatric health care quality and outcomes have different forms for children of different ages, and those involving younger children often direct questions at parents (see, e.g., Hermida et al., 1999; Bradlyn et al., 2003; Beal et al., 2004).
Children’s Active Lifestyles
“It has been quite a while since I have had a [hydrocephalus shunt] revision…. There was a period in time when I had five or six in a row, just back to back. The main reason for that [was that] I was racing wheelchair competitively, at the national level, for a while…. The shunt really couldn’t keep up with the strenuous activity. It couldn’t drain the fluid off my brain fast enough…. Eventually, we found a valve that would drain the fluid quick enough.”
Ben Harder, 2004
Another question considered in this report is whether postmarket surveillance studies are adequate to evaluate how children’s active lifestyles may affect failure rates and longevity for implanted devices (see Chapter 6). Although the term active lifestyle may convey an image of a child in motion
with the potential for colliding with other beings or objects, it has no common clinical or behavioral definition.
The committee interprets children’s active lifestyles as having physical, cognitive, emotional, behavioral, and social aspects that are affected by a child’s stage of development. Physical aspects relevant to device safety include the types of activities engaged in by children of different ages, the environments in which they occur, and their frequency, duration, and intensity.
When FDA granted a humanitarian device exemption for use of a deep brain stimulator with dystonia patients who are 7 years of age or older, it noted that children’s active play and sports participation could damage elements of the implant. It went on to say that “[w]hile some degree of rough play may be unavoidable, children should be advised to avoid games, sports and other pastimes where a strain to the lead/connector assembly or a percussive injury to system components may be likely to occur (e.g., soccer, football/rugby)” (H020007, FDA, 2003, p. 3). (Chronic intractable dystonia is a serious neurological condition characterized by involuntary muscle spasms and abnormal postures or movements.)
The developmental control that children can exert over their physical activity is also relevant to device safety. For example, an infant in a crib and a cognitively intact 14-year-old confined to bed due to illness or injury may both be relatively inactive. The adolescent can, however, be expected to have more awareness of and control over movements such as rolling over that might dislodge or otherwise impair the functioning of a medical device such as a catheter or a breathing tube. Likewise, a 5-year-old and a 25-year-old who have had a cardiac pacemaker implanted may each know that they need to protect the device, but developmental differences in the understanding of risk and causation and in the control of impulses increase the probability of risky behavior by the child, for example, jumping off a porch (see, e.g., Giedd, 2004).
Surgeons may modify their procedures to take children’s activity levels into account. For example, surgeons who perform craniofacial surgery that requires a tracheal or breathing tube may secure the tube by placing wire sutures through the gum because of the high risk of having this tube inadvertently dislodged by the movement of a child and the extreme difficulty of replacing the tube when the facial structures are swollen.
Surgeons implanting a pacemaker in a very young child often will place the pacemaker generator in the abdominal wall, where extra tissue provides more protection than is offered by the usual location near the collarbone. With the usual location, the surgeon connects the generator to the inside of the heart by passing the pacemaker’s leads (small wires) through a vein in the chest. With abdominal placement, surgeons tunnel
the leads through the chest tissue and then stitch them to the outside of the heart. This approach allows the surgeons to avoid placing the leads through young children’s small subclavian veins, where they might cause a thrombosis that would complicate access for pacemaker leads in future years if needed, for example, as the child grows or if problems arise with the original leads.
The social dimensions of children’s lifestyles, especially adolescent lifestyles, are sometimes featured in discussions of medical devices such as insulin pumps and catheters for peritoneal dialysis that require special attention while users are away from home. Websites for children and teens with diabetes provide tips for living with the insulin pump and include discussion of clothing, eating, school physical education activities, and swimming and other sports.
In addition, as the committee heard during its meeting with families, the desire to be or appear “normal” may cause older children and adolescents—with mixed emotions and reactions from their parents—to engage in activities (e.g., playing contact sports while having an implanted pacemaker) that place great stress on implanted or partly implanted devices such as catheters. Some complications associated with pediatric use of medical devices may result from patient activities that have a realistic potential for harm, given the inherent limitations or characteristics of the device. A continuing interest of clinicians, parents, and device manufacturers is strategies for “child-proofing” devices by changing their design or use.
For infants and toddlers, the use of medical devices may also need to take into account another “lifestyle” factor—their lifting, holding, carrying, and other handling by adults. Just as special precautions may be needed to safely secure a device for infant or child activity, so additional precautions may need to be taken with medical devices to accommodate normal child care.
Yet another consideration is that children living at home with complex medical devices often have siblings whose own “active lifestyle” may create safety issues. On the one hand, siblings could endanger an ill brother or sister through play that dislodges or otherwise interferes with a device. On the other hand, playful or curious siblings could encounter electrical and other hazards to themselves. For example, children have electrocuted themselves after inserting partially or completely disconnected electrode wires from a sibling’s cardiorespiratory monitor into wall outlets (Katcher et al., 1986). Family education and parental monitoring are essential safeguards, but thoughtful design or choice of device that considers home environment and other “human factors” also has a role to play in protecting the safety of all members of a family.
FDA GUIDANCE ON ASSESSMENT OF PEDIATRIC MEDICAL DEVICES
As required by the Medical Device User Fee and Modernization Act of 2002 (P.L. 107–250), FDA recently provided guidance on the premarket assessment of pediatric medical devices. The guidance includes a general discussion of developmental considerations and lists a number of factors that should be considered in designing devices or planning clinical studies of devices (FDA, 2004p). The listed factors, which are not mutually exclusive or exhaustive, are
-
height and weight;
-
growth and development;
-
disease or condition;
-
hormonal influences;
-
anatomical and physiological differences from the adult population;
-
activity and maturity level; and
-
immune status.
In a further discussion of “unique host characteristics” of pediatric patients, the guidance offers some illustrations of how these characteristics might figure in the assessment of a medical device. For example, in recommending that assessments consider stage of puberty and other developmental milestones, the guidance suggests that clinicians should consider breast bud development in the placement of certain medical devices in infant or young girls (e.g., placement of chest tube in a tiny infant to relieve air or fluid that has collected in the chest, but outside the lungs).
The guidance also summarized the circumstances when clinical data for pediatric populations are appropriate. These circumstances are when
-
supporting information from sources, such as preclinical bench or animal testing, literature, or adult clinical trials, are inadequate to establish safety and effectiveness for the pediatric indication;
-
adult data are inadequate to predict pediatric risks and adverse events;
-
pediatric data are needed for validation of design modifications; or
-
pediatric data are needed to develop an age-appropriate treatment regimen.
Specific testing requirements will vary depending on the device. FDA, however, stated that its expectations for such tests generally involve the same basic questions and procedures for both adult and pediatric populations.
An article by FDA staff includes additional discussion of pediatric factors as they relate to neurological devices (Pena et al., 2004). With respect
to surgical risks, for example, the authors cite concerns about blood loss for patients with small volumes of blood, possible need for sedation for children who cannot control movement during procedures, and repeat surgeries associated with device replacement or growth-associated migration of a device. They note that of 19 high-risk medical devices involving the central and peripheral nervous system that FDA approved between 1994 and 2003, 8 included indications for use in children as well as adults.
In addition to the guidance on premarket assessment, the agency issued guidance on procedures to ensure that advisory panels that review documents such as applications for premarket approval of medical devices appropriately include or consult with pediatric experts. This guidance, which responds to another provision in the Medical Device User Fee and Modernization Act (21 USC 360(e)(c)), provides for pediatric expertise to be available (through consultation or inclusion in panel deliberations) in a range of situations. These include when
-
there are labeled indications for use that include a pediatric subpopulation or there is a reasonable likelihood that the device would be used in a pediatric subpopulation for the labeled indication;
-
there are data in the study that include a pediatric subpopulation;
-
there is a reasonable likelihood that the data from the study in the adult population may be used by the applicant to subsequently support a pediatric indication;
-
there is a need for advisory panel input on a study design and/or protocol for use of the device in the pediatric population; or
-
there is a reasonable likelihood that the advisory panel may discuss the potential use of the device in the pediatric population.
The next section of this chapter considers how children’s special needs and characteristics may be taken into account in the design and use of medical devices. The descriptive categorizations reflect the committee’s experience and perspectives, reviews of the literature, and information provided during public meetings and other discussions with experts.
DEVICE DESIGN, DEVICE USE, AND DEVELOPMENTAL DIFFERENCES
Children are not small adults—a cliché but true. As described above, children, especially infants and young children, differ from adults in ways that extend beyond the obvious difference in size. These differences may have implications for the design and use of devices and for the methods to evaluate their safety and effectiveness before and after marketing.
Developmental differences between children and adults related to the safe and effective use of medical products have been most extensively analyzed and described for drugs.4 For drugs, scientists and clinicians have constructed a strong rationale for pediatric drug research to assure the safe and effective use of medications with children (see, e.g., Shirkey, 1968; AAP, 1995; Kearns and Winter, 2003; IOM, 2000b, 2004a; Reed and Gal, 2004). Data indicating that some 80 percent of medications listed in the Physician’s Desk Reference lacked any prescribing information for children have also been cited to build the case for such research (AAP, 1995; Steinbrook, 2002).
For medical devices, the committee found nothing equivalent to the pharmacology literature on developmental concerns. With drugs, one is generally considering issues along a spectrum: ingestion, bioavailability, action, untoward actions, metabolism, and disposal of metabolites. This is complex enough. With devices, one might be considering physical interactions (e.g., when a device exerts pressure on skin), metabolic interactions (if a device or component is not inert), and growth (if a device is implanted or connected with a child over an extended period), among other factors. Box 2.1 summarizes some of the developmental considerations for drugs compared to medical devices.
To the extent that pediatric considerations are known for a medical device, the labeling of the device should reflect that knowledge. In some cases, labeling will state that use of a device is not indicated in those under a certain age or those who are not skeletally mature. In other cases, the labeling may describe adaptations or cautions related to pediatric use (see, e.g., the discussion earlier of the deep brain stimulator).
Spectrum of Medical Device Use with Children
The use of medical devices with children spans a wide spectrum, including devices that are used uniquely with children, devices that are reduced in size or otherwise modified for use with children, and devices that do not differ for adult and pediatric use (although some procedures for their use may vary). Use with children is explicitly precluded for some devices.
Box 2.2 and the following discussion illustrate the spectrum of pediatric device use. The use of particular devices as examples does not necessarily imply a committee judgment that the devices have been adequately studied for short- or long-term safety or effectiveness in use with children.
|
BOX 2.1 Drugs
Devices
|
Devices Unique or Nearly Unique to Children
Sometimes children, especially infants, have unique needs or conditions for which specialized devices are developed. The infant incubator is an obvious example.
In addition, children may so dominate the target population in need of a device that the consideration of adult users is secondary rather than primary. One example of a device initially developed for children is the atrial septal occluder. It was originally intended to treat children who have a hole in the wall separating the inflow chambers of the left and right sides of the heart, but it has also been used to treat adults with that condition (Omeish and Hijazi, 2001; Thomson et al., 2002). To cite another example, FDA recently granted limited approval (through a Humanitarian Device Exemption, as discussed in Chapter 4) for a pulmonary valved conduit (a kind of heart valve) that is, again, primarily intended to correct congenital heart defects in children but can be used with adults (H020003, FDA, 2003a). (Although congenital heart disease is sometimes regarded as a condition of children, estimates suggest that there are at least as many adults living with congenital heart disease as there are children [Warnes et al., 2001; Hoffman et al., 2004]).
|
BOX 2.2 Devices unique to children
Devices developed primarily for children but also used with adults
Same core device, different accessories for pediatric use
Variations in device use or technique to accommodate developmental differences
Devices that vary in size for use with small patients
|
Same Device for All Patients
Sometimes the same monitoring, diagnostic, or therapeutic medical device is used for adults and all or most children without requiring pediatric modifications in size, design, or key accessories. For example, ear thermometers do not vary in size for adults and children nor do syringes, although needle sizes vary. Nonetheless, even when devices are identical or very similar, clinical care of children may still differ from care of adults in general ways, for example, by involving physicians, nurses, and others experienced in pediatric care and by providing physical settings that are “child friendly.”
In some cases, adaptations in devices or accessories have followed the identification of adverse outcomes in children. For example, the same size tubing was initially used for all mechanically ventilated patients to connect
the ventilators to the tracheal tube, but the heavy weight of the tubing created problems for infants and children because the tubing tugged on the breathing tube, making it more likely to dislodge. This led to the use of child-appropriate tubing. To cite another example, when problems arose in the use with young children of adult ventilators that had high gas flow rates, companies developed ventilators that provide smaller breaths of air and a slower gas flow rate.
Some innovations—notably, successful miniaturization of device components—may allow a move away from devices made in different sizes toward “one-size-fits-all (or most)” devices. For example, a new left ventricular assist device that is intended to reduce the size and weight of first-generation devices was aimed primarily at adults but the size also allows use with children. (In 2004, FDA approved a humanitarian device exemption for this device for use with children aged 5 to 16 years—before the device was approved for use with adults [H030003, FDA, 2004a].)
Same Core Device, Different Accessories
For some technologies, the core medical device may be the same, but the accessory devices that connect the patient to the device may differ based on patient size or other characteristics. Thus, a dialysis machine can be used for adults and children if the tubing and dialysis coils are reduced in size for small patients. Similarly, a basic pulse oximetry monitor can be used with all age groups, but the sensors that attach the device to the patient vary in size. The sensors may also be attached differently for very young patients (e.g., attached with a gentler adhesive to avoid damaging an infant’s fragile skin or cuffed around an infant’s wrist or ankle rather than attached to a finger).
Another example of a core device with a different accessory for children is the automated external defibrillator for use by first responders outside the hospital. In 2001, FDA approved an external defibrillator for use with children under age 8 (FDA, 2001e). It comes with two sizes of defibrillator pads (which are placed on the chest to deliver the electrical shock). The smaller pads for children attenuate or reduce the shock delivered. The firm that developed the device also collected pediatric heart rhythm information to create new algorithms that determine when a shock is appropriate for a child (Acute Care, no date; Cecchin et al., 2001).5 In
2001, the device was cleared for marketing without testing in children (K003819, FDA, 2001). The sponsor, however, voluntarily agreed to study 50 children for which the device was deployed to gather data on use and results in actual emergency care practice (personal communication, Thomas P. Gross, M.D., Director, Division of Postmarket Surveillance, CDRH, November 7, 2004).
Same Core Device, Adaptations in Programming, Placement, or Operation
When a device does not vary by size of patient, safe and effective applications with children may require other differences or adaptations. Such adaptations may involve changes in the clinical indications for use of a device, the frequency or duration of use for the same indication, the way an implant or its connections are sited in the body, the surgical method used for implantation, or the programming of automated operations such as alarms based on abnormal physiologic signs. For example, many heart rate monitors are used for adults and children, but the alarm settings are adjusted based on the normal range of heart rate for different age groups. As mentioned in the preceding section, the automated external defibrillator approved for use with children under age 8 uses different algorithms for children and adults to determine whether a shock is indicated.
For the deep brain stimulation device described earlier, FDA notes that if two stimulators are used with small patients, their physical placement may need to deviate from that normally used for adults to establish enough separation to minimize electromagnetic interference. For smaller patients, the manufacturer suggests placing one neurostimulator in the abdomen and one in the chest region.6 (See Vidailhet et al., 2005, for a description of a controlled [nonrandomized] prospective study of the device and Greene, 2005, for a commentary on the need for more data, including data on long-term outcomes.)
A different rationale for modifications in device placement applies for central vascular catheters. With infants, it is difficult to identify the physical
landmarks for placing the catheter in the neck (jugular vein) or the upper chest (subclavian vein), and safe placement may require considerable sedation. Therefore, the groin is often selected because landmarks for placement of the catheter can be readily identified and the site can be well anesthetized with less risk of excessive sedation. At the same time, constant moisture, repetitive movement, and other factors make it more difficult to maintain a sterile insertion site in the area. One consequence is that infants with the device tend to show higher rates of infection than adults.
For computer-assisted tomography (CT), several kinds of adaptations have been proposed based on children’s greater vulnerability to the damaging effects of radiation and an increasing recognition that CT scans can deliver significant radiation to children (see, e.g., NRC, 1990; NCI and SPR, 2002). Underscoring the importance of long-term evaluation of medical devices and associated procedures, FDA, the National Cancer Institute, and others have issued guidelines to minimize the harmful effects of repeated diagnostic CT scans on children (Feigal, 2001b; NCI and SPR, 2002; see also Frush and Donnelly, 2001; Frush, 2003). They advise clinicians to
-
take advantage of equipment advances that allow more sensitive dose management;
-
develop and use charts or tables to guide equipment settings based on patient weight or diameter and body area to be scanned;
-
limit repeat scans to what is essential;
-
scan the smallest area of the body possible; and
-
scan at the lowest dose of radiation and lowest level of resolution necessary to achieve needed image quality.
Adjustments in Device Size
Sometimes the physical size of a medical device is the main issue with pediatric use. Infant or child versions exist for many common medical devices such as hospital beds, bandages, and scales.
More complex devices can often be manufactured in sizes to fit all or most pediatric uses without compromising their structure or function. For example, leads for implanted cardiac pacemakers used with children can be made shorter than adult leads without compromising their function, although surgical implantation techniques may vary, especially with infants. To cite another example, a saline-filled testicular prosthetic implant that has been tested in adults and children with a congenitally absent or surgically removed testicle is now available in an extra small size (P020003, FDA, 2002). For this and other implanted devices, replacement with a larger size may be anticipated to accommodate a child’s growth.
Intraocular lens replacement, which has long benefited adults suffering from cataracts, can also help children with certain vision problems. The sizes of the replacement lenses developed for adults are not, however, appropriate for young children. (The mean axial length of a newborn’s eye is 17 mm, whereas that of an adult is 23 to 24 mm.) In addition, the surgical procedure must accommodate developmental considerations such as lower scleral rigidity, greater elasticity of the anterior capsule, and higher vitreous pressure (see, e.g., Dahan, 2000; Ahmadieh and Javadi, 2001; Good, 2001; Pandey et al., 2001).
For certain implanted devices, reductions in size have brought benefits to both children and adults. The cardiac pacemaker is a notable example. Figure 2.1 and Figure 2.2 show the difference in size between an external pacemaker (circa 1957) that was designed to provide short-term life support and a recent implantable pacemaker that offers long-term support.
In some cases, sizing of a device is done at the time of use. For example, cardiac shunts or tubes that are used to connect two blood vessels come in
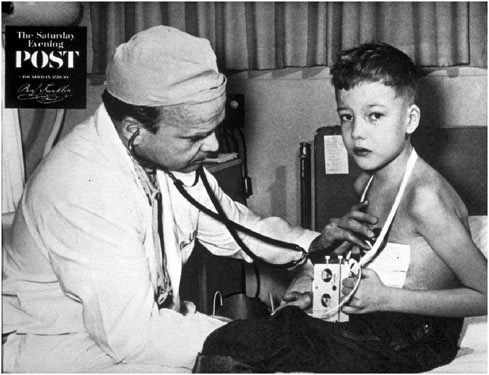
FIGURE 2.1 Child with early external pacemaker, c. 1957 (Used with permission of The Saturday Evening Post).
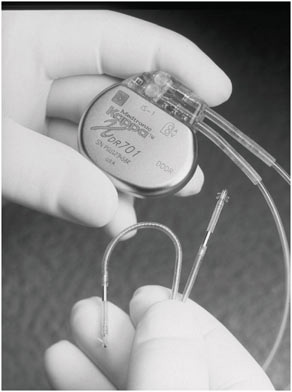
FIGURE 2.2 A modern implantable pacemaker (Medtronic Kappa® 700 series) (Reproduced with permission of Medtronic, Inc.).
different diameters. The appropriate length is determined by the surgeon who cuts them to size just before insertion.
Size adjustments may not be straightforward, even for relatively simple devices. A case in point involves tracheostomy tubes, which are adjusted to fit different size windpipes by creating an array of sizes that vary by increments of 0.5 mm in diameter and simultaneously differ in length. In a child with an abnormally narrow airway, the tube with the appropriate diameter may be too short, which can cause it to become dislodged. Specially constructed tubes can be ordered from the manufacturer, but this option is not feasible in an emergency situation.
Lack of a device in sizes appropriate for the full range of pediatric patients may limit the use of certain interventions. For example, in intracardiac echocardiography (an imaging technique used to guide certain cardiac procedures), the size of the catheter used in the procedure has limited use with very young patients. (The technique, which has not been fully tested in randomized clinical trials, avoids an imaging procedure that requires intu-
bation and general anesthesia [Alboliras and Hijazi, 2004; Zanchetta and Maiolino, 2004].)
Limits to Downsizing or Other Adaptations
Reducing the size of a device that is too large for some or most children may not produce desired results. The reasons may relate to mechanical properties of the device or to characteristics of children other than size. For example, with certain kinds of devices through which blood moves, changes in fluid dynamics in small spaces must be considered. Efforts to shrink left ventricular assist devices (which have been used to support patients awaiting heart transplants) have encountered problems because “blood flow in the smaller version is completely different than in the larger adult heart devices…. [D]ead zones, or low-rate flow zones, can form inside the blood pumps … [and] slow-flowing blood can create clots” (PSU, 2004, unpaged). The miniaturized device mentioned earlier in this chapter, which was not developed specifically for the pediatric population, is designed to minimize such disruptions in blood flow (Bluck and Petty, 2000).
As described above, tracheal tubes are manufactured in different sizes to accommodate the range of airway dimensions of infants, children, and adults, but problems may arise with very small sizes. Although an appropriately sized tube exists to fit in the small airway of the premature infant, the thin wall of the tube has a propensity to kink or buckle, which can cause the tube to become obstructed. Caregivers have developed various means to limit these problems by stabilizing the tube externally (e.g., by wrapping it with tape or taping it to a tongue blade). Also, smaller tubes also are more easily obstructed with mucus and need to be cleared frequently. With a small tube, a small suction catheter is needed, and these small catheters are flimsy and more difficult to use than the larger, thicker suction catheters.
Different limitations arise with mechanical ventilators that are equipped with triggering devices that sense effort by the patient to breathe, which then causes the ventilator to provide a breath. Such triggering devices have not been sensitive enough to detect inspiratory effort in very small babies. Hence, other types of devices, such as a kind of capsule on the chest, have been developed to try to coordinate the effort of the patient and the response of the ventilator.
For sick infants requiring mechanical ventilation, an additional concern is the lack of acceptable face masks. Masks can be made small enough to fit infants, but the pressure needed to create a tight fit and avoid air leakage can bruise or abrade fragile infant skin. Other respiratory devices such as nasal prongs may need to be monitored closely to prevent skin damage and necrosis when used with infants. More generally, the thinness and other
characteristics of children’s skin, especially infant’s, can require care with or modifications of devices that touch the skin to reduce the potential for bruising or skin damage associated with heat, pressure, friction, or other processes.
Skin thickness may also be an issue with certain implants. In a report of pacemaker failures associated with twiddler’s syndrome (when patients deliberately or subconsciously spin the pacemaker’s pulse generator, which can dislodge and damage the pacemaker leads), the authors noted that children might be more susceptible to the problem “because they have thinner subcutaneous tissues, making leads more accessible, and their comprehension of the consequences may be poor” (Abrams and Peart, 1995, p. 190).
Devices That Accommodate Children’s Growth
A unique pediatric problem with the use of certain implanted medical devices is that they either interfere with growth or do not grow as children grow. The approved labeling for a number of orthopedic and other implants describes them as not indicated for individuals with growing bones or skeletal, skull, or other aspect of growth that is less than 90 percent of adult levels (see, e.g., H010002, FDA, 2001; P000057, FDA, 2001; P000058, FDA, 2002; P000013, FDA, 2003).
Some devices or their accessories or the procedures for their use are designed to take children’s growth into account. For example, when surgeons first began to insert the drainage catheter for cerebrospinal fluid shunts into the abdomen, they used tubing just long enough to enter the peritoneal cavity. As children grew these catheters had to be replaced with longer ones. Recent experience suggests that even infants can tolerate a peritoneal catheter long enough to accommodate growth to adulthood (Couldwell et al., 1996). Cardiac pacemaker leads are also implanted so that some significant amount of growth can be accommodated.
Given the risk and discomfort of replacing an implant as a child grows and given the restrictions on the use of certain devices that interfere with (or are compromised by) growth, implants that can “grow” with a child have obvious appeal. Growing children who have bone cancers removed from their limbs and prosthetic devices inserted have faced repeated surgeries to replace or expand the device to accommodate growth. FDA recently approved a device that can be expanded without surgical intervention. As described by FDA, the device employs “a coil that fits around the patient’s leg that produces an electromagnetic field (EMF). The EMF induces an electrical current and subsequent heating of an internal wire [in the implant]. The generated heat softens a polymer locking ring, allowing a slow expansion of an internal compressed spring. The spring expansion pushes the spring housing and femoral housing apart, thus increasing the overall length of the
implant” (FDA, 2003p, unpaged; see also K021489, FDA, 2002). According to the manufacturer’s webpage, FDA has cleared the device for distal femur and proximal tibia implants, but implants for the humerus, proximal femur, and total femur are only available so far under compassionate use guidelines (see Chapter 4) (Wright Medical Technology, Inc., 2004).
To cite another orthopedic example, pediatric orthopedists treating children with leg fractures have increasingly used flexible titanium nails that support the leg as the bone heals but also provide flexibility for growing bones. For children between the ages of approximately 6 and 12, the technique avoids some of the disadvantages of alternative treatments with either a body cast and traction or certain rigid nailing techniques (ECRI, 2004a; Flynn et al, 2004). This technique has not been associated with problems of arrested growth in the trochanter (part of the femur) or osteonecrosis of the head of the femur that have sometimes been reported with rigid nailing techniques (see, e.g., Townsend and Hoffinger, 2000; Alonso and Slongo, 2001; Bartholomew et al., 2001).
Interest in another kind of device, the resorbable implant, is particularly strong among those who treat children with certain craniofacial and orthopedic deformities. These implants are adequately rigid to support repair or reconstruction of a deformity for several months, but they then disappear without requiring removal or replacement and without appreciably interfering with a child’s growth. In a statement to the committee, the American Academy of Pediatrics (AAP) pointed to metal craniofacial fixation devices that create problems with children that are not seen in adults. AAP cited “thinning of scalp leading to annoying prominence of the device … subcutaneous migration of screws … [and] intracranial migration of the devices” (AAP et al., 2004b, p. 8). In the latter process, the device has been engulfed by the child’s growing skull such that “within a few years plates and screws were sometimes found inside the dura resting in the substance of the brain,” a location for which they clearly were not intended.
Until recently, only the results of short-term studies of resorbable implants were available, but investigators have now reported on a combined prospective and retrospective multisite analysis of nearly 2,000 patients under 2 years of age treated over a 5-year period with the same type of device (see, e.g., Eppley et al., 2004). They found a lower rate of device-related complications requiring reoperation than for metal devices and low rates of adverse events (e.g., infections, instability, and foreign-body reaction). Consistent with a characteristic of device innovation, they noted that “the specific types of plates and screws used evolved over the study period from simple plates, meshes, and threaded screws to application-specific plates and threadless push screws whose use varied among the involved surgeons” (Eppley et al., 2004, p. 850).
In an arena that holds potentially broad promise, the emerging field of
tissue engineering is exploring the development of devices such as heart valves or skin that become populated by the patient’s living cells (see, e.g., Rabkin et al., 2002; Stock et al., 2002; Mol et al., 2004; Neuenschwander and Hoerstrup, 2004). Such devices might grow as young patients grow and also avoid or limit immunocompatibility or biocompatibility problems that are often seen with currently used materials.
Other Developmental Concerns
The use with children of implanted heart valves that employ tissue from pigs or cows raises a variety of developmental considerations. When surgeons began implanting such valves in children, they discovered in the course of long-term follow up a more intense immunologic response and more rapid calcification of the valves than had been observed in adults or expected with children (see, e.g., Geha et al., 1979; Schaff and Danielson, 1986; Baskett et al., 2003). The valves, which avoid the risks associated with the anticoagulant therapy required for mechanical heart valves, are still occasionally used with children—with the now familiar risk of calcification factored into decisions about which treatment is best for a particular child.
As noted above, some medical devices that are critically important for certain conditions require cooperation from the patient that may not be possible for infants and very young children. The options in such situations may involve adaptations in the device, development of an alternative device, or foregoing use of the device until the child has matured.
The last two options are both in evidence for patients with cystic fibrosis. Measuring lung function in these patients requires that the patient be capable of certain breathing maneuvers (e.g., taking or expelling a breath upon direction). For infants to age 2, a system has been developed that includes, among other features, a vest that inflates to provide external pressure for the required breathing maneuver (Tillman, 2002). For children between ages 2 and about 6, no satisfactory device has yet been approved (Colin, 2003).
With some devices and therapies, multiple factors, including behavioral factors, may be at work. For example, the design and use of aerosol delivery devices for young patients who have cystic fibrosis and certain other respiratory disorders may be complicated for a combination of reasons, including anatomic (e.g., airway size in relation to drug particle size), physiologic (e.g., highly variable breathing patterns in infants), pathophysiologic (e.g., presence of inflammation, excess mucus), and behavioral (e.g., inability to synchronize breathing patterns) (Cole, 2000). In addition, characteristics of the aerosol device and the aerosolized drug interact, and both must be taken into account in the development and testing of effective interventions.
In a statement to the committee, the American Thoracic Society cited
the lack of guidance “for the average pediatrician pertaining to the multitudes of delivery devices [tested only in adults] … [and] the complexity of treatment of the young infant or toddler…. [In general] the smaller the child, the less the dose of aerosolized drug delivered” (ATS, 2004b, p. 2). The group also noted that studies of nebulized drug deposition in very young children who have tracheostomies are rare in the United States due to concerns about approval of the use of radiolabeled drug markers in such studies. They suggested that such studies could be considered under special regulatory provisions that allow the Commissioner of FDA to approve studies of special importance that could not otherwise be approved under research protection regulations that apply specifically to children (see further discussion of these regulations in Chapter 6).
“Working Around” the Lack of a Child-Appropriate Device
For purposes of this discussion, workarounds are actions devised to cope with a perceived problem without actually fixing it. Although workarounds may involve doing something new, it is useful to reserve the term innovation for a response that is intended not merely to cope with but to solve a problem. Turning off nuisance alarms is a workaround; redesigning an alarm system is an innovation. When workarounds are recognized as insufficient or inefficient responses, they may prompt true innovations.
Common situations that give rise to workarounds include a computer crash or bug (e.g., when a computerized drug order entry system “goes down”), alarms that go off “too” frequently, uncertainty about the appropriate use of equipment (e.g., devices with complex programming procedures), irritating or time-consuming organizational procedures, or unavailability of the right equipment at the right time or at all.
The unavailability of a device properly scaled or otherwise adapted for pediatric use may prompt a workaround. For example, because no stents designed for use in pediatric heart catheterization treatments are commercially available, clinicians use stents in cardiac procedures that were developed for biliary (bile duct) use. This strategy presents some risk of perforation or thrombosis of the targeted vessel (Shaffer et al., 1998). As in this example, workarounds may involve “off-label” or unlabeled uses of medical devices. FDA regulations permit such uses as an element of medical practice and discretion (see Chapter 3).
Another example of a workaround involves the use of a needle designed to aspirate bone marrow to, instead, infuse fluids via infant bone when intravenous access cannot be established during an emergency. The technique has led to different workarounds to stabilize the needle because the needles tend to leak around the entry site unless they are firmly fixed.
Many types of clips or clamps were fashioned to provide secure fixation until recently, when an adjustable “stop” for the needle was devised.
Some workarounds involve the use of devices in ways that are inconsistent with explicit manufacturer instructions because the design of the device makes use consistent with instructions impractical in the real world. For example, alarms for home infusion pumps that signal the presence of air bubbles may be turned off because the alarms respond to small, harmless air bubbles that are common in total parental nutrition fluids. The alarm is then unavailable to detect a larger, potentially fatal air bubble.
Depending on the context, workarounds may be considered either to jeopardize safety (e.g., by disabling alarms or other safeguards) or to enhance safety (e.g., by identifying and compensating for a problem until the problem can be investigated and, when possible, fixed) (Mohr and Batalden, 2002). In some cases, the device characteristic or situation that gives rise to a workaround may be reported as an adverse event or close call. In other cases the problem and the workaround may not be thought of as reportable events within either the context of an institutional patient safety program or FDA’s program for adverse event reports (see Chapter 7). Workarounds devised by parents or medical caregivers for equipment that is used in the home may never be explained or conveyed back to the manufacturer or even to the prescribing clinicians.
Workarounds may be the norm for extended periods. Sometimes they inspire the development of devices or device adaptations to meet children’s needs. For example, the connectors to tracheal tubes add a few milliliters (known as “dead space”) to the volume of tubing between the tracheal tube and the connecting tubing of a ventilator. Although this dead space is trivial for a child or adult, it becomes imposing for a tiny premature baby whose breathing volume may only be a few milliliters. To compensate, caregivers often cut the tracheal tube to make it as short as possible, but this makes fixation of the tube tenuous. Now, newer connectors have been designed to reduce the dead space to less than a milliliter.
IDENTIFYING PROBLEMS OR CONCERNS WITH MEDICAL DEVICES USED WITH CHILDREN
Problems with the potential or actual performance of devices in infants, children, and adolescents may be identified in at least three different ways (Box 2.3). First, they may be identified a priori based on a combination of expert understanding of children’s developmental characteristics and detailed knowledge of the operating characteristics of a particular device derived from theory, bench testing, and, perhaps, animal testing or adult use. For example, engineers identified blood flow problems with downsizing
|
BOX 2.3 A priori identification
Identification through premarket testing involving children
Identification after marketing
|
of mechanical heart valves based on theory, past experience with similar devices, simulations, and laboratory tests (Anderson et al., 2000; Bachmann et al., 2000; PSU, 2004). Some makers of cochlear implants have advised against deep-sea scuba diving based on expectations about the possible effects of severe changes in pressure within the ear (Nussbaum, 2003).To cite another example, because children require higher heart rates than adults (approximately 140 beats per minute for infants versus 70 beats per minute for adults), it is anticipated that battery life may be shorter in certain cardiac devices, which makes the prospect of battery exhaustion and planned, serial replacement of devices a reasonable expectation in the pediatric population rather than an unanticipated adverse event (ACC, 2004).
The same process of a priori reasoning applies to the identification of important concerns related to the effects of children’s growth and development for the number of years that a child has an implant. For example, the developer of the titanium rib built a certain degree of expansion capacity into the device (which requires surgical adjustment) because its purpose is
to facilitate growth in children with severe scoliosis and other conditions that limit chest development. In certain orthopedic repairs, surgeons may remove implanted fixation devices to accommodate children’s growth when they might leave such devices undisturbed in mature patients. For yet other implanted devices such as neurostimulators placed in the brain or bone cements used to repair bone defects, concerns about growth and development lead manufacturers and FDA to advise against use of the device with children who have not completed all or most of their growth in areas (e.g., brain, skeleton) where the device is placed.
In addition, pediatric issues or problems may be revealed during clinical testing of a device with children prior to marketing approval. For example, as described earlier in this chapter, studies of deep brain neurostimulators to treat dystonia revealed that use of the device in children, compared to adults, required adaptations in the placement of the device. If two neurostimulators are implanted, they must be implanted at least 8 inches apart to minimize interference.
Problems may be recognized as experience with a device accumulates following its entry into the market. In some cases, problems are identified or confirmed through systematic postmarket clinical or epidemiological studies. For example, in 2002, a manufacturer of cochlear implants reported to FDA 15 cases of meningitis in implanted patients. Subsequently, other manufacturers reported meningitis cases, mostly in young children. Some clinicians had already become concerned about the risk based on conversations at meetings about their experiences following their patients (Niparko, 2004). Based on a review of the adverse event reports, FDA worked with CDC and health departments in many states and three cities on an epidemiologic study that attempted to assess risk factors for meningitis among implant recipients compared to a control group (Reefhuis et al., 2003).
Some problems with devices may be identified soon after they begin to be used with children. To cite an example, the measurement of transcutaneous partial oxygen pressure in arterial blood (measured with an oxygen electrode) was thought to provide a reliable proxy for arterial blood oxygenation (Huch et al., 1977). Soon, however, clinicians recognized that there was a marked disparity between the two values when an infant’s perfusion (blood flow into tissues) is poor (Peabody et al., 1978a,b). As this problem was becoming more widely recognized, the pulse oximeter was developed to measure arterial blood oxygenation using a different and superior strategy (Jennis and Peabody, 1987).
Other device problems are uncovered only as clinicians follow patients for extended periods. One example involves problems with fracturing of the peritoneal catheters for cerebrospinal fluid shunts (Langmoen et al., 1992; Cuka and Hellbusch, 1995). The experiences cited above with the
calcification of tissue-based heart valves and the migration of craniofixation devices are additional cases in point.
FDA’s regulatory and other activities take each of the modes of problem identification into account. Thus, FDA provides guidance on device design and testing and evaluates information on safety prior to approval or clearance of devices. Through the agency’s adverse event reporting program and requirements for postmarket studies of certain devices, it focuses on safety after devices are marketed. As described in this report, these postmarket strategies have serious limitations. Identification and prevention of problems with devices prior to marketing are, in any case, preferable to postmarket detection.
This chapter has provided a pediatric context for a report that often must focus on policies, practices, and questions that are not specific to children. The next reviews the regulatory framework for FDA’s activities. With a few exceptions, that framework applies to both adults and children.



























