
13
Summary and Research Needs
The research needs stated here relate to the committee’s primary task: “To develop the best possible risk estimate for exposure to low-dose, low-LET [linear energy transfer] radiation in human subjects.”
EVIDENCE FROM BIOLOGY
Molecular and Cellular Responses to Ionizing Radiation
This chapter discusses the biological effects of the ranges of radiation dose that are most relevant for the committee’s deliberations on the shapes of dose-response relationships. Considering the levels of background radiation, the maximal permissible levels of exposure of radiation workers now in effect, and the fact that much of the epidemiology of low-dose exposures includes people who in the past have received up to 500 mGy, the committee has focused on evaluating radiation effects in the low-dose range of <100 mGy, with emphasis on the lowest doses when relevant data are available. Effects that may occur as the radiation is delivered chronically over several months to a lifetime are thought to be most relevant.
At low doses, damage is caused by the passage of single particles that can produce multiple, locally damaged sites leading to DNA double-strand breaks (DSBs). DNA DSBs in the low-dose range can be quantified by a number of novel techniques, including immunofluorescence, comet assay, chromosome aberrations, translocation, premature chromosome condensation, and others. Some of these indicators of DSBs show linearity down to doses of 5 to 10 mGy.
In vitro data on the introduction of gene mutations by low-LET ionizing radiation are consistent with knowledge of DNA damage response mechanisms and imply a nonthreshold low-dose response for mutations involved in cancer development. Experiments that quantified DNA breakage, chromosomal aberrations, or gene mutations induced by low total doses or low doses per fraction suggest that the dose-response over the range of 20 to 100 mGy is linear. Limited data indicate that the dose-response for DNA breakage is linear down to 1 mGy, and biophysical arguments suggest that the response should be linear between zero and 5 mGy.
In vitro studies of gene mutation induction provide evidence for a dose and dose rate effectiveness factor (DDREF) in the range of 2–4. The DDREF has been used in past estimates of risk to adjust data obtained from acute exposures at Hiroshima and Nagasaki to the expected lower risk posed by chronic low-dose exposures that the general population might experience.
Research Need 1. Determination of the level of various molecular markers of DNA damage as a function of low-dose ionizing radiation
Currently identified molecular markers of DNA damage and other biomarkers that can be identified in the future should be used to quantify low levels of DNA damage and to identify the chemical nature and repair characteristics of the damage to the DNA molecule. These biomarkers have to be evaluated fully to understand their biological significance for radiation damage and repair and for radiation carcinogenesis.
Most studies suggest that the repair of ionizing radiation damage occurs through nonhomologous end joining and related pathways that are constitutive in nature, occur in excess, and are not induced to higher levels by low radiation doses.
Data from animal models of radiation tumorigenesis were evaluated with respect to the cellular mechanisms involved. For animal models of radiation carcinogenesis that are dependent on cell killing, there tend to be threshold-like dose-responses and high values of DDREF; therefore, less weight was placed on these data. Once cell-killing dependence is excluded, animal data are not inconsistent with a linear nonthreshold (LNT) dose response, and DDREF values are in the range 2–3 for solid cancers and somewhat higher for acute myeloid leukemia.
Research Need 2. Determination of DNA repair fidelity, especially as regards double- and multiple-strand breaks at low doses, and determination of whether repair capacity is independent of dose
Repair capacity at low levels of damage must be investigated, especially in light of conflicting evidence for stimulation of repair at low doses. In such studies the accuracy of DNA sequences rejoined by these pathways has to be determined, and the mechanisms of error-prone repair of radiation lesions must be elucidated. Identification of critical genetic alterations that can be characteristic of radiation exposure would be important.
Consideration of Phenomena That Might Affect Risk Estimates for Carcinogenesis at Very Low Doses
A number of biological phenomena that could conceivably affect risk estimates at very low radiation doses have been reported. These phenomena include the existence of radiation-sensitive human subpopulations, hormetic or adaptive effects, bystander effects, low-dose hyperradiosensitivity, and genomic instability.
Radiation-Sensitive Subpopulations
Epidemiologic, clinical, and experimental data provide clear evidence that genetic factors can influence radiation cancer risk. Strongly expressing human mutations of this type are rare and are not expected to influence significantly the development of estimates of population-based, low-dose risks. They are, however, potentially important in the context of high-dose medical exposures. Evidence for the complex interaction of weakly expressing genetic factors in cancer risk is growing, but current understanding is insufficient for a detailed consideration of the potential impact on population risk.
Adaptive Response
Adaptive responses have been well documented in bacteria, where exposures to radiation or chemicals induce subsequent resistance to these agents by inducing expression of DNA damage repair genes. This induced expression of repair genes does not occur to a significant extent in human cells, although changes in signal transduction do take place. A type of apparent adaptive response, however, has been documented for the induction of chromosomal aberrations in human lymphocytes stimulated to divide.
In most studies, a priming or adaptive dose of about 10 mGy significantly reduces the frequency of chromosomal aberrations and mutations induced a few hours later by 1000–3000 mGy. Similar effects are sometimes seen with other end points. However, priming doses less than 5 mGy or greater than ~200 mGy generally give very little, if any, adaptation, and adaptation has not been reported for challenge doses of less than about 1000 mGy. To have relevance for risk assessment, the adaptive response has to be demonstrated for both priming and challenging doses of 1–50 mGy.
Furthermore, the induction and magnitude of the adaptive response in human lymphocytes are highly variable, with much heterogeneity demonstrated among different individuals. The adaptive response could not be induced when noncycling lymphocytes were given the priming dose. Although inhibitor and electrophoretic studies suggest that alterations in messenger RNA transcription and protein synthesis are involved in the adaptive response in lymphocytes, no specific signal transduction or repair pathways have been identified. At this time, the assumption that any stimulating effects from low doses of ionizing radiation will have a significant effect in reducing long-term deleterious effects of radiation on humans is unwarranted.
Bystander Effects
The bystander effect that results from irradiated cells’ reacting with nearby nonirradiated cells could influence dose-response relationships. Such an effect might come into play at low-LET doses below 1–5 mGy, where some cells of the body would not be irradiated. Current limitations of low-LET bystander studies include the lack of demonstrated bystander effects below 50 mGy and uncertainties about whether the effect occurs in vivo. Another complication is that both beneficial and detrimental effects have been postulated for bystander effects by different investigators. Until molecular mechanisms are elucidated, especially as they relate to an intact organism, and until reproducible bystander effects are observed for low-dose low-LET radiation in the dose range of 1–5 mGy, where an average of less than 1 electron tracks traverse the nucleus, the assumption should be made that bystander effects will not influence the shape of the low-dose, low-LET dose-response relationship.
Hyperradiosensitivity for Low Doses
In some cell lines, hyperradiosensitivity (HRS) has been reported for cell lethality induced by low-LET radiation at doses less than 100–200 mGy. In this dose range, survival decreases to 85–90%, which is significantly lower that projected from data obtained above 1–2 Gy. It is not known whether HRS for cell lethality would cause an increase in deleterious effects in surviving cells or would actually decrease deleterious effects by increased killing of damaged cells. Until molecular mechanisms responsible for HRS that may or may not play a role in carcinogenesis are understood, the extrapolation of data for HRS for cell lethality to the dose-response for carcinogenesis in the 0–100 mGy range is not warranted.
Genomic Instability
During the last decade, evidence has accumulated that under certain experimental conditions, the progeny of cells surviving radiation appear to express new chromosomal aberrations and gene mutations over many postirradiation cell generations. This feature is termed radiation-induced persistent genomic instability. Some inconsistencies were identified in the data that describe the diverse manifestation of induced genomic instability, and clear evidence of its general involvement in radiation-induced cancer is lacking. Although developing data on the various phenomena classified as genomic instability may eventually provide useful insights into the mechanisms of carcinogenesis, it is not possible to predict whether induced genomic instability will influence low-dose, low-LET response relationships.
Research Need 3. Evaluation of the relevance of adaptation, low-dose hypersensitivity, bystander effects, and genomic instability for radiation carcinogenesis
Mechanistic data are needed to establish the relevance of these processes to low-dose radiation exposure (i.e., <100 mGy). Relevant end points should include not only chromosomal aberrations and mutations but also genomic instability and induction of cancer. In vitro and in vivo data are needed for delivery of low doses over several weeks or months at very low dose rates or with fractionated exposures. The cumulative effect of multiple low doses of less than 10 mGy delivered over extended periods has to be explored further. The development of in vitro transformation assays utilizing nontransformed human diploid cells is judged to be of special importance.
Hormesis
The possibility that low doses of radiation may have beneficial effects (a phenomenon often referred to as “hormesis”) has been the subject of considerable debate. Evidence for hormetic effects was reviewed, with emphasis on material published since the 1990 BEIR V study on the health effects of exposure to low levels of ionizing radiation. Although examples of apparent stimulatory or protective effects can be found in cellular and animal biology, the preponderance of available experimental information does not support the contention that low levels of ionizing radiation have a beneficial effect. The mechanism of any such possible effect remains obscure. At this time, the assumption that any stimulatory hormetic effects from low doses of ionizing radiation will have a significant health benefit to humans that exceeds potential detrimental effects from radiation exposure at the same dose is unwarranted.
Research Need 4. Identification of molecular mechanisms for postulated hormetic effects at low doses
Definitive experiments that identify molecular mechanisms are necessary to establish whether hormetic effects exist for radiation-induced carcinogenesis.
Radiation-Induced Cancer: Mechanism, Quantitative Experimental Studies, and the Role of Molecular Genetics
A critical conclusion on mechanisms of radiation tumorigenesis is that the data reviewed greatly strengthen the view that there are intimate links between the dose-dependent induction of DNA damage in cells, the appearance of gene or chromosomal mutations through DNA damage misrepair, and the development of cancer. Although less well established, the data available point toward a single-cell (monoclonal) origin for induced tumors and suggest that low-dose radiation acts predominantly as a tumor-initiating agent. These data also provide some evidence on candidate, radiation-associated mutations in tumors. These mutations are predominantly loss-of-function DNA deletions, some of which are represented as segmental loss of chromosomal material (i.e., multigene deletions).
This form of tumorigenic mechanism is broadly consistent with the more firmly established in vitro processes of DNA damage response and mutagenesis considered in Chapters 1 and 2. Thus, if as judged in Chapters 1 and 2, error-prone repair of chemically complex DNA double-strand damage is the predominant mechanism for radiation-induced gene or chromosomal mutation, there can be no expectation of a low-dose threshold for the mutagenic component of radiation cancer risk.
One mechanistic caveat explored was that novel forms of cellular damage response, collectively termed induced genomic instability, might contribute significantly to radiation cancer risk. The cellular data reviewed in Chapter 2 identified uncertainties and some inconsistencies in the expression of this multifaceted phenomenon. However, telomere-associated mechanisms did provide a coherent explanation for some in vitro manifestations of induced genomic instability. The data considered did not reveal consistent evidence for the involvement of induced genomic instability in radiation tumorigenesis, although telomere-associated processes may account for some tumorigenic phenotypes. A further conclusion was that there is little evidence of specific tumorigenic signatures of radiation causation, but rather that radiation-induced tumors develop in a tumor-specific multistage manner that parallels that of tumors arising spontaneously.
Quantitative animal data on dose-response relationships provide a complex picture for low-LET radiation, with some tumor types showing linear or linear-quadratic relationships while other studies are suggestive of a low-dose threshold, particularly for thymic lymphoma and ovarian cancer. Since, however, the induction or development of these two cancer types is believed to proceed via atypical mechanisms involving cell killing, it was judged that the threshold-like responses observed should not be generalized.
Radiation-induced life shortening in mice is largely a reflection of cancer mortality, and the data reviewed generally support the concept of a linear dose-response at low doses and low dose rates. Other dose-response data for animal tumorigenesis, together with cellular data, contributed to the judgments developed and the choice of a DDREF for use in the interpretation of epidemiologic information on cancer risk.
Adaptive responses for radiation tumorigenesis have been investigated in quantitative animal studies, and recent information is suggestive of adaptive processes that increase tumor latency but not lifetime risk. However, these data are difficult to interpret, and the implications for radiological protection remain most uncertain.
Research Need 5. Tumorigenic mechanisms
Further cytogenetic and molecular genetic studies are needed to reduce current uncertainties about the specific role of radiation in multistage radiation tumorigenesis; such investigations would include studies with radiation-associated tumors of humans and experimental animals.
The review of cellular, animal, and epidemiologic or clinical studies on the role of genetic factors in radiation tumorigenesis suggests that many of the known strongly expressing cancer-prone human genetic disorders are likely to show an elevated risk of radiation-induced cancer, probably with a high degree of organ specificity. Cellular and animal studies suggest that the molecular mechanisms underlying these genetically determined radiation effects largely mirror those that apply to spontaneous tumorigenesis and are consistent with knowledge of somatic mechanisms of tumorigenesis. In particular, evidence was obtained that major deficiencies in DNA damage response and tumor-suppressor-type genes can serve to elevate radiation cancer risk.
Limited epidemiologic data from follow-up of second cancers in gene carriers receiving radiotherapy were supportive of the above conclusions, but quantitative judgments about the degree of increased cancer risk remain uncertain. However, since major germline deficiencies in the genes of interest are known to be rare, it has been possible to conclude from published analyses that they are most unlikely to create a significant distortion of population-based estimates of cancer risk. The major practical issue associated with these strongly expressing cancer genes is judged to be the risk of radiotherapy-related cancer.
A major theme developing in cancer genetics is the interaction and potential impact of more weakly expressing variant cancer genes that may be relatively common in human populations. The animal genetic data provide proof-of-principle evidence of how such variant genes with functional polymorphisms can influence cancer risk, including limited data on radiation tumorigenesis. Attention was also given to human molecular epidemiology data on associations between functional polymorphisms and cancer risk, particularly with respect to DNA damage response genes.
Given that functional gene polymorphisms associated with cancer risk may be relatively common, the potential for significant distortion of population-based risk was explored with emphasis on the organ specificity of the genes of interest. An interim conclusion was that common polymorphisms of DNA damage response genes associated with organ-wide radiation cancer risk would be the most likely source of major interindividual differences in radiation response.
Research Need 6. Genetic factors in radiation cancer risk
Further work is needed in humans and mice on gene mutations and functional polymorphisms that influence the risk of radiation-induced cancers. Where possible, human molecular genetic studies should be coupled with epidemiologic investigations.
GENETIC EFFECTS OF RADIATION ON HUMAN POPULATIONS
As noted in BEIR V, heritable effects of radiation are estimated using what is referred to as the “doubling dose method” and expressed in terms of increases in the frequencies of genetic diseases in the population over and above those that occur as a result of spontaneous mutations. The doubling dose (DD) is the amount of radiation required to produce as many mutations as those that occur spontaneously in a generation and is calculated as a ratio of the average rates of spontaneous and induced mutations in defined genes. If the DD is small, the relative mutation risk per unit dose (i.e., 1/DD) is high, and if DD is large, the relative mutation risk is low. The DD, therefore, provides a convenient yardstick to express risks and a perspective of whether the predicted increases are trivial, small, or substantial relative to the baseline.
Revision of the Conceptual Basis for Calculating the DD
In the BEIR V report, mouse data on both spontaneous and induced mutation rates were used for DD calculations. A reassessment of the assumptions underlying this procedure revealed that the use of mouse data for spontaneous mutation rates can no longer be considered appropriate and that reverting to the use of human data on spontaneous mutation rates for DD calculations, as was first done in the 1972 BEIR report, is correct. The DD calculated is 1 Gy and is the same as the one based entirely on mouse data.
Revision of the Baseline Frequencies of Mendelian Diseases in Humans
The baseline frequencies of genetic diseases constitute an important quantity in risk estimation. While there is no reason to consider revision of the baseline frequencies of congenital abnormalities (6%) and chronic diseases (65%), these two classes together constitute what are referred to as “mul-
tifactorial diseases” because of the multiple factors involved in their etiology. Advances in human genetics now suggest that the frequencies of Mendelian diseases (i.e., those that are due to mutations in single genes and show simple and predictable patterns of inheritance) have to be revised upwards from the 1.25% used previously (based on estimates made in the mid-1970s) to 2.40% at this time.
Delineation of a New Concept—The Concept of Potential Recoverability Correction Factor
Mouse data on rates of radiation-induced mutations constitute the primary basis for estimating the risk of radiation-inducible genetic diseases in humans. Advances in the molecular biology of human genetic diseases and in studies of radiation-induced mutations in experimental systems show that mouse mutation rates cannot readily be converted into rates of genetic disease in human live births and that a correction factor, the potential recoverability correction factor (PRCF), is required to make the transition from induced mutations in mice to inducible genetic disease in humans. A framework and methods have been developed to estimate PRCFs for Mendelian and chronic multifactorial diseases.
Introduction of the Concept That Adverse Hereditary Effects of Radiation Are Likely to Be Manifest as Multisystem Developmental Abnormalities
The adverse hereditary effects of radiation are more likely to be manifest as multisystem developmental abnormalities than as Mendelian diseases. This concept incorporates elements of current knowledge of the mechanisms of radiation-induced genetic damage, the molecular nature of radiation-induced mutations, the phenotypic manifestations of naturally occurring multigene deletions in humans, empirical observations in mice on the phenotypic effects of radiation-induced multigene deletions, and the enormous number and distribution of genes involved in development in nearly all the human chromosomes. Appropriate mouse data that can serve as a basis for a preliminary estimate of radiation-induced adverse developmental effects have been identified and used.
Risk estimates have been made only for the first two postirradiation generations. The population genetic theory of equilibrium between mutation and selection (i.e., the equilibrium theory) underlies the DD method that is used to estimate genetic risks of radiation. This theory postulates that the stability of mutant gene frequencies (and therefore of disease frequencies) in a population is a reflection of the existence of a balance between the rates at which spontaneous mutations arise in every generation and enter the gene pool and the rates at which they are eliminated by natural selection. When such an “equilibrium population” sustains radiation exposure generation after generation, additional mutations are introduced into the gene pool, and these are also subject to the action of natural selection. The prediction is that a new equilibrium between mutation and selection will be reached. The time it takes in terms of generations to attain the new equilibrium, the rate of approach to it, and the magnitude of increase in mutant (and disease) frequencies are dependent on the induced mutation rate, the intensity of selection, and the type of disease.
Research Need 7. Heritable genetic effects of radiation
Further work is necessary to establish (1) the potential roles of DNA DSB repair processes in the origin of deletions in irradiated stem cell spermatogonia and oocytes (the germ cell stages of importance in risk estimation) in mice and humans and (2) the extent to which large, radiation-induced deletions in mice are associated with multisystem development defects. In humans, the problem can be explored using genomic databases and knowledge of mechanisms of the origin of radiation-induced deletions to predict regions that may be particularly prone to such deletions. These predictions can subsequently be tested in the mouse, these tests can also provide insights into the potential phenotypes associated with such deletions in humans.
With respect to epidemiology, studies on the genetic effects of radiotherapy for childhood cancer, of the type that have been under way in the United States and Denmark since the mid-1990s, should be encouraged, especially when they can be coupled with modern molecular techniques (such as array-based comparative genomic hybridization. These techniques enable one to screen the whole genome for copy number abnormalities (i.e., deletions and duplications of genomic segments) with a resolution beyond the level of a light microscope.
EPIDEMIOLOGIC STUDIES OF POPULATIONS EXPOSED TO IONIZING RADIATION
Atomic Bomb Survivor Studies
The Life Span Study (LSS) cohort of survivors of the atomic bombings in Hiroshima and Nagasaki continues to serve as a major source of information for evaluating health risks from exposure to ionizing radiation, and particularly for developing quantitative estimates of risk. Its advantages include its large size, the inclusion of both sexes and all ages, a wide range of doses that have been estimated for individual subjects, and high-quality mortality and cancer incidence data. In addition, the whole-body exposure received by this cohort offers the opportunity to assess risks for cancers of a large number of specific sites and to evaluate the comparability of site-specific risks.
As an illustration, Figure 13-1 shows estimated ERRs of solid cancer versus dose (averaged over sex and standardized to represent individuals exposed at age 30 at attained
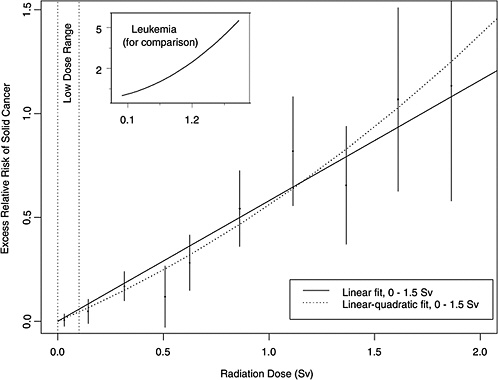
FIGURE 13-1 Excess relative risks of solid cancer for Japanese atomic bomb survivors. The insert shows the fit of a linear-quadratic model for leukemia, to illustrate the greater degree of curvature observed for that cancer.
age 60) for atomic bomb survivors with doses in each of 10 dose intervals less than 2.0 Sv. This plot helps convey the overall dose-response relationship from the LSS cohort and its role in low-dose risk estimation. Specific models are detailed in Chapter 6. It is important to note that the difference between the linear and linear-quadratic models in the low-dose ranges is small relative to the error bars; therefore, the difference between these models is small relative to the uncertainty in the risk estimates produced from them. For solid cancer incidence the linear-quadratic model did not offer statistically significant improvement in the fit, so the linear model was used. For leukemia, a linear-quadratic model (insert in Figure 13-1) was used because it fitted the data significantly better than the linear model.
Plotted points are the estimated ERRs of solid cancer incidence (averaged over sex and standardized to represent individuals exposed at age 30 attained age 60) for atomic bomb survivors with doses in each of 10 dose intervals, plotted above the midpoints of the dose intervals. If R(d) represents the age-specific instantaneous risk at some dose d, then the excess relative risk at dose d is [R(d) − R(0)] / R(0) (which is necessarily zero when dose is zero). Vertical lines are approximate 95% confidence intervals. Solid and dotted lines are estimated linear and linear-quadratic models for ERR, estimated from all subjects with doses in the range 0 to 1.5 Sv. (These are not estimated from the points, but from the lifetimes and doses of individual survivors, using statistical methods discussed in Chapter 6.) A linear-quadratic model will always fit the data better than a linear model, since the linear model is a restricted special case with quadratic coefficient equal to zero. For solid cancer incidence, however, there is no statistically significant improvement in fit due to the quadratic term. It should also be noted that in the low-dose range of interest the difference between the estimated linear and linear-quadratic models is small relative to the 95% CIs.
The full LSS cohort consists of approximately 120,000 persons who were identified at the time of the 1950 census. However, most recent analyses have been restricted to approximately 87,000 survivors who were in the city at the time of the bombings and for whom it is possible to estimate doses. Special studies of subgroups of the LSS have provided clinical data, biological measurements, and information on potential confounders or modifiers.
The availability of high-quality cancer incidence data has resulted in several analyses and publications addressing specific cancer sites. These analyses often include special pathological review of the cases and sometimes include data on additional variables (such as smoking for the evaluation of lung cancer risks). Papers focusing on the following cancer sites have been published in the last decade: female breast cancer, thyroid cancer, salivary gland cancer, liver cancer, lung cancer, skin cancer, and central nervous system tumors. Special analyses have also been conducted of cancer mortality in survivors who were exposed either in utero or during the first 5 years of life.
Health end points other than cancer have been linked with radiation exposure in the LSS cohort. Of particular note, a dose-response relationship with mortality from nonneoplastic disease was demonstrated in 1992, and subsequent analyses in 1999 and 2003 have strengthened the evidence for this association. Statistically significant associations were seen for the categories of heart disease, stroke, and diseases of the digestive, respiratory, and hematopoietic systems. The data were inadequate to distinguish between a linear dose-response, a pure quadratic response, or a dose-response with a threshold as high as 0.5 Sv.
Medical Radiation Studies
Published studies on the health effects of medical exposures were reviewed to identify those that provide information for quantitative risk estimation. Particular attention was focused on estimating risks of leukemia and of lung, breast, thyroid, and stomach cancer in relation to radiation dose for comparison with estimates derived from other exposed populations, particularly the atomic bomb survivors. The possible association between radiation exposure and cardiovascular mortality and morbidity was also reviewed.
For lung cancer, the ERRs per Gy (ERRs/Gy) from the studies of acute high-dose-rate exposures are statistically compatible and in the range 0.1–0.4. It is difficult to evaluate the effects of age at exposure or of exposure protraction based on these studies because only one study (the hemangioma cohort) is available in which exposure occurred at very young ages and protracted low-dose-rate exposures were received. The study of tuberculosis patients, however, appears to indicate that substantial fractionation of exposure leads to a reduction of risk.
For breast cancer, excess absolute risks (EARs) appear to be similar—of the order of 9.9 per 104 person-years (PY) per gray at age 50—following acute and fractionated moderate-to high-dose-rate exposure. Effects of attained age and age at exposure are important modifiers of risk. The excess risks appear to be higher in populations of women treated for benign breast conditions, suggesting that these women may be at an elevated risk of radiation-induced breast cancer. The hemangioma cohorts showed lower risks, suggesting a possible reduction of risks following protracted low-dose-rate exposures.
For thyroid cancer, all of the studies providing quantitative information about risks are studies of children who received radiotherapy for benign conditions. A combined analysis of data from some of these cohorts and data from atomic bomb survivors and from two case-control studies of thyroid cancer nested within the International Cervical Cancer Survivor Study and the International Childhood Cancer Survivor Study provides the most comprehensive information about thyroid cancer risks. For subjects exposed below the age of 15, a linear dose-response was seen, with a leveling or decrease in risk at the higher doses used for cancer therapy. The pooled ERR was 7.7 Gy−1, and the EAR was 4.4 per 104 PY-Gy. Both estimates were significantly affected by age at exposure, with a strong decrease in risk with increasing age at exposure and little apparent risk for exposures after age 20. The ERR appeared to decline over time about 30 years after exposure but was still elevated at 40 years.
Little information on thyroid cancer risk in relation to exposure in childhood to iodine-131 was available. Studies of the effects of 131I exposure later in life provide little evidence of an increased risk of thyroid cancer following 131I exposure after childhood.
For leukemia, ERR estimates from studies with average doses ranging from 0.1 to 2 Gy are relatively close, in the range 1.9 to 5 Gy−1, and are statistically compatible. Estimates of EAR are also similar across studies, ranging from 1 to 2.6 per 104 PY-Gy. Little information is available on the effects of age at exposure or of exposure protraction.
For stomach cancer, the estimates of ERR range from negative to 1.3 Gy−1. The confidence intervals are wide, however, and they all overlap, indicating that these estimates are statistically compatible.
Finally, studies of patients having undergone radiotherapy for Hodgkin’s disease or breast cancer suggest that there may be some risk of cardiovascular morbidity and mortality for very high doses and high-dose-rate exposures. The magnitude of the radiation risk and the shape of the dose-response curve for these outcomes are uncertain.
Research Need 8. Future medical radiation studies
Most studies of medical radiation should rely on exposure information collected prospectively, including cohort studies as well as nested case-control studies. Future studies should continue to include individual dose estimation to the site of interest, as well as an evaluation of the uncertainty in dose estimation. Ideally, where population-based cancer registries do not exist to establish cohorts of cancer survivors, hospital-based registries can be established to identify cohorts of exposed patients whose mortality and morbidity can be followed. If these registries can be linked
to appropriate radiation therapy or diagnostic records, they can be used as a basis for nested case-control studies of specific outcomes, and detailed exposure estimation for the site of interest can be undertaken.
Studies of populations with high- and moderate-dose medical exposures are particularly important for the study of modifiers of radiation risks. Because of the high level of radiation exposure in these populations, they are also ideally suited to study the effects of gene-radiation interactions that may render particular subsets of the population more sensitive to radiation-induced cancer. Genes of particular interest include BRCA1, BRCA2, ATM, CHEK2, NBS1, XRCC1, and XRCC3. These are among the most important genes known to be involved in detection and repair of radiation-induced DNA damage.
Of concern for radiological protection is the increasing use of computed tomography (CT) scans and diagnostic X-rays. Epidemiologic studies of these exposures would be particularly useful if they are feasible, particularly the following: (1) follow-up studies of cohorts of persons receiving CT scans, especially children; and (2) studies of infants who experience diagnostic exposures related to cardiac catheterization, those who have recurrent exposures to follow their clinical status, and premature babies monitored for pulmonary development with repeated X-rays.
The widespread use of interventional radiological procedures in the heart, lungs, abdomen, and many vascular beds, with extended fluoroscopic exposure times of patients and operators, emphasizes the need for recording of dose and later follow-up studies of potential radiation effects among these populations. There is a need to organize worldwide consortia that would use similar methods in data collection and follow-up. These consortia should record delivered doses and technical data from all X-ray or isotope-based imaging approaches including CT, positron emission tomography, and single photon emission computed tomography.
Occupational Radiation Studies
The risk of cancer among physicians and other persons exposed to ionizing radiation in the workplace has been a subject of study since the 1940s, when increased mortality from leukemia was reported among radiologists in comparison to mortality among other medical specialists. Since then, numerous studies have considered the mortality and cancer incidence of various occupationally exposed groups in medicine, industry, defense, research, and aviation industries.
Studies of occupationally exposed groups are, in principle, well suited for direct estimation of the effects of low doses and low dose rates of ionizing radiation. The most informative studies at present are those of nuclear industry workers (including the workers at Mayak in the former USSR), for whom individual real-time estimates of doses have been collected over time with the use of personal dosimeters. More than 1 million workers have been employed in this industry since its beginning in the early 1950s. However, studies of individual worker cohorts are limited in their ability to estimate precisely the potentially small risks associated with low levels of exposure. Risk estimates from these studies are variable, ranging from no risk to risks an order of magnitude or more than those seen in atomic bomb survivors.
Combined analyses of data from multiple cohorts offer an opportunity to increase the sensitivity of such studies and provide direct estimates of the effects of long-term, low-dose, low-LET radiation. The most comprehensive and precise estimates to date are those derived from the U.K. National Registry of Radiation Workers and the three-country study (Canada-U.K.-U.S.), which have provided estimates of leukemia and all cancer risks. Although the estimates are lower than the linear estimates obtained from studies of atomic bomb survivors, they are compatible with a range of possibilities, from a reduction of risk at low doses to risks twice those on which current radiation protection recommendations are based. Overall, there is no suggestion that the current radiation risk estimates for cancer at low levels of exposure are appreciably in error. Uncertainty regarding the size of this risk remains as indicated by the width of the confidence intervals.
Because of the absence of individual dose estimates in most of the cohorts, studies of occupational exposures in medicine and aviation provide minimal information useful for the quantification of these risks.
Because of the uncertainty in occupational risk estimates and the fact that errors in doses have not formally been taken into account in these studies, the committee concluded that the occupational studies were not suitable for the projection of population-based risks. These studies, however, provide a comparison to the risk estimates derived from atomic bomb survivors.
Research Need 9. Future occupational radiation studies
Studies of occupational radiation exposures, particularly among nuclear industry workers, including nuclear power plant workers, are well suited for direct assessment of the carcinogenic effects of long-term, low-level radiation exposure in humans. Ideally, studies of occupational radiation should be prospective in nature and rely on individual real-time estimates of radiation doses. Where possible, national registries of radiation exposure of workers should be established and updated as additional radiation exposure is accumulated and as workers change employers. These registries should include at least annual estimates of whole-body radiation dose from external photon exposure. These exposure registries should be linked
with mortality registries and, where they exist, with national tumor (and other disease) registries. Where national dose registries cannot be set up, cohort studies based on records of nuclear installations are a useful alternative. It is noted that the power of individual cohort studies at the local and even national levels is limited. To maximize the information about the effects of low-dose, protracted exposures from these studies, it is therefore necessary to combine data across cohorts and countries. Most studies published to date have been based on relatively short follow-up periods, and the majority of workers were still young at the end of follow-up. Extended mortality follow-up over the next decades—and, where possible, cancer morbidity follow-up—of these workers, as they enter an age range when cancer incidence and mortality rates increase, will provide useful improvements of the direct cancer risk estimates drawn from these studies of exposure to low-dose, low-LET radiation. It is also important to continue follow-up of workers exposed to relatively high doses, that is, workers at the Mayak nuclear facility and workers involved in the Chernobyl cleanup.
Environmental Radiation Studies
Ecologic studies of populations living around nuclear facilities and of other environmentally exposed populations do not contain individual estimates of radiation dose or provide a direct quantitative estimate of risk in relation to radiation dose. This limits the interpretation of these data.
Several cohort studies have reported health outcomes among persons exposed to environmental radiation. No consistent or generalizable information is contained in these studies. Four ecologic studies of populations exposed to natural background did not find any association between disease rates and indicators of high background levels of radiation exposure. Ecologic studies of children of adults exposed to radiation while working at the Sellafield nuclear facility in Great Britain have suggested some increased risk of leukemia and lymphoma associated with individual dose, but the findings are based on small numbers of cases and the results across studies are not consistent.
Evidence from ecologic studies does not indicate an increased risk of leukemia among persons exposed in utero to radiation from Chernobyl or an increase in rates of childhood leukemia. In contrast to a considerable body of evidence regarding the risk of thyroid cancer in persons exposed to external radiation, there is relatively little information regarding the risk of thyroid cancer in humans exposed internally to 131I. There is some evidence of a small increase in thyroid cancer associated with exposure to 131I from therapeutic and diagnostic uses, but the findings are inconsistent and the small increases in thyroid cancer observed in some studies may be due to the underlying thyroid condition and not radiation exposure.
Results from external environmental exposures to 131I have been inconsistent. The most informative findings are from studies of individuals exposed to radiation after the Chernobyl accident. Recent evidence indicates that exposure to radiation from Chernobyl is associated with an increased risk of thyroid cancer and that the relationship is dose dependent. The quantitative estimate of excess thyroid cancer risk is generally consistent with estimates from other radiation-exposed populations and is observed in both males and females. Iodine deficiency appears to be an important modifier of risk, enhancing the risk of thyroid cancer following radiation exposure.
Ecologic studies of persons exposed to environmental sources of ionizing radiation have not been useful in developing risk estimates. Exposure levels are low, the studies relate to exposure of populations rather than individuals, and there is minimal possibility of follow-up of exposed individuals. The few exceptions to these circumstances are populations where there is unusual exposure because of accidents involving radiation exposure or long-term releases of relatively high levels of ionizing radiation (e.g., Chernobyl, Hanford).
Research Need 10. Future environmental radiation studies
In general, additional ecologic studies of persons exposed to low levels of radiation from environmental sources are not recommended. However, if disasters occur in which a local population is exposed to unusually high levels of radiation, it is important that there be a rapid response not only for the prevention of further exposure but also for the establishment of scientific evaluation of the possible effects of exposure. The data collected should include basic demographic information on individuals, estimates of acute and possible continuing exposure, the nature of the ionizing radiation, and the means of following these individuals for many years. The possibility of enrolling a comparable nonexposure population should be considered. Studies of persons exposed environmentally as a result of the Chernobyl disaster or as a result of releases from the Mayak nuclear facility should continue.
INTEGRATION OF BIOLOGY AND EPIDEMIOLOGY
This chapter highlights the ways in which cellular, molecular, and animal data can be integrated with epidemiologic findings in order to develop coherent judgments on the health effects of low-LET radiation. Emphasis is placed on data integration for the purposes of modeling these health risks. The principal conclusions from this work are the following:
-
Current knowledge on the cellular and molecular mechanisms of radiation tumorigenesis tends to support the application of models that incorporate the excess relative risk projection over time.
-
The choice of models for the transport of cancer risk from Japanese A-bomb survivors to the U.S. population is influenced by mechanistic knowledge and information on the etiology of different cancer types.
-
A combined Bayesian analysis of A-bomb epidemiologic information and experimental data has been employed to provide an estimate of the DDREF for cancer risk.
-
Knowledge of adaptive responses, genomic instability, and bystander signaling between cells that may act to alter radiation cancer risk was judged to be insufficient to be incorporated in a meaningful way into the modeling of epidemiologic data. The same judgment is made with respect to the possible contribution to cancer risk of postirradiation genomic instability and bystander signaling between cells.
-
Genetic variation in the population is a potentially important factor in the estimation of radiation cancer risk. Strongly expressing cancer-predisposing mutations are judged from modeling studies to be too rare to distort population-based estimates of risk appreciably, but they are a significant issue in some medical radiation settings. The position regarding potentially more common variant genes that express only weakly remains uncertain.
-
Estimation of the heritable effects of radiation takes advantage of new information on human genetic disease and on mechanisms of radiation-induced germline mutation. The application of a new approach to genetic risk estimation leads the committee to conclude that low-dose induced genetic risks are very small compared to baseline risks in the population.
-
The committee judges that the balance of evidence from epidemiologic, animal, and mechanistic studies tends to favor a simple proportionate relationship at low doses between radiation dose and cancer risk. Uncertainties in this judgment are recognized and noted.
MODELS FOR ESTIMATING THE LIFETIME RISK OF CANCER
As in past risk assessments, the LSS cohort of survivors of the atomic bombings of Hiroshima and Nagasaki plays a principal role in developing the committee’s recommended cancer risk estimates. In contrast to previous BEIR reports, data on both cancer mortality and cancer incidence (from the Hiroshima and Nagasaki tumor registries) were available to the committee. The cancer incidence data analyzed by the committee included nearly 13,000 cases occurring in the period 1958–1998. In addition, the committee evaluated data on approximately 10,000 cancer deaths occurring in the period 1950–2000, in contrast to fewer than 6000 cancer deaths available to the BEIR V committee.
Although the committee did not conduct its own analyses of data from studies other than the LSS, for most studies with suitable data, results of analyses based on models similar to those used by the committee were available and were evaluated. For cancers of the breast and thyroid, several medically exposed groups offer quantitative data suitable for risk assessment, and the recommended models for these sites are those developed in published combined analyses of data from the relevant studies.
To use models developed primarily from the LSS cohort for the estimation of lifetime risks for the U.S. population, it was necessary to make several assumptions. Because of inherent limitations in epidemiologic data and in our understanding of radiation carcinogenesis, these assumptions involve uncertainty. Two important sources of uncertainty are (1) the possible reduction in risk for exposure at low doses and low-dose rates (i.e., the DDREF), and (2) the “transport” of risk estimates based on Japanese atomic bomb survivors to use in estimating risks for the U.S. population. With regard to the DDREF, the committee concluded that linear risk estimates obtained from the LSS cohort should be reduced by a factor of 1.1 to 2.3 for estimating risks at low doses and low dose rates, and the BEIR VII committee used a value of 1.5 to estimate solid cancer risks. To estimate the risk of leukemia, the BEIR VII model is linear-quadratic, since this model fitted the data substantially better than the linear model. The use of data on Japanese A-bomb survivors to estimate risks for the U.S. population (transport) is problematic for sites where baseline risks differ greatly between the two countries. For cancer sites other than breast and thyroid (where data on Caucasian subjects are available), the committee presents estimates based on the assumption that the excess risk due to radiation is proportional to baseline risks (relative risk transport) and also presents estimates based on the assumption the excess risk is independent of baseline risks. As a central estimate, the committee recommends a weighted estimate of these two results, with the ratio of the two used to reflect the uncertainty in transporting risks. For most sites, a weight of 0.7 is used for relative transport and a weight of 0.3 is used for absolute transport; the weighting is reversed for lung cancer.
The committee provides estimates of lifetime risks of both cancer incidence and mortality for leukemia, all solid cancers, and cancers of several specific sites: stomach, colon, liver, lung, female breast, prostate, uterus, ovary, bladder, and all other solid cancers. The committee’s models provide the basis for sex-specific estimates for exposure scenarios including single exposures at various ages, chronic exposure throughout life, or occupational exposure from age 18 to 65. These models are based primarily on the LSS study, with additional use of medical data for breast and thyroid.
As an example, Table 13-1 shows the estimated number of incident cancer cases and deaths expected to result if a population of 100,000 persons with an age distribution simi-
TABLE 13-1 Committee’s Preferred Estimates of the Lifetime Attributable Risk of Incidence and Mortality for All Solid Cancers and for Leukemia
lar to that of the entire U.S. population were each exposed to 0.1 Gy; also shown are the numbers that would be expected in the absence of exposure. Results are shown for all solid cancers and for leukemia. The estimates are accompanied by 95% subjective confidence intervals that reflect the most important uncertainty sources—namely, statistical variation, uncertainty in the factor used to adjust risk estimates for exposure at low doses and low dose rates, and uncertainty in the method of transport. Additional sources of uncertainty would increase the width of these intervals. Mortality estimates are reasonably compatible with those in previous risk assessments, particularly if uncertainties are considered.
The committee also presents estimates for each of several specific cancer sites and for other exposure scenarios, although they are not shown here. For many cancer sites, uncertainty is very large, with subjective 95% confidence intervals covering more than an order of magnitude.
In general the magnitude of estimated risks for total cancer mortality or leukemia has not changed greatly from estimates provided in past reports such as BEIR V, those of the United Nations Scientific Committee on the Effects of Atomic Radiation, and those of the International Commission on Radiological Protection. New data and analyses have reduced sampling uncertainty, but uncertainties related to estimating risk for exposure at low doses and low dose rates and to transporting risks from Japanese A-bomb survivors to the U.S. population remain large. Uncertainties in estimating risks of site-specific cancers are especially large.
Research Need 11. Japanese atomic-bomb survivor studies
The LSS cohort of Japanese A-bomb survivors has played a central role in BEIR VII and past risk assessments. It is thus important that follow-up for mortality and cancer incidence continue for the 45% of the cohort who remained alive at the end of 2000.
In the near future, an uncertainty evaluation of the DS02 dosimetry system is expected to become available. Dose-response analyses that make use of this evaluation should thus be conducted to account for dosimetry uncertainties.
Development and application of analytic methods that allow more reliable site-specific estimates are also needed. Specifically, methods that draw on both data for the specific site and data on broader cancer categories could be useful.
Research Need 12. Epidemiologic studies in general
Data from the LSS should be supplemented with data on populations exposed to low doses and/or low dose rates, especially those with large enough doses to allow risks to be estimated with reasonable precision. Studies of nuclear industry workers and careful studies of persons exposed in countries of the former Soviet Union are particularly important in this regard.
Studies in non-Japanese populations are also important, especially for estimating risks of cancers in organs where baseline risks vary widely. Studies that elucidate the relationship of radiation and other risk factors (for example, smoking) are needed, possibly by conducting nested case-control studies within cohorts currently under study.
Combined analyses of data from several cohorts have been used successfully in the past and are encouraged to provide a unified treatment of data from the LSS and other studies.
Development and application of analytic methods that take account of dosimetry uncertainties are encouraged for all studies. For the LSS, analyses that make use of the uncertainty evaluation of the DS02 dosimetry system, which is expected to become available in the near future, are needed.
CONCLUSION
The committee concludes that the current scientific evidence is consistent with the hypothesis that there is a linear, no-threshold dose-response relationship between exposure to ionizing radiation and the development of cancer in humans.












