
5
Prospective Innovative Chemical Agent Monitoring Technologies
The challenges of reliably monitoring airborne chemical agents at the very low concentrations required by the airborne exposure limits (AELs) for chemical weapons storage and demilitarization activities are daunting (see Chapter 2). Airborne chemical agent mixing ratios in the range of a few parts per billion (ppb) to hundredths of a part per trillion (ppt) must be measured as quickly as possible, and with high specificity to minimize dangerous false-positive or false-negative detections. The triple constraints of high sensitivity, high specificity, and rapid response place severe demands on any chemical agent monitoring system for demilitarization activities (Hill and Martin, 2002).
Components of the current agent monitoring system (both near-real-time (NRT) alarms and confirmation and historical monitors) described in Chapter 4 achieve high sensitivity by concentrating agent or agent derivatives from large volumes of air onto absorptive cartridges that are subsequently desorbed for analysis. Using gas chromatography, the current systems achieve high selectivity by separating agent or agent-derivative species from potential interferent chemicals. Unfortunately, the concentration and separation steps both take time, so the high sensitivity and selectivity of the current system are achieved at the expense of response rate, which varies from a few minutes for NRT ACAMS and MINICAMS alarms to hours for the DAAMS alarm confirmation and historical measurement system.
As discussed in Chapter 3 in the subsection entitled “Methods of Detection,” advanced monitoring techniques for other trace constituents of the atmosphere are generally based on one of the following:
-
Optical spectroscopy (absorption or scattering of radiation at specific wavelengths),
-
Mass spectrometry (ionization followed by mass selective detection), and
-
Chemical sensing (binding of the analyte at a surface or receptor followed by generation of an optical or electrical signal).
One potential advantage of considering chemical agent measurement techniques based on one or more of these approaches is that they use measurement observables that differ from those exploited by the instruments currently in use. Consequently, these approaches are not subject to the same sources of error or interference, although they may be subject to their own unique sources of error.
A further potential advantage of new chemical agent monitoring technologies that may be developed and deployed to supplement the system currently in use is the possibility of significantly improving response time, which is one of the major weaknesses of the current system. Improvements and enhancements to the components of the current system, as discussed in Chapter 4, promise incremental improvements in sensitivity and selectivity but will do little to improve response time. The goal for any supplementary technology should be to move from NRT (<15 minutes) to real-time (~1 to 10 seconds) measurements at the short-term exposure limit (STEL) or the immediately dangerous to life and health (IDLH) level, without significant loss of sensitivity and specificity.
Over the past two decades, great progress has been made in developing robust, real-time trace chemical measurement systems for deployment at both fixed sites and on mobile platforms for atmospheric chemistry and air quality measurements (Kolb, 2003). Most of these systems are based on optical spectroscopy (in either an open-path or sampling mode), mass spectrometry, or, more recently, molecular-level chemical sensors. These techniques have the potential of providing highly selective, real-time measurements of specific pollutants. In this chapter, techniques that have proven to be effective or promise significant advantages for atmospheric chemistry and air quality measurements are reviewed and evaluated for their potential for innovative airborne chemical agent monitoring.
OVERVIEW OF OPTICAL DETECTION TECHNOLOGIES
Basic Principles
A particularly straightforward approach for monitoring trace gases in the atmosphere is the absorption of infrared, visible, or ultraviolet radiation. The spectrum of a molecule provides a distinctive molecular “fingerprint” for the species being sought, and the amount of radiation absorbed at the specified wavelength is directly proportional to the concentration of that species. This contrasts with techniques such as fluorescence, in which corrections for quenching may be required.
The fraction of light transmitted through a vapor column (transmittance T) is governed by the Beer-Lambert law,
(5-1a)
where I0 and I are the incident and transmitted light intensities, respectively; e is the natural logarithm base defined for all positive real numbers; N is the number density of the absorber (molecules per cubic centimeter); σ is the absorption cross section (square centimeters per molecule); and l is the path length through the absorbing sample in centimeters. Equation (5-1a) is often written as
(5-1b)
where a is the absorptivity and c is the species concentration. The absorbance A is defined as log10T = log 10 (I/I0). The absorptivity is absorbance divided by path length times concentration.
A typical cross section for a strong optical transition is 10−18 cm2. For a linear, single-pass absorption measurement, detector and source noise limit the fractional absorption that can be observed. Using state-of-the-art laser sources, detectors, and signal averaging techniques, a δI/I0 = 1 × 10−5 Hz−1/2 has been obtained (Nelson et al., 2002). This sensitivity permits trace gases to be detected at sub-ppb levels in the ambient atmosphere. Achieving such high sensitivity may be problematic for agent molecules, however, which possess congested, poorly resolved spectra. Furthermore, the required monitoring sensitivity at the current AELs is lower than that which can be achieved by single-pass absorption methods.
A number of sensitivity enhancement strategies exist for attaining the required level of detection. The most straightforward is simply to increase the path length, either by enclosing a sample of the atmosphere in a multiple-reflecting cell or extending the path length a long distance through the atmosphere. Off-axis resonator Herriott cells provide 100 to 300 internal reflections, giving an effective path length up to 210 m (Zahniser et al., 1995). In order to use these cells effectively, a highly collimated light beam (such as that from a laser source) is required. These cells require a fairly large sample of gas (about 0.3 liter) due to geometric constraints, and reflection losses at the mirrors may be as much as 80 percent overall. Long-path measurement through the atmosphere is the basis of differential optical absorption spectroscopy (DOAS) and open-path Fourier transform infrared (FT-IR) spectroscopy. Such measurements integrate over variable concentrations along the optical path, but provide wide coverage in circumstances in which sample collection is difficult or infeasible. DOAS and open-path FT-IR are discussed in more detail below.
An alternative strategy for increasing sensitivity by reducing noise rather than extending path length is frequency modulation (FM) spectroscopy (Werle, 1996). Using FM techniques, δI/I0 can be reduced to a few parts in 107, which allows ppb sensitivity to be achieved. There are several limitations to FM spectroscopy, however. To be effective, the modulation amplitude needs to be greater than the line width of the spectroscopic transition, effectively limiting the technique to small molecules at low pressures. FM spectroscopy is not readily applicable to large molecules with congested (inhomogeneously broadened) spectra, such as chemical agents at ambient atmospheric pressure.
Some of the limitations of FM spectroscopy can be overcome by measuring a signal proportional to δI itself rather than to δI/I0. Two such approaches are photoacoustic spectroscopy (PAS) and cavity ringdown spectroscopy (CRDS). PAS detection uses a sensitive microphone to measure the acoustic response of an absorbing medium to optical energy deposited in the medium. Since the absorbed energy is converted to a temperature increase and thence to a pressure wave by a rapid relaxation process, PAS is well suited to measurements at atmospheric or higher pressures. PAS has been implemented in mobile systems for field monitoring using a variety of laser systems (Sigrist et al., 2001; Nägele et al., 2001; Fischer et al., 2001; Sigrist, 2000), with an equivalent sensitivity of δI/I0 iconid 2 × 10−9/cm, allowing atmospheric trace gases to be detected at concentrations of 70 ppt.
CRDS overcomes some of the limitations of using multiple-reflection gas cells (Busch and Busch, 1999). CRDS is based on the principle that when a pulse of light is injected into an optical resonator (or cavity) formed by highly reflective mirrors, the pulse will circulate within the cavity, and the output of the cavity will decay exponentially with time. The decay rate of the circulating intensity within the cavity is dependent on losses at the mirrors (due to their nonperfect reflectivity) and attenuation by any absorber in the cavity. Since the mirror reflectivity is known (either through an empty cavity measurement or from a measurement made off resonance to any absorbers present in the cavity), measuring the exponential decay allows detection of the absorber in the cavity according to the following equation:
(5-2)
where τ is the measured exponential decay half-life (ringdown time) of the CRDS signal, α is the net absorbance of the analyte molecule, L is the length of the cavity, c is the speed of light, and R is the reflectivity of the mirrors comprising the cavity.
If mirrors of sufficiently high reflectivity (R > 99.99 percent) are available, the light injected into the cavity can make up to 104 round-trip passes, which translates into an effective absorption path length of tens of kilometers for a cavity length on the order of 1 m. Since absorption signals are directly proportional to the path length of the sample, this technique is thus capable of detecting an absorbance due to a very low density of sample. Pulsed CRDS has been shown to be capable of detecting explosive vapors with an absorption sensitivity of 2 × 10−8 per centimeter, which corresponds to detection of 1.2 ppb of TNT vapor (Todd et al., 2002). In order to achieve such sensitivity, multiple absorption wavelengths must be used, and the high-reflectivity cavity mirrors must be kept clean of contamination, greatly limiting their application for monitoring agents in the atmosphere.
Long-Path Optical Measurements
DOAS makes use of an extended optical path through the open atmosphere (Plane and Nien, 1992; Evangelisti et al., 1995; Platt, 1994). In addition to eliminating the need for sample handling, DOAS permits real-time, in situ measurements to be made of the path-averaged atmospheric composition. To achieve the required detection sensitivity, the optical path may need to be extended over a distance of a kilometer or more. As noted earlier, the measurement integrates concentration along the beam path. By deploying a set of retroreflectors at strategic locations, multiple directions or elevations can be sampled in a short period of time from a single monitoring station. The measurement must be carried differentially, that is, at pairs of wavelengths on and off resonance with the absorption feature being monitored, in order to discriminate against background absorption and scattering in the atmosphere, especially by particulates and aerosols. A broadband ultraviolet (UV) source is generally used for DOAS measurements.
Among the advantages of DOAS are that broadband features caused by Rayleigh or Mie scattering are discriminated against, there is no need to estimate the spectral background (as there often is for open-path (OP) FT-IR spectroscopy; see below), and it has fairly good specificity. The biggest disadvantage of DOAS is that only a limited number of molecules have suitable absorption bands in the UV-visible spectral region. The UV absorption spectra of chemical agents (GB, HD, VX, and others) generally show weak and featureless absorption below 250 nm (Rewick et al., 1986), resulting in poor sensitivity and poor discrimination from other UV-absorbing atmospheric components. Atmospheric turbulence can also degrade DOAS detection limits. Finally, certain meteorological conditions (e.g., rain, snow, fog, and clouds) can render the method inoperative.
Fourier Transform Infrared Spectroscopy
Fourier transform infrared (FT-IR) spectroscopy is an instrumental technique that is commonly used for characterizing solid and liquid samples, but it can also be used for detecting and quantifying the presence of gaseous species in the atmosphere. In OP/FT-IR measurements, a beam of infrared radiation from a blackbody radiation source is modulated by a two-beam interferometer, passed over a path length of between 100 m and 800 m, and measured by an appropriate detector. Certain wavelengths are absorbed by molecules in the atmosphere; these molecules may be chemical agents or other constituents of the atmosphere such as water vapor or volatile organic compounds. The intensity of each feature in the absorption spectrum is proportional to the product of the concentration of the molecule and the path length of the beam. Spectral signatures of a number of chemical agents and simulants have been reported by Hoffland et al. (1985) and Sharpe et al. (2003). Figure 5-1 shows the infrared absorptivity of GB and HD vapor in the 700 to 1400 cm−1 region, which corresponds to an atmospheric transmission window.
In principle, FT-IR spectroscopy could measure the concentration of chemical agents at intervals of less than 5 seconds, but the technique is several orders of magnitude less sensitive than the ACAMS is even when the measurement time is increased to several minutes, since detection limits are generally inversely proportional to the square root of the data acquisition time.
The use of FT-IR for chemical agent monitoring at demilitarization facilities has been suggested.1 For perimeter (or fence-line) monitoring by OP/FT-IR spectroscopy, the beam from the interferometer would be expanded by a telescope and passed through the open atmosphere over a distance of typically 100 to 400 m to a retroreflector that returns the beam to the telescope and hence to the infrared detector (see Figure 5-2) (Russwurm and Childers, 2002). For such measurements, the analyte is rarely dispersed uniformly over the beam path, and the path-integrated concentration is measured. Alternatively, the infrared beam may be passed through a multipass gas cell (MPGC) before it reaches the detector (Spellicy and Webb, 2002; Stedman and McLaren, 1996; McLaren and Stedman, 1996). This technique, like ACAMS, is an example of point monitoring, because the contents of the cell are drawn from one specific location. The base path length between the mirrors of the gas
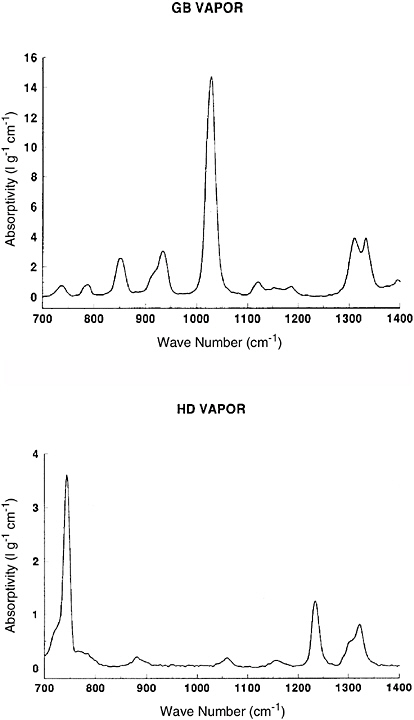
FIGURE 5-1 Infrared spectra of GB vapor (top) and HD vapor (bottom) in the 700 to 1400 cm−1 region. SOURCE: Courtesy of Scott McLaren, Apogee Scientific, Inc., and Donald Stedman, University of Denver.
cell is typically between 2 m and 10 m, with up to 100 passes. See Box 5-1 for an expansion on detection-limit estimates.
OP/FT-IR measurements have advantages and disadvantages relative to the use of MPCGs. The biggest advantage is that the spectrum changes instantly when a plume of the analyte enters the beam path, whereas with MPGC measurements, the contents of the cell are exchanged relatively slowly. The presence of the analyte anywhere in the beam path gives rise to an absorption band in the spectrum. Conversely, for MPGC measurements, if the analyte plume is not at the sampling point, it is not picked up. (This is, of course, a disadvantage with any point monitoring technique.) Since the path-integrated concentration is measured in OP/FT-IR measurements, the actual concentration at any point in the plume is never measured directly. For example, if the path length is 400 m and a plume with a concentration
FIGURE 5-2 Schematic depiction of open-path Fourier transform infrared spectroscopy. SOURCE: Courtesy of H. Yang, University of Idaho.
|
BOX 5-1 Practical detection limits for OP/FT-IR may be estimated using Equation (5-1a) and the vapor spectra shown in Figure 5-1 of this chapter. The absorbance A is defined as where I0 is the incident light intensity and I is the light transmitted through the optical path. The absorptivity is absorbance divided by path length (in centimeters) times concentration (in grams of analyte per liter). For OP/FT-IR, the minimum detectable absorbance change δA is ~1 part in 103, with current instrumentation. A practical path length would be 500 m, or 5 × 104 cm. From Figure 5-1, the most intense GB vapor absorption band has a peak absorptivity of 15 L g−1 cm−1. Thus, the minimum detectable vapor GB concentration is 10−3/(15 L g−1 cm−1 × 5 × 104 cm) = 1.3 × 10−9 g/L or (since 1 cubic meter = 1000 L) 1.3 × 10−6 g/m3, that is, 1.3 × 10−3 mg/m3. These detection limits are further degraded by interfering species, moisture, rain, and atmospheric turbulence. This estimate is to be compared with the 2003/2004 AELs in Table 2-2 in Chapter 2 for GB, which are 10−4 mg/m3, 3 × 10−5 mg/m3, and 1 × 10−6 mg/m3 for the STEL, WPL, and GPL, respectively. It is evident that the sensitivity of OP/FT-IR is at best a factor of 13 to 1300 less sensitive than is required for real-time measurement at these levels. It may also be noted that simply measuring the spectrum at the wavelength where the absorptivity is at a maximum cannot account for the effects of interferents, so most workers in OP/FT-IR prefer to measure several weaker bands in the spectrum along with the strongest band. The AELs for HD mustard are higher than those for GB (Table 2-2), but the infrared detection limit is also less favorable because the maximum absorptivity of the bis(2-chloroethyl sulfide) molecule is about a factor of four smaller than that for GB (Figure 5-1). OP/FT-IR might be barely able to detect HD mustard at the STEL, but is a factor of 12 to 250 too insensitive for detection at the WPL and GPL, respectively. |
of 1 mg/m3 is only present in a 50 m portion of the path, the concentration that would be calculated if it is assumed that the analyte is uniformly distributed over the optical path is 0.125 mg/m3. Thus, the concentration of agent present only in a small segment of the beam path must be very high if the analyte is to be detectable by OP/FT-IR spectroscopy.
A subtle disadvantage of OP/FT-IR spectroscopy is that the sample is at ambient temperature, and hence its absolute temperature can vary by more than ±20°C depending on the time of day and the season. Conversely, the temperature of MPGCs may be controlled fairly accurately. Compensating for the lines in the vibration-rotation spectrum of water vapor is much easier if the sample is always at the same temperature. Similarly, reference spectra of chemical agents are typically measured at ca. 20°C. Since the infrared spectra of all vapor-phase species vary slightly with temperature, quantitative errors may result.
Stedman and McLaren (1996) were able to monitor the concentration of GB in a room at the Chemical Agent Munition Disposal System in Utah using a 96-pass MPGC with a base path length of 6.5 m (total path of 624 m). Using a stored background spectrum, they reported a detection limit of 0.005 mg/m3. Absorption by lines in the spectrum of water vapor was the principal source of interference. By employing a differential technique whereby the previously measured spectrum was used as the background, the detection limit of GB was reduced to 0.0005 mg/m3. However, only changes in the concentration of GB that occur on the time scale of the infrared measurement can be observed with this technique. Thus, it is only useful for detecting catastrophic events in which the concentration of the agent changes very rapidly; it is not likely to be suitable for routine monitoring of agents at the current STELs.
Since lines in the spectrum of water vapor are the main cause of interference, both OP/FT-IR and MPGC approaches would yield poorer detection limits in regions of high humidity (e.g., Anniston, Alabama) than were measured at Tooele in the Utah desert.
McLaren and Stedman (1996) found that detection limits in stack gases increased by at least an order of magnitude because of interference due to the very high partial pressure of water vapor in the stack gases. They also reported that the mirrors in the MPGC became fouled a short time after the flow of the furnace gases was initiated. Single-beam spectra reported by Stedman and McLaren (1996) measured with incinerator exhaust gases (upper trace) and room air (lower trace) flowing through the multipass gas cell are shown in Figure 5-3.
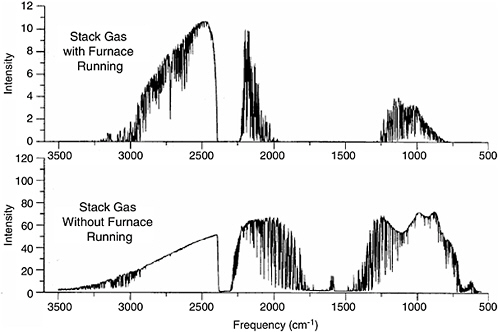
FIGURE 5-3 Single-beam spectra collected when the furnace from which exhaust was being sampled was operating (top) and not operating but drawing ambient facility air (bottom). NOTE: The path length for these spectra was 208 m. Note the 10-fold difference in the ordinate scale and the fact that intense water lines can be seen throughout the entire atmospheric window between about 1250 and 750 cm−1 in the upper spectrum. SOURCE: Stedman and McLaren, 1996.
Finding 5-1. In general, the use of FT-IR spectrometry with either open-path or multi-pass gas cell sampling for monitoring chemical warfare agents (CWAs) at levels below about 0.05 mg m−3 in the atmosphere near the perimeter of chemical agent storage facilities or demilitarization plants is not likely to be effective because of the low sensitivity of this technique. Conversely, FT-IR spectrometry may play an important role in monitoring accidental releases of chemical agents in locations very close to where the agents are stored or incinerated and where the concentration may exceed 0.05 mg m−3. Detecting a catastrophic release within a minute of the event would allow suitable action to be taken.
Surface-Enhanced Raman Scattering
An alternative to absorption-based measurements is based on scattering or re-emission of absorbed light, either as fluorescence or Raman scattering. The measurement of fluorescence from analytes at ppb levels in solution can often be the most sensitive of all quantitative spectroscopic techniques, provided that the molecule of interest has a high quantum efficiency (fluorescence yield) at the analytical wavelength. Unfortunately, most airborne chemical agents do not fluoresce at any easily accessible wavelength and are not present in solution. The UV spectra of the agents are typical of substances that do not possess significant fluorescence yields (Rewick et al., 1986). Not surprisingly, no reports of the successful application of fluorescence for the detection or quantification of airborne chemical agents have been found.
Raman spectrometry exploits the fact that a small portion of the light scattered by a molecule is shifted in frequency by an amount corresponding to a vibrational/rotational transition characteristic of that molecule. While standard Raman spectrometry has nowhere near the sensitivity required to detect very low levels of chemical agents,2 a variant of Raman spectrometry, known as surface-enhanced Raman scattering (SERS), does. In SERS, analytes are deposited on the roughened surface of one of the coinage metals (Ag, Au, Cu). Under the right conditions, the Raman spectrum can be enhanced by four orders of magnitude. Several companies have been developing ways of automatically sampling a variety of analytes, and a few papers reporting on the detection of chemical agents have been published (Alak and Vo Dihn, 1987; Farquharson et al., 2002). These reports deal with the adsorption of agents onto colloidal SERS probes from water and show typical detection limits of tens of ppb. In a system reported by Sylvia et al. (2000), air containing 2,4-dinitrotoluene was passed over a SERS substrate and measured by Raman spectroscopy with a fiber-optic probe. The detection limit was about 5 picograms of 2,4-DNT. The system and spectra that were measured are shown in Figures 5-4 and 5-5, respectively.
While these results are quite promising, the issue of regenerating the roughened gold sensor was not addressed in Sylvia et al. (2000). It should be noted that (1) the concentrations of 2,4-DNT were in the low ppb range, that is, significantly higher than the target AELs for nerve agents and (2) the Raman cross section of nitro-aromatics is much higher than that of phosphonate esters. Several other objections can also be raised to this approach. First and most important, the time resolution will be poor, probably no better than that of ACAMS—that is, these measurements are near real time, not real time. Second, it is very unlikely that any chemical agent will be present as the lone analyte. For example, the air in the region of wooded areas such as Anniston, Alabama, will be loaded with many terpenes and other volatile organic compounds. It is likely that the SERS signal will be masked by the signals from the interferents present at much higher concentrations. The same argument can, of course, be made about OP/FT-IR measurements.
Finding 5-2. Although SERS is the most promising infrared technique of any reviewed by the committee, it is unlikely to be any more sensitive or faster than ACAMS and probably would be less selective and more subject to interference from other airborne molecules.
OVERVIEW OF ION MOBILITY AND MASS SPECTROMETRIC TECHNIQUES
Mass spectrometry and ion mobility spectrometry (IMS) are both highly sensitive analytical techniques involving ions. The instrumentation comes in many forms, and both types of instruments have been used to detect chemical agents (Hill and Martin, 2002). In mass spectrometry, one detects the chemical of interest by measuring the mass of an ion made after ionizing the molecule of interest, while in ion mobility spectrometry, one detects the time that a pulse of ions traverses through a buffer gas, which is related to its mass. However, the IMS signal’s mass relationship is broad, and therefore the technique is not very selective. Inevitably, mass spectrometers involve vacuum systems and ion mobility spectrometers generally do not. This creates a significant difference in the size and cost of the instrumentation. The lack of a vacuum system in an ion mobility spectrometer makes it more suitable for field deployment.
Mass spectrometers are often referred to as the “universal detector,” because some signal can be obtained from essentially any molecule. Electron impact mass spectrometers involve ionizing the sample after it has already entered into the vacuum region. Frequently, this involves 70 eV electrons,
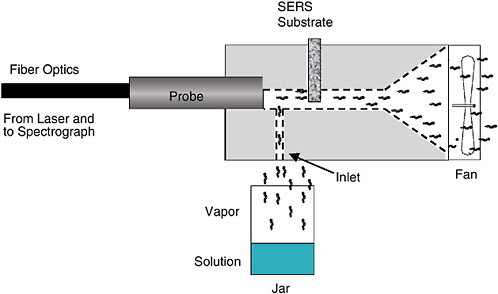
FIGURE 5-4 Schematic diagram of the system used to show the feasibility of surface-enhanced Raman scattering (SERS) measurements of low-concentration explosives in the vapor phase. SOURCE: Reprinted with permission from Sylvia et al., 2000. Copyright 2000 American Chemical Society.
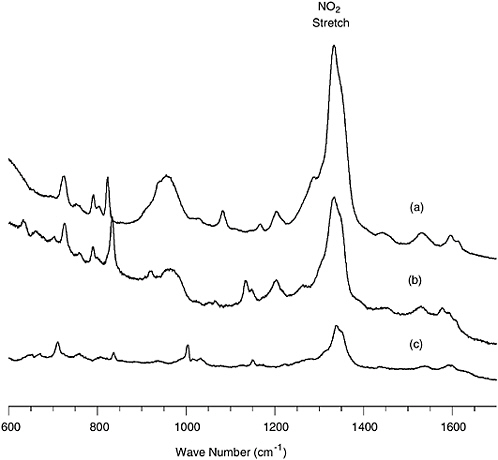
FIGURE 5-5 SERS spectra of (a) 5 pg of TNT, (b) 25 pg of 2,4-DNT, and (c) 50 pg of 1,3-DNB recorded with a 115 mW 785 nm diode laser in 30 s. SOURCE: Reprinted with permission from Sylvia et al., 2000. Copyright 2000 American Chemical Society.
which often leads to fragmentation of the molecule of interest. This fragmentation leads to a loss of selectivity, since a particular mass ion can originate from a molecule having that mass or greater. In order to get both more selectivity and more sensitivity, other ionization schemes are frequently used, including chemical ionization. The latter involves ion-molecule reactions that have exothermicities sufficiently small that fragmentation frequently does not occur. This leaves the molecule of interest intact, thus providing positive mass identification and leading to an increase in selectivity.
As an example of the difference between the two techniques, the electron impact (EI) and chemical ionization (CI) spectra from CH3CNH+ of VX are shown in Figure 5-6. While the electron impact spectrum has many peaks, the

FIGURE 5-6 (Top) Electron impact (EI) and (bottom) acetronitrile chemical ionization (CI) mass spectra of VX agent. The lower trace results from chemical ionization, using CH3CNH+ as the proton transfer medium. By far the dominant peak is protonated VX, with only one other species at m/z = 128 having significant intensity. SOURCE: Rohrbaugh, 1998.
chemical ionization spectrum is dominated by the parent plus one peak. The sensitivity enhancement is due to the ionization occurring at high pressure where the number density is high. The high-pressure ionization advantage applies to ion mobility spectrometry as well.
Figure 5-7 is a schematic representation of a chemical ionization mass spectrometry (CIMS) instrument. Ions are created at the front end of the instrument in an appropriate ion source. After introduction of the sample, the source ions are allowed to react with the sample in a reaction zone. Finally, the ions are sampled into the mass spectrometer and detected. Sensitivity can be increased by producing more ions or by increasing the reaction time. If depletion of the primary ion does not occur, the technique is linear in response to the target molecule. In an ion mobility spectrometer, the ions are pulsed, then transported by an electric field and collected as an ion current on a grid.
The advantage of ion mobility instruments is that they are less complex and therefore cheaper and easier to run than are mass spectrometers. The big drawback is that false positives are common, because compounds with similar weights and molecular shapes are also detected at approximately the same transit time. Several versions of ion mobility spectrometers are used in the field to detect chemical agents (IOM, 1999; Hill and Martin, 2002). These instruments include the Chemical Agent Monitor, Improved Chemical Agent Monitor, Improved Chemical Agent Point Detector System, and Automatic Chemical Agent Detection Alarm. Several of these are small, hand-held units, and false positives are common. They are able to detect concentrations at the IDLH level in 10 s but are not sensitive enough to detect at the STEL. A new technique uses highly asymmetric electric fields but further increases the complexity (Guevremont, 2004). Since use at chemical demilitarization facilities is not constrained by weight or power requirements, and IMS instruments tend to yield many false positives, they will not be considered further.
To increase selectivity further, second-order mass spectrometry, usually termed mass spectrometry/mass spectrometry (ms/ms), can be performed. In this method, the high-energy ions collide with an inert gas at high energy and fragment. Different ions at the same mass often lead to unique fragmentation patterns such that this technique can lead to large increases in selectivity. Additionally, the use of mass spectrometers with high resolving power can unambiguously determine the chemical formula, for example, CO and N2, each of which has a nominal mass-to-charge ratio, m/z = 28, but with peaks separated by a small fraction of a mass unit.
Chemical ionization mass spectrometers use ionization by chemical reaction. Frequently, proton or charge (electron) transfer is used. These instruments can be both extremely sensitive and selective if the proper ion is used in the source. Several studies involving chemical ionization mass spectrometers to detect chemical agents have been reported, all utilizing positive ions (D’Agostino and Provost, 1986; Rohrbaugh, 1995, 1998, 1999, 2000).
Chemical ionization mass spectrometers have been used for many years. Those currently in use in atmospheric chemistry represent a new generation of instrumentation and serve as a guide for sensitivities and selectivities that are currently possible. The atmospheric instruments are both custom built and commercially available, although the latter are not as sensitive as the former. Many of them are used on airplanes and need to be compact. Additionally, some of the airplane platforms have only pilots aboard and therefore require totally automated instrument operation. The automation includes not only the actual measurements but calibrations and subtractions for background species. These instruments are programmed to run at least as long as an airplane flight without user attention. Extending such instrumentation to continuous, autonomous operation is straightforward and would only require ensuring a continuous gas supply for the ionization flow reactor. Instruments used for atmospheric studies require the ability to sample at numerous altitudes and with wind speeds equal to the aircraft speed. These are harsh conditions. The ability of the atmospheric instruments to operate remotely and under a range of stressing conditions should mean that the technology could be adapted for the long periods of reliable operation necessary for use at chemical demilitarization facilities.
A commercial instrument now used extensively by the atmospheric studies community is made by Ionicon Analytik

FIGURE 5-7 Schematic diagram of a generic chemical ionization mass spectrometry (CIMS) instrument. NOTE: IMS, ion mobility spectrometry; HV, high volume. SOURCE: Courtesy of committee member A. Viggiano, Air Force Research Laboratory.
in Austria.3 In this instrument, H3O+ ions are made in a hollow cathode discharge source, which is extremely stable. The H3O+ ions are injected into a drift tube, where they are declustered and allowed to react with trace constituents, mainly by proton transfer. The resulting ions are detected by a quadrupole mass spectrometer. This requires that the proton affinity of the molecule of interest be higher than that of water, in order to ensure that the reaction is exothermic. Work has been done on proton transfer to chemical agents from CH5+ (Rohrbaugh, 2000); HCH3CN+ (Rohrbaugh, 1998); NH4+ (Rohrbaugh, 1995; D’Agostino and Provost, 1986); and CH3OH2+ (Rohrbaugh, 1999). The latter ion was shown to produce little fragmentation. Both proton transfer and clustering were commonly observed with NH4+, and were described as both sensitive and selective. These studies show that protonated species enable sensitive detection of the relevant chemical agents and their degradation products. It should be possible to produce such ions in the Ionicon source by introduction of small amounts of the hydrogen-containing analytes.
The sensitivity of the Ionicon instrument is approximately 0.1 cts/s/ppt, where cts is an abbreviation for counts (de Gouw et al., 2003). Therefore, if the chemistry is clean—that is, there is no background or interference—an accurate measurement of 1 ppt can be made in several minutes. However, interferences at this level are common, since many molecules have proton affinities greater than that of water. Common sensitivities for various chemicals are typically in the tens to hundreds of ppt. Comparing with the IDLH values of 16,000 ppt, 250 ppt, and 100,000 ppt for GB, VX, and HD, respectively, this instrument has the capability to measure these levels in less than 1 s. Measurements at the STEL should be difficult except for HD (423 ppt). A version of the instrument that uses an ion trap mass spectrometer is under development, and this should help in ion identification by reducing interferences, enabling ms/ms measurements to be made.
The custom-built instruments for atmospheric chemistry have much better sensitivity and use a variety of ions as the chemical ionization medium. Generally, these instruments use either proton transfer from H3O+ or a variety of negative ions as the precursor ionization species. The use of negative ions has not previously been explored for use in detecting chemical agents, although it was speculated in a past report that negative ions would be suitable, since there should be no background problem from hydrocarbon interference (Ketkar et al., 1989).
The most sensitive custom-built instrument is the National Center for Atmospheric Research (in Boulder, Colorado) instrument for sulfuric acid vapor detection, in part because there is essentially no background (Eisele and Tanner, 1993). This instrument uses a radioactive ion source that is extremely stable and reliable to make NO3− as the precursor ionization species, and it offers an impressive sensitivity of 1000 cts/s/ppt. This translates into 1, 10, and 60 s detection limits of 0.003, 0.001, and 0.0003 ppt, respectively. This instrument is rather large—it takes up two aircraft racks and weights 400 lb. A version half as sensitive but half this size, achieved mainly through the use of smaller pumps, is under construction. NO3− would not be expected to react with chemical agents, but it may form cluster ions with the agent molecules. Substitution of another ion such as a proton transfer agent should be straightforward. These extremely low detection limits are such that even the GPL of VX (0.05 ppt) could be detected in 1 s. Of course, this relies on finding a suitable chemical ionization species. An instrument at the Georgia Institute of Technology that uses similar chemistry is a factor of three less sensitive and is fully enclosed in a wing pod on the aircraft (Chen et al., 2005). The sensitivity of this instrument is still high enough to be able to detect a compound at the GPL limit of VX.
A different approach is being taken by a group at the National Oceanic and Atmospheric Administration (NOAA) in Boulder, Colorado (Marcy et al., 2005). Various F− transfer agents are used as the CI medium, including SF5−, SF6−, SiF5−, and CF3O−. The instrument uses a beta emitter that is sold as an in-line ionizer (i.e., it does not need a permit). For a variety of compounds, the sensitivity is a few counts per second per ppt, with detection limits on the order of 1 to 30 ppt in 1 s. This is good enough so that it may be possible to allow the STEL for GA/GB (16 ppt) and VX (0.84 ppt) to be detected in real time (1 to 10 s). The AEL limits for HD are much larger than those for the nerve agents, so it may be possible to detect HD at the GPL with a suitable CIMS instrument in real time. Since ion molecule reactions are generally fast when exothermic, it will be worthwhile to use ab initio calculations to determine F− affinities for the chemical agents. If exothermic, F− should be very sensitive and most likely background free, since the molecular weights of the chemical agents are relatively high. Several F− transfer agents are easily made, and the chemistry should easily be transferable to the more sensitive instruments described above.
Two groups (Air Force Research Laboratory (AFRL), Hanscom Air Force Base; and Max Planck Institute (MPI), Heidelberg, Germany) use CO3− as the chemical ionization species. The sensitivity for the AFRL instrument is about 5 cts/s/ppt (Ballenthin et al., 2003), about the same as for the NOAA instrument. The MPI instrument sensitivity is similar. The CO3− is made in a corona discharge, which is another stable, reliable ion source. CO3− may be a good precursor ionization species if the F− transfer agents noted above do not work, since it tends to be quite reactive, often transferring an O atom. For example, preliminary experiments
|
3 |
Additional information is available online at http://www.ptrms.com. Last accessed July 25, 2005. |
show that it is sensitive to TNT.4 One detriment is that detection with CO3− tends to be sensitive to water vapor. This sensitivity would vanish in the Ionicon instrument. The Heidelberg instrument also uses NO3−(HNO3) and H3O+ as chemical ionization species. The most recent version of the MPI instrument uses a quadrupole ion trap as the mass spectrometer. The main advantage of doing so is that collision-induced dissociation experiments can be performed to aid ion identification.
The preceding discussion shows that current state-of-the-art CIMS instrumentation can readily detect certain chemical species at concentrations well below the IDLH limit in real time, that is, 1 s. Real-time detection at the STEL is also likely for several agents. The best instrumentation should be able to detect concentrations for the most stringent monitoring requirement, that is, the GPL of VX, in real time. The problem is in choosing a source ion that is both sensitive and selective. Negative ions hold much promise mainly in terms of selectivity and therefore small background. The ion chemistry is relatively easy to test, both through calculations and through the use of surrogates in ion-molecule reactors such as flow tubes and ion cyclotron resonance instruments. It should be possible to accomplish these feasibility studies in a few months.
A recent development in mass spectrometry related to the detection of surface contamination deserves mention. The Cooks group at Purdue University has recently invented a new technique called desorption electrospray ionization (DESI) (Takats et al., 2004). In this approach, an electrospray source is aimed at a surface, and the ions formed are sampled into an ion trap mass spectrometer through an ion transfer line. The Cooks group has been able to observe the chemical warfare agent simulant dimethyl methylphosphonate (DMMP) on nitrile gloves exposed to DMMP for only 1 s. This was accomplished even after the gloves were washed and dried. The low-volatility explosive cyclotrimethylenetrinitramine (RDX) was also detected on a leather surface. A similar plasma entrainment/ionization technique termed Direct Analysis in Real-Time (DART) has been developed by JEOL USA, Inc. DART has also been used to entrain and ionize a wide variety of chemicals adsorped on surfaces, including VX for most spectrometric analysis (Cody et al., 2005). The DESI and DART techniques, which seem easy to use, show great promise as a means for determining whether materials that may have been exposed to chemical agents are contaminated.
Finding 5-3. Chemical ionization mass spectrometry (CIMS) is a highly sensitive technique that may be able to detect all chemical agents in real time, potentially even at the general population limit. Previous work has focused exclusively on positive ion precursors. Instruments in the atmospheric community extensively use negative ions as a precursor, leading to increased selectivity without sacrificing sensitivity. Commercial CIMS instruments are already available, although with reduced sensitivity.
Recommendation 5-3. The Army should investigate whether present CIMS instrumentation could be immediately used to detect chemical agents at the IDLH limit in real time. The use of negative ions as a precursor should be investigated to improve selectivity. Adaptation of one of the research-grade atmospheric field instruments for real-time detection between the STEL and the GPL for each relevant agent should be considered.
MOLECULAR-LEVEL CHEMICAL SENSORS
A number of innovative technologies exist that have the potential for contributing to the chemical agent monitoring effort. Significant developments are occurring in the area of molecular chemical sensors. These technologies may be able to provide new or supplementary means for worker monitoring, or they may provide the capability for networked sensing outside the fence line of incineration facilities. These methods rely on new sensing paradigms and new chemistries.
Conventional analytical measurements often involve three distinct steps: sampling, pretreatment, and measurement. First, a representative sample is collected. This often entails preconcentrating the sample by passing large volumes of air or water through a filter or adsorbent. Second, to remove interfering substances, the sample is processed by such techniques as chromatography, precipitation, and distillation. In some cases, a label (e.g., dye or electroactive label) is attached to the analyte to make it detectable. Finally, an actual measurement is made on the processed sample.
Chemical sensors integrate all three of these operations. The sampling often consists simply of exposing the sensor to air containing the analyte, and no preprocessing is needed because all the necessary chemistry to impart selectivity and sensitivity is located within the sensing layer. Thus, molecular-level chemical sensors offer significant simplification, and their response times can be rapid. Because of their design, such sensors make measurements continuously.
The chemical sensing technologies discussed below have all been tested with chemical agents or agent simulants and represent the application of state-of-the-art microsensors and microfluidics to chemical agent detection. In addition to the sensors themselves, significant effort is being directed to developing new sensing materials with the requisite specificity and sensitivity to solve challenging detection problems.
Electronic or Artificial Noses
An emerging approach to broadband chemical detection is inspired by the biological olfactory system (Buck and Axel, 1991; Buck, 1996). These systems are based on using an array of semiselective cross-reactive sensors that produce a response pattern which is characteristic of a particular compound or mixture (Gardner and Bartlett, 1999; Albert et al., 2000). The response pattern is used to “train” a computational pattern-recognition system that “memorizes” the pattern and recalls the identity of the analyte upon subsequent exposure. The combination of array detection, complex pattern generation, and computational processing is similar to the way that the olfactory system performs odor recognition.
Conducting Sensor Films
A very large scale integrated circuit (VLSI)-compatible, versatile, low-cost, electronic “nose-chip,” developed at the California Institute of Technology, has enabled arrays of simple, readily fabricated, chemically sensitive resistor films to be produced. An array of sensors that individually respond to vapors can produce a distinguishable response pattern for each separate type of analyte or mixture. Pattern-recognition algorithms and/or neural network hardware are used on the output signals from the electronic nose to classify, identify, and, where necessary, quantify the vapor or odors of concern.
The underlying principle of the “Caltech electronic nose” is extraordinarily simple. When a sorption sensor film is exposed to a gaseous vapor, some of the vapor partitions into the film and causes the film to swell (Figure 5-8). This swelling is probed electrically, because each sensor film consists of a composite that contains conducting particles that have been dispersed into a swellable organic insulator. The composites can either be two-phase systems, consisting of a mixture of conductive particles within an insulating, swellable organic polymer, or they can be single-component composites of nanoparticles in which the sorbent phase is covalently
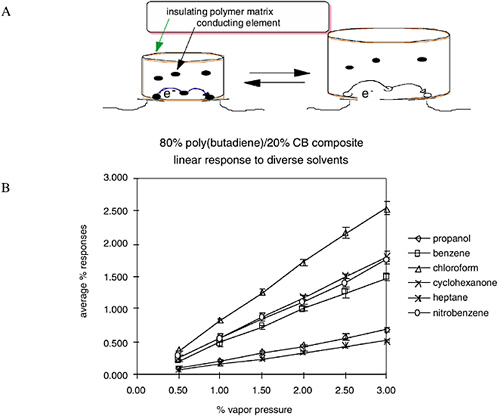
FIGURE 5-8 (A) Swelling occurs as an odorant partitions into the sorption phase. (B) A linear response of an individual sensor signal as a function of concentration is observed for a variety of analytes. SOURCE: Courtesy of N. Lewis, California Institute of Technology.
linked to the nanoparticle through prior chemical reactions (Freund and Lewis, 1995; Lonergan et al., 1996; Doleman et al., 1998; Sotzing et al., 2000). The vapor-induced film swelling produces an increase in the electrical resistance of the film, because the swelling decreases the number of connected pathways of the conducting component of the composite material. Any individual sensor film responds to a variety of vapors, because numerous chemicals will partition into the polymer and cause it to swell to varying degrees. An array of sensors containing different sorption phases yields a distinct fingerprint for each combination of vapors, because the swelling properties over the entire array are different for different vapors (Albert et al., 2000).
The pattern of resistance changes on the array of sensors is diagnostic of the vapor, while the amplitude of the patterns indicates the concentration of the vapor (Severin et al., 2000). Figure 5-9 provides an example of the different patterns produced by various vapors on the electronic nose. Each different odor or analyte produces a different response pattern on an array of such detectors, therefore allowing identification and classification of the target odors through the use of automatic target-recognition techniques (Severin et al., 2000). The swelling/deswelling process is highly reversible in thin polymer films, so typically thousands of cycles can be performed without significant loss of sensor responsivity.
This electronic nose approach to broadband chemical detection has been shown to produce responses in the mid-ppb range to nerve agent simulants DMMP and DIMP either in air or in the presence of much higher concentrations of common interferents such as diesel fuel, lighter fluid, THF, water vapor, and other common solvents (Hopkins and Lewis, 2001). Live-agent testing at the Edgewood Research, Development, and Engineering Center at Aberdeen Proving Ground, Maryland, has shown responses in the low ppb levels for G-class chemical agents. Further improvements in sensitivity are reasonable to expect as the sorbent phase is tailored to be complementary to the chemical properties of the agents of interest (Briglin et al., 2001, 2002). See Figure 5-10.
Optical Sensor Arrays
Tufts University researchers are developing optical sensor arrays. These arrays are prepared by first etching an array of optical fibers. Etching causes wells to form at the end of each optical fiber in the array. The sensors are prepared by attaching fluorescent dyes to microspheres made from various types of commercially available 3 to 5 mm diameter polymer and silica beads or from polymer-coated silica beads (White et al., 1996; Dickinson et al., 1996, 1998, 1999; Albert and Walt, 2000, 2003). Solvatochromic fluorescent indicators (dyes that change their fluorescence according to the polarity of their environment) are incorporated into the beads by entrapment or adsorption. Each dyed bead type represents a stock of ~1.0 × 1011 identically responding bead sensors per gram of material. These sensor stocks can be combined to form a sensor library suited for vapor detection. A sensor array is fabricated by randomly dispersing the sensor bead library into the etched walls on the face of the optical fiber array (Figure 5-11) (Michael et al., 1998). The size of the beads matches the size of the wells such that only one bead assembles into each well. This dispersion of sensors in a random fashion simplifies array fabrication by eliminating the difficulty of positioning each sensor on a defined point within the array. Hundreds to thousands of each type of sensing bead are represented on the array.
A fluorescence imaging system using a charge-coupled device (CCD) detector is used to monitor sensor array responses. Vapor samples are delivered to the array in a pulsatile fashion via a vacuum-controlled sparging apparatus. The CCD camera records the fluorescence responses for all sensor elements during the vapor pulse. By monitoring each sensor’s temporal fluorescence change at a specific wavelength, unique response patterns are collected. The responses are based on how the polarity of the vapor interacts with the sensor bead composition and affects the dye’s emission properties.
As described below, the Tufts University optical nose has addressed one of the most challenging aspects of electronic noses—the requirement for extensive “training,” that is, calibration of the response to a specific type and amount of analyte. During training, cross-reactive arrays are exposed to vapors and the responses from the various sensor types are collected and stored in a database. The responses are used to develop a pattern-recognition algorithm, also called a classifier, for all the vapors. Under optimal conditions, these algorithms are able to differentiate one vapor from another, thereby enabling vapor identification upon subsequent exposure. For various reasons, all sensors in all of the different types of arrays discussed in this subsection on electronic or artificial noses will degrade, requiring that sensor arrays be periodically replaced. To date, no two arrays are the same; consequently, every cross-reactive array to date has required its own training because the responses from one array do not transfer to any other array. This need to replace arrays has reduced the enthusiasm for electronic noses because of the need to train every array. The Tufts University optical nose has demonstrated the ability to transfer training from one array to replicate arrays, because the library of microsphere sensors is made in batches and can be used to prepare many identical arrays. As can be seen in Figure 5-12, the data produced by these bead sensors are highly complex in shape, providing high information content that is useful for selectivity.
The Tufts researchers have also developed a sensor array that is sensitive to chemical agents. The array contains both nonspecific sensors that give it broadband sensitivity and a specific sensor type that responds only to reactive compounds, for example, nerve agents and alkylating agents such as HD. This array has undergone testing with chemical agent simulants.
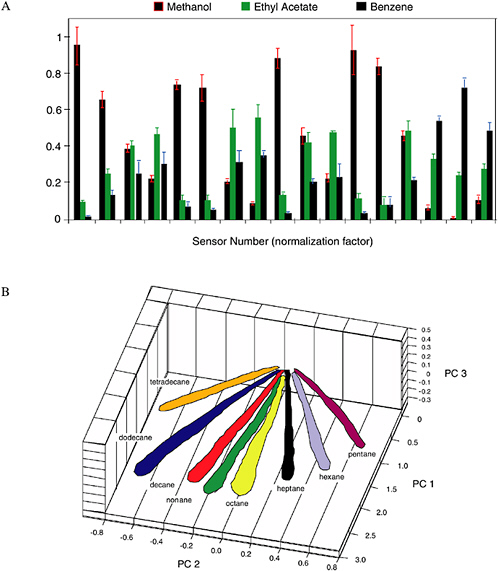
FIGURE 5-9 (A) Response patterns for three different solvents on a 17-element sensor array. (B) Data in principal component space from a 20-detector array exposed to n-tetradecane, n-dodecane, n-decane, n-nonane, n-octane, and n-heptane each at P/P° = 0.005 to 0.03 in air (with P° being the vapor pressure of the analyte at 300 K), showing that the pattern type identifies the vapor, and the magnitude of the pattern signals is linearly proportional to the analyte concentration. Axes labeled PC 1, PC 2, and PC 3 correspond to the first three principal components for the data. SOURCE: Courtesy of N. Lewis, California Institute of Technology.

FIGURE 5-10 Photograph of a “nose-chip,” with 12 columns and 6 rows, each having a different polymeric sensor combination and each pixel having a switch under its pair of contact lines. A single printout provides the output of the sensor pixel being interrogated using a row/column addressing scheme. SOURCE: Courtesy of N. Lewis, California Institute of Technology.

FIGURE 5-11 Microsphere sensors loaded onto the end of an optical fiber array. By averaging the signals of multiple copies of each sensor type in the array, slight differences between beads are eliminated. Furthermore, averaging beads of each sensor type leads to enhanced signal-to-noise ratios. SOURCE: Courtesy of Illumina, Inc.
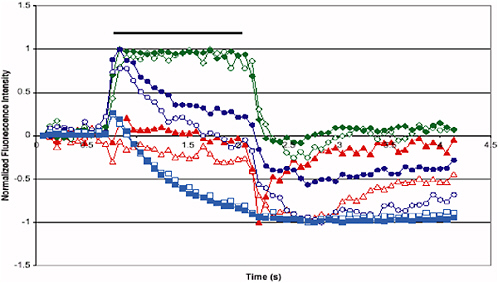
FIGURE 5-12 The average fluorescence response patterns of 12 bead sensors. Benzene (red triangle), toluene (green diamond), 1.3-DNB (●), and 4-NT (■) vapor responses are shown, demonstrating how structurally similar compounds can be distinguished on the basis of differences in their temporal response profiles. The solid black line indicates the time frames during which the vapor pulse (lasting 1.37 s) was delivered to the array. SOURCE: Courtesy of committee member D. Walt, Tufts University.
The Tufts researchers have demonstrated the first artificial nose able to transfer a classifier between two sensor arrays (Stitzel et al., 2001). For commercial applications, transferable classifiers are more efficient because they allow sensor arrays to be replaced without retraining or recalibrating. More importantly, the ability to interchange sensor arrays allows a library of vapor response patterns to be generated, serving as a reference for learned vapors and representing an “odor memory.” This advance could facilitate the investigation of complex problems such as environmental monitoring or clinical diagnostics in which large training sets may be required. Problems of this type are especially difficult because they involve complex mixtures and unknown components that vary from environment to environment or from person to person. A training process alone could be quite extensive and time consuming, requiring hundreds or even thousands of observations to be collected in order to accurately train the array to detect chemical agents in environments with complex backgrounds.
Another type of optical array platform is being developed by the Suslick group at the University of Illinois (Suslick, 2004; Suslick et al., 2004). In this array, a diverse set of chemical indicators, including metalloporphyrins, pH-sensitive dyes, and solvatochromic indicators, are used to generate color patterns that can be read by a color camera and converted into red-green-blue color values for processing (Rakow and Suslick, 2000; Sen and Suslick, 2000). This colorimetric-array sensor technology is based on strong and relatively specific interactions between the analytes and a set of chemoresponsive dyes. The materials cost of the printed arrays is a few cents (the majority of that is the cost of the polyvinylidene fluoride membrane, which in bulk sells for ~$0.04/cm2, and the imaging system is extremely inexpensive (white LED with web camera CCD or even an ordinary flatbed scanner). The arrays have been tested with chemical agents, and the responses to various agents are shown in Figure 5-13. In preliminary studies done in conjunction with ChemSensing (which is commercializing Suslick’s technology) and the Battelle Memorial Institute, excellent sensitivity to several chemical agents was observed, with visual changes in color even at the low ppb level.
New Materials
Another emerging trend is in the development of new sensing materials.
Fluorescent Indicator Detectors
The Swager group at the Massachusetts Institute of Technology has developed a fluorescent indicator that is selective for molecular functional groups. The indicator displays a strong increase in fluorescence in the presence of electrophilic phosphates (Zhang and Swager, 2003). The indicator has differential reactivity to nerve agents (G-agents) relative to the less reactive pesticides, and because it generates fluorescence from a dark state, ultratrace detection of most G-agents is possible. This general method is undergoing further chemical and instrument development by Nomadics, Inc., and working sensor prototypes have been produced that approximate the size of a typical TV/VCR remote (see Figure 5-14). The system gives a reversible response because activated chromophores are readily photobleached, which brings the system back to a stable baseline. Illumination with light removes the color from the activated indicators, thereby returning the signal to baseline. A similar process results in the fading that occurs when a dyed article of clothing is left out in bright sunshine.
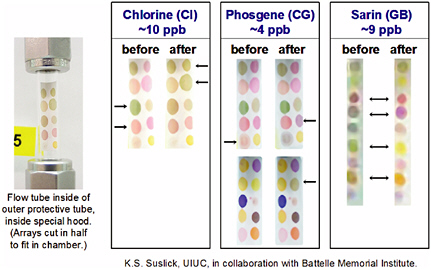
FIGURE 5-13 Color patterns obtained upon exposure to various chemical agents. SOURCE: Courtesy of K.S. Suslick and M.E. Kosal, University of Illinois at Urbana-Champaign.
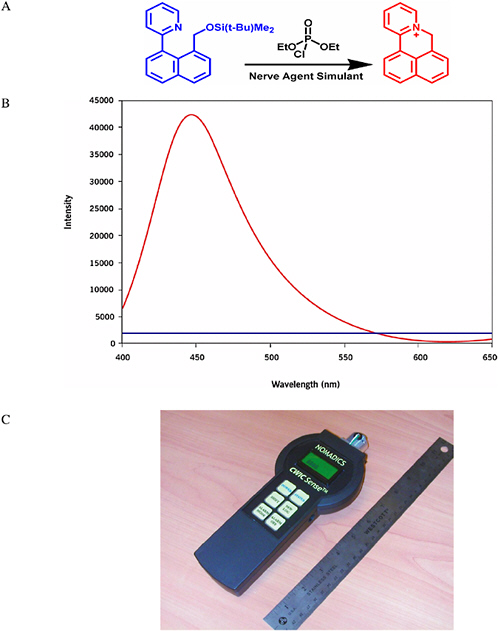
FIGURE 5-14 (A) Reaction of the Chemical Warfare Indicating Chromophore (CWIC) reporter (blue) with a nerve agent simulant produces a fluorescent reporter molecule (red). (B) The fluorescence spectrum of the CWIC before (blue) and after (red) exposure to a nerve agent simulant shows a dramatic increase in intensity. (C) Nomadics, Inc.’s prototype handheld system for chemical detection using the CWIC. SOURCE: Parts A and B: Frye-Mason et al., 2004. Part C: Courtesy of Nomadics, Inc.
The sensitivity of the Nomadics CWIC sensor (Figure 5-14) exceeds the present Department of Defense specifications for the Joint Chemical Agent Detector (JCAD). The CWIC sensor provides a signal-to-noise ratio of 3600 for a 2 second challenge at 0.1 mg/m3. This sensitivity can be compared with JCAD specifications requiring the detection of 1 mg/m3 for <10 s and 0.1 mg/m3 for <30 s. Recent live-agent testing has indicated that the CWIC detection technology can give comparable detection limits of live agents including sarin, tabun, and lewisite (Frye-Mason et al., 2004).
Surface-Enhanced Raman Biosensing
Structured nanomaterials for surface-enhanced Raman-based biosensing are being developed by the Van Duyne group at Northwestern University. In this approach, local fields generated by the structured nanoparticles provide a significant enhancement resulting in high-sensitivity measurements using both Raman (Yonzon et al., 2004; Shafer-Peltier et al., 2003) and surface plasmon resonance mechanisms (Haes and Van Duyne, 2002; Riboh et al., 2003; Haes et al., 2004a, 2004b, 2004c) (see Figure 5-15). The ability to enhance the field by creating nanoscale structures opens up the possibility for preparing sensors and sensing materials with high functionality in a small package without sacrificing sensitivity. Furthermore, these new types of materials may provide completely new transduction mechanisms that could be employed for sensing.
Porous Silicon Technology
The first chemical sensor that utilized the passive optical properties of porous silicon (Si) involved Fabry-Pérot interference from a thin layer (Curtis et al., 1993). The sensing principle of the single-layer Fabry-Pérot films, as well as more complex photonic crystals, is based on what is commonly called the optical thickness of the film. Optical thickness is the product of the refractive index (n) and the thickness (L) of the film. The electrochemical parameters used in the synthesis of porous silicon control both of these parameters precisely and reproducibly: the current density usually controls porosity and hence n, and the length of time that the sample is etched determines L. A high-quality Fabry-Pérot film made of porous silicon has two planar and parallel interfaces and displays high-fidelity fringes in the reflectivity spectrum (see Figure 5-16). A spectral shift in these fringes occurs when the refractive index of the film changes—for example, when a molecule is admitted into the pores—corresponding to a change in optical thickness (Lin et al., 1997). Of the several electrical and optical transduction modes available to porous silicon films, this optical interference method is perhaps the most extensively developed and possibly the most robust. Detection of toxic chemicals (Létant and Sailor, 2000; Sohn et al., 2000), volatile organic compounds (Lauerhaas et al., 1992; Snow et al., 1999; Zangooie et al., 1999), polycyclic aromatic hydrocarbons (Song and Sailor, 1997), explosives (Content et al., 2000; Sailor et al., 2001), DNA (Lin et al., 1997; Chan et al., 2000),
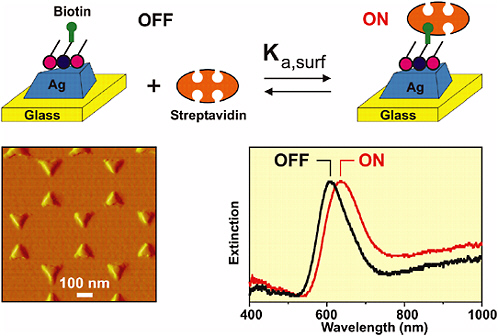
FIGURE 5-15 A nanoscale optical biosensor. Nanoparticle structured surfaces provide field enhancements for chemical detection. SOURCE: Reprinted with permission from Haes and Van Duyne, 2002. Copyright 2002 American Chemical Society.

FIGURE 5-16 Reflectivity spectra from a single-layer porous Si film (left) and from a multilayered (rugate filter) porous Si film (right).
SOURCE: Courtesy of M.J. Sailor, University of California, San Diego.
and proteins have all been reported (Dancil et al., 1999; Janshoff et al., 1998; Zangooie et al., 1998), and detection limits of at least a few ppb have been demonstrated for some of these compounds (Sailor, 1997).
Metal ion catalysts have been used to hydrolyze phosphate esters, including the organophosphate nerve warfare agents. GB (sarin) contains a phosphorus-fluorine bond that is hydrolyzed more rapidly in the presence of such catalysts (see Figure 5-17). When a copper metal ion catalyst is incorporated into the surface oxide of a porous silicon Fabry-Pérot film, a sensor that is highly specific for organofluorophosphonates results. The metal ion catalyst promotes a reaction between the organofluorophosphonate and water to produce hydrofluoric acid that dissolves the thin silicon dioxide film. The reaction results in a shift in the Fabry-Pérot interference fringes that can be monitored with an inexpensive, low-powered diode laser and phototransistor combination (see Figure 5-18). The device is specific for the vapors of sarin and presumably any members of the class of organofluorophosphonates (including diisopropylfluorophosphonate, soman, and cyclosarin (GF)) (Sohn et al., 2000).
Such materials could be used as triggers; they could be used in cheap, low-power devices that could sound an alarm in case of a leak.
Lab-on-a-Chip Technology
Investigators at Arizona State University, the Naval Research Laboratory, and University of California, Riverside, have developed an integrated chip-based sensing device. Field detection of such hazardous substances as chemical warfare agents requires that powerful analytical performance be coupled to miniaturized low-powered instrumentation. “Lab-on-a-chip” devices, in which liquids are manipulated in a microchannel network, offer great promise for converting large and sophisticated instruments into powerful, field-deployable analyzers. Particularly attractive for on-site security applications are microchip devices that offer an analyzer with a very small footprint, a high degree of integration, high performance, fast response, and versatility (Wang, 2004). Such microfluidic devices offer great promise for transporting the forensic laboratory to the sample source, providing an early warning prior to terrorist activity, providing rapid postanalysis “fingerprinting” of a disaster site, and monitoring the cleanup of contaminated/remediated sites. Owing to their small footprint, microchip devices could perform multiple assays simultaneously. Self-contained microchips based on capillary electrophoresis (CE) with electrochemical detection are extremely attractive for on-site security applications, owing to the inherent miniaturization,
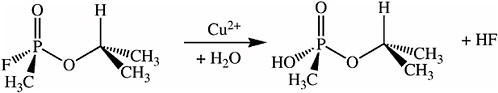
FIGURE 5-17 Metal ion catalysts containing a phosphorus-fluorine bond.
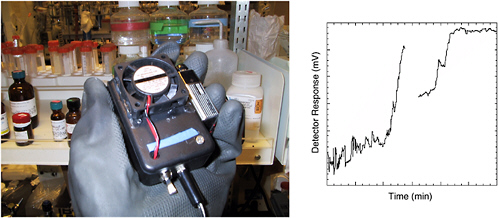
FIGURE 5-18 (Left) Handheld nanosensor device for nerve agent developed for the Micro Unattended Ground Sensors program (program manager: Edward Carapezza) of the Defense Advanced Research Projects Agency. The device contains a porous Si photonic crystal which contains chemistry that allows it to respond specifically to sarin (GB) gas. (Right) Testing run showing response to sarin at 10 ppm within 7 minutes of introduction. The sampling chamber was depleted of agent approximately 10 minutes into the run. SOURCE: Courtesy of M.J. Sailor, University of California, San Diego, and Kwok Ong, Aberdeen Proving Ground/Edgewood Arsenal.
high performance, low cost, and minimal power demands of such detectors.
Several microchip protocols for monitoring chemical agents and their degradation products have been developed (Wang et al., 2004a, 2004b, 2002a). They include the following:
-
Precolumn enzymatic (organic phosphate hydrolase—OPH) reaction of organic phosphate nerve agents coupled to fast CE separation and amperometric detection of the nitrophenol reaction products (Wang et al., 2004a).
-
A chip-based CE/conductivity microfluidic device that was recently developed for fast screening of chemical agent degradation products (Wang et al., 2002a). The miniaturized system relies on an efficient, chip-based separation of alkyl methylphosphonic acids (breakdown products of sarin, soman, and VX nerve agents) followed by sensitive conductivity detection. For example, Figure 5-19 displays an electropherogram for a river sample, spiked with 20 ppm methylphosphonic acid (a), ethyl methylphosphonic acid (b), and isopropyl methylphosphonic acid (c).
-
CE microchip separation and amperometric detection of thiol-containing degradation products of V-type nerve agents (Wang et al., 2004b). The microchip assay relies on the derivatization reaction of 2-(dimethylamino)ethanethiol, 2-(diethylamino)ethanethiol, and 2-mercaptoethanol with o-phthaldialdehyde (in the presence of the amino acid valine) along with amperometric monitoring of the isoindole derivatives. Typical assay times are 2 to 3 minutes.
Analogous microchip monitoring of explosive compounds has also been very successful (Wang et al., 2002b). Current efforts by these investigators are aimed at integrating sampling and preconcentration capabilities and at integrating multiple assays on a single microchip platform.
Summary of Chemical Sensor Technology
The chemical sensor technologies described above represent new research directions. It is important to note that while some of the technologies are being pursued commercially, others are still at the academic laboratory stage. None of the technologies has been tested thoroughly with live chemical agents in diverse background environments. They also have not been tested for their longevity or false-positive/false-negative rates under realistic test conditions. While some of the methods have demonstrated an ability to detect ppb levels of agent, others have only ppm sensitivity. The advantage of most of the systems described here is that they are or could be designed to be small, low-power devices, and they have the potential to be inexpensive and fast. As such, these units
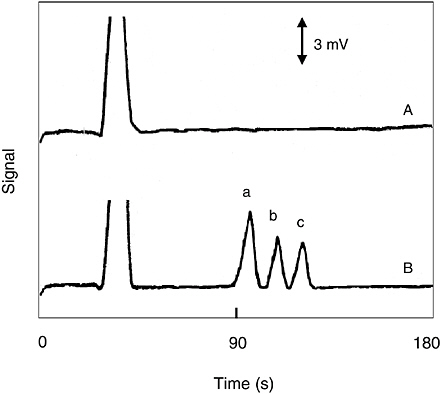
FIGURE 5-19 Peaks exiting the capillary electrophoresis (CE) chip: (a) methylphosphonic acid, (b) ethyl methylphosphonic acid, and (c) isopropyl methylphosphonic acid. SOURCE: Reprinted with permission from Wang et al., 2002a. Copyright 2002 by American Chemical Society.
could be used for detecting leaks at higher levels than those for worker protection (e.g., in storage igloo monitoring). If these sensors can be made small and inexpensive, they could be distributed throughout a facility and networked to provide early alerts for any localized releases of agent at threatening levels. If these advanced concepts are to be used in the time frame of the stockpile demilitarization effort, an accelerated development program will need to be put in place to take them from proofs of concept to commercialization.
Finding 5-4. During the time remaining for the CMA program, new chemical sensor technology is not likely to be useful for demilitarization plant monitoring at the STEL regulatory levels or below. The lead times for developing engineering prototypes and validating the technology with real chemical agents are long and are inconsistent with the program needs.
Finding 5-5. Chemical sensors have the potential to be useful for making rapid measurements at IDLH levels and above, such as for events including spills and leaks.
Finding 5-6. For any of the chemical sensors described, the federal government will be the customer, as there are no significant commercial markets for chemical agent detection. In order to take advantage of these technologies in the requisite time frame, accelerated support mechanisms must be put into place to transition them from research to commercial utility. Chemical sensors developed for active military or homeland defense purposes might be adopted by the Chemical Materials Agency for real-time detection of high agent levels if these sensors become available before the demilitarization program ends.
TECHNICAL MATURITY OF INNOVATIVE MONITORING TECHNOLOGIES
In this chapter, the committee identified real-time or near-real-time technologies that are based on measurement principles that differ from those currently being used for chemical agent monitoring at Army stockpile sites and that have the potential of being applied to agent vapor detection. Table 5-1 summarizes the advanced sensing technologies evaluated and notes sensitivity ranges and current limitations.
While long-path optical detection techniques such as DOAS or FT-IR could in principle possess multiagent capability, they do not appear to possess the requisite sensitivity for real-time monitoring at the new STELs. However, they may provide backup monitoring for large, uncontained releases of agent. In any case, these techniques are severely limited by interference from other atmospheric components, such as volatile organic compounds (from both natural and anthropogenic sources) and rain, fog, or haze. Surface-enhanced Raman spectroscopy may be a promising optical
TABLE 5-1 Summary of Potential Innovative Chemical Agent Monitoring Technologies
|
Methodology |
Sensitivity |
Limitations |
|
UV differential optical absorption spectroscopy (DOAS) |
ppb under optimum conditions |
Lack of suitable UV absorbance; interference by atmospheric components, rain, haze, atmospheric turbulence. |
|
Open-path Fourier transform infrared (OP/FT-IR) spectroscopy |
>0.001 mg agent/m3, i.e., >0.1 ppb under optimum conditions, for measurement times of several minutes |
Interference by atmospheric components, rain, haze, atmospheric turbulence. |
|
Surface-enhanced Raman scattering (SERS) |
tens of ppb |
Not real time; lack of reproducibility and regeneration of SERS substrates. |
|
Chemical ionization mass spectrometry (CIMS) |
sub-ppt under optimal conditions |
Optimizing sensitivity and specificity; product development needed to go from custom instrument to commercial units. |
|
Ion mobility spectrometry (IMS) |
ppb to 10 ppt |
Unacceptably high level of false positives. |
|
Chemical sensors |
“low ppb” levels; mg/m3 (Nomadics) |
Most are laboratory prototypes, not commercially available. |
technique, but it is not real time, and it is unlikely to be any more sensitive than the ACAMS is. Also, it is probably less selective and more subject to interference from other airborne molecules.
Chemical ionization mass spectrometry is a very sensitive real-time technique for measuring concentrations of trace molecules in the atmosphere. The existing instrumentation is complex and expensive (compared with gas chromatographic instruments such as ACAMS or DAAMS). However, requirements for atmospheric monitoring have led to the development of robust, relatively compact, low-power-demand field instruments that could be adapted for chemical agent detection and monitoring at chemical agent demilitarization facilities.
A great deal of ongoing research on chemical sensors is directed toward the development of molecular-level devices that acquire, process, and sense the presence of specific analytes. These sensors are small and inexpensive—attributes that would make them suitable for wide deployment in chemical demilitarization facilities. However, while these sensors have the potential to be useful for making rapid measurements at IDLH levels and above, they are not sensitive enough for GPL or other regulatory-level measurements. In addition, most of these devices are still in the laboratory prototype stage and would require significant investment to make them commercially available.
The introduction of any innovative measurement technology will require intercomparison with accepted measurement protocols and systems (primarily ACAMS and DAAMS) to be able to meet regulatory requirements. Continuing validation and testing of any instrument are also required in order to ensure reliability. This presents an obvious difficulty with proposed open-path measurement techniques such as fence-line FT-IR spectroscopy, since live testing with actual agents in an open atmosphere will not be acceptable to the Army or communities.
REFERENCES
Alak, A.M., and T. VoDinh. 1987. Surface-enhanced Raman spectrometry of organophosphorus chemical agents. Analytical Chemistry 59(17): 2149−2153.
Albert, K.J., and D.R. Walt. 2000. High-speed fluorescence detection of explosives-like vapors. Analytical Chemistry 72(9): 1947−1955.
Albert, K.J., and D.R. Walt. 2003. Information coding in artificial olfaction multisensor arrays. Analytical Chemistry 75(16): 4161−4167.
Albert, K.J., N.S. Lewis, C.L. Schauer, G.A. Sotzing, S.E. Stitzel, T.P. Vaid, and D.R. Walt. 2000. Cross-reactive chemical sensor arrays. Chemical Reviews 100(7): 2595−2626.
Ballenthin, J.O., W.F. Thorn, T.M. Miller, A.A. Viggiano, D.E. Hunton, M. Koike, Y. Kondo, N. Takegawa, H. Irie, and H. Ikeda. 2003. In situ HNO3 to NOy instrument comparison during SOLVE. Journal of Geophysical Research 108(D6): ACH 7−11.
Briglin, S.M., M.S. Freund, B.C. Sisk, and N.S. Lewis. 2001. Array-based carbon black-polymer composite vapor detectors for detection of DNT in environments containing complex analyte mixtures. Pp. 912−921 in Detection and Remediation Technologies for Mines and Minelike Targets VI. Proceedings of SPIE, Volume 4394. Edited by A.C. Dubey, J.F. Harvey, J.T. Broach, and V. George. Bellingham, Wash.: SPIE—The International Society for Optical Engineering.
Briglin, S.M., M.C. Burl, M.S. Freund, B.C. Sisk, and N.S. Lewis. 2002. Array-based carbon black-polymer composite vapor detectors for detection of DNT in environments containing complex analyte mixtures. Paper S4.1 in Symposium S, Combinatorial and Artificial Intelligence Methods in Materials Science. MRS Proceedings Volume 700. Edited by I. Takeuchi, J.M. Newsam, L.T. Wille, H. Koinuma, and E.J. Amis. Warrendale, Pa.: Materials Research Society.
Buck, L.B. 1996. Information coding in the vertebrate olfactory system. Pp. 517−544 in Annual Review of Neuroscience, Volume 19. Palo Alto, Calif.: Annual Reviews.
Buck, L., and R. Axel. 1991. A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell 65(1): 175−187.
Busch, K.W., and M.A. Busch, editors. 1999. Cavity-Ringdown Spectroscopy: An Ultratrace Absorption Measurement Technique. New York, N.Y.: Oxford University Press.
Chan, S., P.M. Fauchet, Y. Li, L.J. Rothberg, and B.L. Miller. 2000. Porous silicon microcavities for biosensing applications. Physica Status Solidi (a) 182(1): 541−546.
Chen, C., L.G. Huey, M. Trainer, D. Nicks, J. Corbett, T. Ryerson, D. Parrish, J.A. Neuman, J. Nowak, D. Tanner, J. Holloway, C. Brock, J. Crawford, J.R. Olson, A. Sullivan, R. Weber, S. Schauffler, S. Donnelly, E. Atlas, J. Roberts, F. Flocke, G. Hübler, and F. Fehsenfeld. 2005. An investigation of the chemistry of ship emission plumes during ITCT 2002. Journal of Geophysical Research.
Christesen, S.D. 1988. Raman cross sections of chemical agents and simulants. Applied Spectroscopy 42(2): 318−321.
Cody, R.B, J.A. Loramee, and H.D. Durst. 2005. Versatile new ion source for the materials in open air under ambient conditions. Analytical Chemistry 77(8): 2297−2302.
Content, S., W.C. Trogler, and M.J. Sailor. 2000. Detection of nitrobenzene, DNT, and TNT vapors by quenching of porous silicon photoluminescence. Chemistry—A European Journal 6(12): 2205−2213.
Curtis, C.L., V.V. Doan, G.M. Credo, and M.J. Sailor. 1993. Observation of optical cavity modes in photoluminescent porous silicon films. Journal of the Electrochemical Society 140(12): 3492−3494.
D’Agostino, P.A., and L.R. Provost. 1986. Capillary column ammonia chemical ionization mass spectrometry of organophosphorus chemical warfare agents and simulants. Biomedical and Environmental Mass Spectrometry 13: 231−236.
Dancil, K.-P.S., D.P. Greiner, and M.J. Sailor. 1999. A porous silicon optical biosensor: Detection of reversible binding of IgG to a protein A-modified surface. Journal of the American Chemical Society 121(34): 7925−7930.
de Gouw, J., C. Warneke, T. Karl, G. Eerdekens, C. van der Veen, and R. Fall. 2003. Sensitivity and specificity of atmospheric trace gas detection by proton-transfer-reaction mass spectrometry. International Journal of Mass Spectrometry, Volume 223−224: 365−382.
Dickinson, T.A., J. White, J.S. Kauer, and D.R. Walt. 1996. A chemical-detecting system based on a cross-reactive optical sensor array. Nature 382(6593): 697−700.
Dickinson, T.A., J. White, J.S. Kauer, and D.R. Walt. 1998. Current trends in “artificial-nose” technology. Trends in Biotechnology 16(6): 250−258.
Dickinson, T.A., K.L. Michael, J.S. Kauer, and D.R. Walt. 1999. Convergent, self-encoded bead sensor arrays in the design of an artificial nose. Analytical Chemistry 71(11): 2192−2198.
Doleman, B.J., R.D. Sanner, E.J. Severin, R.H. Grubbs, and N.S. Lewis. 1998. Use of compatible polymer blends to fabricate arrays of carbon black-polymer composite vapor detectors. Analytical Chemistry 70(13): 2560−2564.
Eisele, F.L., and D.J. Tanner. 1993. Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere. Journal of Geophysical Research 98(D5): 9001−9010.
Evangelisti, F., A. Baroncelli, P. Bonasoni, G. Giovanelli, and F. Ravegnani. 1995. Differential optical absorption spectrometer for measurement of tropospheric pollutants. Applied Optics 34(15): 2737−2744.
Farquharson, S., P. Maksymiuk, K. Ong, and S.D. Christesen. 2002. Chemical agent identification by surface-enhanced Raman spectroscopy. Pp. 166−173 in Spectroscopy-Based Sensor Systems. Proceedings of SPIE, Volume 4577. Edited by S.D. Christesen and A.J. Sedlacek III. Bellingham, Wash.: SPIE—The International Society for Optical Engineering.
Fischer, C., M.W. Sigrist, Q. Yu, and M. Seiter. 2001. Photoacoustic monitoring of trace gases using a diode-based difference frequency laser source. Optics Letters 26(20): 1609−1611.
Freund, M.S., and N.S. Lewis. 1995. A chemically diverse conducting polymer-based “electronic nose.” Proceedings of the National Academy of Sciences of the United States of America 92(7): 2652−2656.
Frye-Mason, G., M. Leuschen, M. la Grone, L. Wald, C. Aker, M. Dock, L.F. Hancock, S. Fagan, and K. Paul. 2004. Reactive chromophores for sensitive and selective detection of chemical warfare agents. Pp. 54−62 in Chemical and Biological Sensing V. Proceedings of SPIE, Volume 5416. Edited by P.J. Gardner. Bellingham, Wash.: SPIE—The International Society for Optical Engineering.
Gardner, J.W., and P.N. Bartlett. 1999. Electronic Noses: Principles and Applications. Oxford, UK: Oxford University Press.
Guevremont, R. 2004. High-field asymmetric waveform ion mobility spectrometry: A new tool for mass spectrometry. Journal of Chromatography A 1058(1−2): 3−19.
Haes, A.J., and R.P. Van Duyne. 2002. A nanoscale optical biosensor: Sensitivity and selectivity of an approach based on the localized surface plasmon resonance spectroscopy of triangular silver nanoparticles. Journal of the American Chemical Society 124(35): 10596−10604.
Haes, A.J., S. Zou, G.C. Schatz, and R.P. Van Duyne. 2004a. A nanoscale optical biosensor: The long range distance dependence of the localized surface plasmon resonance of noble metal nanoparticles. Journal of Physical Chemistry B 108(1): 109−116.
Haes, A.J., S. Zou, G.C. Schatz, and R.P. Van Duyne. 2004b. Nanoscale optical biosensor: Short range distance dependence of the localized surface plasmon resonance of noble metal nanoparticles. Journal of Physical Chemistry B 108(22): 6961−6968.
Haes, A.J., W.P. Hall, L. Chang, W.L. Klein, and R.P. Van Duyne. 2004c. A localized surface plasmon resonance biosensor: First steps toward an assay for Alzheimer’s disease. Nano Letters 4(6): 1029−1034.
Hill, Jr., H., and S.J. Martin. 2002. Conventional analytical methods for chemical warfare agents. Pure and Applied Chemistry 74(12): 2281− 2291.
Hoffland, L., L. Piffath, and J. Bouck. 1985. Spectral signatures of chemical agents and simulants. Optical Engineering 24(6): 982−985.
Hopkins, A.R., and N.S. Lewis. 2001. Detection and classification characteristics of arrays of carbon black/organic polymer composite chemiresistive vapor detectors for the nerve agent simulant dimethylmethylphosphonate and diisopropylmethylphosponate. Analytical Chemistry 73(5): 884−892.
IOM (Institute of Medicine). 1999. Chemical and Biological Terrorism: Research and Development to Improve Civilian Medical Response. Washington, D.C.: National Academy Press.
Janshoff, A., K.-P.S. Dancil, C. Steinem, D.P. Greiner, V.S.-Y. Lin, C. Gurtner, K. Motesharei, M.J. Sailor, and M.R. Ghadiri. 1998. Macroporous p-type silicon Fabry-Perot layers: Fabrication, characterization, and applications in biosensing. Journal of the American Chemical Society 120(46): 12108−12116.
Ketkar, S.N., S. Dheandhanoo, W.L. Fite, J.D. Buchner, and E.P. Hopkins. 1989. New Concepts in Chemical Monitoring, Phase II. Final Report, January 18. Aberdeen Proving Ground, Md.: Program Manager for Chemical Demilitarization.
Kolb, C.E. 2003. Measurement challenges and strategies in atmospheric and environmental chemistry. Pp. 127−136 in The Environment: Challenges for the Chemical Sciences in the 21st Century. Washington, D.C.: The National Academies Press.
Lauerhaas, J.M., G.M. Credo, J.L. Heinrich, and M.J. Sailor. 1992. Reversible luminescence quenching of porous silicon by solvents. Journal of the American Chemical Society 114(5): 1911−1912.
Létant, S., and M.J. Sailor. 2000. Detection of HF gas with a porous silicon interferometer. Advanced Materials 12(5): 355−359.
Lin, V.S.-Y., K. Motesharei, K.-P.S. Dancil, M.J. Sailor, and M.R. Ghadiri. 1997. A porous silicon-based optical interferometric biosensor. Science 278(5339): 840−843.
Lonergan, M.C., E.J. Severin, B.J. Doleman, S.A. Beaber, R.H. Grubbs, and N.S. Lewis. 1996. Array-based vapor sensing using chemically sensitive, carbon black-polymer resistors. Chemistry of Materials 8(9): 2298−2312.
Marcy, T.P., R.S. Gao, M.J. Northway, P.J. Popp, H. Stark, and D.W. Fahey. 2005. Using chemical ionization mass spectrometry for detection of HNO3, HCl, and ClONO2 in the atmosphere . International Journal of Mass Spectrometry 243(1): 63−70.
McLaren, S.E., and D.H. Stedman. 1996. Detection of Chemical Agents by Open-Path FTIR Spectroscopy. Final Report for Stack Monitoring Project, Final Report for U.S. Army Contract DAAM01-94-C-0068.
Michael, K.L., L.C. Taylor, S.L. Schultz, and D.R. Walt. 1998. Randomly ordered addressable high-density optical sensor arrays. Analytical Chemistry 70(7): 1242−1248.
Nägele, M., D. Hofstetter, J. Faist, and M.W. Sigrist. 2001. Low power quantum-cascade laser photoacoustic spectrometer for trace-gas monitoring. Analytical Sciences 17: s497−s499.
Nelson, D.D., J.H. Shorter, J.B. McManus, and M.S. Zahniser. 2002. Subpart-per-billion detection of nitric oxide in air using a thermoelectrically cooled mid-infrared quantum cascade laser spectrometer. Applied Physics B: Lasers and Optics 75(2/3): 343−350.
Plane, J.M.C., and C.-F. Nien. 1992. Differential optical absorption spectrometer for measuring atmospheric trace gases. Review of Scientific Instruments 63(3): 1867−1876.
Platt, U. 1994. Differential optical absorption spectroscopy (DOAS). Pp. 27−84 in Air Monitoring by Spectroscopic Techniques. Edited by M.W. Sigrist. New York, N.Y.: Wiley.
Rakow, N.A., and K.S. Suslick. 2000. A colorimetric sensor array for odour visualization. Nature 406(6797): 710−714.
Rewick, R.T., M.L. Schumacher, and D.L. Haynes. 1986. The UV absorption spectra of chemical agents and simulants. Applied Spectroscopy 40(2): 152−156.
Riboh, J.C., A.J. Haes, A.D. McFarland, C.R. Yonzon, and R.P. Van Duyne. 2003. A nanoscale optical biosensor: Real-time immunoassay in physiological buffer enabled by improved nanoparticle adhesion. Journal of Physical Chemistry B 107(8): 1772−1780.
Rohrbaugh, D.K. 1995. Ammonia Chemical Ionization Mass Spectrometry of Trimethylsilyl Derivatives of Chemical Warfare Agent Degradation Products. ERDEC-TR-242, March. Aberdeen Proving Ground, Md.: Edgewood Research, Development, and Engineering Center.
Rohrbaugh, D.K. 1998. Acetonitrile Chemical Ionization Ion Trap Mass Spectrometry of Chemical Warfare Agents and Their Degradation Products. ERDEC-TR-477, February. Aberdeen Proving Ground, Md.: Edgewood Research, Development, and Engineering Center.
Rohrbaugh, D.K. 1999. Methanol Chemical Ionization Ion Trap Mass Spectrometry of VX Degradation Products. ECBC-TR-017, March. Aberdeen Proving Ground, Md.: Edgewood Research, Development, and Engineering Center.
Rohrbaugh, D.K. 2000. GC/MS Analysis of GA Degradation Products. ECBC-TR-107, August. Aberdeen Proving Ground, Md.: Edgewood Research, Development, and Engineering Center.
Russwurm, G.M., and J.W. Childers. 2002. Open-path Fourier transform infrared spectroscopy. Pp. 1750−1773 in Handbook of Vibrational Spectroscopy, Volume 2. Edited by J.M. Chalmers and P.R. Griffiths. Chichester, UK: Wiley.
Sailor, M.J. 1997. Sensor applications of porous silicon. Pp. 364−370 in Properties of Porous Silicon. Edited by L. Canham. Edison, N.J.: Institution of Electrical Engineers.
Sailor, M.J., W.C. Trogler, S. Létant, H. Sohn, S. Content, T.A. Schmedake, J. Gao, P. Zmolek, J.R. Link, Y. Fainman, F. Xu, and P.E. Shames. 2001. Low-power microsensors for explosives and nerve warfare agents using silicon nanodots and nanowires. Pp. 153−165 in Unattended Ground Sensor Technologies and Applications. Proceedings of SPIE, Volume 4393 . Edited by E.M. Carapezza. Bellingham, Wash.: SPIE—The International Society for Optical Engineering.
Sen, A., and K.S. Suslick. 2000. Shape-selective discrimination of small organic molecules. Journal of the American Chemical Society 122(46): 11565−11566.
Severin, E.J., B.J. Doleman, and N.S. Lewis. 2000. An investigation of the concentration dependence and response to analyte mixtures of carbon black/insulating organic polymer composite vapor detectors. Analytical Chemistry 72(4): 658−688.
Shafer-Peltier, K.E., C.L. Haynes, M.R. Glucksberg, and R.P. Van Duyne. 2003. Toward a glucose biosensor based on surface-enhanced Raman scattering. Journal of the American Chemical Society 125(2): 588−593.
Sharpe, S.W., R.L. Johnson, P.M. Chu, J. Kleimeyer, and B. Rowland. 2003. Quantitative, infrared spectra of vapor phase chemical agents. Pp. 19-27 in Chemical and Biological Sensing IV. Proceedings of SPIE, Volume 5085. Edited by P.J. Gardner. Bellingham, Wash.: SPIE—The International Society for Optical Engineering.
Sigrist, M.W. 2000. Environment: Trace gas monitoring. Pp. 1885−2245 in Applications of Instrumental Methods, Encyclopedia of Analytical Chemistry: Applications, Theory, and Instrumentation. Edited by R.A. Meyers. Chichester, UK: Wiley.
Sigrist, M.W., A. Bohren, T.V. Lerber, and A. Romann. 2001. Environmental applications in laser-based photoacoustic spectroscopy. Analytical Sciences 17: s511−s514.
Snow, P.A., E.K. Squire, P.St.J. Russell, and L.T. Canham. 1999. Vapor sensing using the optical properties of porous silicon Bragg mirrors. Journal of Applied Physics 86(4): 1781−1784.
Sohn, H., S. Létant, M.J. Sailor, and W.C. Trogler. 2000. Detection of fluorophosphonate chemical warfare agents by catalytic hydrolysis with a porous silicon interferometer. Journal of the American Chemical Society 122(22): 5399−5400.
Song, J.H., and M.J. Sailor. 1997. Quenching of photoluminescence from porous silicon by aromatic molecules. Journal of the American Chemical Society 119(31): 7381−7385.
Sotzing, G.A., J.N. Phend, R.H. Grubbs, and N.S. Lewis. 2000. Highly sensitive detection and discrimination of biogenic amines utilizing arrays of polyaniline/carbon black composite vapor detectors. Chemistry of Materials 12(3): 593−595.
Spellicy, R.L., and J.D. Webb. 2002. Atmospheric monitoring using extractive techniques. Pp. 1721−1749 in Handbook of Vibrational Spectroscopy, Volume 2. Edited by J.M. Chalmers and P.R. Griffiths. Chichester, UK: Wiley.
Stedman, D.H., and S.E. McLaren. 1996. Detection of Chemical Agents by Open-Path FTIR Spectroscopy. Final Report for MDM Test Area Project, Final Report for U.S. Army Contract DAAM01-94-C-0068.
Stitzel, S.E., L.J. Cowen, K.J. Albert, and D.R. Walt. 2001. Array-to-array transfer of an artificial nose classifier. Analytical Chemistry 73(21): 5266−5271.
Suslick, K.S. 2004. An optoelectronic nose: “Seeing” smells by means of colorimetric sensor arrays. MRS Bulletin 29(10): 720−725.
Suslick, K.S., N.A. Rakow, and A. Sen. 2004. Colorimetric sensor arrays for molecular recognition. Tetrahedron 60(49): 11133−11138.
Sylvia, J.M., J.A. Janni, J.D. Klein, and K.M. Spencer. 2000. Surface-enhanced Raman detection of 2,4-dinitrotoluene impurity vapor as a marker to locate landmines. Analytical Chemistry 72(23): 5834−5840.
Takats, Z., J.M. Wiseman, B. Gologan, and R.G. Cooks. 2004. Mass spectrometry sampling under ambient conditions with desorption electrospray ionization. Science 306(5695): 471−473.
Todd, M.W., R.A. Provencal, T.G. Owano, B.A. Paldus, A. Kachanov, K.L. Vodopyanov, M. Hunter, S.L. Coy, J.I. Steinfeld, and J.T. Arnold. 2002. Application of mid-infrared cavity-ringdown spectroscopy to trace explosives vapor detection using a broadly tunable (6-8 µm) optical parametric oscillator. Applied Physics B 75(2−3): 367−376.
Wang, J. 2004. Microchip devices for detecting terrorist weapons. Analytica Chimica Acta 507(1): 3−10.
Wang, J., M. Pumera, G.E. Collins, and A. Mulchandani. 2002a. Measurements of chemical warfare agent degradation products using an electrophoresis microchip with contactless conductivity detector. Analytical Chemistry 74(23): 6121−6125.
Wang, J., M. Pumera, M.P. Chatrathi, A. Escarpa, M. Musameh, G. Collins, A. Mulchandani, Y. Lin, and K. Olsen. 2002b. Single-channel microchip for fast screening and detailed identification of nitroaromatic
explosives or organophosphate nerve agents. Analytical Chemistry 74(5): 1187−1191.
Wang, J., G. Chen, A. Muck, Jr., M.P. Chatrathi, A. Mulchandani, and W. Chen. 2004a. Microchip enzymatic assay of organophosphate nerve agents. Analytica Chimica Acta 505(2): 183−187.
Wang, J., J. Zima, N.S. Lawrence, M.P. Chatrathi, A. Mulchandani, and G.E. Collins. 2004b. Microchip capillary electrophoresis with electrochemical detection of thiol-containing degradation products of V-type nerve agents. Analytical Chemistry 76(16): 4721−4726.
Werle, P. 1996. Spectroscopic trace gas analysis using semiconductor diode lasers. Spectrochimica Acta A 52(8): 805−822.
White, J., J.S. Kauer, T.A. Dickinson, and D.R. Walt. 1996. Rapid analyte recognition in a device based on optical sensors and the olfactory system . Analytical Chemistry 68(13): 2191−2202.
Yonzon, C.R., C.L. Haynes, X. Zhang, J.T. Walsh, Jr., and R.P. Van Duyne. 2004. A glucose biosensor based on surface-enhanced Raman scattering: Improved partition layer, temporal stability, reversibility, and resistance to serum protein interference. Analytical Chemistry 76(1): 78−85.
Zahniser, M.S., D.D. Nelson, J.B. McManus, and P.L. Kebabian. 1995. Measurement of trace gas fluxes using tunable diode laser spectroscopy. Philosophical Transactions of the Royal Society of London A 351(1696): 371−381.
Zangooie, S., R. Bjorklund, and H. Arwin. 1998. Protein adsorption in thermally oxidized porous silicon layers. Thin Solid Films 313−314(1−2): 825−830.
Zangooie, S., R. Jansson, and H. Arwin. 1999. Ellipsometric characterization of anisotropic porous silicon Fabry-Pérot filters and investigation of temperature effects on capillary condensation efficiency. Journal of Applied Physics 86(2): 850−858.
Zhang, S.W., and T.M. Swager. 2003. Fluorescent detection of chemical warfare agents: Functional group specific ratiometric chemosensors. Journal of the American Chemical Society 125(12): 3420−3421.


























