4
WORKING GROUPS, DAY 1
WORKING GROUP 1 INFLUENZA VIRULENCE AND ANTIGENIC CHANGE
Chairperson—Robert Webster
Briefer—Peter Palese
Rapporteur—Robert Lamb
Charge: The charge of this working group is to define research priorities associated with understanding influenza virulence, with tracking and predicting antigenic change, and with defining which antigenic changes may be associated with increased virulence, risk of transmission, and pandemic potential.
Potential issues to consider:
-
What studies are needed to define the genetic loci for pathogenicity in avian and human influenza virus strains?
-
What studies are needed to affirm the hypothesis that incremental acquisition of genetic changes can lead to influenza pandemics as compared with the sudden emergence of previous pandemics?
-
What studies are needed to track the rate of antigenic change in avian and human influenza virus strains and to predict changes that may occur?
-
What studies are needed to determine whether pandemic risk can be predicted by virulence factors and/or antigenic characteristics?
REPORT TO PLENARY
Rapporteur—Dr. Robert Lamb
One question concerned how we would define the loci for pathogenicity with influenza virus. We know at the molecular level the importance of the hemagglutinin’s cleavage site. It is required to becleaved to HA1 and HA2 which activates the molecule for fusion..
We know that if we have a multi-basic residue at the HA cleavage site, it has the potential to be cleaved in the trans-Golgi network, and that correlates directly with high pathogenicity of viruses in many cases. If that cleavage site has only a single basic residue, it very often correlates with low pathogenicity. We know that the ability of neuraminidase to bind the protease plasminogen is a determinant of pathogenicity, because binding plasminogen then causes cleavage of the hemagglutinin, such as found for the WSN strain.
We know that the presence of carbohydrate sites around the cleavage site of HA are important for pathogenicity. We know that certain mutations in the NS1 protein affect cytokine production in some viruses. And we know that mutations that affect replication and decrease replication—such as in the cold-adapted virus and the so-called PB2 627 mutation found in many of the H5 viruses—lead to a difference in pathogenicity in the mouse.
One important consideration is that the very same residue makes no difference when the studies are done in ferrets. Researchers are now using mice, ferrets, and chickens as animal models. The use of non-human primates for influenza virus studies is somewhat questionable. Although mice are obviously the smallest and cheapest animal, there are major questions about the use of inbred mice, particularly the popular strains that lack the MX1 gene, as those do not produce the induction of a normal antiviral state. The best model animal system is probably the ferret: it requires no adaptation for human viruses. However, we do need to agree internationally on one strain of ferret, because British ferrets and Japanese ferrets and American ferrets are not all the same.
Another problem is that very few immunological reagents exist for the ferret. How would we study a cytokine response? We need a ferret DNA sequence genome project, like those under way for another 11 or so organisms, from zebra fish to mouse, and of course humans before that. We also need to increase the number of ferrets. That doesn’t just mean ordering a few more. We need a very large breeding colony—the approach taken with the breeding of woodchucks for hepatitis-B studies. We should also determine if other small animal models would work, such as hamsters and mini-pigs. An underlying need is for biological containment facilities for both tissue-culture work and animal work.
We need to determine the genes needed for both transmission and pathogenicity, which we would do by making reassortments using reverse genetics—the so-called 6 plus 2, 5 plus 3, etc combinations of 8 genes. We would then study the genetic basis underlying transmission, virulence, and pathogenicity in terms of the function of proteins, the structure of proteins at the atomic level, and how proteins go together to make complexes such as polymerase. We do not completely understand the components of what can be reassorted from one strain versus another. The suggestion is there are incompatibilities among proteins—that not all P proteins can go together.
We were asked whether incremental changes in the genome lead to pandemics. We know that from 1997 to 2005 the H5N1 virus gained the ability to kill ducks, and that it has transferred to cats and killed them. We need studies where a series of viruses have changed in virulence pathogenicity.
We need to obtain the genome sequences of all these viruses and post them on public databases. The new NIAID-NIH–sponsored public database with The Institute of Genome Research (TIGR) sequencing genomes of human viruses is a start, but we need such an approach for other viruses as well. We also need reverse genetics working for all of these viruses. And when we have all those components in place, we can then show that mutations are sufficient and necessary for the property of pathogenicity being examined.
The third question we were asked was whether studies are needed to track the rate of antigenic change in avian and human strains, and to predict the changes that occur. As background, note that only three hemagglutinin subtypes—H1, H2 and H3—cause major human disease
We know, as Dr. Palese has often pointed out, that the 1918 virus caused a W-shaped death rate. That is, if we plot the number of deaths on the Y-axis and the age groups that died on the X-axis, we find that young people died early on, and then there was a gap, and older people died much later. People between 30 and 60 years of age presumably had prior exposure to an H1-like virus. We should add that it is thought that H3 and H1 both circulated before 1918, and that there is no evidence that H5, H7, H9, or any of the highly pathogenic viruses in avian species were found in humans in those earlier times.
A question about viral archeology inevitably comes up—the sort of studies that Dr. Jeff Taubenberger has done. Such investigation is very difficult because tissues were not preserved before 1900. His work was done on formalin fixed tissues. The question was raised whether we could examine tombs in churches for examples of antibodies, but we felt that would be extremely difficult.
At the theoretical level we can continue to predict changes and the effect on antigenic epitopes. An example is the paper in the Proceedings of the National Academies of Science by evolutionary biologist Dr. Simon Levin and his group, who predicted HA clusters and antigenic evolution. Such studies should continue.
Surveillance of influenza viruses—now occurring throughout the world—should also continue, as should nucleotide sequencing of viral genomes. Ideally, we need better immunological markers. We point out that hemagglutinin inhibition tests are insensitive, and that they don't work on H5 viruses.
Question 4 addressed the studies needed to determine pandemic risk associated with antigenic characteristics. We need to determine the extent of antigenic variation that yields a pandemic strain. We need both human and animal studies to follow how much variation can occur in a strain and still lead to disease and pandemic potential.
We would like to know whether preexisting antibody to one subtype can have an effect on infection with another subtype. That is, we can look at the ability of viruses of different subtypes to infect animals such as ferret that already have antibody levels to common human viruses. If we already have some antigenic determinants to a component of any one of those viruses, might that provide some benefit against infection with another influenza virus?
Lastly, Dr. Purnell Choppin and others asked whether we need studies that ask whether human genetic changes increase or decrease susceptibility to influenza virus infection. Could such studies be coupled to the NCI cancer genome project? For the mouse, such work could easily be coupled with the huge forward genetics projects occurring at five centers in the United States for mutagenizing the mouse on a total random basis and mapping any genetic changes. Could we use those leftover mice to determine if their susceptibility to influenza had increased or decreased? Although the mouse may not be a very good model, those projects provide an avenue of research.
Our workshop’s first priority is to determine the sequences of human, animal, and avian isolates within an epidemiological framework. We want to stress the latter, because just having the sequences doesn’t help. We need to know the clinical data from human cases to accompany the sequences.
Our second major recommendation is to determine the genes and their function for transmission and pathogenicity in ferrets using qualified reagents. That one sentence
encompasses several needs. Such an effort will require ferrets—lots of them. We are talking about determining not just which genes are involved but also their function. That will require a lot of basic research.
|
Research Recommendations
|




WORKING GROUP 2
CONTROLLING ANIMAL INFLUENZA AND DECREASING ANIMAL-TO-HUMAN TRANSMISSION
Chairperson—David Halvorson
Briefer—David Swayne
Rapporteur—Bruce Innis
Charge: The charge to this group will be to define research needs related to strategies for controlling the spread of influenza among animals and decreasing the risk of spread from animals to people. Specific research areas may include assessing the role and impacts of vaccine in animals; environmental modifications and precautions to decrease the spread of infection within and between farms; optimal approaches to culling animal populations; and prevention or management of exposure to infected animals by people.
Specific Questions:
-
What studies are needed to determine whether the circulating avian influenza H5N1 virus in Southeast Asia is likely to cause a human pandemic?
-
What studies are needed to assess the effectiveness of strategies to control animal influenza?
-
What studies are needed to assess optimal approaches to use and to assess impact of avian influenza vaccines?
-
What effective cross-cutting technologies for control of animal influenza are translatable to human pandemic scenarios?
Report to Plenary
Rapporteur: Dr. Bruce Innis
We selected several objectives for the intermediate 5-to-10-year horizon. The highest priority is to focus resources on country-specific epidemiological studies targeted at understanding both disease and transmission in animals and in humans.
We focused principally on avian influenza. The countries now affected in Southeast Asia do not have a unitary system of poultry production. Thus it is important to look not only at industrial settings but also at village farms, where much bird raising occurs, and also at live markets, and to remember that animals are raised for recreational uses and captivity.
It is also important to investigate the epidemiology of humans who care for these animals—we are dismayed that more is not being done on that front. We would like to know where transmission is taking place. Why are people who apparently are exposed not becoming sick? And in many cases, why are they not becoming infected?

We recognize that the serological method is a barrier to such investigation: no easily accessible tool such as an ELISA test is available. Most such investigation is based on microneutralization, and even that is not yet standardized.
We would like to complement epidemiological studies by evaluating pathogenesis in the affected bird species, looking at both domestic and captive birds, and at wild birds as a lower priority.
We would also like to perform reverse genetic studies to identify avian influenza genes from specific strains with the greatest interspecies transfer potential. Of course, caution needs to be exercised to contain these strains, and there would still be a gap once we developed these resorted viruses, as we lack methods to realistically assess their person-to-person transmission potential. A tool for doing so needs to be developed in parallel.

We also prioritized identifying control measures optimized for conditions in developing countries, although some U.S. communities raise birds under the conditions used in Asian villages. As many as 100,000 people in Southern California are raising small poultry flocks with 10 birds or more in their backyards, for example. We would like to see an assessment of creative
solutions for such small flock holders. What kinds of educational approaches would work? What economic incentives can be used? What changes in agricultural systems should be employed?

We view studies of knowledge, attitude, and practice at recreational, village, and industry levels of bird rearing as very important. We need to assess what interventions are being practiced now, and leverage local pathways of communication and public education systems to introduce better practices and ensure that the people who own these animals and their children are aware of the objectives and risks of control. As a complement, operational research needs to assess the impact of interventions such as education. That is predicated on baseline surveys, which should be done now.
Although many animals are vaccinated, we need improved standards for vaccine purity, safety, and especially potency, particularly for avian influenza. Research needs to improve procedures for lot release that assess potency. We need to have to better understand vaccine potency and how to measure it, and we need greater regulatory oversight. Although that is usually a national responsibility, international dialogue is also important. This isn't a topic for research, but it is certainly a topic for discussion.

Besides improving animal vaccines, field studies need to assess their effectiveness, particularly in Asia. If vaccines will be used to control avian influenza, then studies need to
assess their impact. Although domestic ducks may be a very important reservoir, we know almost nothing about the effectiveness of these vaccines or their suitability for ducks and geese. We need to develop vaccines for these waterfowl.
In the long-term, various efforts could improve veterinary vaccines. Cell culture might be used to produce influenza vaccine antigens to be combined with new adjuvant systems. The usual adjuvant system for veterinary vaccines is an oil and water emulsion. Birds that have been vaccinated and retain oil are not considered suitable for sale, so we need oils that serve as an adjuvant that can be more rapidly metabolized. We also need to develop vaccines that not only maintain a flock’s economic viability but also reduce transmission. The need for techniques for mass-delivery of vaccines is also great.
Lastly, we prioritized the development of animals to predict and understand the potential for circulating animal influenza viruses, including H5N1 Asian viruses, to infect humans, and to be transmitted from human to human.







WORKING GROUP 3
INFLUENZA DIAGNOSTICS FOR SURVEILLANCE
Chairperson –Richard Webby
Briefer – Nancy Cox
Rapporteur – Alan Hay
Charge: The charge of this working group is to define research needed to improve influenza surveillance through improved diagnostics and surveillance methods in clinical settings and in animals.
Specific Questions to address:
-
What studies are needed to assess and improve the specificity, sensitivity, and robustness of current diagnostic methods in influenza surveillance?
-
What studies are needed to assess diagnostic methods that are more rapid, cheaper, easier to handle and transport or that have additional capabilities such as detecting subtype or antiviral resistance?
-
What studies are needed to define or assess new epidemiological approaches to surveillance that will be more sensitive in detecting novel influenza strains and their extent in a region to provide data on possible pandemic candidates as early as possible?
-
What studies are needed to assess diagnostic approaches that may be of value in resource poor areas where laboratory facilities are not readily available and issues of specimen handling and transport are potential barriers to surveillance?
-
What studies are needed to address diagnostics in animal populations?
Report to Plenary
Rapporteur-Dr. Alan Hay
As you heard this morning, influenza is a very networked enemy, and combating it also requires a network approach. The issues are complicated by different circumstances in different parts of the world, such as how we would apply diagnostics to emergencies such as that in Southeast Asia.
|
Research Priorities—Immediate (1-2 years)
-Ensuring resources for the availability and distribution of diagnostic tests -Public health and private sectors should be involved -Network would provide financial and technical capacities required -Organization and management structure -WHO and other organizations would play a significant role |
Our first priority is the urgent need to gain much more information about the viruses circulating in avian and animal populations in Southeast Asia, the extent of infections in human beings, the relationship in terms of sporadic infections, and the nature of infections that occur in clusters. Are the latter also sporadic avian-to-human infections, or do they represent a greater degree of human-to-human transmission?
We obviously need to know much more about the progression of H5N1 disease in individuals, and how to apply diagnostic tests most efficiently. But the highest priority is to strengthen and extend the WHO global network, with an immediate emphasis on establishing an effective network of national and regional laboratories in Southeast Asia. We also need to ensure that resources are available for distributing diagnostic tests. These clearly need to be inexpensive and simple to use, and much effort should go into studies to evaluate the tests in underdeveloped settings. We further need interaction between public health and private-sector interests regarding the avian situation, and effective dialogue between public health and veterinary people.
The network will clearly need enough financial and technical capacity as well as an effective organization and management structure. WHO and other organizations would play a significant role.
|
Research Priorities—Immediate (1-2 years)
|
Our other priority concerned research to develop rapid, inexpensive, uncomplicated, and sample-stable diagnostic tests for typing and subtyping. We need tests that will actually diagnose H5N1 infections and distinguish them from other influenza infections. We also need research on optimizing sample collection and distribution. Many point-of-care diagnostic tests have been developed. We heard this morning about the limitations of those, and we clearly need an environment that encourages both small and larger private interests to develop those tests so they would be available now but also in a pandemic situation. We also need to develop high-throughput analytical procedures for diagnosing viral infections and developing therapies. An effective microarray technology is particularly important.
We need to establish an epidemiological strategy and create the tools—including clinical case definitions and syndrome surveillance—to support those studies, to understand the nature of the infections in Southeast Asia and the risks they pose. In a five-year timeframe, we considered integrating studies in humans and animals and developing the most suitable sampling strategies to determine the populations to focus on. We also suggested developing a real-time database of all available information. We further need to develop strategies to diagnose disease, so we can understand the nature of an infectious agent as quickly as possible. That entails developing syndromic surveillance techniques.
|
Research Priorities-Intermediate term (5 Years)
|
Our 10-year priorities include understanding the biological and epidemiological dynamics of respiratory pandemics, focusing on flu but also the nature of influenza in relation to other agents. We would also like to understand multi-pathogen population-based systems so we can better evaluate the most suitable interventions.
|
Research Priorities- Long Term (10 years)
|





WORKING GROUP 4
ANTIVIRALS AND NON-SPECIFIC APPROACHES, TREATMENTS AND IMMUNOTHERAPIES
Chairperson – Richard Whitley
Briefer – Fred Hayden
Rapporteur – Charles Hackett
Charge: This working group will identify research priorities related to the development, evaluation, and use of antiviral drugs and immunotherapies. Issues to consider include studying the impacts of currently available antiviral drugs and strategies to increase their effectiveness or efficiency; studies of other licensed products that may have benefit in treatment of prevention of influenza; studies of drugs in the development pipeline; and studies of immune therapies and biologicals. Research needed to predict, identify, evaluate, and decrease antiviral resistance also is relevant
Specific Questions:
-
What studies are needed to identify the optimal strategies for use of currently available antiviral drugs?
-
What studies are needed to evaluate licensed agents that may moderate the occurrence or severity of influenza?
-
What new antiviral agents are being developed and what research is needed to evaluate these agents or to identify new drug treatment or prevention options?
-
What potential immunotherapeutics could be developed and what research is needed for their evaluation?
-
What studies can help in prediction, identification, assessment, and prevention of antiviral drug resistance?
-
What studies on the pathophysiology of influenza can offer insights for prevention?
Report to Plenary
Rapporteur: Dr. Charles Hackett
At the top of our list are clinical trials for antivirals and immunotherapeutics. One of the main priorities is to develop pandemic protocols now. We reflected on experience with SARS, where public health authorities faced difficult decisions on which of many different possible courses of action they could take unless they had determined ahead of time which protocols to put in place.
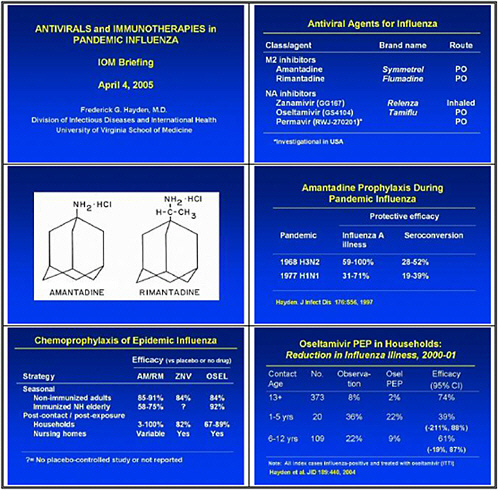
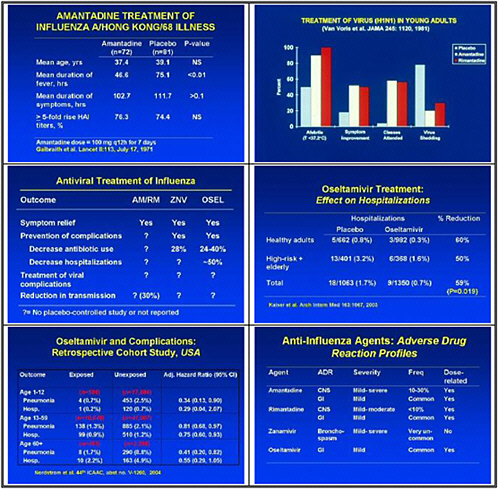
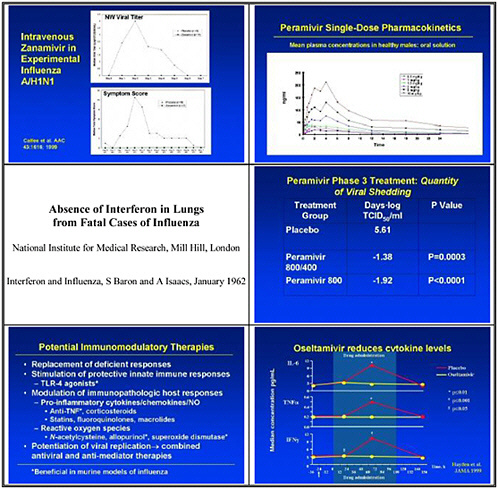
We need to obtain data on the virologic course and immune responses following pandemic influenza infection, as well as response to therapy following human H5 infections and other potential pandemic infections. A major priority is safety and tolerability of available drugs—oseltamivir pharmacokinetics (PK)—especially in infants less than 1 year old. We also need to determine the PK and tolerability of parenteral drug use, and to assess the long-term (12–20 weeks) tolerability of oseltamivir in inhaled zanamivir prophylaxis.
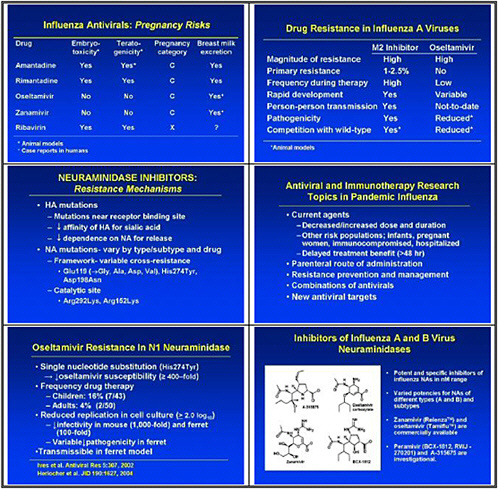
We also need to look at high-priority/high-risk populations, such as pregnant women and immuno-compromised hosts. We need data on these populations, because we need to resolve questions about safety and efficacy of treatments before using them in a pandemic.
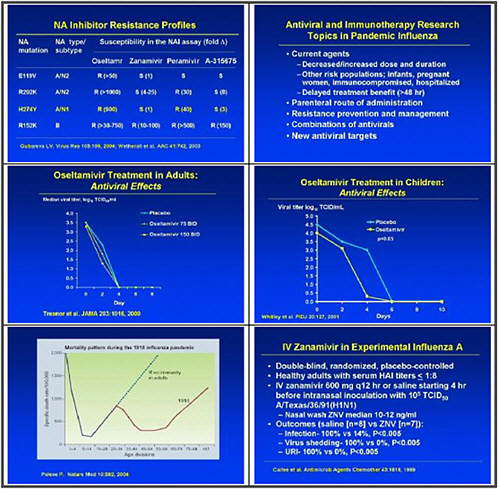
We also need to study emergence of H5N1 resistance in animal models, and strategies for preventing it. Animal models seem to be the best vehicle for obtaining good data. We need to assess the probability of licensure for orally inhaled zanamivir for disease prophylaxis, because oseltamivir resistance was uncovered and because well-controlled studies show that this approach may work.
The short-term priority is to accelerate drug development and discovery programs, including assessment of orphan drugs. We want to support operational infrastructure research. We decided that the strength of our group is to recommend research that will go hand-in-hand with policy development. Thus we see the need for clinical trials of drugs administered more than 48 hours after viral infection, and data on differences in sites of care and drug deployment and response time.
We need research on physician prescription of antiviral drugs, because many physicians are not geared up to prescribe such drugs. That would have to change if antiviral drugs were a major cornerstone of a pandemic treatment. That effort should start now, and would dovetail with policy development.
We need to test systematic approaches to influencing inflammatory expression and disease. We drew that lesson from looking at SARS, where inflammation damages lungs and steroids were used for treatment, even though steroids have unintended and longer-term consequences. We need to look at other ways of controlling inflammation as a priority.
In the medium term, we pointed to accelerated clinical trials of antivirals. We need to test oseltamivir monotherapy versus combination with an M2 inhibitor or ribavirin or other novel therapies in high-risk populations. We also need to test therapeutic efficacy of parenteral peramivir in hospitalized influenza patients, and to test prophylactic efficacy and tolerability of topical long-acting neuraminidase inhibitors.
We need to develop a contemporary virus challenge pool or pools for studies of experimental human influenza. This is an important piece of the puzzle, but obtaining the appropriate challenge virus that will provide the characteristics of the disease that we need to test takes time. We also need to use those to test candidate immunomodulators and antivirals in healthy challenge patients.
We need to develop immune-based therapies such as monoclonal antibodies, polyclonal antibodies, and polyclonal antibodies for therapy and prophylaxis, including monoclonal antibodies that target multiple proteins, so the virus will not escape by mutation of the targeted epitope.
We also need to consider polyclonal antibodies. New developments in polyclonal antibodies from animals with humanized immune systems might provide a passive therapy for pandemic influenza.
This slide shows examples of potent and specific inhibitors of neuraminidase of various viruses, influenzas A and B. They have different potencies according to the viral subtypes. Some are available and some are investigational. Thus there is a pipeline of long-acting neuraminidase
inhibitors, including conjugated sialidase, HA inhibitors, polymerase inhibitors including siRNA, and protease inhibitors. However, the pipeline must be developed.

The longer-term goal is to support ongoing small molecule discovery programs. One with promise is siRNA as a systemic or topical antiviral; siRNA holds potential as a novel approach toward new antiviral targets, such as polymerases. We also need to address incentives for industrial partners, as it is very difficult for a drug company to suddenly pursue another antiviral.
Over the longer-term, innate immune effector molecules need development both as specific antiviral molecules, and also for general innate immune activation as a strategy for broad prophylaxis. We need to promote the development of innate-immune system based modulators of inflammatory cascades as alternatives to steroids.
What should we be doing now? One suggestion was to create a clinical trial infrastructure for therapeutics, including a uniform protocol for development and data capture. Some trials could be done in Southeast Asia. To develop a public health policy, we also need research on transmission and treatment factors. Such an effort would include operational research to define the optimal infrastructure for developing, stockpiling, and distributing efficacious antiviral agents as quickly as possible.