

Clinical Challenges for Bioinformatics
GeorgeL GaborMiklos
Secure Genetics Pty Limited and Human Genetic Signatures Pty Limited, Sydney, Australia
The clinical validation of mathematically and statistically rigorous bioinformatic models in the prognostic contexts of human diseases, and in the evaluation of methylation signatures, is a difficult endeavor. However, if transcriptomic and proteomic data are to significantly enhance therapeutic protocols, they must provide an improvement on current treatments, where for example, adjuvant systemic administration of anti-cancer drugs generally yields net gains in survival of only a few months, and where in the case of breast cancers, only 10% or so of patients benefit from such treatments.
Genome wide analyses, exemplified by microarrays, have become a backbone of molecular research, but difficulties are emerging in their applications to cancers and complex diseases (Ein-dor et al., 2004, Bioinformatics, in press; Miklos and Maleszka, 2004, Nature Biotechnology, 22, 615; Yeung et al., 2004, Genome Biology, 5, R48). For example, analyses of the same lung cancer data by different bioinformatic pipelines, implemented by computer experts, statisticians and bioinformaticians from academia and the pharmaceutical sector, have found almost no commonalities between the gene sets that are claimed to be of prognostic significance to patient survival (Critical Assessment of Microarray Data Analysis meeting 2003). In addition, these new analyses revealed little overlap with the genes that were considered to be of most importance in the original studies. A similar situation holds from microarray data on leukemias. The use of different commonly used preprocessing packages, such as MAS5.0, RMA and GCRMA, (Bumgarner, 2004), on data from smokers versus non-smokers, also yields largely non-overlapping gene sets. Furthermore, analysis of breast cancer survival datasets demonstrates that prognostic gene cohorts are not unique and that equally predictive lists can be produced from the same data. Thus, the “top” genes cannot be considered as the main candidates for anti-cancer treatment, since there are many different groups of “top” genes. Similarly, the genes that have been prioritized in neuropsychiatric disorders such as the schizophrenias, barely overlap with those that have emerged from clinical, in situ, SNP, drug perturbation, knockout and association studies. Finally, data from Saccharomyces, where genome-wide knockouts and phenotypic data have been compared to expression data, have shown that no simple relationship exists between genes selected on the basis of their expression level changes and the biology of the perturbed system (Birrell et al., 2002, PNAS, 99, 8778).
At the clinical level, continuing impediments to therapeutic progress are the ill-defined boundaries of most diseases at the level of the individual and the extensive phenotypic variation of human diseases; for example, different samples from the same tumor are molecularly highly heterogeneous. We are faced with broad categories such as the dementias, the cancers and AIDS, all of which are heterogeneous collections of perturbed biological systems that have undoubtedly reached their phenotypic endpoints by different trajectories in different individuals. Despite the implications of this heterogeneity, ultra-sensitive transcriptomic and proteomic technologies are nevertheless enthusiastically applied to human tissue samples, in many cases, inappropriately chosen ones. This is particularly dangerous when the etiology and the clinical symptoms are separated by decades, or when genomes, and hence cellular networks and attractor basins are massively imbalanced or rerouted owing to aneuploidogenic processes. A major challenge therefore, is to derive a robust mapping between the phenotypic space defined by physicians and the perturbed networks which occur at different levels from the molecular, developmental and neuroanatomical through to the cognitive. A second challenge involves deconvoluting the dynamics of change; how does the initial perturbation set in motion the
cascade of events which percolate and modify the various levels until some form of altered trajectory becomes clinically recognizable?
Finally, at the epigenetic level, each cell type has a different methylation signature that characterizes that cell type. However, there is a semi-fluid modulation of methylation signatures that characterizes each cell which is a result of all the epigenetic changes that have occurred since fertilization and the current tissue niche in which that cell resides. The unique cellular signature of an individual can alter owing to diet, age, stress, drugs and so forth. Hence, at the methylation coalface, we face a far more interesting and complex clinical situation than the current emphasis on hardware changes such as mutations, SNPs, and gross genomic imbalances, since methylation signatures are dynamic and context dependent. They provide snapshots of the current network status and hence of our current cellular operating systems. The rewards of rigorous bioinformatic analyses in this sphere are likely to be profound.
Human Genome Annotation
Z Zhang, P Harrison, Y Liu, N Carriero, D Zhang, P Bertone, J Karro, D Milburn, N Echols, J Rinn, M Snyder, M Gerstein
Yale University
A central problem for 21st century science will be the analysis and understanding of the human genome. My talk will be concerned with topics within this area, in particular annotating pseudogenes (protein fossils) in the genome. I will discuss a comprehensive pseudogene identification pipeline and storage database we have built. This has enabled use to identify >10K pseudogenes in the human and mouse genomes and analyze their distribution with respect to age, protein family, chromosomal location. One interesting finding is the large number of ribosomal pseudogenes in the human genome, with 80 functional ribosomal proteins giving rise to ~2,000 ribosomal protein pseudogenes. At end I will talk broadly about pseudogenes, in terms of their composition and mutation rates and I will compare pseudogenes in the human with those in a number of other model organisms, including worm, fly, yeast, and various prokaryotes. I will also talk about the problem of identifying pseudogenes in relation to the overall problem of finding genes in genome.
Comparative analysis of processed pseudogenes in the mouse and human genomes.
Z Zhang, N Carriero, M Gerstein (2004) Trends Genet 20:62-7.
Identification of pseudogenes in the Drosophila melanogaster genome.
PM Harrison, D Milburn, Z Zhang, P Bertone, M Gerstein (2003) Nucleic Acids Res 31:1033-7.
Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome.
Z Zhang, PM Harrison, Y Liu, M Gerstein (2003) Genome Res 13:2541-58.
Patterns of nucleotide substitution, insertion and deletion in the human genome inferred from pseudogenes.
Z Zhang, M Gerstein (2003) Nucleic Acids Res 31:5338-48.
Studying genomes through the aeons: protein families, pseudogenes and proteome evolution.
PM Harrison, M Gerstein (2002) J Mol Biol 318:1155-74.
Identification and analysis of over 2000 ribosomal protein pseudogenes in the human genome.
Z Zhang, P Harrison, M Gerstein (2002) Genome Res 12:1466-82.
Digging for dead genes: an analysis of the characteristics of the pseudogene population in the Caenorhabditis elegans genome.
PM Harrison, N Echols, MB Gerstein (2001) Nucleic Acids Res 29:818-30.
Using Evolution to Explore the Human Genome
David Haussler
University of California, Santa Cruz
The reference sequence of the human genome was recently produced, along with drafts of the chimp, mouse, rat, dog, chicken and other genomes. Data and analysis of these is available on the genome browser at http://genome.ucsc.edu, a site that now averages more than 5,000 distinct users per day. This is the site where the first publicly accessible working draft of the human genome was posted. The site currently features an interactive “microscope” on the human genome and its evolution, via cross-species comparative genomics.
In 2002, a statistical estimate based on a simple measure of similarity between short orthologous segments in the human and mouse genomes suggested (very roughly) that about 5% of the human genome shows signs of being under purifying selection. Purifying selection occurs in the most important functional segments of the genome, where random mutations are mostly deleterious and hence are rejected by natural selection, leaving the orthologous segments in different species more similar than would be expected under a “neutral” mutation model. With more genomes now available, we find that this rough estimate of the fraction of the genome under purifying selection is holding up, and we are better able to pinpoint specific segments in the human genome that are evolving under this type of selection.
We are using new “context dependent” models of molecular evolution to find regions of the human genome that are not only under purifying selection, but are specifically evolving like protein-coding regions in genes. Here evolutionary analysis leads to a functional prediction. These methods have led to the prediction of many new human genes. Related analysis led to the discovery of a host of previously unexplored non-coding elements in the human genome that are under extremely strong purifying selection as well. We call these “ultraconserved” elements. Their function is currently unknown.
This work brings up an interesting information theoretic question: how well can we use the genomes of our present day animal relatives to reconstruct the evolutionary history of the individual bases of our genome, or, in other words, how much information about the ancestral state of our DNA bases was irrevocably lost? This depends a lot, but not exclusively, on how far back in time you want to go. Via simulation, we estimate that most of the DNA sequence of the common ancestor of all placental mammals can be predicted with 98% accuracy. This placental ancestral genome can in fact be reconstructed better than that of some more recent human ancestors because of the favorable phylogenetic tree topology present in the rapid radiation of species of placental mammals in the last part of the Cretaceous period. The full theory of the reconstructability of ancestral DNA bases, via a mutual information analysis, appears to be non-trivial.
References: see http://genome.ucsc.edu/goldenPath/pubs.html
Transforming Men into Mice (and into Cats, Dogs, Cows, Rats, Chimpanzees, etc): Evolutionary Lessons from Mammalian Sequencing and Comparative Mapping Projects
Pavel Pevzner
University of California at San Diego
In a pioneering paper, Nadeau and Taylor, 1984 estimated that surprisingly few genomic rearrangements have happened since the divergence of human and mouse 80 million years ago. Every genome rearrangement study involves solving a combinatorial puzzle to find a series of genome rearrangements to transform one genome into another. I will briefly describe some genome rearrangements algorithms and show how these algorithms shed light on previously unknown features of mammalian evolution. In particular, they provide evidence for extensive re-use of breakpoints from the same relatively short regions and reveal a great variability in the rate of micro-rearrangements along the genome. Our analysis also implies the existence of a large number of very short “hidden” synteny blocks that were invisible in comparative mapping data and were ignored in previous studies of chromosome evolution. These results suggest a new model of chromosome evolution that postulates that breakpoints are chosen from relatively short fragile regions that have much higher propensity for rearrangements than the rest of the genome.
This is a joint work with Glenn Tesler.
Genome Evolution and Protein Networks
Peer Bork
European Molecular Biology Laboratory, Heidelberg
Although with the availability of many completely sequenced metazoan genomes our understanding of functionality encoded therein increases, there are still numerous features in the genomes that need to be exploited for functional and evolutionary purposes. Here, I start off by describing an emerging, so far undescribed, new gene family in human that appears to drive the shaping of up to 10% of human chromosome II. Then I illustrate more generally the dynamics of gene content in metazoan genomes and how it correlates with various other measurements of genome evolution such as intron content, protein architecture or synteny. All of these measures indicate that the speed of evolution differs in some lineages. Over larger time scales some of the genomic features such as gene neighborhood indicate functional constraints that can be used for function prediction and for the construction of protein interaction networks with remarkable accuracy. Analysis of such networks reveals the functional modularity therein and how it changes in time.
Signal and Noise in Genome Sequences
Phil Green
Howard Hughes Medical Institute and University of Washington
Interpreting genome sequences requires distinguishing ‘signal’, i.e. encoded functional elements, from ‘noise’, i.e. non-functional, neutrally evolving sequence. We are working towards an improved understanding of both sides of this dichotomy.
The characteristics of non-functional sequence reflect underlying mutational processes, which remain poorly understood. Previous studies of mammalian pseudogene data by several investigators have revealed that transitions occur more frequently than transversions, G:C mutates to A:T at a higher rate than the reverse, and rates depend significantly on the flanking nucleotide context, with methylated C’s in CpG dinucleotides being notable hotspots. Studies of synonymous coding substitutions have suggested a ‘generation time effect’ consistent with the idea that most mutations occur in conjunction with DNA replication. Recent work of Duret and others points to an important role for recombination in the substitution process, likely reflecting the effects of biased gene conversion.
Using data from the NISC project (www.nisc.nih.gov/), we have discovered (Nat Genet 33, 514-517 (2003)) a mutational asymmetry associated with transcribed regions that we believe reflects an asymmetry in DNA polymerase errors that is unmasked by transcription-coupled repair. This mutational asymmetry has acted over long evolutionary periods to produce a compositional asymmetry within transcribed regions of mammalian genomes. In more recent work, Dick Hwang in my lab has developed a powerful Bayesian Markov Chain Monte Carlo approach that allows systematic exploration of variation in context-dependent rates and mutational asymmetry with respect to position within an evolutionary tree and within a sequence. We have applied this to investigate variation in mutational patterns in mammalian evolution, finding in particular that CpG mutations show a reduced generation time effect relative to other mutation types (Hwang and Green, PNAS in press). I will discuss ongoing work investigating the extent to which context-dependent mutations and biased gene conversion can explain the compositional characteristics of non-functional DNA in mammals.
Our work on signals currently focuses on the computational identification of coding sequences and splicing-related motifs. We have recently begun systematic large-scale experimental testing of gene predictions (via sequencing of RT-PCR products) in selected regions of eukaryotic genomes, with the goal of determining all gene structures in these regions. The results are then used to improve our computational models. I will describe our initial work in C. elegans.
Protein Interactions
David Eisenberg, Peter Bowers, Michael Strong, Huiying Li, Lukasz Salwinski, Robert Riley, Richard Llwellyn, Einat Sprinzak, Todd Yeates
UCLA-DOE Institute of Genomics and Proteomics, UCLA
Protein interactions control the life and death of cells, yet we are only beginning to appreciate the nature and complexity of their networks. We have taken two approaches towards mapping these networks. The first is the synthesis of information from fully sequenced genomes into knowledge about the network of functional interactions of proteins in cells. We analyze genomes using the Rosetta Stone, Phylogenetic Profile, Gene Neighbor, Operon methods to determine a genome-wide functional linkage map. This map is more readily interpreted when clustered, revealing groups of proteins participating in a variety of pathways and complexes. Parallel pathways and clusters are also revealed, in which different sets of enzymes operate on different substrates or with different cofactors. These methods have been applied genome-wide to Micobacterium tuberculosis and R. Palustris, as well as to more than 160 other genomes. Many results are available at: http://doe-mbi.ucla.edu/pronav The outcome is increased understanding of the network of interacting proteins, and enhanced knowledge of the contextual function of proteins. The information can be applied in structural genomics to find protein partners which can be co-expressed and co-crystallized to give structures of complexes. These inferred interactions can be compared to directly measured protein interactions, collected in the Database of Interacting Proteins: http://dip.doe-mbi.ucla.edu/. These observed networks constitute a second approach to detailing protein networks.
References
Visualization and interpretation of protein networks in Microbacterium tuberculosis based on hierarchical clustering of genome-wide functional linkage maps . M. Strong, T.G. Graeber, M. Beeby, M. Pellegrini, M.J. Thompson, T.O. Yeates, & D. Eisenberg (2003). Nucleic Acids Research, 31, 7099-7109 (2003).
Prolinks: a database of protein functional linkages derived from coevolution. P.M. Bowers, M. Pellegrini, M.J. Thompson, J. Fierro, T.O. Yeates, and D. Eisenberg (2004). Genome Biology, 5:R35.
In silico simulation of biological network dynamics. L. Salwinski & D. Eisenberg (2004), Nature Biotechnology, 22, 1017-1019.
From Cellular Networks to Molecular Interactions and Back
Hanah Margalit
The Hebrew University of Jerusalem
The recent large-scale functional genomic and proteomic experiments provide various types of genome-scale information, such as binding sites of transcription factors and protein-protein interactions. Representing these various types of data as networks of molecular relations opens new ways for exploring the mechanistic modules and the underlying evolutionary forces that shape the cellular circuitry.
To study the mechanistic modules responsible for the switching (on or off) of a variety of cellular processes we integrated the networks of protein-protein interaction and transcription regulation in Saccharomyces cerevisiae. Recent studies have focused on either regulatory or proteomic interactions. Yet, analyzing each of these networks separately hides the full complexity of the cellular circuitry, as many processes involve combinations of these two types of interactions. To this end we developed a new algorithm for detecting composite motifs in networks comprising two or more types of connections. Analysis of the integrated network of protein-protein interaction and transcription regulation in S. cereviaiae revealed several composite network motifs that may constitute the functional building blocks of various cellular processes.
Network methodology can be used to study other aspects of the cellular circuitry. By representing chromosomal adjacency of genes as a network and integrating it with the transcriptional regulatory network we revealed links between transcription regulation and chromosomal organization in both Escherichia coli and S. cerevisiae. Our findings suggest that in both organisms transcription regulation has shaped the organization of transcription units on the chromosome. Differences found between the organisms reflect the inherent differences in transcription regulation between pro- and eukaryotes.
Detailed examination of the networks provides insight into the characteristics of the molecular interactions comprising them. In turn, the knowledge gained from known interactions can be used in the development of predictive algorithms for identifying novel interactions. Application of these algorithms genome-wide will enrich the repertoire of molecular interactions and provide a more complete picture of the cellular networks. I will describe our algorithm for predicting target genes of novel transcription factors, based on their amino acid sequence and on knowledge of the binding pattern of other proteins in their family. It is possible that in the future such approaches may enable the determination of the regulatory networks in the cell based on genomic sequence data alone.
Protein-Protein Interactions: The Challenge of Predicting Specificity
Shoshana J. Wodak
Hospital for Sick Children, Toronto Ontario, Canada
Protein-protein interactions are probably amongst the most ubiquitous types of interactions and play a key role in all cellular processes. Determining the interaction network of whole organisms has therefore become a major theme of functional genomics and proteomics efforts. Computational methods for inferring protein interactions are likewise attracting much interest. Particularly remarkable has been the setup of CAPRI (Critical Assessment of PRedicted Interactions), a community-wide experiment analogous to CASP (Critical Assessment of Structure Predictions), but aimed at assessing the performance of protein-protein docking procedures. To this day seventeen complexes offered by crystallographers as targets prior to publication, have been subjected to structure prediction by docking their two components. Hundreds of predictions for these complexes were submitted by an average of 20 predictor groups and assessed by comparing their geometry to the X-ray structure and by evaluating the quality of the prediction in the regions of interaction. Over the four years of CAPRI’s existence progress in the prediction quality was clearly observed, but major challenges remain. One is the ability to handle conformational flexibility, which often plays a major role. Another key challenge is to single out specific from non-specific association modes, a problem for which computational analyses are still seeking solutions. Various aspects of these challenges will be discussed and possible avenues for future progress will be outlined.
The Modern RNA World: Computational Screens for Noncoding RNA Genes
Sean R. Eddy
Washington University, St. Louis
Some genes produce RNAs that function directly as RNA rather than encoding proteins. The diversity of noncoding RNAs in nature is largely unknown, because RNA genes have been difficult to detect systematically, and most current genefinding approaches focus exclusively on protein coding genes. Genome sequence analysis, functional genomics, and new computational algorithms have enabled several recent experiments that have begun to show that RNA genes and RNA-based regulatory circuits are more prevalent that we suspected.
Towards an RNA Splicing Code
Christopher Burge
Massachusetts Institute of Technology
Most human genes are transcribed as precursors containing long introns that are removed in the process of pre-mRNA splicing. The specificity of splicing is defined in part by splice site and branch site sequences located near the 5' and 3' ends of introns. However, even considering transcripts with only very short introns, these sequences contain only about half of the information required for accurate recognition of exons and introns in human transcripts. Indeed, it is well known that human transcripts contain a vast excess of sequences that match the consensus splice site motifs as well as authentic sites yet are virtually never used in splicing – so-called ‘decoy’ splice sites and pairs of decoy splice sites known as ‘pseudoexons’. The ability of the splicing machinery to reliably distinguish authentic exons and splice sites from a large excess of these imposters implies that sequence features outside of the canonical splice site/branch site elements must play important roles in splicing of most or all transcripts. Prime candidates for these features are exonic or intronic cis-elements that either enhance or silence the usage of adjacent splice sites. My lab is using a combination of computational and experimental approaches to understand the elements that control the specificity of splicing. Recently completed efforts have focused on: (i) improved modeling of the classical splice site motifs using constrained maximum entropy models(1); (ii) predictive identification and SNP-based validation of exonic splicing enhancers (ESEs) in human genes(2,3); and (iii) studies of variations in the sequence and organization of splicing regulatory elements between different vertebrates(4). I will briefly summarize this work and related work from other labs and describe in more detail the results of a screen for exonic splicing silencers (ESSs) and the development of a first-generation RNA splicing simulation algorithm(5). To systematically identify ESSs, an in vivo splicing reporter system was developed and used to screen a library of random decanucleotides. Screening of cells representing between one- and two-fold coverage of the ~ one million possible decanucleotides yielded 141 ESSs, 133 of which were unique. The silencer activity of over a dozen of these sequences was also confirmed in a heterologous exon context and in a second cell type. Of the unique ESS decamers, 21 pairs differed by only a single nucleotide, and most could be clustered into groups to yield seven putative ESS motifs. Some of these motifs resemble known motifs bound by the hnRNP proteins H and A1, while others appear novel. Motifs derived from the ESS decamers are enriched in pseudoexons and in alternatively spliced exons, suggesting roles in suppressing pseudoexon splicing and in regulating alternative splicing. Potential roles of ESSs in constitutive splicing were explored using an algorithm, ExonScan, which simulates splicing based on known or putative splicing-related motifs. ExonScan analysis suggests that these ESS motifs play important roles in both suppression of pseudoexons and in splice site definition. Synergistic combinations of computational and experimental approaches appear most promising for making further progress towards complete understanding of the RNA splicing code.
1. Yeo, G. and Burge, C. B. (2004). J. Comp. Biol. 11, 377-394.
2. Fairbrother, W., Yeh, R.-F., Sharp, P. A. and Burge, C. B. (2002). Science 297, 1007-1013.
3. Fairbrother, W. G., Holste, D., Burge, C. B. and Sharp, P. A. (2004). PLoS Biol. 2, e268.
4. Yeo, G., Hoon, S., Venkatesh, B. and Burge, C. B. (2004). Proc. Natl. Acad. Sci USA (in press).
5. Wang, Z., Rolish, M., Yeo, G., Tung, V., Mawson, M. and Burge, C. B. (2004). (unpublished data).
Discovering Evolutionary Mechanisms from Multiple Metrics of Molecular Evolution
Christopher Lee
University of California, Los Angeles
The availability of multiple genome sequences is the first essential ingredient for obtaining a detailed history of the evolutionary mechanisms that have constructed modern organisms. A second key ingredient is the development of multiple metrics of rates for different evolutionary processes, and of different types of selection pressure. We have used metrics for a wide variety of evolutionary processes—exon creation and loss; splice site movement; protein reading frame preservation; point substitution rates and selection pressures; premature termination codons, and conditional selection pressures—to examine the role of alternative splicing in the evolution of mammalian genomes. These data show that alternative splicing can produce a striking acceleration in evolution of a single exon of a gene, by reducing negative selection pressure against changes to that exon. This acceleration in the evolution of a specific protein subsequence shows clear independent evidence of adaptive benefit, that has been strongly selected for during recent evolution. Human genome data suggest that up to half of recently created exons may have been introduced through such an alternative splicing mechanism. We have also used new metrics of selection pressure to automate discovery of drug resistance mutations in HIV, and to analyze the evolutionary pathways of the viral population.
Probing The PDB
Helen M. Berman
Protein Data Bank; Research Collaboratory for Structural Bioinformatics, aRutgers, the State University of New Jersey
The RCSB Protein Data Bank (PDB; www.pdb.org) is a publicly accessible information portal for researchers and students interested in structural biology. At its center is the PDB archive – the sole international repository for the 3-dimensional structure data of biological macromolecules.
This talk will focus on the tools provided by RCSB PDB to browse and explore these structures. Structures can be searched and reviewed using a variety of parameters. Data from related resources, including Gene Ontology, EC, KEGG Pathways, and NCBI are mapped to structures and loaded into the database. Structures are also linked to their corresponding entry in other databases, including Swiss-Prot, SCOP, and PubMed.
The RCSB PDB is managed by three RCSB members - Rutgers, The State University of New Jersey; the San Diego Supercomputer Center at the University of California, San Diego; and the Center for Advanced Research in Biotechnology/UMBI/NIST. Support is from the NSF, NIGMS, the Office of Science, DOE, NLM, NCI, NCRR, NIBIB, and NINDS. The RCSB PDB is a member of wwPDB.
Structural Alignment and Classification of all Known Protein Structures
Rachel Kolodny, Patrice Koehl and Michael Levitt
Stanford University
We have carried out the largest and most comprehensive comparison of protein structural alignment methods. Specifically, we evaluate six publicly available structure alignment programs: SSAP, STRUCTAL, DALI, LSQMAN, CE and SSM by aligning all 8,581,970 protein structure pairs in a test set of 2,930 protein domains specially selected from CATH v.2.4 to ensure sequence diversity. Our own method STRUCTAL has also been run on SCOP v. 1-65.
Here we use this data to discuss the importance of having an objective different geometric match measures with which to evaluate an alignment. With this improved analysis we show that there is a wide variation in the performance of different methods; the main reason for this is that it can be difficult to find a good structural alignment between two proteins even when such an alignment exists. Methods that do best in our study are neither the most popular nor those that are generally accepted to work well. We find that STRUCTAL and SSM perform best, followed by LSQMAN and CE. Our focus on the intrinsic quality of each alignment allows us to propose a new method, called ‘Best-of-All’, which combines the best results of all methods. Some commonly used methods miss almost half of the good ‘Best-of-All’ alignments.
We discuss the differences between a set of structural alignments and a classification of structures. We compare the most common classifications of protein structures (CATH, SCOP and DALI/FSSP) and show that they are really quite different. We also present preliminary results on an objective method that derives a classification of structures from a series of pair-wise alignments.
Comparative Plant Genomics: Evaluation of the Model Genome Concept
Volker Brendel
Iowa State University
The first plant genome was made available to near completion in 2000. Arabidopsis thaliana continues to be an important model system for studying genome content and organization and for functional genomics. Four years later, the rice genome is essentially finished, representing the first monocot genome and a size scale-up of threefold compared to the Arabidopsis genome. The fast expansion of the number of prokaryotic and animal genomes over a short period of time appears to have jumped over into the plant genome research field: the genome of Medicago truncatula and Lotus japonicus are also soon to be finished, sequencing projects for tomato and Physcomitrella patens have been announced, and a request for proposals is out for sequencing the maize genome (which is approximately the size of the human genome). I will discuss efforts of my group to catch up with the computational analysis of all these data. While the excitement is always with respect to the novel projects, how well do we actually understand the current genome data? How many gene models are solidly established? How large is the error rate in computational gene structure predictions? In view of inevitable transitive gene structure annotation when comparing genomes, assessments of accuracy are of paramount importance. I will discuss various tools to facilitate gene structure annotation and evaluation and present arguments for the feasibility and necessity of community-based annotation.
Lessons from the Arabidopsis Genome: Decoding Evidence for Novel Transcription
Terry Gaasterland
The Rockefeller University and Scripps Institution of Oceanography, Genome Research Center, University of California, San Diego
Several recent surveys of gene expression indicate that genome transcription activity extends well beyond mRNA, tRNA and rRNA gene expression. Large scale studies that completely tiled human chromosomes 21 and 22 onto 2-color or 1-color microarrays and hybridized with total RNA found considerable transcriptional activity in intergenic, intronic, and UTR antisense regions (Rinn et al 2003; Kapranov et al 2002). Studies that used chromatin immuno-precipitation to isolate DNA bound to selected transcription factors followed by hybridization on DNA microarrays (“ChIP-chip” studies) have found unexpected binding events in regions annotated as intergenic (Euskirchen et al 2004; Martone 2003; Kampa et al 2004; Cawley et al 2004). In contrast, ChIP-chip studies of POL-II binding sites have tended to identify primarily previously annotated coding regions (Ren & Dynlacht 2004). Other data sources include SAGE-like surveys of gene expression using the Massively Parallel Signature Sequence (MPSS) technology, which invariably yield substantial evidence for transcription outside of previously annotated genes as well as quantitative gene expression levels for annotated genes.
Some of the additional transcription is explained by the presence of small non-coding RNA genes in intergenic regions. In the case of microRNAs, transcripts from these ~150-350 nucleotide (nt) genes fold into secondary structures with long, imperfect hairpins that are processed by a protein complex that recognizes and cleaves double-stranded RNA to release short ~19-23 nt single-stranded RNA molecules. These microRNAs are complementary to mRNA transcripts and suppress protein expression by repressing translation or by triggering mRNA degradation. In plants, microRNAs tend to bind within coding regions; in animals, they bind to the 3'UTR.
This talk presents observations about control of gene and protein expression in Arabidopsis thaliana based on the following data sources: tissue specific MPSS data and Affymetrix gene expression data on stress response, genome-wide prediction of binding site clusters for known transcription factors, evaluation of alternative splicing evident in cDNA and EST sequences, and microRNA identification and mRNA target prediction (Hoth et al 2003). These data have all been combined to yield a model of microRNA regulation of gene and protein expression in plants.
1. Rinn JL, Euskirchen G, Bertone P, Martone R, Luscombe NM, Hartman S, Harrison PM, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M. The transcriptional activity of human Chromosome 22. Genes Dev. 2003 17(4):529-40.
2. Kapranov P, Cawley SE, Drenkow J, Bekiranov S, Strausberg RL, Fodor SP, Gingeras TR. Large-scale transcriptional activity in chromosomes 21 and 22. Science. 2002 296(5569):916-9.
3. Euskirchen G, Royce TE, Bertone P, Martone R, Rinn JL, Nelson FK, Sayward F, Luscombe NM, Miller P, Gerstein M, Weissman S, Snyder M. CREB Binds to Multiple Loci on Human Chromosome 22. Mol Cell Biol. 2004 24(9):3804-14.
4. Martone R, Euskirchen G, Bertone P, Hartman S, Royce TE, Luscombe NM, Rinn JL, Nelson FK, Miller P, Gerstein M, Weissman S, Snyder M. Distribution of NF-kappaB-binding sites across human chromosome 22. Proc Natl Acad Sci U S A. 2003 100(21):12247-52.
5. Kampa D, Cheng J, Kapranov P, Yamanaka M, Brubaker S, Cawley S, Drenkow J, Piccolboni A, Bekiranov S, Helt G, Tammana H, Gingeras TR. Novel RNAs identified from an in-depth analysis of the transcriptome of human chromosomes 21 and 22. Genome Res. 2004 14(3):331-42.
6. Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, Piccolboni A, Sementchenko V, Cheng J, Williams AJ, Wheeler R, Wong B, Drenkow J, Yamanaka M, Patel S, Brubaker S, Tammana H, Helt G, Struhl K, Gingeras TR. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004 116(4):499-509.
7. Ren B, Dynlacht BD. Use of chromatin immunoprecipitation assays in genome-wide location analysis of mammalian transcription factors. Methods Enzymol. 2004;376:304-15.
8. Hoth S, Ikeda Y, Morgante M, Wang X, Zuo J, Hanafey MK, Gaasterland T, Tingey SV, Chua NH. Monitoring genome-wide changes in gene expression in response to endogenous cytokinin reveals targets in Arabidopsis thaliana. FEBS Lett. 2003 554(3):373-80.
Building Genotype Phenotype Data Resources
Russ Altman
Stanford University
In the post-genome era, one of the major challenges to informatics is to support the association of genotype with phenotype. In particular, methods are required to represent and analyze data in standard formats using agreed semantics, in order to build a useful public database. Such standards may require that methods for collecting certain types of data (particularly high-throughput data) be standardized to allow for integration of data across multiple conditions. The best progress has been made in standardizing exchange of genotype and some types of phenotype data, most notably microarray expression data. However, there are significant challenges in representing other phenotype data, because the experimental methods used to collect this data are diverse, and because many biologists are not willing to constraint their scientific programs in order to be compatible with standards. This can lead to individually powerful data sets that stand alone, difficult to integrate with other data sources.
We are building the Pharmacogenomics Knowledge Base (PharmGKB, http://www.pharmgkb.org/) as an initial example of a diverse post-genome database. Pharmacogenomics is the study of how variation in the genome leads to variation in the response to drugs. The PharmGKB contains information about genotypic variation in a set of populations, and associates these with variation in phenotypes at molecular, cellular, organ and organisms levels. The PharmGKB currently contains genotyping information from more than 5000 individuals and phenotype information from more than 3000 individuals.
The goals of PharmGKB include the development of new tools for pharmacogenomics, and the mining/integration of existing databases. We have developed a method for defining haplotype tagging SNPs, and have shown that these htSNPs can be used to efficienctly recover the full genotype. We have also developed text mining algorithms to catalog all published gene-drug interactions, in order to provide comprehensive coverage of the literature in PharmGKB. The current challenges to PharmGKB include 1) defining standards for exchange of phenotype information, 2) supporting association studies for finding genotype-phenotype correlations, 3) defining and supporting the definition of drug-related pathways, 4) linking high-throughput data sources with genes, drugs and diseases of interest, and 5) linking molecular structural and cheminformatics information to pharmacogenetic variation.
Highly Expressed Genes Based on Codon Usage Biases in Archaeal and Eukaryotic Genomes
Samuel Karlin and Jan Mrázek
Stanford University
Based on rRNA sequence criteria, life has been broadly divided into the three domain: bacteria, archaea and eukaryotes, which are believed to reflect phylogenetic relationships. The archaea are further classified into Crenarchaea and Euryarchaea and recently possibly nano-archaea. For most bacterial organisms during exponential growth, ribosomal proteins (RP), transcription/translation processing factors (TF), and the major chaperone/degradation genes (CH) functioning in protein folding and trafficking tend to be highly expressed. The gene classes (RP, CH, and RF) serve as representative of highly expressed genes, and our method specifies genes with rather similar codon usages as PHX genes. These assignments are reasonable under fast growth conditions, where there is a need for many ribosomes, for proficient transcription and translation, and for many CH proteins to ensure properly folded, modified, and translocated protein products. The codon usage difference of the gene group F with respect to the gene group G is calculated by the formula 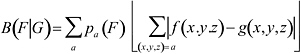 , where
, where ![]() are the average amino acid frequencies of the genes of F. Predicted expression levels with respect to individual standards can be based on the ratios
are the average amino acid frequencies of the genes of F. Predicted expression levels with respect to individual standards can be based on the ratios
where C is the totality of all genes of the genome. Using these gene classes as standards, a gene is Predicted Highly eXpressed (PHX) if its codon usage is rather similar to at least two of the RP, TF, and CH gene classes and deviates strongly from the average gene of the genome. An overall estimate of the expression level of the gene g is ![]() defined by the equation
defined by the equation ![]() .
.
The criterion ![]() and where at least two of the values
and where at least two of the values ![]() ,
, ![]() ,
, ![]() , exceed 1.05 provides an excellent benchmark in reflecting high protein molar abundance in a rapid growth environment.
, exceed 1.05 provides an excellent benchmark in reflecting high protein molar abundance in a rapid growth environment.
In all currently available archaeal genomes, the thermosome chaperonin genes rank among the top PHX. DnaK (HSP70) is found PHX virtually only in archaeal mesophiles or in archaeal moderate thermophiles. The Lon protease is absent from the Crenarchaea but usually PHX among the euryarchaea. Archaea genomes are also pervasive with proteasome units. Other distinctive proteins of archaea generally PHX and absent from bacteria highlight PCNA (proliferating cell nuclear complex) a replication auxiliary factor (sliding clamp subunit) responsible for tethering the catalytic subunit of DNA polymer to DNA during high-speed replication. The ribosomal protein P0 (acidic, regulatory) whereas the ribosomal machinery in eukaryotes contains P0,P1,P2 featuring a hyperacidic run at its carboxyl end. Other distinctive PHX genes found in all archaea: Cdc48 Cell division control protein 48; Cdc6 Replication initiation; RadA DNA repair and recombination protein in archaea.
In prokaryotes, the maximum E(g) level correlates negatively with the doubling time of the organisms. Compared to bacterial genomes, relatively few RP genes of archaea are PHX and many are expressed as average genes. A clear exception is M. maripaludis. The yeast genome parallels E. coli in PHX genes plus the addition of actin, cofilin and related genes. The most PHX genes of Drosophila encode the cytoskeletal proteins.






















