
2
Overview of the Drug Development, Regulation, Distribution, and Use System
CHAPTER SUMMARY
The drug system encompasses four main stages—research and development; regulatory review; medication manufacturing, distribution, and marketing; and medication use—that each contain multiple critical control points at which quality, safety, and efficacy can be addressed, and at which breakdowns can occur. This chapter provides an overview of the major components of the drug system and the points that might lead directly or indirectly to errors as well as opportunities for learning, recovery, and improvement.
As noted in two previous Institute of Medicine (IOM) reports—To Err Is Human: Building a Safer Health System (IOM, 2000) and Crossing the Quality Chasm: A New Health System for the 21st Century (IOM, 2001), redesigning health care to improve quality and safety requires definitive action by all stakeholder groups interacting with the health system. Applied to this report, stakeholders of the drug system associated with research, innovation, regulation, clinical practice, payment, education, legislation, and reporting should be assessed according to how well quality and safety are (or can be) achieved, among other factors. Advancing this concept requires that the disciplines of human factors engineering, organizational psychology, sociology, and informatics must become the basic sciences of quality just as molecular biology, pharmacology, and genetics are the basic sciences of medicine (Brennan et al., 2005). Quality and safety in medication use depends directly on the extent to which the principles of these
sciences are built into the overall drug system (Califf et al., 2002). Integrating the sciences of quality with the biomedical and health sciences will ultimately facilitate the translation of safety and quality in medication use from theory to clinical practice.
As a first order of business, the points at which safety and quality can be compromised must be identified. Currently, the potential for harm is present throughout the system. Harm can be due to any number of factors, many of which are now in the national spotlight, including undisclosed harmful side effects of a drug for specific patient populations; lax follow-through on regulatory responsibility after product approval; human error in prescribing, dispensing, administering, and monitoring effects in patients; and inadequate patient activation and education. This chapter identifies the key issues of the overall drug system that affect safety and quality in medication use. Subsequent chapters in this report provide recommendations for improvement, many of which incorporate the “sciences of quality” mentioned above.
STRUCTURE OF THE OVERALL DRUG SYSTEM
Currently more than 10,000 prescription drugs and biologics (FDA, 1999) and more than 300,000 over-the-counter (OTC) products are on the market in the United States (RSW, 2001). In 2004, 215 prescription and 71 OTC drugs were recalled because of manufacturing and distribution problems or serious adverse reactions (FDA, 2004a).
The regulatory element of the drug system evolved over the past century from being focused on regulating interstate transport and misbranded products to being built on an infrastructure with the goal of reliable standards, processes, and laws to ensure some degree of safety and efficacy in medicinal agents. The result is a sophisticated, comprehensive drug system encompassing four stages that interact with, support, and reinforce each other to varying degrees (see Figure 2-1): (1) research and development (R&D), where ideas for new drugs are conceived and candidates are clinically tested; (2) regulatory review by the Food and Drug Administration (FDA) to validate or counter the research findings and ensure proper labeling; (3) manufacture, distribution, and marketing of products that have received regulatory approval; and (4) use of medications available either through a prescription or OTC. Prescription drugs, biologics, and some OTCs follow this model. The product development and regulatory review stages are abbreviated for other OTCs and for generics.
Each element of the drug system is governed by its own set of standards and methods for scientific analysis to advance the safety, quality, and efficacy of products and their use. As the chief protector of the public health, the FDA has responsibility for developing and enforcing the standards in all
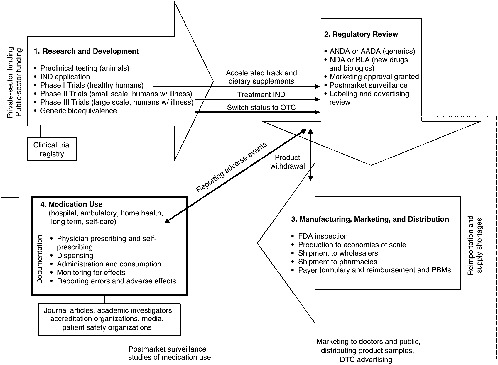
FIGURE 2-1 Four stages of the drug system.
NOTE: AADA = Abbreviated Antibiotic Drug Application; ANDA = Abbreviated New Drug Application; BLA = Biologic Licensing Application; DTC = direct to consumer; FDA = U.S. Food and Drug Administration; IND = Investigational New Drug Application; NDA = New Drug Application; OTC = over-the-counter; PBM = Pharmacy Benefits Manager.
areas except clinical practice, which is governed by state boards of medicine, nursing, and pharmacy; professional societies; and accreditation organizations.1 Compliance with FDA regulatory standards is the responsibility of the manufacturers who promote their products in the marketplace. Safe and effective use of medications is the responsibility of providers who prescribe the medications and patients who take them.
Standards2 for each component of the drug system act as links in a chain of events that have an important bearing on the competence and effectiveness of drug therapies in medical care. The key points at which important interventions can be implemented are identified in Figure 2-1. Building safety and quality into the system starts with rational ideas for new drug products, followed by sound scientific research; reliable clinical testing; rigorous regulatory reviews; appropriate labeling; use of good manufacturing processes; proper distribution techniques; adequate supplies; ethical marketing practices; competent prescribing, dispensing, and administration of medications; and finally suitable monitoring of the patient, reporting of errors, and measurement of outcomes (Martin, 1978). If standards do not exist, are inadequate, have not been met, or are not enforced at any point along this chain, patient safety and quality of care can be compromised. For example, restriction on the publication of a drug’s side effects can affect a prescriber’s ability to choose the best drug for a patient or to identify and respond to an adverse reaction in a timely manner; lax enforcement of regulatory requirements for drug labeling can result in product confusion in a high-stress, fast-paced clinical setting; formulary restrictions can force a switch to a medication that may be less appropriate for a patient than the one initially prescribed; or the failure to document all of the medications a patient is taking (including OTCs and dietary supplements) can cause a drug–drug interaction that could have been prevented.
In the first three of these examples, problems in the drug development, regulation, and distribution systems contribute to medication-use errors that should be corrected. Yet most links or components of the drug system operate in a quasi-silo state with less-than-ideal means of sharing important
information or responding to safety-related problems. In the last example, the problem results from an error within the medication-use system itself (e.g., insufficient information). Most often this is the case: medication errors are the result of a problem incurred during the prescribing, dispensing, administration, or monitoring phases of the medication-use system.
Nevertheless, both aspects of causation—how the drug is prepared (developed, regulated, distributed) and how it is used in clinical practice or self-care—must be addressed if errors in the medication-use system are to be reduced and prevented. The remainder of this section addresses the former (research and development; regulatory review; and manufacture, distribution, and marketing). The second section of the chapter addresses medication use.
Research and Development
The R&D process involves more than the development of new products; it encompasses the overall generation and disclosure of high-quality data that can be used with confidence by providers and patients in medical care, by providers and technology vendors to populate knowledge bases and clinical decision-support systems, by regulators in assessing benefit/risk balances for protection of the public health, and by researchers for continued innovation and advancement of science and medicine (Califf, 2004). Issues related to study design, data quality, and disclosure can have direct bearing on the development of the medication knowledge base needed to support clinicians and pharmacists in clinical decision making and prescribing; preparation and administration of appropriate dosages; and monitoring of patient response (positive and negative) to a medication, particularly the ability to discern symptoms of disease from effects of the drug. Public availability of information from trials also is necessary to support consumers in their self-care, disease management, and medication self-management. Data quality can be compromised by poor clinical study designs, less-than-optimal methods of data analysis, and/or conflicts of interest that affect the objectivity of investigators (Califf and DeMets, 2002a,b; Strom, 2004; March et al., 2005). The failure to disclose negative study results (e.g., serious adverse side effects) can have fatal effects on patients (Bodenheimer, 2000; Moore et al., 1998).
Current State of R&D
Pharmaceutical R&D for new drugs and biologics aims to meet a medical need in a specified patient population by creating medications with characteristics of high activity, low toxicity, and relatively few side effects. Fundamentally, approval for marketing a drug is based on an assessment of
the balance of the benefit and risk of using the drug in the specified population. The ability to separate toxic and side effects from therapeutic effects on the basis of preclinical evaluation is an ongoing challenge. Sizable amounts of time and effort are spent on trying to increase this margin, but ultimately the balance of benefit and risk cannot be defined until clinical trials have been conducted in relevant populations (Martin, 1978; Califf, 2004). Trends in drug development over the past few decades have led to significant improvements in study designs, reducing the incidence of incorrect conclusions concerning dosage, efficacy, and safety while deepening understanding of how the molecular structures of potential new drugs interact with specific human cellular structures. More recent scientific discoveries in the areas of genomics, biotechnology, and informatics are expected to increase significantly the number of new molecular targets and the ability to develop medicines with greater specificity and fewer side effects (NRC, 2004), although this promise has not been realized, and the time frame for pragmatic advances remains unclear (Califf, 2004).
Clinical Study Design
Traditionally, the R&D process has been performed in sequential stages. After discovery of potential compounds for new medicines and preclinical testing in the laboratory and in animals for safety and biological activity against the targeted disease, the manufacturer (i.e., sponsor) submits an Investigational New Drug Application (IND) to the FDA or other international regulatory authority for review.3 The IND contains plans for clinical studies in humans (Phases I, II, and III), all data from preclinical testing, and complete structural and manufacturing information. At any time after the IND has been submitted, the sponsor may request an accelerated development and approval track (“fast track”) for drugs that promise substantial benefit over existing therapies for serious or life-threatening illnesses. Granting of fast track status is based on the case that the drug would fulfill a critical unmet health need, early evidence of the drug’s effects on a surrogate end point,4 commitments to undertake postmarket studies, and/or agreement to restrict distribution and use after approval (FDA, 1999).
Most Phase I studies use healthy volunteers to test the drug’s actions, both metabolic (pharmacokinetics [PK]) and pharmacologic (pharmacody-
namics [PD]);5 side effects associated with increasing doses; and if possible, early evidence of efficacy (FDA, 1998). Phase II studies use a small group of patients with the condition in well-controlled circumstances to evaluate the dose that optimally affects the chosen biological target, the method of delivery (e.g., oral, intravenous), the dosing interval, and short-term side effects, and to extend the preliminary evidence of safety from Phase I (Walters, 1992; Leonard, 1994; FDA, 1998). A substantial number of drug trials are discontinued after both Phases I and II because of ineffectiveness, safety problems, or intolerable side effects. If the Phase I and II trials are successful, the sponsor may apply for Treatment IND status to provide promising drugs to patients with a life-threatening disease (e.g., AIDS) if no comparable therapy exists or the patients cannot participate in clinical studies.
Phase III trials are the most critical in the determination of a drug’s approval for labeling by the FDA and international regulatory authorities. Typically, Phase III trials are structured as randomized controlled trials involving enough patients carefully selected, often across multiple sites, to obtain data on the drug’s overall benefit/risk relationship so that regulators, often guided by expert panels, can be comfortable that the balance is favorable for the defined population (Nies, 2001). While such studies typically can last from 1 to 4 years and commonly include from 1,000 to 10,000 patients, generally only a few hundred patients are treated for more than 3 to 6 months with the drug, regardless of the duration of treatment required in clinical practice. As a result, only the most profound and overt risks and side effects that occur immediately after taking a drug can be detected if the occurrence rate is 1 in 100 administrations. Risks that are medically important but delayed, less frequent than 1 in 1,000 administrations, or not evenly distributed across the population may not be revealed prior to marketing (Nies, 2001). In particular, serious adverse effects for a specific patient population (e.g., pediatric, geriatric, those with renal dysfunction or multiple comorbidities) usually will not be known, as those groups are not well represented in the trials (Lee et al., 2001; Klein et al., 2002). Accordingly, postmarket surveillance and evaluation studies (Phase IV) are often requested for further evaluation of safety issues (e.g., adverse effects) after approval.
During a January 2005 meeting on drug development science sponsored by the FDA and the Association of American Medical Colleges (AAMC), participants from academia, industry, and government identified
crucial problems with the current model and opportunities for improvement (AAMC, 2005). The participants found that study designs often are not tailored to the pharmacology of potential new drugs and the patient populations that will use them, and frequently are not structured to allow adequate evaluation of a broad range of doses.
Each of the above factors can contribute to issues of patient safety and quality of care in the medication-use process. For example, drugs can produce very different effects in elderly patients and younger adults. The elderly are more likely to have impaired kidney and renal function, to be taking other medications, or to have other medical conditions. Few clinical studies include substantial numbers of elderly patients, however, even though the elderly are a growing proportion of the general population (FDA, 1999; Noah and Brushwood, 2000; Boyd et al., 2005).
Data Quality
While randomized controlled trials are considered the gold standard for assessing efficacy, they rarely provide all the information needed in clinical practice (Teutsch et al., 2005). Drugs are usually compared with a placebo, and studies frequently use surrogate or intermediate measures of efficacy, such as blood pressure, low-density lipoprotein cholesterol, or tumor shrinkage, rather than tangible patient outcomes, such as mortality, morbidity, and quality of life. Placebo-based comparisons serve regulatory requirements, leaving long-term studies comparing treatments to post-approval. Without data on health outcomes, extrapolation from the carefully selected patient populations used in clinical trials to patient populations seen in typical practice settings and from the patient population used in a trial to another patient population introduces uncertainty (Teutsch et al., 2005).
A variety of leaders have voiced concern about the threat posed to scientific integrity by conflicts of interest among industry and academic researchers, private-sector investigators, and regulators (Bodenheimer, 2000; Chopra, 2003; Fontanarosa et al., 2004; Psaty et al., 2004). There is evidence that research has tended to overemphasize drug benefits while downplaying risks (Rochon et al., 1994; Rothman and Michels, 1994; Bero and Rennie, 1996; Bekelman et al., 2003).
Disclosure of Results
Currently, public disclosure of results through registration is required only for clinical gene-transfer trials registered with the National Institutes of Health (NIH) and studies conducted under INDs (FDA, 2004b). Nondisclosure (failure to register) of all clinical trials from start to completion and
|
BOX 2-1 Summary of Key Problems with the Research and Development Process Affecting Safety and Quality in the Medication-Use System
|
failure to report results (both positive and negative) in a public database have left sizable gaps in the knowledge base that can affect decision making by regulators and clinicians, as well as the work of researchers and editors of medical journals (Steinbrook, 2004; IOM, 2006). (See Box 2-1 for a summary of key problems with the research and development process.)
Regulatory Review
Prior to marketing in the United States, all new prescription drugs (including generics), OTC drugs, and biologics are subjected to formal regulatory review and approval by the FDA’s Center for Drug Evaluation and Research (CDER). The primary objectives of the regulatory review are to evaluate a drug’s safety and effectiveness and to determine whether its benefits outweigh its risks. Regulatory review also verifies that industry has taken the appropriate measures to prepare the products properly for the market.
The balance of benefit and risk is influenced significantly by intended use, and varies from drug to drug and from one patient group to another (FDA, 1999; University of Utah, 2006). For example, greater risk may be tolerated for a drug designed to treat a life-threatening illness than for one designed to treat the common cold. Likewise, lower risk may be required for drugs intended for geriatric patients, who are more likely to have renal or hepatic impairment and multiple conditions (FDA, 1994). As genomics and proteomics enable drug development to become increasingly individualized, it will be possible to establish more specific benefit and risk assessments for particular patient populations with certain clinical or genetic characteristics. This capability will necessitate reexamination of the current benefit/risk model used for regulatory approval (Califf, 2004).
Review of Clinical Data for New Drugs and Biologics
Assessment of new drugs (i.e., new molecular entities [NMEs]) is based on the New Drug Application (NDA) or the Biologic Licensing Application (BLA)—dossiers submitted by the drug sponsor that include all data from preclinical and clinical studies on safety and efficacy, proposed labeling, and manufacturing details. A team from CDER’s Office of New Drugs reviews the dossiers; communication with the sponsor occurs throughout the process to address scientific, medical, and procedural issues. The FDA uses advisory committees of external scientific experts for advice and opinions to broaden its basis for decision making on an NDA/BLA or regulatory issue.
For a drug to win approval, the FDA does not require that it be better than products already available, only that it be effective (better than nothing [i.e., placebo]) and fairly safe (Deyo, 2004). A drug is determined to be effective if it achieves a “surrogate outcome” (e.g., lowers cholesterol) without its effects on life expectancy being known. The FDA does not approve every use for which a drug may be prescribed by a clinician, only the use evaluated during its clinical trial.
Postmarket Surveillance of New Drugs
Some of the risks associated with a new drug are not known at the time of regulatory review because the data from clinical trials are limited in terms of patient population, study size, and/or duration. Consequently, drugs must continue to be evaluated as they are used in clinical settings to detect less frequent but significant adverse side effects, long-term effects, or effects in different patient populations. Two mechanisms are available for this purpose: (1) postmarket surveillance studies, and (2) the FDA’s adverse event reporting systems (see later in the chapter). Both approaches rely on manufacturers to collect, evaluate, and report data on their own products (Fontanarosa et al., 2004).
Postmarket studies can be designed to observe a drug’s effects in a larger, more heterogeneous population over 3–4 years (Berndt et al., 2005). The FDA requires postmarket studies as a condition for approval in only two product categories—drugs granted fast track status and drugs for which the manufacturer desires a pediatric indication (Fontanarosa et al., 2004). Such studies are optional for other product categories, although strongly encouraged. Manufacturers complete fewer than half of the postmarket studies they commit to undertaking as a condition for approval (FR, 2004a; Fontanarosa et al., 2004). At the request of the FDA, the IOM Committee on Assessment of the U.S. Drug Safety System is evaluating the agency’s postmarketing surveillance. More detail on surveillance systems is given in
the section on adverse event reporting and surveillance systems later in the chapter.
Review of Clinical Data for Generics and OTCs
The FDA uses a process similar to that for NMEs to review new generic drugs and OTCs. Sponsors of generics file an Abbreviated New Drug Application (ANDA) or Abbreviated Antibiotic Drug Application (AADA) that provides information supporting equivalence to an FDA-approved brand-name drug in terms of active ingredients, dosage, safety, strength, administration, quality, performance, and intended use. Generic manufacturers are not required to replicate the extensive clinical trials of the original drug, but must demonstrate bioequivalence; this can be done by measuring bioavailability (e.g., rate and extent of absorption) of the generic in 24 to 36 healthy subjects (FDA, 1999).
For OTCs the FDA has established drug monographs for each OTC product class, covering acceptable ingredients, doses, and formulations (FDA, 1998). An FDA team assesses a product’s conformance to the monograph, as well as to OTC labeling guidelines.
Product Labeling
After deciding to approve a drug for a specific indication, the FDA evaluates the product labeling. Labeling is a broad term that encompasses a number of materials developed by pharmaceutical companies, including the professional product label (also known as the package insert); medication guides (for drugs posing a serious public health concern); patient package inserts (with content often used in media advertisements); product packaging (which pertains to the external package labeling of the drug); and any written, printed, or graphic material used for marketing (Kenny, 2001).
Professional product labels (package inserts) are developed by companies on the basis of Phase III data. They are evaluated by the FDA for compliance with federal regulations, rather than for usefulness6 to health care professionals and consumers. Medication guides and patient package inserts are written for consumers in a more user-friendly language. However, problems with the design and content of all labeling materials affect their readability, comprehensibility, and usefulness (FR, 2006; Hubal and Day, 2006). The FDA’s recently published new rule on drug labeling is an
improvement (FR, 2006), but additional work is still needed on better incorporating the principles of cognitive and human factors engineering to address remaining issues concerning information presentation and nomenclature (http://www.fda.gov/cder/regulatory/physLabel/default.htm).
FDA Risk and Safety Communication
As a drug is used in clinical practice, new information and precautions for safety may be needed. The FDA can require the manufacturer to revise some of the information in the product labeling materials, although it can take close to 2 years to reach agreement with the manufacturer and incorporate labeling changes. Changes may include the addition of a “black box warning”—the strongest warning on a label, highlighting serious adverse reactions or special problems that could lead to injury or death (Wagner et al., 2006). Black box warnings are not easy for consumers to access as they are applied to the label (i.e., package insert and external package); most consumers do not read the insert and do not receive their prescriptions in the manufacturers’ packaging (Szefler et al., 2006). Furthermore, companies tend to resist adding such a warning to a drug’s label (Weatherby et al., 2002; Wagner et al., 2006). Of note, there are virtually no black box warnings on OTC products even though serious errors in administration occur with these products, and such warnings could greatly benefit consumers, particularly parents who must administer OTC medications to infants and children (Presecky, 2006). The FDA also distributes “dear doctor letters” to communicate new risk information directly to providers, yet these communications are relatively ineffective in changing prescribing behavior unless they are widely publicized (Smalley et al., 2000; Weatherby et al., 2001, 2002).
Recently, the FDA began developing and posting on its website supplemental emerging safety information derived from its reporting system (the MedWatch program; see later in the chapter) in an effort to improve the quality of postmarket information about prescription drugs for health care providers and consumers. Also, there is renewed interest in earlier efforts to improve the design and content of consumer drug information distributed through the pharmacy (i.e., pharmacy leaflets) (see Chapter 4).
Review of Product Packaging
Poor labeling on product packaging has contributed to serious medication errors (see Chapter 6) (Cohen, 2000). For example, packaging-related problems can make it easy for busy clinicians to misread poorly presented drug dosing units (e.g., concentration and strength) or to confuse drugs with names that sound similar (e.g., Lamictal, for seizure disorders, and
Lamisil, an antifungal) (Cohen, 2005). For all drugs, inserts and packaging that lack highly visible, easy-to-read instructions, warnings, and contraindications presented in layman’s terms (versus complex medical jargon) can lead to incorrect perceptions and poor retention by prescribers and patients alike.
To address labeling or packaging errors that occur after approval, the FDA sends a request for changes to the manufacturer. If the manufacturer has failed to respond to requests for labeling changes and patient harm recurs as a result of related errors, the FDA seeks to bring about the required changes through negotiation. Labeling for a generic must be identical to that for the reference drug. And recent requirements for the labeling of OTC drugs have created more consumer-friendly labels. The uniform labeling requirements standardized the presentation of “Drug Facts” on the outside of the OTC package in an easy-to-follow format using simpler language and clear visual markings. The FDA recommends, but does not require, manufacturers to include a phone number if more information is needed or if an adverse reaction occurs.
Monitoring of Marketing Materials
Labeling for marketing and advertising purposes is reviewed by CDER’s Division of Drug Marketing, Advertising and Communications to ensure that product claims are truthful and not misleading. Promotional materials (i.e., advertisements) are submitted for review at the time of their initial use, but the FDA does not evaluate these materials before they are used by companies in the marketplace. (See section on marketing practices later in this chapter.) (See Box 2-2 for a summary of key problems with the regulatory review process.)
|
BOX 2-2 Summary of Key Problems with the Regulatory Review Process Affecting Safety and Quality in the Medication-Use System
|
Manufacture, Distribution, and Marketing
Plans for the manufacture, distribution, and marketing of drugs are developed by manufacturers and evaluated by the FDA. Although by this time products have passed the regulatory approval process, including validation of the data from clinical trials, issues affecting the medication-use system can arise during these processes as well. For example, drug shortages or discontinuations in certain dosages force patients to switch their prescription to another drug that may not be as appropriate for them or to resort to potentially unsafe practices, such as manipulating doses manually (e.g., tablet cutting) or purchasing from unknown Internet vendors. Restrictive formularies or lack of drug coverage for prescribed medications can lead to prescription sharing among family and friends. Marketing practices and campaigns that overemphasize the benefits of a drug to providers and consumers without appropriate disclosure of its risks can lead to inappropriate prescribing and adverse drug effects.
Manufacturing Controls
During the last stages of regulatory review for new drugs and generics, the FDA evaluates the adequacy of the sponsor’s plans/controls for manufacturing to ensure the product’s identity, strength, quality, and purity. The agency may even inspect a sample of clinical trial locations to verify the accuracy of the data in the NDA, as well as to inspect manufacturing and repackaging facilities to confirm compliance with international standards known as Current Good Manufacturing Practices (CGMP) (FDA, 2003). Inspections are a significant step in the review process, aimed at minimizing consumers’ exposure to adulterated drug products. The inspections demonstrate a company’s ability to manufacture a drug within tight parameters from batch to batch, day to day, year to year, and to prove that the same controls that received regulatory approval are being applied in the actual manufacture of the product (FDA, 1999).
Distribution to Pharmacies and Consumers
Once products have been produced to standards, they are ready to enter the distribution system that transfers drug products from manufacturers to pharmacies or retail outlets. Traditionally, wholesalers have functioned as the key intermediaries, providing services for storage and delivery to pharmacies. However, the rising cost of health care and prescription drugs, as well as other factors, has prompted the use of other methods to bring drug products to pharmacies, consumers, and patients. Some pharmacies, both provider- and community-based, now receive drug supplies directly from the manufacturer, delivered through the company’s own ser-
vices. A growing method of bringing drugs to consumers is through mail order pharmacies (such as those established by pharmacy benefits managers [PBMs]), Internet pharmacies, and pharmacies of general (usually large) retail outlets (e.g., Walmart, Target).
Mail Order and Internet Pharmacies
As the demand for and cost of prescription medications have increased, so, too, has the demand for more cost-efficient models for distributing drugs to consumers through mail order systems. Such systems include both the businesses of PBMs and the Internet. PBMs are third-party entities that evolved from claims administration and mail order pharmacies into organizations that also provide a range of drug benefit and clinical-based services (HPA, 2003).
Use of PBMs has grown considerably over the past decade with the expansion of their services to utilization management, disease management, and, more recently, medication safety for individuals with chronic diseases and associated polypharmacy-related issues. PBMs generally make pharmacists available to assist consumers with questions about their medications. Nonetheless, a substantial portion of consumers continue to prefer the convenience of their local pharmacy and personal contact with the community pharmacist.
The Internet has emerged as a growing marketplace for the purchase of drugs (GAO, 2004). It offers consumers the benefit of being able to shop from home at any time, and the ability to compare prices of multiple vendors and purchase from a wide range of drug categories (GAO, 2004). Although the Internet pharmacy market is subject to the same laws that govern traditional pharmacies, it is global and difficult to regulate. A recent report by the Government Accountability Office (GAO, 2004) notes that many Internet pharmacies do not comply with state pharmacy laws; for example, they sell drugs that are improperly packaged, counterfeit, or unapproved. Most important from a consumer safety standpoint, in some instances, prescription drugs can be purchased without a prescription.
Marketing to Consumers, Providers, and Payers
Most stakeholders in the drug system are introduced to drug products for the first time through marketing and advertising campaigns. The FDA’s Division of Drug Marketing, Advertising and Communication estimates that industry spends $25 billion annually to promote drug products in the marketplace (Abrams, 2005). Marketing can take the form of visits by company representatives to physicians’ offices to discuss new drugs in person and provide sample packs and gifts7; rebates to health plans and PBMs
for preferential formulary placement; industry-sponsored continuing medical education (CME) programs focused on new drugs; funding of disease management programs; direct payment of travel expenses to attend medical association conferences; and direct-to-consumer advertising that promotes new drugs to the public at large in print, broadcast, and electronic media (Chung et al., 2003; Blumenthal, 2004).
A body of evidence confirms that these strategies have an influence on physicians’ objectivity and behaviors, especially prescribing practices, formulary choices, and assessment of medical information (Levy, 1994; Wilkes et al., 2000; Carney et al., 2001; Goodman, 2001; NIHCMREF, 2002; Blumenthal, 2004; Chimonas and Rothman, 2005). Wazana’s (2000) extensive literature review on physician–pharmaceutical industry interactions revealed that some positive outcomes were identified (for example, an improved ability to identify the treatment for complicated illnesses), but most studies found negative outcomes, although no study evaluated the impact on patient outcomes. The impact of physician–pharmaceutical industry interactions is particularly concerning since these strategies are employed even for new drugs that may have little or no discernable advantage over existing drugs or other treatment options (Avorn, 2004), and for which there may be only limited data from short-term clinical trials that may not have uncovered serious adverse effects (Califf and DeMets 2002a,b). In some cases, drugs attain preferential placement in formularies because of company financial incentives (e.g., discounts, rebates) rather than quality and evidence-based decision making (Chung et al., 2003). Thus many groups within the medical community are calling for changes in the way the industry interacts with the medical community (Katz et al., 2003; Blumenthal, 2004; Studdert et al., 2004; Brennan et al., 2006).
Distribution of Free Samples
The primary promotional tool for new drugs is the distribution of free samples to providers. In 2003, companies distributed about $16 billion worth of free samples (although this figure represents retail value, only 20– 30 percent of which is the actual value) (IMS Health, 2004). While making samples broadly available to patients, particularly those with lower incomes, may be well intentioned, there is growing evidence that the provision of free samples directly affects physician’s patterns in selecting and prescribing medications and in addressing issues of medication safety (Chew et al., 2000; Maguire, 2001; Petersen, 2000). Free samples are frequently taken by patients without a prescription and without documentation in
|
BOX 2-3 Summary of Key Problems with the Manufacturing, Distribution, and Marketing Processes Affecting Safety and Quality in the Medication-Use System
|
health records, thus bypassing the safety check on drug–drug interactions that may otherwise flag a potential error (Chew et al., 2000; Groves et al., 2003; Taira et al., 2003). Furthermore, free samples are most often the newest, least well tested drugs, and patients are thus being encouraged to take these drugs when others might, in fact, be safer for them (Avorn, 2004). (See the discussion in Chapter 6.)
Formularies
Companies also interact with insurance payers and PBMs to secure listing and reimbursement pricing in drug formularies. A formulary is a payer’s list of covered drugs, designed to restrict the listing of drugs and/or the level of coverage in each therapeutic class for cost-saving purposes (Husakamp et al., 2003). Unlike other nations that use formularies to determine access, payers in the United States maintain an open system to accommodate the broadest population and its potential medication needs; formularies for prescription drugs are used solely to determine tiered copayment and reimbursement structures, not access. For example, the Veteran Health Administration (VHA), private-sector health maintenance organizations (HMOs), private-sector payers, and now the Centers for Medicare and Medicaid (CMS) with the new prescription drug benefit use open formularies, although coverage is tiered (also called incentive based) (Thomas, 2003; Landon et al., 2004; Shrank et al., 2005). HMOs tend to have more restrictive formularies (prohibiting payment for certain drugs), but many have also moved to tiered structures (Shrank et al., 2005). (See Box 2-3 for a summary of key problems with the manufacturing, distribution, and marketing process.)
STRUCTURE AND FUNCTION OF THE MEDICATION-USE SYSTEM
The steps described above provide the basic foundation for safety in producing and distributing medications that meet consumers’ medical and
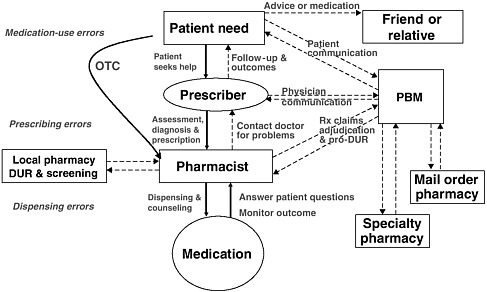
health needs. The medication-use system that is built on that foundation encompasses the continuum of (1) prescribing by the clinician (or self-prescribing), followed by transcribing; (2) preparing and dispensing by the pharmacist; (3) administering by the provider or consumer (self-care); and (4) monitoring for therapeutic and adverse effects (by nurse, surrogate, or self). Each of these steps includes critical control points at which decisions and actions can contribute to safety or errors. Figures 2-2, 2-3, and 2-4 outline these critical control points for the different health care settings.
The primary stakeholders involved in the medication-use system are patients/consumers and their families, providers, payers, regulators, employers, manufacturers, distributors, and policy makers. Secondary stakeholders include accrediting, patient safety, and quality improvement organizations; medical journal editors; and the general media. The dynamics of the system for medication delivery are shown, along with relevant stakeholders, in Figure 2-5.
Achieving safe and effective use of medications requires coordinated efforts by all stakeholders, with mutual recognition that each has unique perspectives on what constitutes appropriate or rational medication use (Knowlton and Penna, 2003). Patients/consumers and their families have an interest in maintaining their personal health and safety at a reasonable cost, as do their employers. Health care providers (physicians, nurse practitioners, physician assistants, nurses, pharmacists) have an interest in addressing patient problems effectively and achieving therapeutic objectives. Regulators have an interest in ensuring the safety of the general public and taking disciplinary action when necessary. Pharmaceutical manufacturers have an interest in developing and marketing new drugs in the service of society and their stockholders. Payers have an interest in providing their enrollees with insurance coverage at a reasonable cost (Knowlton and Penna, 2003). Community pharmacies and PBMs have an interest in providing patients and consumers with useful information about their medications and averting potential errors. Accrediting organizations have an interest in assessing health care providers’ compliance with medical safety standards and best practices. Patient safety reporting organizations have an interest in collecting data on events and developing protocols to improve safety. Medical journal editors have an interest in publishing comprehensive and accurate information about medications and their use. And the general media have an interest in writing newsworthy stories about health care and exposing any problems.
Unfortunately, the complex and diverse interests of the primary stakeholders have resulted in a medication-use system that is disjointed and inefficient in terms of manpower and resource consumption. Errors in medication delivery are the largest single category of medical errors in health care (IOM, 2000). Errors occur with all types of medications (e.g., pre-
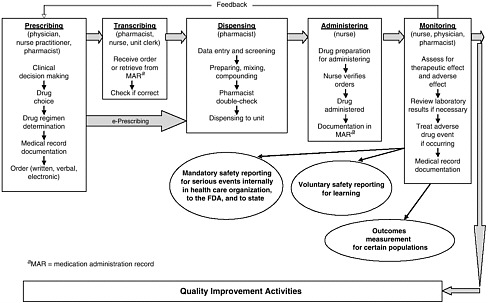
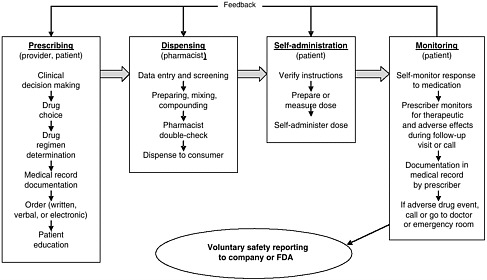
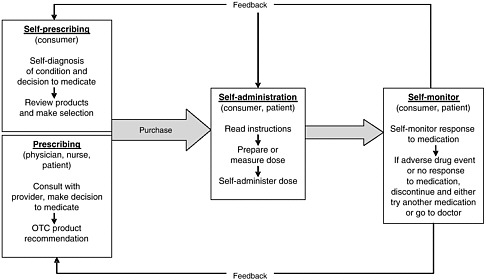
scription, generic, OTC) and in all health care settings (e.g., hospital care, ambulatory care, nursing home care, and home self-care). Errors can be those of commission (e.g., prescribing the wrong dosage) or omission (e.g., failing to prescribe a medication that would likely benefit a patient). Errors can occur at any point along the medication-use continuum as a result of multiple factors in the health system, including those associated with the patient, the provider (e.g., experience, expertise, and overall human factors such as fatigue and stress), the care team (e.g., lack of communication between clinicians, shifts, and settings), the work environment (e.g., lack of clinical decision support, product labeling), and the department/institution (e.g., absence of a culture of safety) (Vincent, 2001). This section provides an overview of the points along the continuum of the medication-use system in community and inpatient care settings at which medication errors can occur: prescribing and ordering, self-prescribing, transcribing, preparing and dispensing, administering and consuming, self-administering, monitoring for effects, and self-monitoring.
Prescribing and Ordering
The prescribing domain in community, hospital, and long-term care settings involves clinical decision making, selection of a drug and drug regimen, medical record documentation, and ordering. The clinician has the responsibility to engage the patient in discussion about the appropriateness of a prescription drug as part of the treatment plan and about how to design the regimen to meet the patient’s needs. Although the patient should participate in the decision making on whether to use medication therapy, the clinician retains responsibility for ensuring medically appropriate prescribing and accuracy in medical record documentation and prescription ordering.
Quality and safety in the medication-use system require good clinical decision making about patient care and therapeutic options. As stated in the Quality Chasm report (IOM, 2001), the best care results from the conscious, explicit, and judicious use of current best evidence and knowledge of patient values by well-trained, experienced clinicians. Thus, effective clinicians rely on best practices as appropriate for a given patient, yet they maintain the freedom to make choices that science cannot guide, such as those based on relationships and observation. These clinicians understand and respect the patient’s special circumstances, preferences, and values, knowing they are vital to patient-centered care. They also are attuned to the patient’s economic circumstances (e.g., uninsured, underinsured) and formulary restrictions as they may affect drug selection, especially if the patient must pay for the medications out of pocket.
Good decision making need not always be based on the results of randomized controlled trials for two reasons: (1) such results are not always available, and (2) other forms of knowledge may be definitive. For example, few drug products used for neonatal and pediatric patients have been tested in randomized controlled trials in these populations. When a drug exists that has been tested in the adult population, pediatricians must use their medical expertise and overall knowledge of therapeutics to make decisions regarding the “off-label” use of the drug to meet the needs of their patients (see Box 2-4).
Moreover, all illnesses do not require drug therapy. The need for a drug should be evaluated and weighed against alternative treatments to avoid overuse or inappropriate uses of medications (IOM, 2001). For instance, antibiotics are contraindicated for treating the common cold or a viral infection but are often requested and prescribed nonetheless, thus contributing to problems of antibiotic resistance. As another example, certain drugs, particularly antidepressants, analgesics, and muscle relaxants, are commonly and inappropriately prescribed for elderly patients, contributing to adverse drug events that necessitate health care services, physician contact, hospitalization, and emergency department visits (Golden et al., 1999; Hanlon et al., 2000a,b; Fick et al., 2004; Fialova et al., 2005).
When the decision is made to select a medication, care is necessary to screen the drug regimen for potential drug–drug and drug–food interactions; age- or gender-related metabolic or pharmacologic considerations; incidence and severity of side effects; tolerance effects over time; relationship to placebo effects; and comparability to other, nonmedication-related treatments (Nies, 2001). Poor decision making can result in prescribing that fails to help the patient or causes harm. Even if the correct decisions are made in determining the medication regimen, poor communication of prescription orders in any format (written, oral, electronic) can lead to serious adverse drug events (Cohen, 2000; USP, 2004).
A number of studies have cited prescribing as a principal source of overall medication errors, estimating incidence rates of 18.9 to 58.4 percent (Bates et al., 1995; Lesar et al., 1997; Gurwitz et al., 2000; USP, 2004). The numerous types of prescribing errors identified in the literature include the following (Lesar et al., 1990):
-
Failure to alter drug therapy in patients with impaired renal or hepatic function.
-
Failure to notice a patient’s history of allergy to the prescribed drug class or missing critical information about a patient’s known drug allergies.
-
Use of the wrong drug name (e.g., sound-alike or look-alike names), wrong dosage form (e.g., intramuscular versus intravenous injection), or
|
BOX 2-4 Off-Label Use of Medications Unlicensed use of medications is common when a health care need for a patient or patient population is not met by currently available therapies. A licensed medication (i.e., one that has received regulatory approval) is used off-label when prescribed by clinicians in a manner that they deem medically appropriate, but that is outside the agreed-upon statement of the medication’s efficacy (Dick et al., 2003). Examples include administration by a different route, use outside a defined age range, use of a higher or more frequent dose, and prescription for a different indication. Unlicensed use includes modifications to a licensed medication, such as dispensing in a different formulation (e.g., crushing tablets to prepare a suspension); new medications available under a special manufacturing license; use of chemicals as medications (e.g., chemotherapy agents); medications used before a license is granted (e.g., those under an IND); and imported unlicensed medications (Dick et al., 2003). Although the FDA does not regulate off-label use, it can regulate the promotion of such uses (Loder and Biondi, 2004). Federal law and state insurance commissioners have attempted to define acceptable off-label use to prevent insurers from refusing to pay for such treatment. In 1990, the Omnibus Budget Reconciliation Act (OBRA) confirmed that “medically accepted indication” includes off-label use and should be supported by one of the following:
Estimates of off-label medication use range from 20 to 60 percent, depending on the drug and patient population (Dick et al., 2003; NCI, 2004; Murphy, 2005). Off-label prescribing occurs most frequently with pediatric, oncology, obstetric, and |
-
wrong abbreviation (e.g., “qd” [every day] instead of “qid” [4 times per day]).
-
Incorrect dosage calculations, including wrongly placed decimal point and wrong rate, frequency, unit of measure, or route of administration.
-
Wrong patient (i.e., faulty patient identification checking).
-
Failure to prescribe when there is an indication (e.g., omission of beta-blockers post–acute myocardial infarction) or prescribing without an indication (e.g., use of antibiotics to treat the common cold).
-
Other factors, such as failure to assess drug–drug or drug–food interactions or duplicative therapies (Lesar et al., 1997; Dean et al., 2002; Bobb et al., 2004; USP, 2004).
Prescribing errors are attributed chiefly to the provider’s insufficient knowledge about the medication and its correct use or about the patient
|
AIDS patients, although it is by no means limited to these populations. The highest rate of such uses is with pediatric patients (Murphy, 2005). Up to 90 percent of these patients (especially neonates) are prescribed at least one drug off-label (Jong et al., 2001; Lifshitz et al., 2001) based on the modification of adult formulations, dosage strengths, and dosage levels (Jong et al., 2001). Published information from pediatric trials of other drugs and the clinical experiences of other physicians also are relied upon for decision making. While necessary, such methods can underscore the important differences between adults and children in development and the metabolism and excretion of a particular drug, increasing the risk of an adverse drug event (Christensen et al., 1999; Jong et al., 2001). Some drugs are now tested in pediatric populations, but significant ethical concerns about such testing mean that off-label use of drugs will continue to be necessary to meet the needs of these patients. The second-largest rate of off-label use occurs with oncology patients (Poole and Dooley, 2004; Kos, 2005). A survey of oncologists found that 60 percent of these clinicians prescribe off-label for cancer patients who may require drugs approved for a different type of cancer or a different disease, or at different dosages, frequencies, or duration from those approved (NCI, 2004). One study found that about one-third of oncology prescriptions were off-label, and more than 50 percent of oncology patients received at least one drug off-label (NCI, 2004). Off-label use is common in oncology because cancer drugs rarely receive generalized approval, but are approved for a specific biological target or a particular type of tumor. Once the drug is on the market, however, further research and off-label use may demonstrate its action on different targets present in other types of cancer (NCI, 2004). Conversely, the side effects of cancer drugs can vary depending on the type of cancer being treated, such that the risk of an adverse event or reaction can increase when the side effects of an off-label use are unknown. Because medical needs of certain patient populations cannot be met with approved uses of many medications, off-label prescribing will continue to be an important part of clinical practice. |
(e.g., incomplete medical history), miscommunication among providers (e.g., illegible handwriting on written orders, misunderstanding of verbal orders, mistakes using electronic ordering), lapses in provider performance (e.g., nonadherence to policies and procedures, slips or memory lapses), and lapses in documentation (e.g., incomplete charting) (Cohen, 2000; IOM, 2000, 2004c; Phillips et al., 2001; USP, 2004). More specifically:
-
Errors related to medication knowledge may be the result of gaps in timely access to drug information at the point of care, in understanding of the complexities of the use of specific drugs, and in access to comprehensive knowledge bases needed to build expertise in drug therapy (Lesar et al., 1997). Also, in some cases information that would be useful in preventing errors (e.g., the correct dose of aspirin to prevent coronary heart disease) does not exist.
-
Incomplete medical histories contribute to prescribing errors. Many patients fail to list all the medications they are taking (e.g., OTCs, dietary supplements), or the provider may forget to ask about known drug allergies or to review laboratory data that would further improve prescribing for the patient (Lesar et al., 1997; Smith et al., 2005).
-
Poorly handwritten prescription orders are the chief culprit in miscommunications among prescribing clinicians, nurses, and pharmacists, and have often resulted in serious injury or death due to incorrect understanding of the drug or its dosage, route, or frequency (Cohen, 2000).
-
Although widespread use of computerized provider order entry or electronic prescribing systems has the potential to reduce errors associated with poorly handwritten prescriptions, errors can still occur in the interaction between the clinician and the technology as a result of issues in such areas as usability, readability, training, and suboptimal system safeguards (Horsky et al., 2005). Indeed, one study found that computerized provider order entry systems facilitated 22 different types of errors8 (Koppel et al., 2005).
-
Oral orders (e.g., those given over the phone to a pharmacy or between clinicians involved in emergency care) can result in an error if, for example, product names sound alike; dosages are unclear (e.g., “two 50 milligrams,” which can be interpreted as 250 milligrams instead of 100 milligrams); or concentrations are not specified (Cohen, 2000).
-
Performance lapses, or slips, can occur when a provider sets out to do one thing and actually does something else. Such lapses can be due to a lack of focus on the task at hand, distracting conversations (e.g., talking and listening to others talk about things unrelated to the task), interruptions, a poor working environment (e.g., high noise levels, low lighting), poor workflow (e.g., workflow that is not logical), and uneven workload (e.g., too little or too much) (Davis, 1996).
Self-Prescribing
During self-care with OTC products and dietary supplements, the consumer (or a family member) is responsible for prescribing based on his or her own (or the family member’s) assessment and diagnosis of the condition. Determining which medication or supplement to take can be based on a review of labels and comparison of products deemed appropriate; the
suggestion or experience of a family member, friend, community pharmacist, or community provider; and/or advertisements. Primary sources of information for selecting OTCs and dietary supplements vary according to the population group. A 2001 survey by the Consumers Healthcare Products Association, the industry trade group, found that consumers seek advice in treating minor ailments from family and friends first (27 percent), followed by physicians (20 percent), medical reference books (10 percent), pharmacists (7 percent), and the Internet (7 percent) (RSW, 2001). Generally, older Americans are much more likely than their younger counterparts to rely on their health care provider, regardless of the seriousness of their health condition (RSW, 2001). Individuals with lower incomes are more likely to rely on providers for recommendations, while those with higher incomes are more likely to turn to the Internet. Likewise, individuals who use dietary supplements (and alternative medicines) for minor ailments are significantly more likely than those who do not use dietary supplements to seek information from family and friends (25 percent), medical reference books (13 percent), and newspapers/magazines (8 percent) (RSW, 2001). Although pharmacists did not rank as the first choice for health-related information, an overwhelming majority of Americans (84 percent) agree that they are a good source of information for treatment of minor ailments.
A debatable issue concerns the ability of consumers to understand and use product labels when deciding to take an OTC or dietary supplement. The pharmaceutical industry asserts that an overwhelming majority of consumers take the necessary precautions, such as reading directions before using a product for first time use (95 percent), examining labels to help choose medications (89 percent), and reviewing possible side effects and interactions (91 percent) (RSW, 2001). This is an improvement over figures cited in an earlier poll by Harris Interactive (NPSF, 1997) that found only 20 percent of consumers read the label for side effects before making an OTC purchase, and 77 percent do not read the dosage instructions at all (Cropper, 2005). Language and literacy barriers exacerbate problems with consumers’ understanding and use of drug labels (IOM, 2004a).
Transcribing
Once the drug regimen has been determined, orders from the prescribing clinician are sent to the pharmacy and, in the hospital setting, the nurses’ station for processing. Transcription is the official term used to describe the complex set of tasks involved in interpreting and processing orders. Many medication errors are associated with the transcription process, particularly if a drug name looks or sounds like that of another drug or is illegible. In the order, the clinician must provide complete details on the drug regimen (patient name, drug name, dosage, formulation, route,
frequency, units, flow rates, duration, reconstitution information) (Manasse and Thompson, 2005). Prior to processing, both the pharmacist and assigned nurse must communicate directly with the ordering clinician if there is even the slightest question concerning any aspect of the drug regimen or its clinical appropriateness (Cohen, 2000).
Because health care institutions and pharmacies vary widely in the extent to which they have implemented information technology, varying different methods are used to send medication orders to the pharmacy in the inpatient setting. Some pharmacies receive orders in written or typed form via fax, scanned image, a vacuum tube system, or carbon copies of the original; others may receive orders through a state-of-the-art computerized provider order entry system (Manasse and Thompson, 2005). Community pharmacies generally receive orders via fax or handwritten prescription or orally over the phone. Oral orders warrant greater caution given the ease with which miscommunication can occur, and should be read back, with spelling of the drug name and dosage, to the clinician (Cohen, 2000; Allinson et al., 2005). All health care organizations and pharmacies should have guidelines (e.g., readback of all verbal orders) in place to reduce the possibility of errors occurring in the transcription process (Cohen, 2000). While computerized provider order entry systems have been promoted as the primary method for reducing errors in transcription because they eliminate handwritten prescriptions, other factors, such as improved processes for drug naming to minimize look-alike, sound-alike names, also could improve the transcription process (see Chapter 6).
Preparing and Dispensing
Following transcription, the pharmacist begins the preparation and dispensing process. Entry of orders into the pharmacy database system allows for automated screening of orders for therapeutic duplications, drug interactions, allergies, or doses that are not within an acceptable range; if therapeutically appropriate, screening the order against the patient’s laboratory test results can avert potential adverse events (Manasse and Thompson, 2005). About 91 percent of hospital or health system pharmacies have a computerized database system, and 87 percent have access to patient admission, discharge, and transfer data through links in the database (Ringold et al., 1999). If changes need to be made for therapeutic reasons or in response to a supply shortage, the pharmacist may do so only with the approval of the prescriber, and all of the initial steps in processing must be repeated. If pharmacists do not know all the drug-related products (i.e., prescription, OTC, dietary supplements) a consumer is taking, however, their ability to perform drug interaction checking is inhibited in both the inpatient and community setting. Interaction check-
ing can be particularly difficult when consumers use multiple pharmacies to fill their prescriptions and fail to communicate this to each pharmacy.
Depending on the specific order and patient, preparation of the medication may require counting, measuring, or compounding (mixing of ingredients); repackaging (e.g., unit doses); and labeling. Activities associated with preparation present the greatest opportunity for error within the pharmacy (Manasse and Thompson, 2005). Most inpatient errors involve selection/ dispensing of an incorrect drug (e.g., because of sound-alike, look-alike names or packaging), dosage strength (e.g., incorrect dilution), formulation (e.g., tablet versus intravenous), or dosage calculation (e.g., incorrect calculation of flow rate for intravenous medication) (Cohen, 2000; Phillips et al., 2001). Outpatient errors tend to center on incorrect drug labeling information (e.g., use or administration of the drug) (Buchanan et al., 1991; Flynn et al., 2003).
During preparation, a large percentage of oral and injectable medications used in the inpatient setting require further manipulation (compounding and/or repackaging) prior to administration, increasing the risk of error. Most drugs are licensed for adult use; reformulation and compounding are most often necessary to treat neonates or pediatric patients who cannot swallow tablets or capsules and require dosage concentrations and formulations tailored to their age, body weight, and body surface area (Nunn, 2003). Adult patients with a rare condition for which an orphan drug9 is no longer manufactured may require the pharmacist’s expertise to compound and formulate a medication from chemicals and ingredients available only in bulk (Kastango, 2003). Also, repackaging is common for inpatient facilities so as to provide medications in unit doses and thereby minimize dose manipulation and errors at the bedside. Currently, 79 to 99 percent of hospital pharmacists repackage oral medications, and 29 percent repackage injectables (Cohen, 2000; Pedersen et al., 2003). However, new federal regulations to go into effect in 2007 require manufacturers (or third-party repackagers) to provide all products to hospitals in unit dose form with bar codes (FR, 2004b). In a further effort to decrease errors, some hospitals also use decentralized automated dispensing systems (e.g., ward-based cabinets) for storing certain medications that are in unit dose form (e.g., narcotics, as-needed drugs, limited floor stock) (Cohen, 2000). These systems can be accessed by nurses with “swipe cards” or personal identification numbers. Finally, a growing number of community-based and most mail order pharmacies are using automated dispensing systems, including centralized systems that can produce unit doses (e.g., via strip or envelope packag-
ing),10 or that use bottle-filling machines; others are experimenting with decentralized systems that rely on prefilled bottles and manufacturer-packaged items (Cohen, 2000).
Patient counseling in a pharmacy provides an opportunity for the pharmacist to inform the patient about his/her medications, encourage medication adherence, and answer any questions the patient may have. In the OBRA of 1990 (P.L. 101-508), Congress required that pharmacists counsel Medicaid patients. Since then, boards of pharmacy in most states have come to require some type of counseling for all patients (NABP, 2004). For many reasons (e.g., low patient demand, lack of cost-effectiveness data, time constraints, lack of reimbursement), however, pharmacies often offer counseling only as requested by a consumer. A study of 100 prescription orders dispensed in 1994 in community pharmacies in New Jersey, New York, and Florida found that oral counseling had been provided to 64 patients, covering on average 3 of the 14 categories11 (i.e., dosage, frequency of administration, drug or food interactions) of drug information required by OBRA 1990 (Allan et al., 1995). Similar results were observed in a more recent, larger eight-state study. In this study, about two-thirds of consumers had been given oral information—on average 2.3 items from a 5-item list (Svarstad et al., 2004). The study also found that higher levels of pharmacist counseling were associated with younger pharmacists, less busy pharmacies, and more demanding state regulations. In terms of mail order pharmacies, counseling is generally available as requested by telephone.
Dispensing errors account for an estimated 6–12 percent of all medication errors (Buchanan et al., 1991; Allan et al., 1995; Flynn et al., 2003). Research suggests that the main causes of such errors are issues concerning workload and staffing, distractions during processing, suboptimal packaging and labeling, poorly designed work areas, and outdated or incorrect drug reference information (Cohen, 2000; Phillips et al., 2001). A review of the literature reveals that:
-
Failure to double-check orders, medication, and labels is a common cause of dispensing errors.
-
High workload/low staffing is the primary contributing factor to medication errors associated with preparing and dispensing medications in both community and institutional settings (Davis and Cohen, 1994; Roberts et al., 2002).
-
Interruptions (temporary cessation of prescription filling) and distractions (external stimulus without cessation of prescription filling) are highly correlated with dispensing errors (Flynn et al., 1999). Error rates per half hour of 6.65 percent for interruptions and 6.55 percent for distractions were found, with incorrect instructions to the patient being the most common error. About 26 percent of pharmacists’ time is spent dealing with issues (interruptions) related to third parties and miscellaneous administrative tasks (NACDS, 1999).
-
Product labels are often read under less-than-ideal conditions, and the way a medication is packaged and labeled can have a significant impact on error rates. Problematic aspects of packaging and labeling include look-alike packaging, obscure placement of critical safety information, and print that is too small and lacks sufficient distinctions in contrast or boldness (Cohen, 2000; Phillips et al., 2001; IOM, 2000, 2004c; USP, 2004).
-
Improper lighting, inadequate counter space, poor placement of telephones, and uncomfortable temperature and humidity create a work area that can negatively affect workflow from one task to another and contribute to errors caused by clutter or contamination (Cohen, 2000).
-
Drug reference files, texts, and/or database systems may not be current, resulting in errors associated with outdated and incorrect information (Cohen, 2000). Constant updating of drug information is particularly critical to patient safety given the limited data available when medications enter the market and the amount of new data on medications already in use among the population.
Administering and Consumption
Nurses have primary responsibility for administering medications in acute care hospitals, in long-term care facilities, and during home care. In certain instances, a nursing assitant/technician may be permitted to administer selected medications (Munroe, 2003; Castle and Engberg, 2005). In many of these settings, the environment for nurses is demanding, characterized by long work hours, staffing shortages, high patient and staff turnover, and constant interruptions (O’Shea, 1999; IOM, 2004b; Jenkins and Elliott, 2004; Suzuki et al., 2005). Accurate administration of medications can be challenging in this environment.
Tasks associated with preparing medications for administration can range from simple retrieval of a unit dose from a ward-based automated dispensing system to reconstitution of a powder with a sterile diluent
(Cohen, 2000; Kastango, 2003; IOM, 2004b). Most medications are now administered in unit dose form to minimize the amount of compounding by nurses. Ideally, medications should be kept in the dispensing container and in their individual packages until they reach the bedside so as to decrease the risk of their being confused with another patient’s drug. In addition, it is standard practice for the drug label to be read three times prior to administration—when obtaining the drug from the storage area; when preparing the dosage at the bedside; and after administration, when discarding the package (Cohen, 2000; Manias et al., 2005)—although there is some support for registered nurses’ competence to perform single-checking (Jarman et al., 2002). Averting errors also requires careful attention to dosage and route when preparing medications. For example, pediatric and chemotherapy doses should indicate milligrams per kilogram (mg/kg) or milligrams per meter squared (mg/m2) in order to leave little margin for error (Cohen, 2000). Nurses must also ensure that drug infusion or administration devices are functioning properly and programmed accurately to ensure that the dose and infusion rate are correct (Smetzer, 2001; Fields and Peterman, 2005; Nicholas and Agius, 2005). These nursing activities are indispensable to patient safety.
Perhaps most important, the nurse is often the last professional to evaluate the appropriateness of the medication that has been prescribed. In fact, a study of medication errors found that nurses were responsible for intercepting 86 percent of all errors made by physicians, pharmacists, and others involved in providing medications for patients (Leape et al., 1995). Nurses’ involvement and vigilance during the preparation process is thus central to accurate medication administration.
Medication administration is founded on what are termed the “five rights”—the right drug, in the right dose, by the right route, at the right time, to the right patient (Manias et al., 2005; Nicholas and Agius, 2005; Schull, 2005; Manasse and Thompson, 2005). While achieving the five rights is essential to safe medication administration, more complex factors must also be considered to ensure positive outcomes. First, medications can be administered via a number of different routes and formulations—oral tablet, capsule, or liquid; intravenous solution; intramuscular injection; inhalant; eye/ear drops; topical cream or solution; transdermal patch; or other means—depending on the patient, drug, and condition. Without attention to this issue, for instance, a liquid intended for oral dosing might be administered intravenously. Excessive variations in dosing regimens (e.g., multiple sliding scales for insulin dosing, as needed), use of high-risk drugs (e.g., anticoagulants, narcotics), and the proliferation of new drugs and devices add significantly to the intricacies of the administration process (Greengold et al., 2003; USP, 2003). In addition, the severity of a patient’s medical condition and the presence of comorbidities further increase the
challenges to evaluating the safety and appropriateness of a medication that has been ordered (IOM, 2001). Relative to the other points along the medication-use continuum, the administration process has the fewest safeguards and fewest support mechanisms, and it often relies on a single health care professional for perfection (Cohen, 2000; IOM, 2003, 2004b).
Several inpatient facilities are beginning to implement bar code medication administration systems to increase assurance that the five rights are being achieved (IOM, 2002; Patterson, 2003; FR, 2004b; Burke et al., 2005). The bar codes placed on unit doses of medications are encoded with the patient’s name, drug, dose, route, and time of administration. Bar code scanners (placed in each patient’s room) are linked to computerized databases containing the patient’s drug regimen. The database may be crosslinked to other health information systems, such as a patient identification master file, an order entry system, and/or the pharmacy database (FR, 2004b; Nicholas and Agius, 2005). The nurse scans the bar code on the medication package and the patient’s identification wristband, allowing the system to determine whether there is a match. Following a confirmation signal, the nurse administers the medication. If there is an alert, the nurse stops the process from going forward, preventing a potential medication error. Because medication administration is a high-volume activity, bar code medication administration systems can provide needed support to nurses during clinical care. They also generate data for the medication administration record (MAR).
Maintenance of an accurate MAR is essential to safety and quality of care (IOM, 2004b). This record serves as a log of all medication-related activities for each patient. Entries are made immediately after a dose has been administered to minimize errors of omission (Cohen, 2000). MARs also document that medications were given in a timely manner for the correct indications. Expanded records are usually reserved for high-risk drugs (e.g., anticoagulants, cardiac drugs, insulin) so as to record important variables affecting administration (e.g., international normalized ratio [INR], used to measure prothrombin time). All medications are typically documented consistently in one place for ease of reference by the team of health care providers that may be caring for a patient. The MAR also serves as a reference in the event of a medication error (Gladstone, 1995; Wakefield et al., 1999). In some cases, third-party payers have reviewed the MAR to look for inconsistencies and gaps in treatment and to find evidence for denying payment.
The types of errors associated with administration-related mortality include (1) dosing errors (40.9 percent, 36.4 percent of which were overdoses); (2) incorrect drug (16 percent); and (3) incorrect route (9.5 percent) (Phillips et al., 2001). Causes of administration errors include miscommunications, miscalculations, workload/staffing problems, interruptions, rapid
increases in knowledge and technology demands, and incomplete documentation (IOM, 2004b).
-
Miscommunications during medication administration generally result from errors in transcribing oral or written orders (e.g., prescriber fails to insert a zero before a decimal point), reading product names (e.g., look-alike, sound-alike), or labeling (e.g., similar or misleading container labels) (Donohue and Needleman, 1998; Phillips et al., 2001). Commonly used abbreviations for drug names, dosage units, and references to timing of administration cause many medication errors (e.g., the abbreviation “U” for units of insulin can be read as a zero, leading to an overdose) (Cohen, 2000). Also, only the metric system should be used in the MAR, and apothecary symbols and terms that can easily be misinterpreted should be avoided.
-
Miscalculations of medication dosages are often due to the complexity of drug protocols (e.g., for cancer chemotherapy), the need for speedy action in emergency situations, marketing of multiple concentrations of drug products, and the availability of highly concentrated drug products on nursing units (e.g., those that are intended only for compounding infusions but that might be given undiluted) (Phillips et al., 2001; Fields and Peterman, 2005).
-
As noted above, the work environment for nurses can contribute to medication errors (O’Shea, 1999; IOM, 2004b; Jenkins and Elliott, 2004). As the numbers of available hospital beds and lengths of stay have decreased, patient turnover rates have risen (some by 40–50 percent in an 8-to 10-hour period), increasing the workload of hospital nurses even as funding reductions and resulting work environment dissatisfaction have led to inadequate staffing (Norrish and Rundall, 2001). High rates of nursing staff turnover (21.3 percent per year for hospitals and 56 percent for long-term care facilities) have adverse consequences for staffing levels, quality of care, and patient safety (AHCA, 2002; The HSM Group, 2002). Although most nursing shifts are 8–12 hours, mandatory overtime and double shifts contribute to nursing-related medication administration errors (IOM, 2004b).
-
Distractions and interruptions as nurses carry out their primary patient care responsibilities increase the potential for adverse events, such as errors in patient identification as a nurse prepares doses for more than one patient. Many distractions and interruptions are associated with added tasks that nurses undertake during staffing shortages, such as delivering and receiving food trays, performing housekeeping tasks, transporting patients, and performing ancillary services (e.g., delivery of medical supplies, blood products) (IOM, 2004b). Distractions also result from the fact that patients hospitalized today have less stable health conditions than they did,
-
on average, when longer hospital stays were the norm. Thus, nurses often must respond to the health crises of some patients, which distract them from timely and thoughtful medication administration to others.
-
The growth of a rapidly expanding knowledge base in clinical care, drugs, devices, and health information technology is forcing changes in the work nurses are asked to perform (IOM, 2004b; Nicholas and Agius, 2005). Appropriate levels of training, continuing education, reconditioning of workflows, and support mechanisms are necessary to minimize medication-related errors (Gladstone, 1995). This includes improved familiarity with less common medications, attention to commonly used medications to which many patients are allergic (e.g., antibiotics, nonsteroidal anti-inflammatory drugs), and more vigilant follow-through on medications that require monitoring to ensure proper dosing (e.g., warfarin, lithium, digoxin) (Woods and Johnson, 2002). Technologies that provide ready access to this information are essential.
Self-Administration
From the perspective of consumers, the most common types of medication errors are associated with administration of wrong dosages; unnecessary medicating; adverse drug reactions, including drug–drug interactions; and nonadherence. Errors occur from overdosing or underdosing as a result of inadequate instructions and use of inconsistent or improper measuring devices. For example, the household teaspoon is the device used most frequently for measuring liquid medication for home administration, instead of a dosing syringe. Common errors also include misinterpreting instructions, confusing teaspoons with tablespoons on a medicine cup, and misreading a dosage chart when the weight is not typical for a particular age group (Madlon-Kay and Mosch, 2000). One study found that acetaminophen (Tylenol) dosing by parents was inaccurate 73 percent of the time, resulting in ineffective fever control and increased emergency room visits in two-thirds of cases (Gribetz and Crunley, 1987). A recent article reported that two infants died from suspected overdoses of an OTC cold medicine (Presecky, 2006). The cold medicine had been administered with a 1 mg eyedropper provided in the product package. The dosage for the infants was 0.2 mg (two-tenths of one dropper) but was misunderstood to mean 2 droppers full of medicine. The probability of medication dosing errors is greatly increased with high-risk medications that have complex dosing regimens, such as oral chemotherapy agents, oral anticoagulants, opioids, and insulin (Watzke et al., 2000; Grissinger et al., 2003; Hartigan, 2003). These drugs have narrower therapeutic indices, meaning there is less margin for error, and the consequences of error may be more devastating (Cohen, 2000). Many dosing errors could be avoided with the use of more accurate
devices, such as oral dosing syringes; color coding of age–weight dosing zones, particularly for liquid medications administered to children; and better presentation of use information and safety warnings (Frush et al., 2004).
Other types of dosing errors are associated with the frequency or duration of treatment. One study found that only 38 percent of patients correctly administered medications when instructed to do so every 6 hours; most thought they were to consume the medication every 6 hours when awake and thus to take three rather than four doses (Madlon-Kay and Mosch, 2000). In other cases the prescriber may write “q6h,” the abbreviation for every 6 hours, when intending that the patient take the medication three times per day. Additionally, unnecessary use of antibiotics for the wrong infections or when no infection is present and not taking all doses through the prescribed treatment duration are important factors contributing to antimicrobial resistance (Davey et al., 2002).
Monitoring for Effects
Monitoring (also referred to as assessment, evaluation, observation, and surveillance) involves obtaining and evaluating clinical indicators and other relevant information to determine a drug’s effect in an individual patient (Knowlton and Penna, 2003). Monitoring for desired and undesired effects is a crucial step in the care process and in the prevention or detection of adverse drug events. In every setting in which care is delivered—ambulatory care sites, hospitals, schools, workplace health sites, home health care, and nursing homes—assessment and monitoring is a primary responsibility of licensed nurses (IOM, 2004b). Pharmacists also may play a role in assessing beneficial or adverse effects during inpatient care, as may patients (including family members) in ambulatory and self-care.
At its best, monitoring is individualized, taking into consideration that different patients may experience different therapeutic results and outcomes, and it is responsive, correcting the regimen if an adverse effect is found (Knowlton and Penna, 2003). Assessing the effect of medications can be accomplished through direct observation of the patient, use of monitoring devices, and/or information technology (e.g., predefined triggers in a laboratory database) (Forester et al., 2004; Manasse and Thompson, 2005). The type and frequency of patient monitoring activities vary by care setting, clinical condition, and other characteristics of the patient (IOM, 2004b).
In acute care hospitals, bedside monitoring of the patient’s condition prior to, during, and following medical procedures such as initiation of new medications, surgery, or a course of medical therapy typically includes monitoring vital signs (i.e., temperature, heart rate and rhythm, breathing rate and character, blood pressure), airway, risk/presence of infection, fluid
intake and output, electrolytes, and pain (Bulechek et al., 1994). In intensive care units, monitoring is more frequent, more often invasive, and technologically complex (IOM, 2004b). In long-term and home care, other patient characteristics are observed and evaluated to determine the response to medications, including cognition, communication, vision, mood and behavior patterns, psychosocial well-being, and ability to perform daily care activities (e.g., grooming, bathing). Results of patient monitoring and any adverse effects are documented in the patient’s medical record. For nursing homes and home health services, these assessments must be completed by licensed nurses according to federally prescribed guidelines to comply with federal regulatory and reimbursement requirements (i.e., Medicare) (ANA, 1998; IOM, 2004b).
Medical devices designed for patient monitoring range from small, wearable devices that monitor a single physiological parameter, such as blood pressure, to complex devices (e.g., respiratory oximeters, electrocardiograms) that measure a variety of parameters and transmit them electronically to a central monitoring station. Changes in physiological responses detected with these devices can signal a nurse that the patient may be experiencing an adverse drug reaction. For example, a heart monitor may detect an inappropriate change in heart rate or rhythm after administration of a cardiac drug. Medication infusion devices, such as smart pumps and patient-controlled analgesia machines, go a step further and maintain a record not only of medications administered, but also of errors that may have occurred. In addition, telemedicine and remote patient monitoring devices that are connected to specialized computer modems and can reliably measure and transmit physiological data (e.g., blood pressure, heart rate, blood glucose level) are a growing method of care management supporting providers and patients in rural settings (inpatient, ambulatory, home/self-care) (Field and Grigsby, 2002).
The ability of pharmacists to monitor the effectiveness of drug therapy through computerized pharmacy and laboratory database systems has been an important advance in assessing patient responses to medications, especially in inpatient settings (Knowlton and Penna, 2003). Linkage of these systems enhances opportunities for improved monitoring through evaluation and review of drug appropriateness, drug dosages, drug–drug interactions, drug–allergy conflicts, drug blood serum concentrations, and metabolic responses, particularly for potent medications with narrow therapeutic indices (Armstrong, 2000; Knowlton and Penna, 2003; Schiff et al., 2003). Electronic medical records with event-driven surveillance systems are able to monitor patients around the clock and have been shown to detect some adverse drug events early enough to prevent their progression from mild or moderate to severe (Classen et al., 1991; Evans et al., 1991, 1994; Jha et al., 1998; Bates and Gawande, 2003). These systems monitor specific signals
from laboratory results, medication orders, information on vital signs, drug levels, and text reports to identify patients experiencing possible adverse drug events. Pharmacists then follow up on high-risk patients. Patient monitoring could be further improved through the incorporation of linked pharmacy and laboratory data into electronic order entry and real-time decision-support technologies, although issues concerning alert mechanisms must be resolved if this potential is to be realized (Schiff et al., 2003). In addition, recognition of the benefits of the pharmacist’s involvement in medication monitoring is extending beyond the inpatient setting to home health care, nursing homes, and community care (Knowlton and Penna, 2003).
It should be noted that susceptibility to adverse reactions is greatly increased in patients with multiple health conditions taking multiple medications. This is a growing problem for the elderly and others with chronic illnesses receiving care from several clinicians who may fail to coordinate medication treatment. On average, Medicare enrollees with chronic conditions are seen by eight different physicians or other providers during the course of a single year (Anderson and Knickman, 2001). Studies of medication errors among the elderly (Gurwitz et al., 2003, 2005) found that in both hospital and ambulatory care settings, monitoring errors were attributable to inadequate laboratory monitoring of drug therapies, or to a delayed response or failure to respond to signs and symptoms of drug toxicity (36 percent) or laboratory evidence of drug toxicity (37 percent) (Gurwitz et al., 2003, 2005). The most common preventable adverse effects from these monitoring errors were electrolyte/renal, gastrointestinal tract, hemorrhagic, and metabolic/endocrine events. Patient adherence was a contributing factor in 20 percent of the cases studied (Gurwitz et al., 2003). Another study of four primary care practices found that 25 percent of patients experienced an adverse drug event over a 3-month period (13 percent of these events were serious, 39 percent were preventable, and 6 percent were both serious and preventable) (Gandhi et al., 2003). The events were attributed to poor communication—the physician’s failure to respond to symptoms reported by the patient or the patient’s failure to report symptoms to the physician.
Using the methods noted above, the overarching goal of monitoring is the early detection of a downturn in a patient’s health status or the occurrence of an adverse event and the initiation of activities to restore the patient’s health (IOM, 2004b). Drugs may cause adverse effects in patients for a variety of reasons. For example, a drug may be highly potent and toxic at therapeutic doses, a drug may interact in an unforeseen way with another drug or a food product, a patient may have a particular sensitivity to a drug, a wrong drug or improper dosage may be administered, or a drug may be improperly manufactured (Noah and Brushwood, 2000). The signs and symptoms of an adverse reaction may be unpredictable (e.g., a skin
rash or anaphylaxis); foreseeable (e.g., nausea with chemotherapy); or unanticipated because they arise from errors in prescribing, dispensing, or administering (Noah and Brushwood, 2000). Regardless of the cause, as discussed above, nursing surveillance is critically important in preventing, identifying, and recovering from adverse events (IOM, 2004b). In summary, medication safety and monitoring depend on the following (Noah and Brushwood, 2000):
-
Knowledge of results of laboratory tests that affect drug dosages.
-
Knowledge of previously unrecognized adverse reactions.
-
Knowledge of adverse reactions that were previously recognized and were thought to be preventable, but are in fact not being prevented.
-
Knowledge of previously recognized, unpreventable adverse reactions that were thought to occur at acceptably low rates in light of the drug’s anticipated benefit, but occur more frequently in practice than anticipated.
Health care providers can enhance patient safety by welcoming the involvement of patients and families, especially in monitoring care and responses to medications. There should be no place in the health care system where a surrogate is prevented from being present whenever a patient without full faculties is receiving medications.
Self-Monitoring
Self-monitoring of responses to OTCs, prescription medications, and dietary supplements is an important aspect of good self-care and self-management. Since most patients do not discuss their use of OTCs with their primary care provider, they rely on their own judgment and product labels for prescribing, administering, and monitoring their consumption of these products (Simaon and Winkle, 1997; Frank et al., 2001). The greatest concern with the use of OTCs is the possibility of interactions with other products, mainly prescription medications, that can produce an adverse reaction. Warnings about such interactions are often listed on product labels, but difficulty in understanding the labels can increase the probability of an error or adverse effect (Patel et al., 2002). Likewise, a growing literature documents interactions of complementary and alternative medicines (including dietary supplements) with OTCs and prescription drugs (D’Arcy, 1993; Calis and Young, 2004). Thus, active self-monitoring is necessary for those consuming these products to identify and prevent serious events.
Self-monitoring of physiological and psychological responses to prescription drugs is even more critical to the identification of adverse events. Insufficient self-monitoring and nonadherence to drug regimens are well-
noted problems that can contribute to poor health outcomes, adverse events, and emergency room visits (Sawicki, 1999; Cummings et al., 2000). Some prescription medications have a particularly high propensity to drug and food interactions; examples are warfarin, an anticoagulant, and monamine oxidase (MAO) inhibitors, an (older) class of antidepressant. One study found an abundance of adverse interactions between warfarin and commonly used medications and foods such as anti-infective agents, lipid-lowering drugs, nonsteroidal anti-inflammatories, certain antidepressants, anabolic steroids, fish oil, mango, green tea, and grapefruit juice, to name a few (Holbrook et al., 2005). In total, 34 reports of major interactions were confirmed in the study, as were 41 highly probable and 38 probable causations. Likewise, a wide range of drug and food interactions have been reported with the use of MAO inhibitors (Livingston and Livingston, 1996; NLM, 2005). Dangerous reactions such as sudden high blood pressure may result when these agents are taken with certain drugs, foods, or drinks, such as antihypertensives, asthma medicines, other antidepressants, cheese, poultry, fish, sausage, overripe fruit, alcoholic beverages, and high amounts of caffeine (NLM, 2005). Individuals taking these medications have a difficult time adhering to their regimens without adequate education and support mechanisms.
A number of factors affect individuals’ ability to engage in illness self-management such as their particular illness and life circumstances. Barriers to self-management generally fall into three categories: knowledge deficits (e.g., insufficient information, literacy issues); practical barriers (e.g., physiological, functional, or financial constraints); and attitudinal factors (e.g., personal beliefs, culture, values, and experiences). These barriers are discussed extensively in Chapter 4.
Conversely, several studies have noted certain individuals’ ability, given adequate education, to participate successfully in self-care and disease management for various health conditions, including diabetes, which requires frequent self-monitoring of blood glucose levels to make adjustments in self-administered insulin therapy; depression, which requires self-assessment of changes in psychosocial affect resulting from prescribed medications; and cancer, which requires self-monitoring of adverse reactions to powerful chemotherapy agents (Grissinger et al., 2003; Ikesue et al., 2004; Schroeder et al., 2004). For example, self-monitoring of glucose levels and strict adherence to insulin therapy or oral hypoglycemic agents, along with extensive patient education, lead to major improvements in medical outcomes and substantial decreases in long-term complications of diabetes (Tamada et al., 1999). Similar positive results were found in initial studies of patients receiving oral anticoagulation therapy (Sawicki, 1999). Self-management support programs that emphasized use of portable capillary whole-blood analyzers for regular testing of prothrombin time, together
with structured patient education, improved the accuracy of medication management and overall quality of life. However, additional studies are needed to assess the impact of such self-monitoring on bleeding and thromboembolic complications. Given the growing prevalence of chronic conditions in the U.S. population, investment in the development of well-designed programs to assist patients with self-monitoring is essential to achieving improvements in medication safety. (See Box 2-5 for a summary of key problems with provider processes.)
ADVERSE EVENT REPORTING AND SURVEILLANCE SYSTEMS
Health care providers and safety agencies use error and adverse event reporting programs to learn about potential safety risks and the circumstances of individual errors (Smetzer and Cohen, 2006). Currently, a wide range of external reporting programs are available (IOM, 2004c; Smetzer and Cohen, 2006). Some of the systems are voluntary, and others are mandatory; some receive data only on adverse events, while others receive reports on all medication errors. These reporting programs include the following:
-
Institutional error reporting programs such as the Veterans Administration Patient Safety Reporting System and the U.S. Pharmacopeia’s MedMARx Program.
-
Mandatory state reporting programs, for which almost half of states require mandatory reporting of certain serious adverse events (Rosenthal and Booth, 2005).
-
Voluntary national reporting programs, such as the U.S. Pharmacopeia–Institute for Safe Medication Practice Medication Errors Reporting Program and the FDA’s MedWatch Program.
Reportable events include those due to practice-based errors (e.g., misadministered drug dosage), product safety issues (e.g., an adverse reaction to a drug), or hazardous situations (e.g., confusing labeling). All of these reporting systems currently utilize their own reporting formats. For a serious adverse event that occurs in a hospital, this means that several reports must be completed to meet the requirements for different reporting systems. The lack of a common reporting format and terminology is a significant factor that not only inhibits the reporting process, but also prevents comparison of data across health care organizations and pooling of data from different reporting programs. The IOM report, Patient Safety: Achieving a New Standard for Care, discussed extensively the need for common standards for patient safety reporting systems.
|
BOX 2-5 Summary of Key Problems with Provider Processes Affecting Safety and Quality in the Medication-Use System Prescribing
Transcribing
Preparing and Dispensing
Administration and Monitoring
Reporting
Self-Care
|
Institutional Reporting Systems
Many health care organizations have an internal incident reporting program, frequently managed by a patient safety officer or equivalent, who carries out analyses of the errors and determines the external programs to which the incident reports should be sent. Often, reports are made directly to one or more of the external programs by the patient or provider, bypassing the patient safety office.
Rates of reporting of events have been quite low (Leape, 2002). Providers’ fear of discoverability during litigation and professional disciplinary action has been a major factor affecting their willingness to report (IOM, 2004c). This particular concern should be alleviated with the recent passage of the Patient Safety and Quality Improvement Act of 2005 (P.L. 109-41). The act promotes the establishment and use of voluntary patient safety reporting systems, peer review protection from report disclosure during legal proceedings, and protection of providers who report from professional retaliation. However, the legislation failed to set measurable goals and criteria for evaluating the reporting program’s success or failure.
Evidence on the effectiveness of reporting programs is limited. There is anecdotal evidence that the Joint Commission on Accreditation of Health Care Organizations’ Sentinel Event Reporting System and the U.S. Pharmacopeia–Institute for Safe Medication Practice Medication Errors Reporting Program have led to important safety improvements (Leape, 2002). The only reporting program whose effect on safety has been demonstrated by a controlled trial is the National Nosocomial Infection Survey (Haley et al., 1985). Many reporting programs distribute newsletters or advisories to alert providers of hazardous situations and possible preventive measures. The impact of these materials is not known (Leape, 2002). However, performance measurement is an important component of quality improvement programs, which include activities directly related to patient safety (IOM, 2001). The new legislation mentioned above is expected to increase physician participation in adverse event reporting systems, and subsequently the translation of findings into learning and improvement. This will facilitate assessment of the positive, neutral, or negative impact of reporting systems in the near term.
State Reporting Systems
About half of all states have passed legislation or regulations related to hospital reporting of adverse events; almost all of these reporting systems are mandatory. The development of state reporting systems is tracked by the National Academy for State Health Policy (NASHP, 2006). Each state reporting system takes a different approach as to what events must be
reported and what type of information must be provided (Leape, 2002; IOM, 2004). The main reason for many of these reporting systems is to ensure accountability, although a number of state reporting systems also have a learning component (Rosenthal and Booth, 2005). Each system has the potential to improve patient safety through analysis of event reports and dissemination of best practices and lessons learned. For example, the Pennsylvania Patient Safety Authority regularly issues patient safety advisories (PPSA, 2006).
Evidence that mandatory reporting systems have led hospitals to introduce changes is largely anecdotal (Leape, 2002). Despite interest in analyzing the data from reporting systems and providing feedback that can be used to improve patient safety, states have found barriers to analysis and feedback (Rosenthal and Booth, 2005). Rates of reporting are low because hospitals fear the consequences of disclosure. Further, reporting is discouraged by the cumbersome and time-consuming nature of reporting systems (Leape, 2002). In addition, states often lack the clinical expertise to analyze the data. To address these barriers, in May 2005 the National Academy for State Health Policy convened a meeting of stakeholders in reporting systems to identify mechanisms and tools for improving reporting and feedback (Rosenthal and Booth, 2005).
Federal Reporting and Surveillance Systems
The FDA’s spontaneous reporting system (the MedWatch program) collects information about adverse events associated with all marketed drugs. The system depends on voluntary reports submitted by clinicians and mandatory reports submitted by manufacturers (comprising patient/ clinician reports forwarded to the company) (Fontanarosa et al., 2004). While about 250,000 such reports are received annually, several shortcomings of the system have been described in government, academic, and press publications (FDA, 1999). The reliance on voluntary reports and the factors that discourage reporting (e.g., time pressures, fear of liability, and lack of perceived benefit) result in significant underreporting of adverse outcomes and thus the inability to calculate true rates of such events (Fontanarosa et al., 2004). The reports also suffer from poor data quality, often including inadequate documentation and detail, which limits the ability to establish causal relationships in the analysis of the events. The consumer version of the system, MedWatch Plus, is designed to collect direct reports from consumers who have experienced an adverse drug reaction (Behrman, 2005). A number of activities are under way to improve the reporting systems, the quality and interpretation of the data gathered, and the use of postmarket surveillance studies, thus helping to ensure the safety and efficacy of marketed medicines.
|
BOX 2-6 Centers for Disease Control and Prevention’s Active Surveillance Systems for Clinical Care The CDC’s adverse event surveillance systems include the National Healthcare Safety Network (the successor to the National Nosocomial Infections Surveillance System, the Dialysis Surveillance Network, and the National Surveillance System for Health Care Workers) (Tokars et al., 2004) and the National Electronic Injury Surveillance System-Cooperative Adverse Drug Event Surveillance project (NEISS-CADES). Through NEISS-CADES, the CDC conducts nationally representative surveillance for adverse drug events (ADEs) treated in hospital emergency departments. The program is aimed at controlling or preventing injury by identifying and describing the public health burden of outpatient ADEs, generating hypotheses about risk factors for these events, and helping to design interventions for reducing medication errors in the outpatient setting. Estimates for 2004 indicate that approximately 700,000 patients were treated in emergency rooms for an ADE, and approximately 100,000 were admitted or transferred to another facility (Budnitz, 2005). Early data indicate that unintentional overdoses were the most common cause of ADEs (39 percent), and that two drugs (i.e., warfarin and insulin) were associated with 16 percent of all ADEs and 33 percent of ADEs in patients over age 50 (Budnitz et al., 2005). NEISS-CADES has several important limitations. First, the system is limited to ADEs occurring outside the hospital and to those that result in emergency room visits. Second, the system may fail to capture some serious outpatient ADEs (those treated in a care setting other than an emergency department) and may include nonserious events (as patients may use emergency departments for primary health care) (Walls et al., 2002). Third, the system is designed for national surveillance and not for quality improvement by individual hospitals. Nonetheless, given the importance of monitoring the national health burden of ADEs as one aspect of medication safety and quality improvement, the continued operation and enhancement of NEISS-CADES could play an important role in monitoring the nation’s progress toward reducing medication-related harm in the outpatient setting. The system’s usefulness would be enhanced by identifying appropriate measures of drug exposure, ensuring continued data quality, and developing mechanisms for timely data dissemination. |
The Centers for Disease Control and Prevention (CDC) performs both active and passive surveillance of safety-related morbidity and mortality associated with vaccines, treatment of infectious diseases, and other aspects of health care. Adverse events involving vaccines are reported passively through the Vaccine Adverse Event Reporting System (VAERS), maintained by the CDC jointly with the FDA. Reporting is mandatory for manufacturers, and for health professionals for specified adverse events (see the Reportable Events table posted at vaers.hhs.gov) associated with the following vaccines: tetanus; pertussis; measles, mumps, and rubella; rubella; inacti-
vated polio; hepatitis B; hemophilus influenzae type B (polysaccharide); hemophilus influenzae type B (conjugate); varicella; and pneumococcal conjugate (IOM, 2004c). From its establishment in 1990 through the end of 2001, VAERS had received over 128,000 reports (CDC, 1999).
Gaps in scientific knowledge about the possible adverse effects of vaccines and in the capacity to evaluate such effects scientifically prompted the CDC to initiate the Vaccine Safety Datalink (VSD) project in 1990 (Medstat, 2002). This project involves partnerships with several large HMOs to conduct high-quality scientific evaluations of important safety questions related to immunization. The CDC also has a number of reporting and surveillance systems for evaluating the prevalence of adverse events in clinical settings (see Box 2-6).
REFERENCES
AAMC (Association of American Medical Colleges). 2005. Drug Development Science: Obstacles and Opportunities for Collaboration Among Academia, Industry, and Government. Washington, DC: FDA.
Abrams T. 2005. FDA, Division of Drug Marketing, Advertising, and Communications. Submission to the IOM Committee on Identifying and Preventing Medication Errors. Washington, DC: FDA.
Allan EL, Barker KN, Malloy MJ, Heller WM. 1995. Dispensing errors and counseling in community practice. American Pharmacy NS35(12):25–33.
Allinson TT, Szeinbach SL, Schneider PJ. 2005. Perceived accuracy of drug orders transmitted orally by telephone. American Journal of Health System Pharmacists 62(1):78–83.
AHCA (American Health Care Association). 2002. Results of the 2001 AHCA Nursing Position Vacancy and Turnover Survey. Washington, DC: AHCA.
ANA (American Nurses Association). 1998. Standards of Clinical Nursing Practice. Washington, DC: ANA.
Anderson G, Knickman JR. 2001. Changing the chronic care system to meet people’s needs. Health Affairs 20(6):146–160.
Angell M. 2004. The Truth About Drug Companies. New York: Random House.
Armstrong EP. 2000. Electronic prescribing and monitoring are needed to improve drug use. Archives of Internal Medicine 160(18):2713–2714.
Avorn J. 2004. Powerful Medicines: The Benefits, Risks, and Costs of Prescription Drugs. New York: Vintage Books, Random House.
Bates DW, Gawande AA. 2003. Improving safety with information technology. New England Journal of Medicine 348(25):2526–2534.
Bates DW, Cullen DJ, Laird N, Petersen LA, Small SD, Servi D, Laffel G, Sweitzer BJ, Shea BF, Hallisey R, Vander Vliet M, Nemeskal R, Leape LL. 1995. Incidence of adverse drug events and potential adverse drug events. Implications for prevention. ADE Prevention Study Group. Journal of the American Medical Association 274(1):29–34.
Behrman RE. 2005. Adverse Reactions: Information in, Information out. [Online]. Available: http://www.iom.edu/CMS/3740/24155/29378.aspx [accessed February 6, 2006].
Bekelman JE, Li Y, Gross GP. 2003. Scope and impact of financial conflicts of interest in biomedical research. Journal of the American Medical Association 289(4):454–465.
Berndt ER, Gottschalk AHB, Strobeck MW. 2005. Opportunities for Improving the Drug Development Process: Results from a Survey of Industry and the FDA. Cambridge, MA: National Bureau of Economic Research.
Bero LA, Rennie D. 1996. Influences on the quality of published drug studies. International Journal of Technology Assessment in Health Care 12(20):209–237.
Blumenthal D. 2004. Doctors and drug companies. New England Journal of Medicine 351(18):1885–1890.
Bobb A, Gleason K, Husch M, Feinglass J, Yarnold PR, Noskin GA. 2004. The epidemiology of prescribing errors. Archives of Internal Medicine 164(7):785–792.
Bodenheimer T. 2000. Clinical investigators and the pharmaceutical industry. New England Journal of Medicine 42(20):1540–1543.
Boyd CM, Darer J, Boult C, Fried LP, Boult L, Wu AW. 2005. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: Implications for pay for performance. Journal of the American Medical Association 294(6): 716–724.
Brennan TA, Gawande A, Thomas E, Studdert D. 2005. Accidental deaths, saved lives, and improved quality. New England Journal of Medicine 353(13):1405–1409.
Brennan TA, Rothman DJ, Blank L, Blumenthal D, Chimonas SC, Cohen JJ, Goldman J, Kassirer JP, Kimball H, Naughton J, Smelser N. 2006. Health industry practices that create conflicts of interest: A policy proposal for academic medical centers. Journal of the American Medical Association 295(4):429–433.
Buchanan TL, Barker KN, Gibson JT, Jiang BC, Pearson RE. 1991. Illumination and errors in dispensing. American Journal of Hospital Pharmacy 48(10):2137–2145.
Budnitz D. 2005. The CDC’s National Electronic Injury Surveillance System: Cooperative Adverse Drug Event Surveillance Project. Washington, DC: IOM Committee on Identifying and Preventing Medication Errors.
Budnitz DS, Pollock DA, Mendelsohn AB, Weidenbach KN, McDonald AK, Annest JL. 2005. Emergency department visits for outpatient adverse drug events: Demonstration for a national surveillance system. Annals of Emergency Medicine 45(2):197–206.
Bulechek G, McCloskey J, Titler M, Denehey J. 1994. Nursing interventions used in practice. American Journal of Nursing 94(10):59–66.
Burke KG, Mason DJ, Alexander M, Barnsteiner JH, Rich VL. 2005. Making medication administration safe: Report challenges nurses to lead the way. American Journal of Nursing 28(Suppl. 2):2–3.
Califf RM. 2004. Defining the balance of risk and benefit in the era of genomics and proteomics. Health Affairs 23(1):77–87.
Califf RM, DeMets DL. 2002a. Principles from clinical trials relevant to clinical practice: Part I. Circulation 106:1015–1021.
Califf RM, DeMets DL. 2002b. Principles from clinical trials relevant to clinical practice: Part II. Circulation 106:1172–1175.
Califf RM, Gibbons RJ, Brindis RG, Smith SC. 2002. Integrating quality into the cycle of therapeutic development. Journal of the American College of Cardiology 40(11):1895– 1901.
Calis KA, Young LR. 2004. Clinical analysis of adverse drug reactions: A primer for clinicians. Hospital Pharmacy 39(7):697–712.
Carney SL, Nair KR, Sales MA, Walsh J. 2001. Pharmaceutical industry-sponsored meetings: Good value or just a free meal? Internal Medicine Journal 31(8):488–491.
Castle NG, Engberg J. 2005. Staff turnover and quality of care in nursing homes. Medical Care 43(6):616–626.
CDC (Centers for Disease Control and Prevention). 1999. FDA on VAERS. [Online]. Available: http://www.cdc.gov/nip/vacsafe/concerns/hepB/fdatest.htm#Limitations [accessed January 11, 2006].
Chew LD, O’Young TS, Hazlet TK, Bradley KA, Maynard C, Lessler D. 2000. A physician survey of the effect of drug sample availability on physicians’ behavior. Journal of General Internal Medicine 15(7):478–483.
Chimonas S, Rothman DJ. 2005. New federal guidelines for physician-pharmaceutical industry relations: The politics of policy formation. Health Affairs 24(4):949–960.
Chopra SS. 2003. Industry funding of clinical trials: Benefit or bias? Journal of the American Medical Association 290(1):113–114.
Christensen ML, Helms RA, Chesney RW. 1999. Is pediatric labeling really necessary? Pediatrics 104(3 Pt. 2):593–597.
Chung RS, Taira DA, Noh C. 2003. Alternate financial incentives in multitiered formulary systems to improve accountability for outcomes. Journal of Managed Care Pharmacy 9(4):360–365.
Classen DC, Pestotnik SL, Evans RS, Burke JP. 1991. Computerized surveillance of adverse drug events in hospital patients. Journal of the American Medical Association 266(20):2847–2851.
Cohen MR. 2000. Medication Errors: Causes, Prevention, and Risk Management. Sudbury, MA: Jones and Bartlett Publishers.
Cohen MR. 2005. Overview of the Institute for Safe Medication Practice and Reported Adverse Drug Events. Huntingdon Valley, PA: Institute of Safe Medication Practice.
Coyle SL. 2002. Physician–industry relations: Part 1: Individual physicians. Annals of Internal Medicine 136(5):396–402.
Cropper CM. 2005. Counter intelligence. BusinessWeek. [Online]. Available: http://www.businessweek.com/@@1N2NgIUQOPQ*bx0A/magazine/content/05_21/b3934126_mz070.htm [accessed June 7, 2006].
Cummings SR, Palermo L, Browner W, Marcus R, Wallace R, Pearson J, Blackwell T, Eckert S, Black D. 2000. Monitoring osteoporosis therapy with bone densitometry: Misleading changes and regression to the mean. Journal of the American Medical Association 283(10):1318–1321.
D’Arcy PF. 1993. Adverse reactions and interactions with herbal medicines. Part 2. Drug interactions. Adverse Drug Reactions and Toxicological Reviews 12:147–162.
Davey P, Pagliart C, Hayes A. 2002. The patient’s role in the spread and control of bacterial resistance to antibiotics. Clinical Microbiology and Infectious Disease 8(Suppl. 2):43–68.
Davis NM. 1996. Performance lapses as a cause of medication errors. Hospital Pharmacy 31:1524–1527.
Davis NM, Cohen MR. 1994. Ten steps for ensuring dispensing accuracy. American Pharmacy NS34(7):22–23.
Dean B, Schachter M, Vincent C, Barber N. 2002. Prescribing errors in hospital inpatients: Their incidence and clinical significance. Quality Safety Health Care 11(4):340–344.
Deyo RA. 2004. Gaps, tensions, and conflicts in the FDA approval process: Implications for clinical practice. Journal of the Board of Family Practice 17(2):142–149.
Dick A, Keady S, Mohamed F, Brayley S, Thomson M, Lloyd BW, Heuschkel R, Afzal NA. 2003. Use of unlicensed and off-label medications in pediatric gastroenterology with a review of the commonly used formularies in the U.K. Aliment Pharmacology and Therapeutics 17:571–575.
Donohue SM, Needleman SM. 1998. Potential cause of medication administration error. Anesthiology 89(3):800–803.
Evans RS, Pestotnik SL, Classen DC, Bass SB, Menlove RL, Gardner RM, Burke JP. 1991. Development of a computerized adverse drug event monitor. Proceedings of the Annual Symposium on Computer Applications in Medical Care 23–27.
Evans RS, Pestotnik SL, Classen DC, Horn SD, Bass SB, Burke JP. 1994. Preventing adverse drug events in hospitalized patients. The Annals of Pharmacotherapy 28(4):523–527.
FDA (U.S. Food and Drug Administration). 1994. Specific Requirements on Content and Format of Labeling for Human Prescription Drugs. Revision of Pediatric Use Subsection in the Labeling. Final Rule Edition. CFR Part 201 [Docket No. 92N-0165]. Rockville, MD: FDA.
FDA. 1998. The CDER Handbook. Rockville, MD: U.S. Department of Health and Human Services.
FDA. 1999. From Test Tube to Patient: Improving Health Through Human Drugs. Rockville, MD: FDA.
FDA. 2003. FDA’s Review Process for New Drug Applications: A Management Review. Rockville, MD: U.S. Department of Health and Human Services.
FDA. 2004a. 2004 Report to the Nation: Improving Public Health Through Human Drugs. Rockville, MD: FDA.
FDA. 2004b. Guidance for Industry Information Program on Clinical Trials for Serious or Life-Threatening Diseases and Conditions. Rockville, MD: FDA.
Fialova D, Topinkova E, Gambassi G, Finne-Soveri H, Jomsson PV, Carpenter I, Schroll M, Onder G, Sorbye LW, Wagner C, Reissigova J, Bernabei R. 2005. Potentially inappropriate medication use among elderly home care patients in Europe. Journal of the American Medical Association 293(11):1348–1358.
Fick DM, Maclean JR, Rodriguez NA, Short L, Vanden Heuvel R, Waller JL, Rogers RL. 2004. A randomized study to decrease the use of potentially inappropriate medications among community-dwelling older adults in a Southeastern managed care organization. American Journal of Managed Care 10(11):761–768.
Field MJ, Grigsby J. 2002. Telemedicine and remote patient monitoring. Journal of the American Medical Association 288(4):423–425.
Fields M, Peterman J. 2005. Intravenous medication safety system averts high-risk medication errors and provides actionable data. Nursing Administration Quarterly 29(1):78–87.
Flynn EA, Barker KN, Gibson JT, Pearson RE, Berger BA, Smith LA. 1999. Impact of interruptions and distractions on dispensing errors in an ambulatory care pharmacy. American Journal of Health System Pharmacists 56(13):1319–1325.
Flynn EA, Barker KN, Carnahan BJ. 2003. National observational study of prescription dispensing accuracy and safety in 50 pharmacies. Journal of the American Pharmaceutical Association 43(2):191–200.
Fontanarosa PB, Rennie D, DeAngelis CD. 2004. Postmarketing surveillance-lack of vigilance, lack of trust. Journal of the American Medical Association 292(21):2647–2650.
Forester AJ, Halil RB, Tierney MG. 2004. Pharmacist surveillance of adverse drug events. American Journal of Health System Pharmacists 61(14):1466–1472.
FR (Federal Register). 2004a. Report on the Performance of Drug and Biologics Firms in Conducting Postmarketing Commitment Studies. Washington, DC: U.S. Department of Health and Human Services.
FR. 2004b. Bar Code Label Requirements for Human Drug Products and Biological Products: Final Rule. Washington, DC: National Archives and Records Administration.
FR. 2006. Requirements on Content and Format of Labeling for Human Drug and Biological Products and Draft Guidances and Two Guidances for Industry on the Content and Format of Labeling for Human Prescription Drug and Biological Products: Final Rule and Notices. Washington, DC: National Archives and Records Administration.
Frank C, Godwin M, Verma S, Kelly A, Birenbaum A, Seguin R, Anderson J. 2001. What drugs are our frail elderly patients taking? Do drugs they take or fail to take put them at increased risk of interactions and inappropriate medication use? Canadian Family Physician 47:1198–1204.
Frush KS, Luo X, Hutchinson P, Higgins JN. 2004. Evaluation of a method to reduce over-the-counter medication dosing error. Archives of Pediatric Medicine 158:620–624.
Gandhi TK, Weingart SN, Borus J, Seger AC, Peterson J, Burdick E, Seger DL, Shu K, Federico F, Leape LL, Bates DW. 2003. Adverse drug events in ambulatory care. New England Journal of Medicine 348(16):1556–1564.
GAO (Government Accountability Office). 2004. Internet Pharmacies: Some Pose Safety Risks for Consumers. Washington, DC: GAO.
Gladstone J. 1995. Drug administration errors: A study into the factors underlying the occurrence and reporting of drug errors in a district general hospital. Journal of Advanced Nursing 22:628–637.
Golden AG, Preston RA, Barnett SD, Llorente M, Hamdan K, Silverman MA. 1999. Inappropriate medication prescribing in homebound older adults. Journal of the American Geriatrics Society 47(8):948–953.
Goodman B. 2001. Do drug company promotions influence physician behavior? Western Journal of Medicine 174:232–233.
Greengold NL, Shane R, Schneider P, Flynn E, Elashoff J, Hoying CL, Barker K, Bolton LB. 2003. The impact of dedicated medication nurses on the medication administration error rate. Archives of Internal Medicine 163(19):2359–2367.
Gribetz B, Crunley SA. 1987. Underdosing of acetaminophen by parents. Pediatrics 80: 630–633.
Grissinger M, Kroon L, Prenna P. 2003. Misadventures in insulin therapy: Are your members at risk? Journal of Managed Care Pharmacy 9(Suppl. 3).
Groves KEM, Sketris I, Tett SE. 2003. Prescription drug samples: Does this marketing strategy counteract policies for quality use of medicines? Journal of Clinical Pharmacy and Therapeutics 28:259–271.
Gurwitz JH, Field TS, Avorn J, McCormick D, Jain S, Eckler M, Benser M, Edmondson AC, Bates DW. 2000. Incidence and preventability of adverse drug events in nursing homes. American Journal of Medicine 109(2):87–94.
Gurwitz JH, Field TS, Harrold LR, Rothschild J, Debellis K, Seger AC, Cadoret C, Fish LS, Garber L, Kelleher M, Bates DW. 2003. Incidence and preventability of adverse drug events among older persons in the ambulatory setting. Journal of the American Medical Association 289(9):1107–1116.
Gurwitz JH, Field TS, Judge J, Rochon P, Harrold LR, Cadoret C, Lee M, White K, LaPrino J, Mainard JF, DeFlorio M, Gavendo L, Auger J, Bates DW. 2005. The incidence of adverse drug events in two large academic long-term care facilities. American Journal of Medicine 118(3):251–258.
Haley RW, Culver DH, White JW, Morgan WM, Emori TG, Munn VP, Hooton TM. 1985. The efficacy of infection surveillance and control programs in preventing nosocomial infections in U.S. hospitals. American Journal of Epidemiology 121(2):182–205.
Hanlon JT, Fillenbaum GG, Schmader KE, Kuchibhatla M, Horner RD. 2000a. Inappropriate drug use among community-dwelling elderly. Pharmacotherapy 20(5): 575–582.
Hanlon JT, Shrimp LA, Semla TP. 2000b. Recent advances in geriatrics: Drug-related problems in the elderly. Annals of Pharmacotherapy 34:360–365.
Hartigan K. 2003. Patient education: The cornerstone of successful oral chemotherapy treatment. Clinical Journal of Oncology Nursing 7(6):21–24.
Holbrook AM, Pereira JA, Labris R, McDonald H, Douketis JD, Crowther M, Wells PS. 2005. Systematic overview of warfarin and its drug and food interactions. Archives of Internal Medicine 165(10):1095–1106.
Horsky J, Kuperman GJ, Patel VL. 2005. Comprehensive analysis of a medication dosing error related to CPOE. Journal of the American Medical Informatics Association 12(4): 377–382.
HPA (Health Policy Alternatives, Inc.). 2003. Pharmacy Benefit Managers: Tools for Managing Drug Benefit Costs, Quality, and Safety. Washington, DC: Pharmaceutical Care Management Association.
The HSM Group. 2002. Acute Care Hospital Survey of RN Vacancy and Turnover Rates. Chicago, IL: American Organization of Nurse Executives.
Hubal R, Day RS. 2006. Understanding the frequency and severity of side effects: Linguistic, numeric, and visual representations. In: Bickmore T, Green N, Editors. Argumentation for Consumers of Healthcare: Papers From the 2006 Spring Symposium. Technical Report SS-06-01. Menlo Park, CA: American Association of Artificial Intelligence.
Husakamp HA, Deverka PA, Epstein AM, Epstein RS, McGuigan KA, Frank RG. 2003. The effect of incentive-based formularies on prescription-drug utilization and spending. New England Journal of Medicine 349(23):2224–2232.
Ikesue H, Ishida M, Uchida M, Harada M, Haro T, Mishima K, Itoh Y, Kotsubo K, Yoshikawa M, Oishi R. 2004. Monitoring for potential adverse drug reactions in patients receiving chemotherapy. American Journal of Health System Pharmacy 61(22):2366– 2369.
IMS Health. 2004. Total U.S. Promotional Spending by Type, 2003. [Online]. Available: http://www.imshealth.com/ims/portal/front/articleC/0,2777,6599_44304752_44889690,00.html [accessed June 7, 2006].
IOM (Institute of Medicine). 2000. To Err Is Human: Building a Safer Health System. Washington, DC: National Academy Press.
IOM. 2001. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press.
IOM. 2002. Leadership by Example: Coordinating Government Roles in Improving Health Care Quality. Washington, DC: The National Academies Press.
IOM. 2003. Priority Areas for National Action: Transforming Health Care Quality. Washington, DC: The National Academies Press.
IOM. 2004a. Health Literacy: A Prescription to End Confusion. Washington, DC: The National Academies Press.
IOM. 2004b. Keeping Patients Safe: Transforming the Work Environment of Nurses. The National Academies Press.
IOM. 2004c. Patient Safety: Achieving a New Standard for Care. Washington, DC: The National Academies Press.
IOM. 2006. Developing a Registry of Pharmacologic and Biologic Clinical Trials. Washington, DC: The National Academies Press.
Jarman H, Jacobs E, Zielinski V. 2002. Medication study supports registered nurses’ competence for single checking. Internal Journal of Nursing Practice 8(6):330–335.
Jenkins R, Elliott P. 2004. Stressors, burnout and social support: Nurses in acute mental health settings. Journal of Advanced Nursing 48(6):622–631.
Jha AK, Kuperman GJ, Teich JM, Leape L, Shea B, Rittenberg E, Burdick E, Seger DL, Vander Vliet M, Bates DW. 1998. Identifying adverse drug events: Development of a computer-based monitor and comparison with chart review and stimulated voluntary report. Journal of the American Medical Informatics Association 5(3):305–314.
Jong GW, Vulto AG, de Hoog M, Schimmel KJM, Tibboel D, van den Anker JN. 2001. A survey of the use of off-label and unlicensed drugs in a Dutch children’s hospital. Pediatrics 108(5):1089–1093.
Kastango ES. 2003. Compounding Sterile Preparations. Bethesda, MD: American Society of Health System Pharmacists.
Katz D, Caplan AL, Merz JF. 2003. All gifts large and small: Toward an understanding of the ethics of pharmaceutical industry gift-giving. American Journal of Bioethics 3(3):39–46.
Kenny R. 2001. Introduction to Compliance with FDA Labeling and Advertising Requirements. [Online]. Available: http://www.ehcca.com/presentations/PharmaReg1/203_1.pdf [accessed June 30, 2005].
Klein DF, Thase ME, Endicott J, Adler L, Glick I, Kalai A, Leventer S, Mattes J, Ross P, Bystritsky A. 2002. Improving clinical trials. Archives of General Psychiatry 59(3):272–278.
Knowlton CH, Penna RP. 2003. Pharmaceutical Care. 2nd ed. Bethesda, MD: American Society of Health System Pharmacists.
Koppel R, Metlay JP, Cohen A, Abaluck B, Localio AR, Kimmel SE, Strom BL. 2005. Role of computerized physician order entry systems in facilitating medication errors. Journal of the American Medical Association 293(10):1197–1203.
Kos M. 2005. Satisfaction with pharmacotherapy for approved and off-label indications: A Delphi study. Annals of Pharmacotherapy 39(4):649–654.
Landon BE, Reschovsky JD, Blumenthal D. 2004. Physicians’ views of formularies: Implications for Medicare drug benefit design. Health Affairs 23(1):218–226.
Leape LL. 2002. Reporting of adverse events. New England Journal of Medicine 347(20): 1633–1638.
Leape LL, Bates DW, Cullen DJ, Cooper J, Demonaco HJ, Gallivan T, Hallisey R, Ives J, Laird N, Laffel G, Nemeskal R, Petersen L, Porter K, Servi D, Shea B, Small S, Sweitzer B, Thompson B, Vander Vleit M. 1995. Systems analysis of adverse drug events. Journal of the American Medical Association 274(1):35–43.
Lee PY, Alexander KP, Hammill BG, Pasquali SK, Peterson ED. 2001. Representation of elderly persons and women in published randomized trials of acute coronary syndromes. Journal of the American Medical Association 286(6):708–713.
Leonard EM. 1994. Quality assurance and the drug development process: An FDA perspective. Quality Assurance 3:178–186.
Lesar TS, Briceland LL, Delcoure K, Parmalee JC, Masta-Gornic V, Pohl, H. 1990. Medication prescribing errors in a teaching hospital. Journal of the American Medical Association 263(17):2329–2334.
Lesar TS, Briceland L, Stein D. 1997. Factors related to errors in medication prescribing. Journal of the American Medical Association 277(4):312–317.
Levy R. 1994. The role and value of pharmaceutical marketing. Archives of Family Medicine 3:327–332.
Lifshitz M, Gavrilov V, Gorodischer R. 2001. Off-label and unlicensed use of antidotes in pediatric patients. European Journal of Clinical Pharmacology 56:839–841.
Livingston MG, Livingston HM. 1996. Monamine oxidase inhibitors. An update on drug interactions. Drug Safety 14(4):219–227.
Loder EW, Biondi DM. 2004. Off-label prescribing of drugs in specialty headache practice. Headache 44(7):636–641.
Loucks M. 2003. Department of Justice Investigations and the Pharmaceutical Industry. Washington, DC: Fourth Annual Pharmaceutical Regulatory and Compliance Congress and Best Practices Forum.
Madlon-Kay DJ, Mosch FS. 2000. Liquid medication dosing errors. Journal of Family Practice 49(8):741–744.
Maguire P. 2001. Samples: Cost-Driver or Safety Net? [Online]. Available: http://www.acponline.org/journals/news/jan01/drugsamples.htm [accessed August 21, 2005].
Manasse HR, Thompson KK. 2005. Medication Safety: A Guide for Health Care Facilities. Bethesda, MD: American Society of Health System Pharmacists.
Manias E, Aitken R, Dunning T. 2005. How graduate nurses use protocols to manage patients’ medications. Journal of Clinical Nursing 14:935–944.
March JS, Silva SG, Compton S, Shapiro M, Califf R, Krishnan R. 2005. The case for practical clinical trials in psychiatry. American Journal of Psychiatry 162(5):836–846.
Martin EW. 1978. Hazards of Medications. Philadelphia, PA: J.B. Lippincott Company.
Medstat. 2002. Implementation Planning Study for the Integration of Medical Event Reporting Input and Data Structure for Reporting to AHRQ, CDC, CMS, and FDA. Final Report: Volume 2–Appendixes. Rockville, MD: AHRQ.
Moore TJ, Psaty BM, Furberg CD. 1998. Time to act on drug safety. Journal of the American Medical Association 279:1571–1573.
Moynihan R. 2003. Who pays for the pizza? Redefining the relationships between doctors and drug companies. 2: Disentanglement. British Medical Journal 326:1193–1196.
Munroe DJ. 2003. Assisted living issues for nursing practice. Geriatric Nursing 24(2): 99–105.
Murphy D. 2005. Pediatric Drug Development and Medication Errors. Washington, DC: IOM Committee on Identifying and Preventing Medication Errors.
NABP (National Association of Boards of Pharmacy). 2004. Survey of Pharmacy Law. Mount Pleasant, IL: NAPB.
NACDS (National Association of Chain Drug Stores). 1999. Pharmacy Activity Cost and Productivity Study. Alexandria, VA: NACDS.
NASHP (National Academy for State Health Policy). 2006. Patient Safety Toolbox for States. [Online]. Available: http://www.pstoolbox.org/_docdisp_page.cfm?LID=6BC2AB7D-6F1E-4DF2-AD20DAE18001147B [accessed, June 7, 2006].
NCI (National Cancer Institute). 2004. Q&A: Off-Label Drugs. [Online]. Available: http://www.nci.nih.gov/clinicaltrials/learning/approval-process-for-cancer-drugs/page5 [accessed June 29, 2005].
Nicholas PK, Agius CR. 2005. Toward safer IV medication administration. American Journal of Nursing 105(3):25–30.
Nies AS. 2001. Principles of therapeutics. In: Hardman JG, Limbird LE, Gilman AG, Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 10th ed. New York: McGraw Hill.
NIHCMREF (National Institute for Health Care Management and Research and Educational Foundation). 2002. Prescription Drug Expenditures in 2001: Another Year of Escalating Costs. Washington, DC: NIHCMREF.
NLM (National Library of Medicine). 2005. MedlinePlus: Antidepressants, Monamine Oxidase Inhibitors (MAO) (Systemic). [Online]. Available: http://www.nlm.nih.gov/medlineplus/druginfo/uspdi/202054.html [accessed August 30, 2005].
Noah BA, Brushwood DB. 2000. Adverse drug reactions in elderly patients: Alternative approaches to postmarket surveillance. Journal of Health Law 33(3):383–454.
Norrish B, Rundall T. 2001. Hospital restructuring and the work of registered nurses. Milbank Quarterly 79(1):55.
NPSF (National Patient Safety Foundation). 1997. National Patient Safety Foundation at the AMA: Public Opinion of Patient Safety Issues Research Findings. Louis Harris and Associates.
NRC (National Research Council). 1995. Standards, Conformity Assessment, and Trade. Washington, DC: National Academy Press.
NRC. 2004. Health and Medicine: Challenges for the Chemical Sciences in the 21st Century. Washington, DC: The National Academies Press.
Nunn AJ. 2003. Making medicines that children can take. Archives of Disease in Childhood 88(5):369–371.
O’Shea E. 1999. Factors contributing to medication errors: A literature review. Journal of Clinical Nursing 8:496–504.
Patel VL, Branch T, Arocha JF. 2002. Errors in interpreting quantities as procedures: The case of pharmaceutical labels. International Journal of Medical Informatics 65(3): 193–211.
Patterson ES. 2003. Addressing human factors in bar code medication administration systems. Hospital Pharmacy 38(11):S16–S17.
Pedersen CA, Schneider PJ, Scheckelhoff DJ. 2003. ASHP national survey of pharmacy practice in hospital settings: Dispensing and administration—2002. American Journal of Health System Pharmacists 60(1):52–68.
Petersen M. 2000, November 15. Growing opposition to free drug samples. New York Times. Business.
Phillips J, Beam S, Brinker A, Holquist C, Honig P, Lee LY, Pamer C. 2001. Retrospective analysis of mortalities associated with medication errors. American Journal of Health System Pharmacists 58:1835–1841.
Poole SG, Dooley MJ. 2004. Off-label prescribing in oncology. Supportive Care in Cancer 12(5):302–305.
PPSA (Pennsylvania Patient Safety Authority). 2006. Patient Safety Authority. [Online]. Available: http://www.psa.state.pa.us/psa/site [accessed June 7, 2006].
Presecky W. 2006, February 25. FDA joins probe after 2 infants die in Kane. Chicago Tribune.
Psaty BM, Furberg CD, Ray WA, Weiss NS. 2004. Potential for conflict of interest in the evaluation of suspected adverse drug reactions: Use of cerivastatin and risk of rhabdomyolysis. Journal of the American Medical Association 292(21):2622–2631.
Ringold DJ, Santell JP, Schneider PJ. 1999. ASHP national survey of pharmacy practice in acute care settings: Dispensing and administration. American Journal of Health System Pharmacists 57(19):1759–1775.
Roberts DE, Spencer MG, Burfield R, Bowden S. 2002. An analysis of dispensing errors in U.K. hospitals. Internal Journal of Pharmacy Practice 10(Supplement):R6.
Rochon PA, Gurwitz JH, Simms RW, Fortin PR, Felson DT, Minaker KL, Chalmers TC. 1994. A study of manufacturer-supported trials of nonsteroidal anti-inflammatory drugs in the treatment of arthritis. Archives of Internal Medicine 154:157–163.
Rosenthal J, Booth M. 2005. Maximizing the Use of State Adverse Event Data to Improve Patient Safety. Portland, ME: National Academy for State Health Policy.
Rothman K, Michels K. 1994. The continuing unethical use of placebo controls. New England Journal of Medicine 331(6):394–398.
RSW (Roper Starch Worldwide). 2001. Self-Care in the New Millennium. Washington, DC: Consumer Healthcare Products Association.
Sawicki PT. 1999. A structured teaching and self-management program for patients receiving oral anticoagulation: A randomized controlled trial. Journal of the American Medical Association 281(2):145–150.
Schiff GD, Klass D, Peterson J, Shah G, Bates DW. 2003. Linking laboratory and pharmacy: Opportunities for reducing errors and improving care. Archives of Internal Medicine 163(8):893–900.
Schondelmeyer S. 2005. Community Pharmacy Perspectives on Preventing Medication Errors at the July 6, 2005, Meeting of the IOM Committee on Identifying and Preventing Medication Errors, Washington, DC.
Schroeder K, Fahey T, Ebrahim S. 2004. How to improve adherence to blood pressure-lowering medication in ambulatory care? Archives of Internal Medicine 164(7): 722–732.
Schull PD. 2005. Five Rights Still Resound. Nursing Spectrum. [Online]. Avaliable: http:// community.nursingspectrum.com/MagazineArticles/article.cfm?AID=17801 [accessed June 7, 2006].
Shrank WH, Young HN, Ettner SL, Glassman P, Asch SM, Kravitz RL. 2005. Do the incentives in 3-tiered pharmaceutical benefit plans operate as intended? Results from a physician leadership survey. American Journal of Managed Care 11(1):16–22.
Simaon HK, Winkle DA. 1997. Over-the-counter medications: Do parents give what they intend to give? Archives of Pediatrics and Adolescent Medicine 151(7):654–656.
Smalley W, Shatin D, Wysowski DK, Gurwitz J, Andrade SE, Goodman M, Chan KA, Platt R, Schech SD, Ray W. 2000. Contraindicated use of cisapride: Impact of Food and Drug Administration regulatory action. Journal of the American Medical Association 284(23):3036–3039.
Smetzer J. 2001. Safer medication management. Nursing Management 32(12):44–48.
Smetzer J, Cohen MR. 2006. Medication Error Reporting Systems in Medication Errors. 2nd ed. Washington, DC: American Pharmacists Association.
Smith PC, Araya-Guerra R, Bublitz C, Parnes B, Dickinson LM, Van Vorst R, Westfall JM, Pace WD. 2005. Missing clinical information during primary care visits. Journal of the American Medical Association 293(5):565–571.
Steinbrook R. 2004. Public registration of clinical trials. New England Journal of Medicine 351(4):315–317.
Strom BL. 2004. Potential conflict of interest in the evaluation of suspected adverse drug reactions. Journal of the American Medical Association 292(21):2643–2646.
Studdert DM, Mello MM, Brennan TA. 2004. Financial conflicts of interest in physicians’ relationships with the pharmaceutical industry: Self-regulation in the shadow of federal prosecution. New England Journal of Medicine 351(18):1891–1900.
Suzuki K, Ohida T, Kaneita Y, Yokoyama E, Uchiyama M. 2005. Daytime sleepiness, sleep habits and occupational accidents among hospital nurses. Journal of Advanced Nursing 52(4):445–453.
Svarstad BL, Bultman DC, Mount JK. 2004. Patient counseling provided in community pharmacies: Effects of state regulation, pharmacist age, and business. Journal of the American Pharmaceutical Association 44(1):22–29.
Szefler SJ, Whelan GJ, Leung DY. 2006. Black box warning: Wake-up call or overreaction? Journal of Allergy and Clinical Immunology 117(1):26–29.
Taira DA, Iwane KA, Chung RS. 2003. Prescription drugs: Elderly enrollee reports of financial access, receipt of free samples, and discussion of generic equivalents related to type of coverage. American Journal of Managed Care 9(4):305–312.
Tamada JA, Garg S, Jovanovic L, Pitzer KR, Fermi S, Potts RO. 1999. Noninvasive glucose monitoring: Comprehensive clinical results. Journal of the American Medical Association 282(19):1839–1844.
Teutsch SM, Berger ML, Weinstein MC. 2005. Comparative effectiveness: Asking the right questions, choosing the right method. Health Affairs 24(1):128–132.
Thomas CP. 2003. Incentive-based formularies. New England Journal of Medicine 349(23): 2186–2188.
Tokars JI, Richards C, Andrus M, Klevens M, Curtis A, Horan T, Jernigan J, Cardo D. 2004. The changing fact of surveillance for health care-associated infections. Clinical Infectious Diseases 39:1347–1352.
University of Utah. 2006. Chapter 1: Testing and Marketing: Drug Development. [Online]. Available at: http://www.pharmacy.utah.edu/pharmtox/common_meds/ICM1.html [accessed June 5, 2006].
USP (U.S. Pharmacopeia). 2003. Summary of Information Submitted to MedMarx in the Year 2002. Rockville, MD: USP.
USP. 2004. MedMarx 5th Anniversary Data Report: A Chartbook of 2003 Findings and Trends 1999–2003. Rockville, MD: USP.
Vincent C. 2001. Clinical Risk Management: Enhancing Patient Safety. 2nd ed. London, UK: BMJ Books.
Wagner AK, Chan KA, Dashevsky I, Raebel MA, Andrade SE, Lafata JE, Davis RL, Gurwitz JH, Soumerai SB, Platt R. 2006. FDA drug prescribing warnings: Is the black box half empty or half full? Pharmacoepidemiology and Drug Safety 15(6):369–386.
Wakefield DS, Wakefield BJ, Borders T, Uden-Holman T, Blegen M, Vaughn T. 1999. Understanding and comparing differences in reported medication administration error rates. American Journal of Medical Quality 14(2):73–80.
Walls CA, Rhodes KV, Kennedy JJ. 2002. The emergency department as usual source of medical care: Estimates from the 1998 National Health Interview Survey. Academic Emergency Medicine 9(11):1140–1145.
Walters PG. 1992. FDA’s new drug evaluation process: A general overview. Journal of Public Health and Dentistry 52:333–337.
Watzke HH, Forberg E, Svolba G, Jimenez-Boj E, Krinninger B. 2000. A prospective controlled trial comparing weekly self-testing and self-dosing with the standard management of patients on stable oral anticoagulation. Thrombosis and Haemostasis 83(5): 661–665.
Wazana A. 2000. Physicians and the pharmaceutical industry: Is a gift ever just a gift? Journal of the American Medical Association 283(3):373–380.
Weatherby LB, Walker AM, Fife D, Vervaet P, Klausner MA. 2001. Contraindicated medications dispensed with cisapride: Temporal trends in relation to the sending of “Dear doctor” letters. Pharmacoepidemiology and Drug Safety 10:210–218.
Weatherby LB, Nordstrom BL, Fife D, Walker AM. 2002. The impact of wording in “Dear doctor” letters and in black box labels. Clinical Pharmacology and Therapeutics 72(6):735–742.
Wilkes MS, Bell RA, Kravitz RL. 2000. Direct-to-consumer prescription drug advertising: Trends, impact, and implications. Health Affairs 19:110–128.
Woods A, Johnson SD. 2002. Executive summary: Toward a taxonomy of nursing practice errors. Nursing Management 33(10):45–48.























































