
3
The Ecology of Pathogenesis
OVERVIEW
Over the course of the last century, the identification of increasing numbers of microbial pathogens and the characterization of the diseases they cause has begun to reveal the extraordinary complexity and individuality of host-pathogen relationships. The vast majority of microbes does not produce overt illness in their hosts, but instead act as persistent colonists. Genetic changes in either host or microbe may disrupt this equilibrium and shift the relationship toward pathogenesis, resulting in illness and possibly death for the host. These considerations are reflected in the contributed papers collected in this chapter, which explore how pathogens coexist within host-microbial communities, placing infectious disease within an ecological and evolutionary context.
In this relatively recent, dynamic model of pathogenesis, it has become exceedingly difficult to identify what makes a microbe a pathogen. This challenge is taken up at the beginning of the chapter by Stanley Falkow, who describes the variety of circumstances that lead to pathogenesis. Recognizing that a considerable amount of infectious disease is caused by “accidents” of transmission, susceptibility, and host response to microbes, Falkow illustrates how primary pathogens pursue an adaptive strategy that produces disease in normal hosts. This perspective informs an ecological model of pathogenicity as a product of ongoing evolution between pathogen and host.
The chapter continues with a detailed exploration of a single human pathogen, the bacterium Helicobacter pylori, which is strongly associated with increased risk for peptic ulcer disease and gastric cancer. Martin Blaser considers this example of amphibiosis—a term coined decades ago by microbial ecologist
Theodore Rosebury to describe a relationship between two life forms that is either symbiotic or parasitic, depending on the context—as representative of most relationships between humans and their indigenous organisms. The intricate signaling that occurs between humans and H. pylori has provided important insight on the effects of indigenous microbes on normal human physiology, as well as on disease. It also raises questions about the consequences of the disappearance of H. pylori (and other less-detectable indigenous bacteria) from the human gastrointestinal tract, a trend apparently underway in the industrialized world.
In contrast to the sole human-microbe interaction known to produce peptic ulcers, a broad range of normal luminal bacteria can induce and perpetuate intestinal inflammation (and possibly extraintestinal inflammatory conditions such as arthritis) in genetically susceptible hosts. Balfour Sartor’s contribution to this chapter describes bacterial factors and genetically programmed host responses that influence whether the host’s response to commensal bacteria is one of coexistence or of aggressive defense via inflammation, as occurs in idiopathic inflammatory bowel disease (IBD), Crohn’s disease, and ulcerative colitis. Greater understanding of the mechanisms of induction and perpetuation of intestinal inflammation may indicate how these responses could be inhibited in order to restore mucosal homeostasis, and how therapies for these conditions might be tailored to individual patients.
The chapter concludes with further reflections on the human microbiome by Maria Dominguez-Bello, who notes two promising areas for continued research on host-microbe ecology. The first is the rumen, which she portrays as a model system of host-microbe mutualism and the subject of seminal studies on digestive processes in humans and other animals. The second research area—which follows from Blaser’s aforementioned observation that modern life is changing the human microbiota—is the comparative study of indigenous microbes in human populations outside the industrialized world. To this end, Dominguez-Bello describes her own work among indigenous Venezuelan Amerindian tribes that examines the association between microbiome diversity and human health.
THE ZEN OF PATHOGENICITY
Stanley Falkow
Stanford University
The following remarks are meant to present a human’s idea of the microbe’s “point-of-view” and the various ways that a microorganism might cause disease. In so doing, I will offer a view of host-pathogen relationships that is in keeping with the goal of this workshop of replacing the war metaphor. As a first example of the intricacies of such relationships, consider a host macrophage engulfing the plague bacillus (Yersinia pestis), as shown in Figure 3-1. To many, this apparently defensive moment represents the essence of the host-parasite relationship.
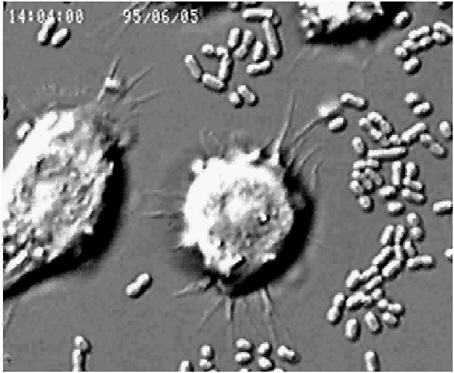
FIGURE 3-1 Host macrophage engulfing the plague bacillus (Yersinia pestis).
SOURCE: Falkow (2005).
In the end, the ingested bacteria are killed, but so is the host cell. There are more bacteria than host cells, and often the microbe has the last laugh.
The impact of microbes on humans extends to our culture (Cockburn, 1971; McNeill, 1976). Figure 3-2 shows variations in the human population over several centuries and notes the factors driving these changes, which are dominated by disease and war. The graph reveals an interesting trend: peaks of human population, followed by die-offs due to infectious diseases, followed by renaissance periods of peak human productivity in the resulting population valleys. If this pattern continues to hold, then we are approaching a time of epidemics, of which HIV/AIDS may be considered a harbinger.
Throughout most of our history, people have lived in small groups. Most known epidemic infections of humans require populations of 50,000 to 100,000 to spread; therefore, the diseases that are strictly adapted to our species are relatively recent in the evolutionary sense. The development of pathogens specific to humans was further encouraged by the crowding, defective hygiene, and poor nutrition associated with the transition to living in larger communities. In addition, the domestication of animals has permitted the evolution of infectious diseases such as measles (from canine distemper and rinderpest), diphtheria (from
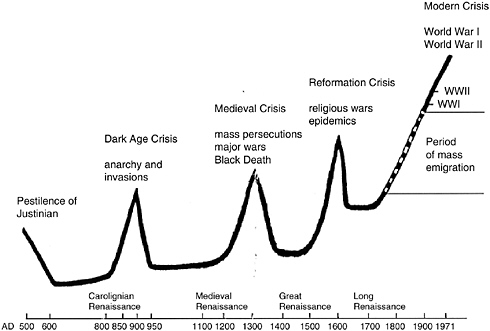
FIGURE 3-2 Infectious diseases were, and still are, the most common cause of death worldwide.
SOURCE: Falkow (2005).
cows), and influenza (from birds). Humans over time have become the preferred hosts of several species of bacteria, viruses and, in many parts of the world, worms. We really are a fertile landscape of opportunity for microbes.
What Is a Pathogen?
Once it was understood that microbes caused disease, the term pathogen was coined to describe any disease-causing organism, and this is the term still employed in medicine. Although this definition is certainly practical, it may not be entirely accurate from a biological—that is, the microbe’s—standpoint. As I see it, humans play host to the following categories of microbes:
-
Transients: Microbes that we ingest and with which we come in contact, but which do not persist on us. They are just “passing through.”
-
Commensals: (to eat from the same table): The hundreds of microbial species growing within each of us. Many of them are with us from the moment of birth, when we are first colonized by microbes, and remain with us to the grave. Some commensal species play essential roles in the development of our gas-
-
trointestinal tract and immune system. Their last role is to participate in our decomposition (think of it as recycling) after we die.
-
Pathogens: I use the term primary pathogen (which is not a universally accepted term) to describe microbes that cause disease in apparently normal human hosts. By contrast, opportunistic pathogens cause disease only in compromised human hosts. Commensals can be opportunists, and an opportunist in one host can be a primary pathogen in another host.
Differentiating between pathogens and commensals is important to devising new ways to thwart off attacks by pathogens. However, this is not an easy distinction to make because of the considerable similarities between the colonization strategies used by both pathogens and commensals. Successful microbes of both classes must find ways to enter their hosts (us), locate a niche to colonize, interact intimately with the host, replicate, persist (although this may not be necessary for pathogens), disseminate to new hosts, and, ultimately, evolve.
I believe that the major difference between pathogens and commensals is that pathogens have the inherent ability to cross anatomic and biochemical barriers in the host that serve to limit colonization by commensals. Most pathogens establish themselves in niches devoid of other microorganisms. For example, E. coli that cause diarrheal disease and urinary tract infections do not occupy the same niche as the commensal strains of E. coli in the colon. Instead, enterohemorrhagic E. coli specifically colonize a special niche within the colon near the lymphatic centers, while the urinary tract pathogens leave the colon altogether for another mucosal surface. Pathogens have the inherent ability to colonize these host-protected niches, and the invasive properties pathogens use to get there are essential to their survival in nature and are often host specific. This adaptive strategy, or lifestyle, is specific to primary microbial pathogens and absent from opportunists.
A considerable amount of human infectious disease is not caused by primary pathogens. Some infectious diseases—such as Lyme disease, rabies, and botulism—occur in humans through accidents of transmissibility, when we are infected by microbes that are specific to other host species. Most potential bioterrorism agents fit this general description. In these cases, disease is a disadvantage to both the host and the microbe, because the host often dies before the microbe can be transmitted to another host. Other human infections are accidents of susceptibility, which result from noninherent defects in host immunity. These defects could be either mechanical or biochemical, but in either case allow commensals that were previously held in place by normal host defense mechanisms to cross the barriers that contain them. Finally, there are human diseases that represent accidents of host response, or heritable defects in host immunity like that seen in cystic fibrosis.
Although we can get disease from a variety of microorganisms, I would argue that only some of them are primary pathogens. This subset of disease-causing
microorganisms has evolved specifically to cross host barriers in order to survive, and in crossing host barriers they often cause injury. The virulence factors that allow pathogens to pursue this strategy are equivalent to the fur, claws, and fangs of higher animals: adaptations to an organism’s environment that favor survival. Pathogenicity in this instance reflects the ongoing evolution between a parasite and host, and disease is the product of a microbial adaptive strategy for survival. Moreover, what we recognize clinically as disease is very often our human response to microbial invasion. In some cases, disease is a host-microbe experiment gone wrong, and when that happens, the host is as likely to be “at fault” in causing disease as the microbe.
Host Defenses
Animals have evolved both innate and adaptive immune mechanisms to avoid microbial intrusion or traumatic insult. The main function of the innate immune system is to keep commensal or transient organisms in their place—that is, to limit their ability to colonize host tissues beyond the mucosal surface. Immune signal cascades mediated by toll-like receptors (TLRs), which specifically recognize molecules characteristic of prokaryotes and other kinds of parasites, alert the host to microbial intrusion. The features recognized by TLRs are not unique to pathogens, but they set off an elaborate cascade that can summon cells with antibacterial characteristics, such as the macrophages that are recruited to ingest the Yersinia in Figure 3-1. Invading microbes without specific mechanisms to avoid the innate immune system are destroyed. Pathogens must possess inherent ways to avoid or subvert these innate host defense mechanisms.
Adaptive immunity, or the ability to produce antibodies that can neutralize microbes, occurs in higher animals. The capacity of humans to make antibody is of course critical to the effectiveness of vaccines. If we know how to stimulate production of antibody against a particular antigen, it is relatively easy to develop a vaccine against an organism that invades the bloodstream. However, it is much more difficult to make vaccines directed against microbes that inhabit the mucosal surfaces (e.g., the gut). Thus, the development of mucosal vaccines has been a great challenge, particularly when one considers the huge differences in individual susceptibility to infection between, for example, a normal neonate and one that has a low birth weight, is preterm, or is otherwise physically compromised. Some pathogens have devised ways to get around the adaptive immune system by constantly changing their surface or by camouflaging their appearance.
Steps to Successful Pathogenesis
The following section describes the challenges faced by pathogens, and the various mechanisms they have evolved to address them.
Entry
There are 9 natural portals of microbial entry in humans (10, if the umbilicus is included); along with injured tissues, these are the typical routes of entry for pathogens. The skin and the mucosal surfaces present formidable physical barriers to microbes: tight cell junctions; the mucus “stream” that flows through the gut, capturing organisms washing them out of the body; and antimicrobial substances. All of these obstacles, along with competition from commensals, stand in the way of the microbe.
Colonization
Following entry, the microbe must find a niche to inhabit. This process is often called colonization, and the microbe usually achieves it by attaching itself to a unique host cellular target, or by adapting to host physiology. Bacteria have hairlike structures called pili that have evolved to recognize and adhere to a specific receptor on the surface of the host cell. The various forms of pili have been studied extensively in the uropathogenic strains of E. coli. Genomic analysis of Salmonella reveals at least eight different kinds of pili that the bacterium uses to find targets for attachment in different contexts, both inside and outside the host’s body.
Microbes are very simple creatures, but they can perform the tasks of recognition at their surfaces through either proteins or proteinaceous appendages. The bacterial strain that causes salmonellosis, for example, recognizes specific kinds of cells in the terminal ileum.1 Protein-based adherence mechanisms are also found in viruses; herpesviruses, for example, have proteins on their surfaces that recognize specific receptors on host cells.
Persistence
Many common pathogens are organisms such as Pneumococcus, group A Streptococcus, Meningococcus, and Hemophilus influenzae that all humans carry at some point in their lives. These microbes occasionally cause clinically apparent disease, because they have an adaptive strategy of persistence achieved through mechanisms that avoid, circumvent, or subvert host defense mechanisms. One of the common mechanisms bacteria use is encapsulation, as occurs in Streptococcus pneumoniae (the most common cause of bacterial pneumonia) and Ba-
cillus anthracis (anthrax). The capsule, made of carbohydrate or protein, surrounds the microorganism and interferes with phagocytosis, much as the slimy coating on a wet bar of soap makes it hard to grab. Without a capsule, S. pneumoniae is essentially avirulent; it can live in the human host, but it is very unlikely to cause disease.
Another means to persistence is to breach the tight junctions between epithelial cells or to get inside the cells themselves. Salmonella and Shigella can stimulate human epithelial cells and cause them to extend cytoplasmic ruffles that act to trap the bacteria and literally pull them into the cell. Other bacteria harness the cytoskeleton; the organism that causes enterohemorrhagic E. coli reorganizes the cytoskeletal actin of its host cell in the colon epithelium into a sort of pedestal. The bacterium sticks avidly to this pedestal, where it is bathed with host cell nutrients that the microbe uses for growth. The attached bacteria replicate to form microcolonies on the surface of the host cell.
Yet, other pathogens persist in their hosts by circumventing the intracellular trafficking that allows host phagocytes to take up commensal bacteria or other kinds of particles, put them into vacuoles, and digest them. In some cases, the digested products trigger the production of antibodies by the host’s immune system. Some pathogenic bacteria divert normal trafficking by phagocytes; others (e.g., Yersinia) kill the phagocyte quickly after they attach or have been ingested.
Enzymes and toxins allow pathogenic bacteria to spread through local tissues and perturb host immune function. Group A Streptococci secrete substances that disintegrate the molecular “cement” between cells—hyaluronic acid—so that the bacteria slip easily along the tissue plane. Other secreted streptococcal toxins kill host cells. The released host cell contents form a viscous mass that might impair the ability of bacteria to spread through the tissues, but the bacteria make a DNase that dissolves this barrier. Thus, the microbe releases a series of substances exquisitely timed to permit the microbe to spread, replicate, and persist in an environment that is normally lethal for other microorganisms.
Replication
What do microbes gain from host-pathogen relationships? Replication. The successful microbe is one that can replicate sufficiently to be transmitted to a new susceptible host. What does the host gain from its associations with microbes? Usually immunity sufficient to clear the immediate threat and, in the best of circumstances, forevermore.
Some pathogens persistently colonize their hosts and continuously transmit small numbers of their kind into the host’s environment. Under these conditions, disease represents an option, not a goal, for a pathogen. It is merely one means to the end of replication, not a necessary outcome of colonization. When an organism causes disease, it is but a byproduct of its survival strategy. And, disease is often as much a factor of the state of the host defenses as it is of the microbes’ need to breach host barriers.
For example, while many people become ill in influenza outbreaks, even more people escape disease. In seeking to understand host-pathogen relationships, disease can distract us from understanding the actual mechanisms that underlie these associations. However, we must understand how pathogens operate in order to devise new ways to protect ourselves from their ill effects.
Dissemination
Bacteria need an exit strategy from their hosts. Microbes are conveyed between humans in a variety of ways: through respiratory droplets or saliva, fecaloral transmission, and even in our lovemaking. Zoonotic infections, which rarely spread person-to-person, are transmitted through animal bites, feces, and insect vectors (ticks, fleas, etc.).
Even organisms such as Salmonella typhi, Mycobacterium tuberculosis, and Helicobacter pylori, which cause lifelong infections, must eventually find new hosts. The ease and the frequency with which a pathogen leaves its host may influence its virulence.
The Roles and Origin of Bacterial Toxins
A particularly fascinating aspect of bacterial evolution is the existence of toxins. Vibrio cholerae, for example, produces an extraordinarily powerful and well-known toxin that causes patients with cholera to pass copious amounts of watery stools. These patients survive only if the number of liters of fluid they lose can be balanced with the same volume of fluid going in. Humans do not carry the cholera bacterium; its reservoir is probably in marine estuaries.
Toxins appear to perform a variety of functions for bacteria, including nutrient acquisition, the breakdown of anatomic barriers, facilitation of exit and transmission, and the modulation of immune function. It seems unlikely that the original purpose of toxins was to poison mammals. Thus, if we can put aside our desire to avoid cholera or botulism or other toxic bacterial infections, and instead consider what advantages toxins confer upon bacteria, we may come up with a better way to neutralize toxin-producing pathogens.
I suspect that bacterial toxins first evolved in compost heaps when bacteria came in contact with predatory amoeba and especially with nematodes. Pound for pound, nematodes eat more bacteria than any other organism. To avoid becoming prey, bacteria harmed their predators. Thus, the toxins that bacteria initially evolved to avoid phagocytosis by amoebae became—after millions of years of evolution—the same molecules that stop the human phagocyte from engulfing bacteria.
Finally, it has been said of bacterial toxins that we incriminate the microbe for the sins of the viruses (Hayes, 1968). Many toxins are encoded by accessory genetic elements, and the advantage they confer upon the organism that possesses
them is unknown. We might be able to get answers to such questions by comparing bacteria such as Bacillus anthracis, which carries a DNA insert known as a pathogenicity island (further discussed below), and its close relative Bacillus cereus, which does not carry a pathogenicity island. Such studies could also provide new insights on protecting ourselves from anthrax.
Corollaries of Pathogenicity
A better understanding of the previously described steps that pathogens must take in order to live successfully in their hosts could result in the search for new ways to protect humans from infectious diseases. Such efforts should also address the following “corollaries” of pathogen behavior, as revealed through microbiological research.
Bacteria Have Keen Senses
Pathogens and other bacterial specialists (almost all bacteria are specialists) use elaborate regulatory mechanisms based on environmental and biochemical cues. Bacteria have extremely sensitive chemical sensory systems that measure environmental variables such as oxygen and carbon dioxide concentration and pH. Significant changes in these measures signal the microbe to alter the products it makes to suit its environment. Such adjustments allow simple organisms to survive transitions.
When a bacterium in a bit of feces on the ground moves into a human’s mouth, it undergoes an enormous change in pH and comes in contact with lysozyme and a variety of other antibacterial chemicals. Then the ingested bacterium is plunged into the acid vat of the stomach and quickly passes into the small bowel, where it is bathed in bile and an impressive array of digestive enzymes, all along being propelled by the forces of peristalsis. The bacterium takes all of this in stride and responds to each change in environment by rapid changes in its own structure and metabolism. These constantly changing environmental cues permit many microbes to anticipate when they will reach their optimal niche. Thus, Salmonella uses changes in pH, the viscosity of intestinal mucus, and the concentration of oxygen adjacent to host epithelial cells to determine when it will encounter its target cell in the terminal ileum. The bacterium quickly readies itself for infection during its transition through the gut. Clearly, it can respond far faster to us than we can to it.
Pathogens Are Opportunists
As Walt Kelly’s comic strip hero, Pogo, said, “We have met the enemy, and he is us.” Pathogens can respond to a host’s biological and social behavior (Falkow, 1998). Many pathogens that coexist uneventfully with other hosts cause
disease when they encounter humans; for example, the Lyme bacterium (Borrelia burgdorferi) persists in mice, deer, and ticks, and only causes disease when it colonizes humans or dogs. These hosts represent new “opportunities” for such microbes, and unfortunately may result in new diseases for hosts that (unlike mice, deer, and ticks) have not evolved a relationship with a given pathogen.
Similar scenarios involving human behavior underlie several recent infectious crises, including Legionnaire’s disease and toxic shock syndrome. The increasing presence of aerosols in our environment—created by taking showers rather than sitting demurely in a bathtub, by air conditioning, by misting of produce in supermarkets—has provided new opportunities for organisms to spread in a “modern” world. The bacterium Legionella, for example, lives primarily in amoebae in an aquatic environment. When amoebae are dispersed in aerosols that are inhaled by humans, they—along with their bacterial hitchhikers—are taken up by macrophages in the lung. Once inside a human cell, the bacterium replicates and causes a clinical pneumonia we call Legionnaire’s disease. Likewise, the response of American industry to women’s demand for more absorbent tampons, which gave them greater freedom during menstruation, resulted in a product that unexpectedly encouraged growth of certain staphylococci that typically exist in small numbers in women’s genital tracts. The regrettable result was the emergence of a new disease entity: toxic shock syndrome.
Bacteria Evolve Rapidly
The previous 20 years of research on bacteria reveal that they become pathogenic through a horizontal exchange of genetic information. In some ways microbial genomes comprise a genetic information network, operating much like the Internet. Mobile genetic elements such as plasmids, phages, and transposons are constantly streaming among microorganisms as they contact each other or enter into new environments. This elementary form of gene transfer enables microbes—often quite diverse ones—to share information with one another (Dobrindt et al., 2004).
For example, as described by Martin Blaser and colleagues (see paper in this chapter), there are two types of H. pylori: one that is associated with gastritis, and another that is associated with peptic ulcer and gastric cancer. The difference between the two types is that the more pathogenic type contains an insertion of DNA that is called a pathogenicity island. This extra DNA encodes a protein that leads to cancer and ulcers in some human hosts, although exactly how it does that remains to be determined. It is known that such islands of genes often allow bacteria to synthesize surface proteins that function much like a hypodermic needle (type III secretion). This structure makes contact with host cells and permits the bacteria to deliver other proteins, called effectors, directly into the host cell. The injected effector proteins have the ability to change or capture or manipulate normal host functions to the microbe’s favor. This apparently ancient and relatively common pathogenic mechanism is present in plant pathogens as
well as in those infecting animals and humans, but this remarkable microbial attribute has only been revealed and appreciated within the past decade or so.
In the larger context of host-microbe interactions, the exchange of genetic information in packets of DNA—transferred by direct contact, or through bacterial viruses, or even through the uptake of naked DNA—has enabled bacteria to experiment with their environment. In some cases, these experiments have led microbes to develop survival mechanisms that we see as pathogenicity and disease. However, it is also clear from genomics and other research that this genetic process has produced a variety of nonpathogenic microbial adaptations; for example, nitrogen fixation benefits microbes as well as crop plants (and therefore, also humans).
Conclusion
We have tried to control infectious disease with antibiotics and vaccines. Every time we attempt to control microbes by killing them, it provides them with a strong genetic selection for resistant organisms. There has been a continuous dialog over the past 50 years in regard to the problem of antimicrobial resistance. If we are going to address the problem of antibiotic resistance, I believe we will need to take a new, more sensible approach to the problem. Moreover, we will need to address the fact that pathogenicity in all microbes—not just bacteria—can occur in big genetic jumps, rather than through slow, adaptive evolution; HIV is a case in point. We also must reflect upon the lesson of bacterial genomics: many pathogens adapted to humans are relatively recent in evolution. We have to understand that the host-pathogen interaction is a dynamic process, and there are likely to be surprises in our future.
The mapping of the human genome and of bacterial genomes provides the means to better understand host-microbe relationships. In the context of infectious diseases, it is likely to be just as important to learn about the host as the microbe. For example, a single gene makes a dramatic difference in the susceptibility of mice to Salmonella; those that have the gene are resistant, and those that do not have it die upon infection (Govoni and Gros, 1998). Also, recent findings associate different human lymphocyte antigen (HLA) types in humans with susceptibility or resistance to infectious diseases (Segal and Hill, 2003). In the future, I expect that it will be possible to quickly genotype patients with infectious disease and understand how to moderate and mitigate therapy according to an individual’s genetically determined risk. This ability should also lead to new ideas for therapies for infectious diseases.
I will conclude with a story about my experience researching plague many years ago. One summer, the fleas we used to study Yersinia were not doing well. When we inspected the fleas under the microscope, we discovered that they had mites. Then we examined the mites and found that they themselves had little mites, upon which there were microbes (see Figure 3-3).
FIGURE 3-3 Illustration of the old rhyme (based on a poem by Jonathan Swift, author of Gulliver’s Travels2), “Big fleas have little fleas upon their backs to bite them, and little fleas have lesser fleas and, so on, ad infinitum.” We need to keep that idea in mind today, along with the fact that, in the end, microbes always have the last laugh.
SOURCE: Falkow (2005).
|
2 |
Swift actually wrote, “So, naturalists observe, a flea Has smaller fleas that on him prey; And these have smaller still to bite ’em; And so proceed ad infinitum.” Swift J. 1733. Poetry, a Rhapsody. [Online]. Available: http://www.online-literature.com/quotes/quotation_search.php?author=Jonathan%20Swiftl. |
Acknowledgments
I wish to thank Alison Mack and Eileen Choffnes, who made a rambling talk into prose. Many of the ideas presented here were previously published in several review articles that contain references to the following primary literature:
Falkow S. 1997. What is a pathogen? American Society for Microbiology News 63:359–365.
Falkow S. 1998. The microbe’s view of infection. Annals of Internal Medicine 129(3):247–248.
Finlay BB, Falkow S. 1997. Common themes in microbial pathogenicity revisited. Microbiology and Molecular Review 61(2):136–139.
Monack DM, Mueller A, Falkow S. 1994. Persistent bacterial infections: The interface of the pathogen and the host immune system. Nature Reviews Microbiology 2(9):747–765.
PATHOGENICITY AND SYMBIOSIS: HUMAN GASTRIC COLONIZATION BY HELICOBACTER PYLORI AS A MODEL SYSTEM OF AMPHIBIOSIS
Martin J. Blaser3
Amphibiosis, a term coined by the microbial ecologist Theodore Rosebury about 50 years ago (Rosebury, 1962), is the biological condition in which the relationship between two life forms is either symbiotic or parasitic, depending on the context. This is a more precise term than commensalism and describes a more complex relationship than symbiosis. It is my hypothesis that amphibiosis best describes most relationships between humans and their indigenous organisms. A second hypothesis is that human gastric colonization by Helicobacter pylori is a model system for amphibiosis.
H. pylori as a Model of Our Indigenous Biota
H. pylori are curved gram-negative bacteria that live in the mucous layer of the human stomach (Blaser and Atherton, 2004). Most individuals acquire H. pylori early in life. It does not invade the gastric tissue, but lives in the mucous layer, persisting for the lifetime of the host in most cases (Hazell et al., 1986). Its very persistence is one of the hallmarks of amphibiosis. Although these organisms were first seen in the human stomach about 100 years ago, they were not isolated in pure culture by Warren and Marshall until in 1982 (Warren and Marshall, 1983). However, there is now extensive evidence that H. pylori has long been part of the human biosphere. From the population biology studies examining more than 300 H. pylori isolates from around the world (Falush et al., 2003), modern H. pylori populations are not clearly delineated (Figure 3-4A). H. pylori are naturally competent for transformation (Israel et al., 2000), and populations of organisms show evidence for extensive recombination (Suerbaum

FIGURE 3-4 Modern and ancient H. pylori populations, determined by applying the STRUCTURE algorithm to sequences in eight conserved H. pylori genes in > 300 contemporary isolates (Falush et al., 2003).
SOURCE: Blaser (2005).
et al., 1998). Using an algorithm to assign ancestry to each nucleotide, Falush et al. showed that all or essentially all modern H. pylori populations derive from five ancestral populations. It is likely two were originally African populations and two were strongly associated with Eurasia and one with East Asia (Figure 3-4B). Relating such data to known human migrations (Ghose et al., 2002), it has become clear that H. pylori has been present in human populations for at least
60,000 years. Indirect evidence also suggests that an ancestral H. pylori strain has been present in humans, and our predecessors, for millions of years (Blaser, 1998).
These studies, suggesting that we have shared much history, are consistent with a different model of human identity; the human body is home to more microbial cells than human cells (Savage, 1977) (Figure 3-5A). Further, since our indigenous and transient microbes are varied, they have been considered by Lederberg as our “microbiome” (Lederberg, 2000). I propose an integration of the human 46 chromosome genome and the extremely large microbiome to produce a metabolome (Figure 3-5B). In this conception, we, along with our major constituents, have been selected for coexistence, and our metabolic circuitry is comingled and synergistic. This idea can help us understand both normal physiology and disease, and our relationship with H. pylori provides an important probe (and test) of the underlying biology.
Whether the normal biota are useful or harmful has been debated for more than a century (Mackowiak, 1982). Pasteur, the “father” of microbiology, considered that the normal biota (flora) are essential to life. Metchnikoff, one of the founders of immunology, believed that normal flora are antagonistic and compete with the host. There is evidence that supports both view points (Mackowiak, 1982); in its aggregate, this is amphibiosis.
Signaling
In the relationship between host and colonizing microbes, an important interface is the signaling between host and microbe. A prototypic microbe on a mucosal surface, subject to being washed away by flow, is multiplying, producing metabolites and toxins, and possibly directly adhering to the epithelium (Blaser, 1997). Each of these activities may be considered as signals to the host. The host produces a physical and chemical milieu, its own metabolites, and defense molecules that also may be considered as signals to the microbes.
Signaling between microbe and host may be unlinked. This signaling pattern would be a mirror of warfare, with an arms race between microbe and host. Acute infection by a pathogen best fits this paradigm. The alternative model is that there is a linkage between microbial and host signals that have coevolved due to selective pressure on both host and microbe to coexist. This is the model that may be most appropriate for the indigenous biota, and the cross-signaling must be dynamic, reflecting variations induced by microbial diversity, host development and aging, and other environmental cofactors.
Our interactions with H. pylori illustrate several aspects of these issues (Figure 3-6). H. pylori colonization is mostly acquired early in life (Oliveira et al., 1994; Pérez-Pérez et al., 2003), with an early population bloom leading to the gradual development of immunity. The host recognizes and responds to the organism, causing the microbial population to diminish in number, and then ensues stable equilibrium lasting decades in which effective H. pylori multiplication and
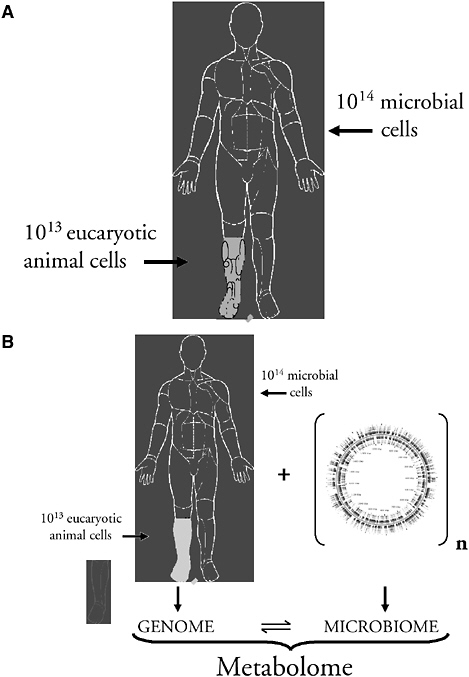
FIGURE 3-5 Who are we? A: Microbial cells outnumber all host cells in the human body. B: An integrated view of host-microbial interactions in the human body, constituting the metabolome. In this model, our indigenous microbes are as much a part of human physiology as is a recognized human organ, such as the liver.
SOURCE: Blaser (2005).
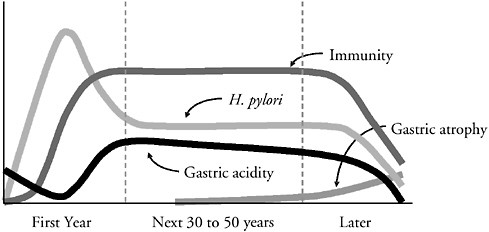
FIGURE 3-6 Schematic of the natural history of H. pylori populations and host characteristics in colonized individuals over their lifetime. This model is divided into an initial (transient) period, a long equilibrium, and a late decline. Host recognition and response (immunity) limits H. pylori populations, but the ongoing interaction progressively damages gastric tissues, leading to localized and then generalized gastric atrophy. As this continues, gastric acidity is reduced, H. pylori populations decline, and as they decline, host responses diminish. In this formulation, H. pylori may be a model for other persistent colonizers that both elicit host responses and induce tissue injury.
SOURCE: Blaser (2005).
host effector mechanisms are balanced. It is a stalemate, but with effects on gastric physiology (Calam, 1995; Moss and Calam, 1993). Over decades, the consequence of this slow inflammatory interaction is the gradual development of gastric atrophy (Kuipers et al., 1995a; Kuipers et al., 1995b). As atrophy increases, H. pylori populations fall, sometimes to zero, and the immune response subsequently falls as well (Karnes et al., 1991) (Figure 3-6).
Thus, there is an initial transient state, a long period of equilibrium, and then a late transitional state. A model for persistence includes both adherent and mucosal (free-living) H. pylori populations, nutrients derived from the host permeating the mucous layer, and a number of H. pylori-produced effectors that induce the flow of nutrients to feed the organism and allow them to multiply (Kirschner and Blaser, 1995). The essential feature of the model is a negative-feedback loop; this is the only way persistence can be modeled within appropriate parameters (Blaser and Kirschner, 1999; Kirschner and Blaser, 1995). The model is robust, encompassing a wide range of host, microbial, and interactive variation, but in the absence of feedback, no equilibrium can be achieved; microbial populations (MP) go to infinity or to zero. This linked signaling between coevolved species could represent a model for other persistent indigenous organisms as well.
A more specific version of the model is that H. pylori, the coevolved microbe living in the gastric lumen (Figure 3-7), is subject to a variety of stresses,
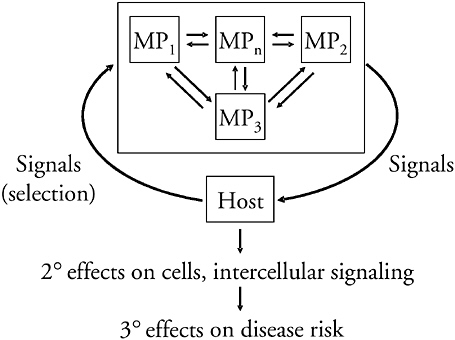
FIGURE 3-7 Equilibrium relationships between coevolved persistent microbes and their hosts. The colonizing microbe is considered as a group of highly related MP that differ in their expression of “contingency” genes, living in equilibrium with one another, with the exact relationships determined by the relative fitness of the MPs in relation to the (selecting) host signals. H. pylori serves as the model organism, and there are primary, secondary, and tertiary consequences of the relationship.
SOURCE: Blaser (2005).
including pH, nutrient limitation, and peristalsis. Organisms have been selected that signal specific host cells to produce effectors that abrogate the stress. Such a model represents an equilibrium relationship, but since both hosts and their H. pylori populations are unique (El-Omar et al., 2000; Israel et al., 2000; Kuipers et al., 2000; Machado et al., 2003), the exact relationships form unique equilibria, and in changing contexts, they form dynamic unique equilibria. My hypothesis is that this primary equilibrium affects secondary events (Blaser, 1992), including epithelial cell cycle, inflammatory responses, and neuroendocrine pathways (Blaser and Atherton, 2004), and that these in turn affect risk of disease (Figure 3-7).
The Disappearance of H. pylori
In developing countries, H. pylori is most often acquired in childhood; by the time adulthood is reached, virtually everyone is colonized by the organism
(Oliveira et al., 1994; Pérez-Pérez et al., 2003) (Figure 3-8), often with multiple strains (Ghose et al., 2005). In contrast, in developed countries such as the United States, H. pylori prevalence is much lower, reflecting a birth cohort phenomenon (Parsonnet, 1995; Rehnberg-Laiho et al., 2001). When current U.S. adults were children, there was much more H. pylori in circulation than now exists for our children. Since all developed countries were formerly developing countries, over time, the incidence of acquisition has gradually fallen (Parsonnet, 1995; Rehnberg-Laiho et al., 2001), and continues to fall (Pérez-Pérez, 2002). In fact, H. pylori is disappearing in developed countries. This seems puzzling. How can an organism that probably has been with humans for tens of thousands of years, if not longer, now be disappearing? In turn, this phenomenon raises two questions: why is H. pylori disappearing, and what are the consequences of its disappearance?
Why is H. pylori disappearing? Important factors probably include its less efficient transmission, based on cleaner water and smaller family size (Goodman and Correa, 2000; Klein et al., 1991). The transmission of microbes is a function of the population structure of human communities; one of the large demographic changes associated with recent socioeconomic development is that families are becoming smaller. Older siblings play an important role in H. pylori transmission (Goodman and Correa, 2000; Goodman et al., 1996), and there are now fewer children per family. Host survival advantage also may have been removed. If H. pylori protected against lethal diarrheal disease (Rothenbacher, 2000), and
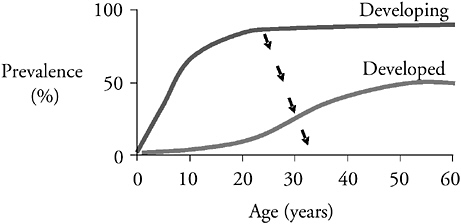
FIGURE 3-8 Prevalence of H. pylori in human populations in developing and developed countries. Because all developed countries were formerly developing countries, the prevalence of colonization should have shifted downward (↓) over the course of development, and much evidence supports this hypothesis. As socioeconomic development is proceeding, H. pylori is disappearing, especially from children.
SOURCE: Blaser (2005).
such illnesses are themselves diminished due to improved hygiene, then any positive selection for the presence of H. pylori may be removed. As overall H. pylori populations fall, there also is diminished ability for gene exchange, making the colonizers of an individual host less robust. Furthermore, we are more than 60 years into the antibiotic era. A course of amoxicillin may eliminate H. pylori 10–20 percent of the time (Rauws et al., 1988). This is insufficient for planned therapy to eradicate H. pylori, but if all the antibiotics that are given in childhood are summated, there is enormous pressure, not only on the development of resistance, but on the presence of H. pylori in individual hosts.
Consequences of H. pylori Disappearance
Regardless of why H. pylori is disappearing, probably for the first time in human history there are large numbers of adults who do not have the organism; thus, through clinical and epidemiologic comparisons, we can examine the consequences of colonization vis a vis its lack.
For persons colonized with H. pylori, within the gastric mucosa the lamina propria contains numerous lymphocytes and macrophages. Pathologists call this “chronic gastritis” (Harford et al., 2000; Tham et al., 2001). An alternative nomenclature is that these cells represent the “physiologic response to an indigenous organism.” This phenomenon may be considered an analogy to the lamina propria in the colon. We consider the colon to be normally colonized by bacteria and the colonic lamina propria is replete with inflammatory/immunologic (defense) cells. A germ-free colon shows few or no such cells in the lamina propria, but when the colon is “conventionalized,” there is infiltration of the lamina propria with host phagocytic and immunologic cells. Regardless of what we call the phenomenon, it is clear that the cells in the lamina propria reflect the presence of microbes in the mucous layer, clearly in the colon, and now apparently in the stomach as well (Blaser, 1990).
Why is that important? In 1975, Peleyo Correa developed a model of the pathway for the most important type of gastric carcinogenesis, involving adenocarcinoma of the stomach (Correa et al., 1975). In his model, there is a progression over decades, from normal gastric mucosa to chronic gastritis, atrophic gastritis, metaplasia, dysplasia, and then to cancer (Correa et al., 1975). A long series of investigations supports Correa’s hypothesis, but it was not known what caused the initial transition from normal gastric mucosa, to chronic gastritis. As indicated above, there is overwhelming evidence that the acquisition of H. pylori is the main cause for this transition (Blaser, 1990). Therefore, it was a reasonable hypothesis that the presence of H. pylori could be a risk factor for development of gastric cancer.
On the basis of both epidemiologic and animal challenge studies, it is now clear that colonization by H. pylori is an important risk factor for gastric cancer (IARC, 1994; Peek and Blaser, 2002). Thus, one of the biological costs of
Helicobacter pylori gastric colonization is that some hosts will develop gastric cancer. In certain ethnic groups, such gastric cancers might affect up to 5 percent of the population, mostly at older ages, in their 60s, 70s, and beyond, in a log-linear relationship with age. Clearly, as with other “environmental” carcinogens (e.g., tobacco smoke), the cost of carrying H. pylori is not absolute; not everyone gets gastric cancer. It is a risk relationship, with the greatest impact late in life.
In parallel studies, gastric colonization with H. pylori tripled or quadrupled the risk of peptic ulcer disease (Nomura et al., 1994; Nomura et al., 2002a). There are many data from retrospective (treatment) studies that indicate the same kind of relationship (Hentschel et al., 1993). The magnitude of the risk relationship between H. pylori and gastric cancer is greater than the relationship with ulcer disease.
Differences Among H. pylori Strains
H. pylori strains can be divided by the presence or absence of the cag island. First discovered based on the presence of the gene, cagA (Covacci et al., 1993; Cover et al., 1990; Crabtree et al., 1991; Tummuru et al., 1993), a marker for the presence of the rest of the island, this is a region of about 40 kilobases on the H. pylori chromosome (Tummuru et al., 1995; Censini et al., 1996; Akopyants et al., 1998). The cag island is flanked by direct DNA repeats, and includes multiple direct DNA repeats (Aras et al., 2003a; Tomb et al., 1997); it is a metastable genetic element subject to partial or total deletion events (Censini et al., 1996), but through recombination the island also can be restored to a negative strain (Kersulyte et al., 1999). The cag island contains type IV secretion system (TFSS) genes that translocate the CagA protein from Helicobacter cells into gastric epithelial cells (Hatakeyama, 2004; Odenbreit et al., 2000). There are sites on the CagA protein that undergo tyrosine phosphorylation by host cell Src-like kinases, and then this phosphoprotein interacts with several cellular signal transduction pathways (Amieva et al., 2003; Higashi et al., 2001; Naumann et al., 1999). I had previously postulated that there might be strong signals from H. pylori cells to the host (Blaser, 1992), and that these signals provide the substrate for the feedback relationship (Blaser and Kirschner, 1999; Kirschner and Blaser, 1995). In fact, the injection of the CagA protein is a very strong signal that H. pylori provides to host cells; recent data indicate that much of the interaction between H. pylori and host cells involves cag island genes (Segal et al., 1999; Viala et al., 2004).
Because we can differentiate between two groups of helicobacters, cag positive and cag negative, it is next reasonable to ask whether there is differential risk of disease in persons carrying one or the other strain. Studies now have shown 50 to 300 percent increases in risk of gastric cancer in persons with cag-positive strains compared to those with cag-negative strains (Blaser et al., 1995a; Nomura et al., 2002b; Parsonnet et al., 1997). How do cag-positive strains increase risk? There have been well over 100 studies that show effects of cag-
positivity on proinflammatory cytokines, tissue infiltration with inflammatory cells, epithelial cell cycle events, and intracellular signaling pathways (Blaser and Atherton, 2004; Hatakeyama, 2004; Peek and Blaser, 2002).
One of the hypotheses of how H. pylori colonization leads to gastric cancer risk is that the organism induces a variety of host responses that are affected by strain, host, or cofactor differences; there is evidence that each of these factors affects cancer risk (Blaser et al., 1995b; El-Omar et al., 2000; Machado et al., 2003). For example, host characteristics affect risk; particular proinflammatory genotypes drive the interactions toward atrophic gastritis, leading to effects on cell cycle and mutation leading to adenocarcinoma (El-Omar et al., 2000; Machado et al., 2003). Although this conception is the major thrust of current investigation, there is an alternative hypothesis (Figure 3-9). Atrophic gastritis, per se, alters the gastric environment, allowing for changes in gastric microecology. The development of atrophic gastritis can permit new organisms to colonize the stomach; it may be that these microecologic changes are driving toward adenocarcinoma. Thus, whether the H. pylori-induced increases in cancer risk are due to direct effects on the tissue or indirect, due to replacement of the biota, are two testable hypotheses.
Since H. pylori is a risk factor for gastric cancer (IARC, 1994; Peek and Blaser, 2002), and H. pylori is disappearing, gastric cancer should be disappearing. In fact, in virtually every developed country studied, 20th century gastric cancer rates are declining (Howson et al., 1986); it now is clear that the disappearance of H. pylori is responsible for at least a part, if not the major part, of this phenomenon. This provides a strong justification for medical attempts to make all humans Helicobacter free.
Esophageal Diseases
However, new cancers continue to arise. Adenocarcinoma of the esophagus has dramatically increased in the United States since at least 1970 (Devessa et al., 1998); there has been a six-fold rise over the past 25 years (Pohl and Gilbert, 2005). The evidence is clear that this is not due to a surveillance artifact. In fact, adenocarcinoma of the esophagus is the most rapidly increasing cancer in the United States, and there are data from England, Scandinavia, and Australia that confirm the rapid rise in the incidence of this tumor, and of the closely related, if not identical, adenocarcinoma of the gastric cardia. It is likely that these gastroesophageal (GE) junction tumors represent a single disease.
Why are these cancers rising at the same time that gastric cancer is falling? It is clear that the pathway for the development of these cancers relates to a condition called gastroesophageal reflux disease (GERD) (Lagergren et al., 1999), which can then lead to a metaplastic process called Barrett’s esophagus, which can lead to dysplasia and adenocarcinoma; this process may take 20 to 40 years.
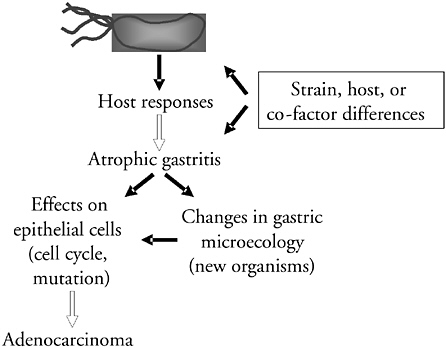
FIGURE 3-9 Potential pathways by which H. pylori colonization increases risk of gastric cancer. The mainstream of current research suggests that the interaction of H. pylori and host cells per se, as affected by strain, host, and environmental differences, leads to atrophic gastritis, and then to cancer. An alternative model is that the critical dimension of the H. pylori-host interactions is that they lead to the development of atrophic gastritis, and that with atrophic gastritis, there is expansion of other MPs, or replacement by microbes that cannot survive well in the normal high-acid H. pylori-rich stomach. It may be these other microbes that, through greater tissue injury and a more enhanced proinflammatory interaction, provide the direct oncogenic signals.
SOURCE: Blaser (2005).
Reflux esophagitis was first described in the medical literature in the 1930s, and Barrett’s esophagus was first described by Dr. Norman Barrett, a surgeon, in 1950.
Are these diseases related to the disappearance of H. pylori? In a series of clinical and epidemiologic studies, we and others asked two questions: what is the state of the esophagus, and how does that relate to the presence of H. pylori? A number of studies now have shown that people who have esophageal disease are less often colonized with H. pylori than those with a normal esophagus (Chow et al., 1998; Loffeld et al., 2000; Vaezi et al., 2000; Vicari et al., 1998). Because we showed that cagA-positive strains are more interactive than cagA-negative strains, we particularly looked at cagA status. In a study of Dutch patients, we asked who carries a cag-positive strain. The results showed that for persons with a normal esophagus, it was 33 percent, for hiatal hernia, 14 percent; esophagitis,
16 percent; and Barrett’s, 6 percent (Loffeld et al., 2000). In these cross-sectional studies, as the groups are moving nosologically from normal esophagus toward malignancy, cag-positive strains were becoming less prevalent. Consequently, we performed studies to examine directly whether there is a relationship between the presence of Helicobacter, especially cag-positive strains, and cancers of the GE junction. In a study of 130 cancer cases, and 220 controls, compared to the reference Helicobacter-negative group, the odds ratio for cag-negative strains was 1.1, but the odds ratio was 0.4 for cag-positive strains (Chow et al., 1998). Thus, an inverse association was observed for the presence of cag-positive strains and adenocarcinoma of the esophagus, the disease that is rising so rapidly. There have been several independent confirmations of these phenomena, concerning GERD, Barrett’s, and adenocarcinoma (de Martel, 2005; Queiroz et al., 2004; Warburton-Timmsa et al., 2001; Ye et al., 2004).
A study from Sweden by Ye and colleagues showed an odds ratio of 0.2 for cag-positive strains (Ye et al., 2004). Recently de Martel and colleagues examined the Kaiser cohort of 130,000 people. For H. pylori positive persons, the odds ratio associated with developing esophageal adenocarcinoma was 0.37 (de Martel, 2005). Again, there was an inverse relationship between esophageal adenocarcinoma and H. pylori in this prospective study, although they did not show any differential effect for cagA. There now have been several epidemiologic studies showing that the presence of Helicobacter has an inverse association with adenocarcinomas involving the GE junction and its precursor lesions (Chow et al., 1998; de Martel, 2005; Peek et al., 1999; Ye et al., 2004); one study that did not show a significant association may have had technical artifacts (Wu et al., 2003).
H. pylori Strain Type and Risk of Disease by Location
The data suggest that in relation to diseases of the lower stomach and esophagus, such as ulcer disease and gastric cancer, carrying a cag-positive strain increases risk of disease (Blaser, 1999). Carrying a cag-negative strain enhances risk but to a much lower extent (Parsonnet et al., 1997). However, considering diseases of the upper stomach and esophagus, such as GERD, Barrett’s, and adenocarcinoma, having a cag-positive strain appears to be protective, and cag-negative strains are relatively neutral (Chow et al., 1998; Loffeld et al., 2000; Queiroz et al., 2004; Vaezi et al., 2000; Vicari et al., 1998; Warburton-Timmsa et al., 2001; Ye et al., 2004). As such, most of the disease relationship concerns cag-positive strains, which is not surprising, since they are the more interactive with the host (Amieva et al., 2003; Hatakeyama, 2004; Higashi et al., 2001; Naumann et al., 1999; Odenbreit et al., 2000; Segal et al., 1999).
Although the mechanisms involved may be complex, Yamaji and colleagues studied more than 6,000 patients who underwent endoscopy in Japan (Yamaji et al., 2001). The investigators categorized the subjects by both Helicobacter and pepsinogen status to create a scale ranging from a relatively normal stomach to
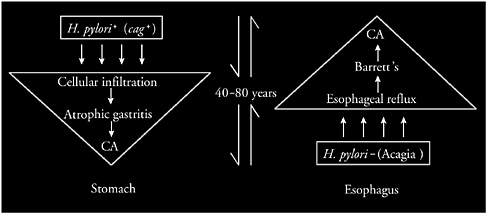
FIGURE 3-10 Proposed reciprocal relationship between adenocarcinomas of the stomach and esophagus. H. pylori colonization initiates a multidecade sequence of events leading to atrophic gastritis and ultimately to gastric cancer. Conversely, the absence of H. pylori, especially of cag-positive strains (a phenomenon that I have termed acagia), predisposes to GERD and sequelae, increasing risk of esophageal adenocarcinoma, a process that also requires multiple decades.
SOURCE: Blaser (2005).
atrophy. For individuals in categories progressively related to atrophy, gastric cancer prevalence went up, and GERD went down. Conversely, moving from atrophy to normal on the scale was associated with increased reflux and decreased gastric cancer prevalence.
As such, I propose that there is a reciprocal relationship between adenocarcinomas of the stomach and the esophagus (Figure 3-10). The data suggest that over several decades persons who have H. pylori, especially cag-positive strains, have increased risk of cellular infiltration of their stomach, and atrophic gastritis, leading to gastric cancer. In contrast, persons who are not colonized with H. pylori, especially not having cag-positive strains (a condition I have called acagia in 1998) (Wu et al., 2003), have increased risk for GERD, Barrett’s, and esophageal cancer.
H. pylori Protection Against Esophageal Disease
How might H. pylori protect against esophageal disease? There could be direct or indirect effects on the esophagus, and the work of Yamaji (Yamaji et al., 2001) and others cited above (Kuipers et al., 1995a, 1995b) suggest there may be indirect effects operating through gastric physiology.
We also are interested in direct effects of H. pylori (or its absence) on the esophagus, involving either esophageal tissue or the esophageal biota.
Dr. Zhiheng Pei found that bacteria can be visualized in the distal esophagus in biopsies that he examined histopathologically (Yamaji et al., 2001). Consequently, studies were performed on four healthy persons to examine 16S ribosomal RNA (rRNA) clones from their esophageal biopsies (Pei et al., 2004). In these subjects, 95 species-level operational taxonomic units (SLOTU) were identified; diversity estimations predicted about 130 different SLOTU in the healthy esophagus. The 95 SLOTU belonged to 6 phyla and 41 genera; 59 SLOTU were homologous with culture-defined species, 34 were 16S clones, and 2 were not homologous with any known clones. Fourteen SLOTU were shared by all 4 individuals, accounting for 64 percent of the clones. These preliminary observations provide evidence that there is a conserved esophageal biota, and there are host-specific organisms. Ongoing studies support these observations and suggest that differences exist in the esophageal biota present in individuals with normal esophagus and with GERD (Lu et al., 2005; Pei et al., 2005).
H. pylori, Gastric Hormones, and Metabolic Effects of Colonization
The stomach may be considered an endocrine organ, since it produces the hormones gastrin and somatostatin (Calam, 1995; Moss and Calam, 1993; Moss et al., 1992). Several years ago, investigators found that the stomach produces leptin (Sobhani et al., 2002). Leptin is a hormone, first described in adipose tissue, involved in energy homeostasis; among its many effects, leptin signals the hypothalamus that appetite is sated (Matson et al., 2000). It is not surprising that the stomach was found to produce leptin since it is an organ involved in eating. Several years ago, investigators found that the stomach produces another hormone related to metabolism, called ghrelin (Lee et al., 2002). In many ways, ghrelin is a physiologic antagonist of leptin, because one activity of ghrelin is to signal the hypothalamus to eat (Horvath et al., 2001).
Recently, several investigators have examined the relationship of H. pylori status and gastric hormonal expression. Azuma and colleagues explored gastric leptin levels prior to and after treatment to eradicate H. pylori in 40 subjects; in 33, the organism was successfully removed, and in 7, treatment was unsuccessful). For pretreatment, gastric leptin RNA levels were about the same in the two groups. For those in whom treatment was not successful (i.e., H. pylori remained), the posttreatment leptin levels did not substantially change. However, when H. pylori was removed (treatment success), leptin expression diminished significantly. Interestingly, about 20 percent of the successfully treated subjects gained substantial weight during the six months subsequent to treatment (Azuma et al., 2001).
Nishi and colleagues examined plasma ghrelin levels by whether subjects were H. pylori-positive or not; those who were H. pylori-negative had significantly higher ghrelin levels than those who were positive (Isomoto et al., 2004).
Nwokolu studied the effect of H. pylori eradication on gastric physiology in 10 subjects (Nwokolo et al., 2003). After H. pylori eradication, ghrelin levels rose, leptin declined, gastrin declined, and gastric acidity rose (Nwokolo et al., 2003). The gastrin and acidity results confirmed prior studies and directly illustrate the effects of H. pylori on local (gastric) physiology. In total, the above studies provide evidence that in the hormonally active stomach, H. pylori colonization has an effect on local hormone production, which has both local and systemic effects. This is an important confirmation of the metabolome concept.
Variation in H. pylori Genotypes Within an Individual Host
There now is extensive evidence that H. pylori populations within a host are changing over the course of colonization (Israel et al., 2001; Kuipers et al., 2000). There is competition between strains as well as recombination (Falush et al., 2001; Kersulyte et al., 1999). However even for a single strain, variation is ongoing. For example, even in a population of cag-positive strains, there will be strains in which all or part of the cag island is deleted, so that the H. pylori population of an individual’s stomach is heterogeneous with regard to cagA presence (Occhialini et al., 2001; Wirth et al., 1999).
The 3′ region of cagA is highly polymorphic due to repetitive DNA (Azuma et al., 2002; Yamaoka et al., 1998). In a single individual, biopsies from the antrum and the corpus may show size differences in this region because of the direct DNA repeats (Aras et al., 2003b). Translation of these regions shows heterogeneity in the presence of the tyrosine phosphorylation domain (Aras et al., 2003b). H. pylori has evolved into a system using deletion and recombination, based on repetitive DNA motifs, so that cells can change phenotype (Aras et al., 2001, 2002, 2003a). In the important cag signaling moiety, this variation is biologically relevant; in any individual with a cag-positive strain, there can be variants that produce and inject the protein-containing phosphorylation sites, as well as variants without the sites or that are unable to inject or do not have the island. Each of these organisms signals the host differently; the cag island is a model for other loci in the genome.
Conclusions
I have presented a model in which H. pylori evolves within a single host (Figure 3-11). The population of strains is undergoing genetic change through a variety of mechanisms, including point mutation and intragenomic and intergenomic recombination. There is much evidence to suggest that H. pylori becomes a quasispecies, similar to HIV or hepatitis C (Kuipers et al., 2000). Two strains can recombine into a broader quasispecies that is being selected for fitness in a microniche. The microniches are in communication with one another, and the
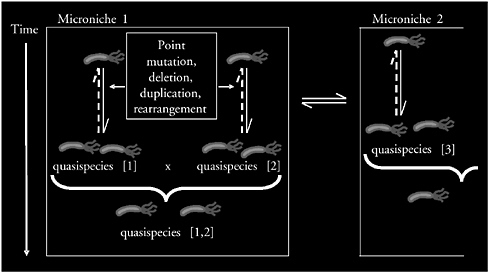
FIGURE 3-11 Schematic of H. pylori variation in a single host. The genomes of H. pylori cells are highly plastic, subject to point mutation and intra- and intergenomic variation. A model is proposed in which related microbial individuals, a quasispecies, compete with one another in a microniche. Multiple quasispecies may compete or recombine. Local selection determines the population structure in that niche, and by competing and cooperating, populations from adjacent niches influence the overall H. pylori composition.
SOURCE: Blaser (2005).
sum of the microniches determines the macroniche. The presence of microniches permits multiple independent strains to colonize one individual’s stomach (Blaser and Kirschner, 1999).
In conclusion, there is increasing evidence that H. pylori are part of our ancient indigenous biota, but that they are disappearing due to modern lifestyles. As such, that part of human physiology related to its presence is changing, which has consequences for both health and disease and may not be limited to the proximal gastrointestinal tract. H. pylori populations in each host are varied and continue to diversify over the course of the colonization of that host. Carriage of H. pylori clearly has biological costs and benefits to humans. Perhaps most importantly, the disappearance of H. pylori may be a marker for the extinction of other less easily detectable indigenous bacteria. The data suggest that human microecology is changing, and that H. pylori is an indicator organism. I propose that the activities of the human genome together with our indigenous microbial genomes (to the nth power) determine both the instantaneous and long-term status of the metabolome (Figure 3-5B). It is the interactions between the host and its microbes that participate in human physiology and homeostasis.
INDUCTION OF PATHOGENIC IMMUNE RESPONSES IN SUSCEPTIBLE HOSTS BY COMMENSAL ENTERIC BACTERIA
R. Balfour Sartor4
We coexist with an exceedingly complex mass of interacting bacteria and fungi in the distal ileum and colon. These predominantly anaerobic organisms are in intimate contact with the intestinal epithelium where reciprocal regulation of microbial and host gene expression is evident, as eloquently discussed by Jeff Gordon (Bäckhed et al., 2005). These resident intestinal bacteria profoundly influence both pathogenic and regulatory mucosal and systemic immune responses (MacDonald and Gordon, 2005; Strober et al., 2004). Strong experimental evidence supports the hypothesis that homeostasis versus chronic intestinal inflammation is determined by the host’s genetically determined innate and adaptive immunologic responses to luminal commensal microbial antigens and adjuvants (Sartor, 2004). This review discusses the bacterial components and the genetically programmed host responses that determine coexistence or aggressive responses to commensal bacteria. These concepts help explain the pathogenesis of chronic immune-mediated intestinal inflammation such as the idiopathic IBD, Crohn’s disease, and ulcerative colitis, as well as differential host responses to enteric microbial pathogens.
Normal Host Responses
The normal host with appropriate regulated immune responses develops tolerance when confronted with commensal bacteria (Figure 3-12A). This state of relative nonresponsiveness is mediated by regulatory T cells, dendritic cells, and epithelial cells that produce transforming growth factor β (TGF β), interleukin 10 (IL-10), protective prostaglandins and PGJ2, interferon (IFN) α, and intracellular inhibitory molecules such as PPARγ and A20 (Strober et al., 2004). Some regulatory cells, such as CD4+CD25+ T cells, may display membrane-bound TGF β. In contrast, genetically susceptible hosts with dysregulated immune responses develop chronic relapsing intestinal inflammation mediated by macrophages, TH1, and possibly natural killer (NK) T cells that secrete IL-1β, tumor necrosis factor (TNF), IL-12, IL-13, IL-17, IL-23 and IFNγ. Homeostasis is mediated not only by the net immunosuppressive tone of the mucosal immune response, but also by the controlled uptake of luminal microbial antigens and adjuvants. This is accomplished by an intact mucosal barrier that consists of epithelial tight junctions, secreted immunoglobulin A (IgA), α and β defensins secreted by Paneth cells and activated epithelial cells, respectively, as well as by a complex biofilm composed of mucosally adherent mucus, intestinal trefoil factor, and bacteria that are adapted
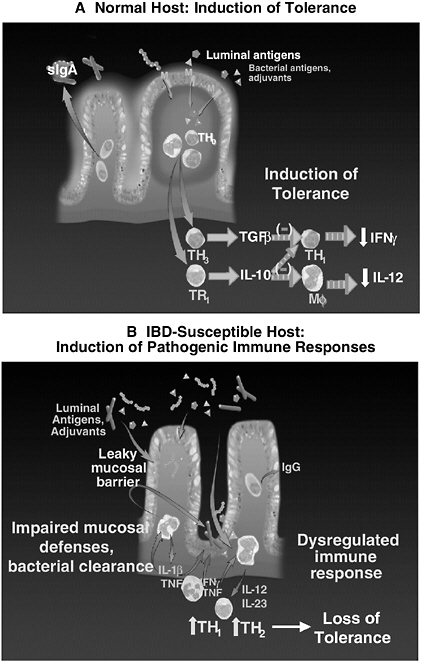
FIGURE 3-12 Induction of homeostatic or pathogenic immune responses by commensal bacteria. A: Normal host. Luminal bacteria are excluded by an impermeable epithelial barrier and biofilm. Controlled uptake of luminal antigens leads to regulatory immune responses in organized lymphoid aggregates. B: Susceptible host. A break in the mucosal barrier leads to activation of innate and acquired immune responses that progress to pathogenic TH1 or TH2 cell activation.
SOURCE: Sartor (2005).
to this ecologic niche. The net effect of this diverse mucosal shield is resistance to colonization and epithelial adherence by pathogens and a barrier to bacterial translocation. If this barrier is transiently broken by an acute infection or toxin, the epithelium of normal hosts almost instantly restores continuity through restitution, which is a rapid migration of epithelial cells along the intact basement membrane. This process is dependent on TGF β, other growth factors, and intestinal trefoil factor, and is independent of cellular proliferation. Furthermore, translocating microbial organisms are rapidly phagocytosed and killed by cytokine, NFκB-stimulated reactive oxygen metabolites, and nitric oxide. These multilayer defenses allow the distal small intestine and colon not only to maintain a relatively quiescent immunologic state while coexisting with commensal enteric bacteria, but also to appropriately respond to a variety of pathogens.
Pathogenic Mucosal Immune Responses to Bacteria in Genetically Susceptible Hosts
Hosts with a variety of genetic defects in mucosal barrier function, immune regulation, or bacterial killing can develop chronic relapsing intestinal inflammation when confronted with the same microbial environment present in normal hosts (Figure 3-12B). Either an intrinsic, genetically determined defect in the mucosal barrier, a transient breach of mucosal defenses by a self-limited pathogen, or exposure to an epithelial toxin such as a nonsteroidal anti-inflammatory drug (NSAID) can lead to increased bacterial translocation or uptake of antigens and adjuvants from commensal bacteria. Resident mucosal macrophages are relatively unresponsive to microbial adjuvants due to down regulated TLRs and CD14 (Smythies et al., 2005), but they can respond to pathogenic amounts of microbial stimulants. Liberation of chemokines induces immigration into the lamina propria of more highly responsive monocytes and neutrophils with full expression of TLRs and CD14. Activation of these cells by bacterial adjuvants through pattern recognition receptors such as TLR2, TLR4, and TLR5, which bind to bacterial peptidoglycan and lipoteichoic acid, then lipopolysaccharide (LPS) and flagellin, respectively, activate NFκB and a host of proinflammatory molecules including TNF, IL-1β, IL-6, chemokines, IL-12 and IL-23 (Sartor and Hoentjen, 2005). IL-12 activates TH1 cells to secrete IFNγ, while IL-23 induces IL-17. In concert with TNF, IFNγ lyses epithelial cells to cause mucosal ulceration. Similarly, IL-13, which increases in ulcerative colitis, can then increase mucosal permeability by interfering with epithelial tight junctions, inducing epithelial apoptosis and interfering with epithelial restitution (Heller et al., 2005). Tissue damage is further intensified by liberation of metalloproteases such as collagenase, elastase, and stromolysin that degrade the extracellular matrix. Normally, such pathogenic responses are rapidly down-regulated by appropriate induction of immunosuppressive molecules such as IL-10 and TGF β. However, hosts with genetic defects in immune regulation, bacterial killing, or mucosal barrier repair develop
inflammatory responses that are perpetuated by continued uptake of microbial antigens and ligands of pattern recognition receptors. This leads to loss of tolerance to commensal bacteria and chronic immune-mediated inflammation.
The best evidence that commensal enteric bacteria have a role in the pathogenesis of chronic intestinal inflammation is provided by studies in gnotobiotic rodents (Sartor, 2004). In at least 10 different diverse models ranging from genetically engineered mice and rats to nonhuman primates, there is no immune activation and no colitis in the absence of bacterial stimulation (Figure 3-13). However, exposure of these susceptible hosts to nonpathogenic resident bacteria activates macrophage and effector T lymphocytes that cause colitis. For example, IL-10 knockout mice on a 129 SvEv background raised in a germ-free (sterile) environment exhibit no colitis, but develop histologic evidence of mild colitis within one week of colonization with specific pathogen-free (SPF) bacteria (Sellon et al., 1998). This progresses to moderate colitis after two weeks of bacterial colonization and to severe transmural TH1-mediated colitis by 3–4 weeks. The onset of histologically detectible colitis is preceded by colonic secretion of IL-12 p40 and enteric-specific IFNγ secretion by CD4+ T cells (Kim et al.,
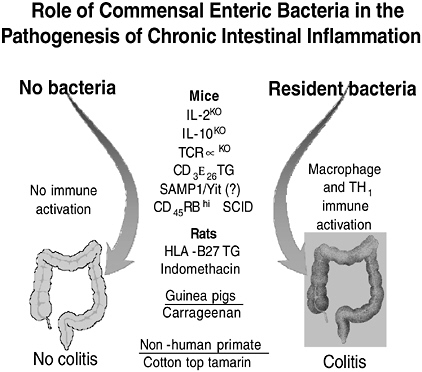
FIGURE 3-13 Immune activation and chronic intestinal inflammation depends on the presence of commensal luminal bacteria in multiple animal models.
SOURCE: Sartor (2005).
2005b). Although the timing is different in the other listed models, the net result is quite similar. In these models, broad spectrum antibiotics prevent the onset and treat established disease while more selective antibiotics that suppress a narrow bacterial spectrum of commensal bacteria can attenuate disease in prophylactic protocols but not reverse established colitis (Hoentjen et al., 2003; Rath et al., 2001).
A single model, colitis induced by dextran sodium sulfate (DSS),provides an exception to this rule. DSS is an epithelial toxin that induces rapid colonocyte apoptosis with shortening of crypts and eventually mucosal ulceration of the colon within one week of exposure to 3–5 percent DSS in drinking water. Initial studies by Axelsson showed enhanced rather than attenuated colitis in germ-free mice fed DSS (Axelsson et al., 1996). More recently, Rakoff-Nahoum et al. (2004) reported that MyD88-deficient mice showed enhanced mortality and mucosal inflammation after exposure to DSS relative to wild-type congenic mice. MyD88 is a key adapter protein that is necessary for all TLR signaling to NFκB. Depletion of commensal microbiota by broad-spectrum antibiotics did not alter the results in knockout mice and potentiated DSS-induced colonic injury in wild-type mice (Araki et al., 2005).
Resolution of these seemingly disparate data requires an understanding of the complexity of pro- and anti-inflammatory signals induced by intestinal bacteria. DSS induces colitis that is mediated by innate immune mechanisms that selectively target the epithelium. Dieleman et al. (1994) demonstrated in severe combined immune deficient (SCID) mice that the absence of T lymphocytes did not affect acute DSS-induced colitis. In an elegant series of studies, Greten et al. (2004) demonstrated that NFκB activation in epithelial cells versus myeloid cells led to differential effects on colitis. Targeted deletion of IKKβ in enterocyte villin-CRE/IKKβF/F mice treated with the procarcinogen azoxymethane plus three cycles of DSS lost significantly more weight than wild-type controls and had increased mucosal injury. However, when IKKβ was deleted in myeloid cells (lysM-CRE/IKKβF/F), this led to markedly decreased colitis with the same dose of DSS. These results suggest that NFκB activation on epithelial cells, presumably by commensal enteric bacteria, induces homeostatic responses while myeloid cell NFκB activation induces colitis. This conclusion is supported by observations using the same epithelial and myeloid-targeted NFκB in activation through a radiation model (Egan et al., 2004) and ischemia/reperfusion (Chen et al., 2003).
We conclude from these gnotobiotic studies that normal luminal bacteria can induce and perpetuate chronic T cell-mediated colitis and gastroduodenitis and associated extraintestinal inflammation such as peripheral arthritis in genetically susceptible hosts. However, although luminal bacterial and genetic factors are essential, neither is sufficient to cause chronic inflammation. Colitis does not develop in the absence of either microbial stimulation or genetic susceptibility. Induction of chronic immune-mediated inflammation requires the interaction of
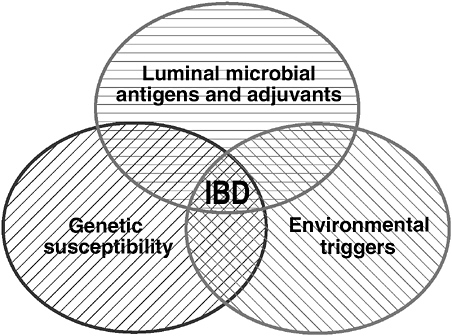
FIGURE 3-14 Interaction of genetic, environmental, and microbial factors in the pathogenesis of IBD.
SOURCE: Sartor (2005).
genetic and environmental factors (Figure 3-14). Finally, luminal bacteria can induce both detrimental and protective responses, with epithelial activation of NFκB by commensal bacteria being predominantly beneficial.
Microbial and Host-Specific Pathogenic Responses
We have demonstrated in gnotobiotic rodents that all bacterial species are not equal in their ability to induce colitis. Both bacterial and host specificity is evident in these studies. HLA B27 transgenic rats raised in a germ-free environment exhibit no colitis but develop active TH1-mediated inflammation in the colon, distal stomach, and duodenum within one month of being colonized with SPF (free of Helicobacter species and all demonstratable pathogens) enteric bacteria derived from feces of wild-type normal rats (Rath et al., 1996). Selective colonization (monoassociation) of transgenic rats with Bacteroides vulgatus leads to the onset of moderate T cell-mediated colitis within one month of colonization. However, equal luminal levels of E. coli do not cause colitis (Rath et al., 1999). Cocolonization with Lactobacillus GG and SPF bacteria attenuated colonic inflammation (Dieleman et al., 2003). These results indicate that some common enteric commensal bacteria are aggressive, some are neutral, and yet others are
protective. Bacteroides vulgatus is not a traditional pathogen since colonization of nontransgenic (wild-type) littermates have no detectible inflammation despite equal luminal concentrations of B. vulgatus (Rath et al., 1999). We have demonstrated species specific responses within the Bacteroides genus. Bacteroides vulgatus, Bacteroides thetaiotaomicron, and Bacteroides fragilis cause colitis in HLA B27 transgenic—but not nontransgenic—controls. However, Bacteroides distasonis did not cause inflammation (Mann et al., 2003).
We then demonstrated host-specific responses to selective enteric commensal bacterial species. As mentioned, HLA B27 transgenic rats monoassociated with B. vulgatus develop a cecal-predominant colitis, but no disease with E. coli nor with Enterococcus faecalis (Kim et al., 2005a; Kim et al., 2005b). Exactly the opposite results were found in IL-10 deficient mice, in which B. vulgatus caused no inflammation and E. coli and E. faecalis induced chronic colitis. Yet another model, the bone marrow-transplanted CD3ε transgenic mouse, showed no inflammation when colonized with any of these three organisms. Of particular clinical interest, we demonstrated different phenotypes of disease in a single host colonized with two different bacterial species. E. faecalis induced slow onset (10–12 weeks) distal colitis in IL-10 deficient mice and duodenal inflammation leading to obstruction and gastric outlet obstruction with protracted monoassociation (26 or more weeks) (Kim et al., 2005b). In contrast, E. coli monoassociation induced a predominantly right-sided colitis three weeks after colonization. Dual association of germ-free IL-10 knockout mice with E. coli plus E. faecalis caused an aggressive pancolitis with onset one week after colonization (Kim et al., 2004).
These results indicate that certain commensal bacterial species can selectively induce disease in hosts with different genetic backgrounds and cause different phenotypes of disease in a single genetically susceptible host. Some bacterial species are aggressive, some protective, and some neutral, but no single species yet identified can induce as aggressive a colitis as the complex microflora of the distal intestine. These results have obvious therapeutic implications in that antibiotics, probiotics, and prebiotics with selective spectra of activity can be used to treat IBD patients, but each subset of patients may respond selectively to various agents. Thus, we will need to individualize treatments for each patient.
Mechanisms of Genetic Susceptibility to Intestinal Inflammation
These results clearly indicate that host susceptibility is a dominant determinant of experimental colitis. Likewise, IBD patients show increased concordance in monozygotic versus dizygotic twins. The concordance rate in Crohn’s disease is 44–58 percent compared to 3–5 percent in dizygotic twins (Halfvarson et al., 2003). Ulcerative colitis exhibits less of a genetic influence with concordance in monozygotic twins at 6–18 percent. The search for IBD-related genes has consisted of two approaches. A genomewide search using single nucleotide polymorphism probes have found association in many chromosomes, with chromosome
1, 5, 6, 12, and 16 being replicated (Newman and Siminovitch, 2005; Schreiber et al., 2005). In 2001, two independent groups simultaneously identified NOD2/CARD 15 as the gene responsible for susceptibility to Crohn’s disease (Hugot et al., 2001; Ogura et al., 2001). A single polymorphism in this gene enhances susceptibility 1.5 to 3 fold over incident ranges, but a homozygous polymorphism, or two independent (compound heterozygous), abnormalities enhances susceptibility approximately 30 fold. Similarly, polymorphisms in the organic cation transporter (OCTN) genes 1 and 2 on chromosome 5 have been associated with Crohn’s disease (Peltekova et al., 2004). Genes responsible for susceptibility on the other identified chromosomes have not yet been recognized.
A second approach is to search for candidate genes in multiplex families. These types of studies have implicated the HLA region, polymorphisms in TLR4 and TLR5, as well as in IL-10, PPARγ, and IL-1 receptor antagonists (Newman and Siminovitch, 2005). The genes most commonly associated with IBD are listed in Table 3-1.
It is quite interesting, and potentially important, to note that the genes studied to date fall into several categories relating to either epithelial barrier function or response to bacteria. The first gene implicated in Crohn’s disease, NOD2/CARD 15, is an intracellular bacterial sensor whose leucine-rich repeat (LRR) region binds to the bacterial peptidoglycan component muramyl dipeptide (Girardin et al., 2003). The three most common single nucleotide polymorphisms associated with Crohn’s disease lie within, or very near, the LLR region. Several groups have demonstrated diminished NFκB activation in mutant NOD2/CARD 15, in-
TABLE 3-1 Genes Associated with IBD and Experimental Colitis
|
Gene |
Chromosome (Human) |
Function |
|
|
A. |
Crohn’s disease |
|
|
|
|
NOD 2 (CARD 15) |
16 |
NFκB activation/regulation, killing intracellular pathogens, Paneth cell function (α defensin production) |
|
OCTN 1/2 |
5 |
Organic cation, carnitine transporter, transports xenobiotic substances |
|
|
DLG 5 |
10 |
Epithelial scaffolding protein |
|
|
PPARγ |
|
Intracellular inhibitor of NFκB and cellular activation |
|
|
B. |
Ulcerative colitis |
|
|
|
|
Mdr-1 |
7 |
Efflux transporter drugs and possibly xenobiotic compounds |
dicating that these common mutations lead to loss of function (Ogura et al., 2001). Recent data indicate that Crohn’s related polymorphisms and NOD2/CARD 15 lead to decreased intracellular killing after epithelial invasion by Salmonella (Hisamatsu et al., 2003), decreased α defensin production (Wehkamp et al., 2004), and defective suppression of NFκB activation after peptidoglycan ligation of TLR2 (Watanabe et al., 2004). Decreased α defensin production is particularly intriguing in light of the knowledge that NOD2/CARD 15 is constitutively expressed in Paneth cells that are selectively found in the small intestine (Lala et al., 2003). NOD2 polymorphisms are highly associated with ileal Crohn’s disease. In addition, Swidsinski et al. (2002) have demonstrated genetically enhanced mucosally associated bacterial populations in active Crohn’s disease patients versus normal controls. Recent observations in NOD2-deficient mice indicate defective clearance of oral Listeria monocytogenes with defective cryptdin-like defensin 4 and 10 production (Kobayashi et al., 2005). These investigations support the concept that NOD2/CARD 15 protects the ileal mucosa by several mechanisms and is an important contributor to mucosal homeostasis of the ileum in response to commensal bacteria and possibly pathogens.
Conclusions
Mammals have involved multiple redundant mechanisms to protect themselves from the aggressive microbial milieu in the distal ileum and colon. Normally, the host lives in peaceful coexistence with these commensal organisms because of net tolerogenic mucosal immune responses mediated by production of immunosuppressive TGFβ and IL-10. However, intermittent exposure to pathogens and other environmental triggers, such as NSAIDs, can initiate acute injury in the intestines of all patients. Normal hosts rapidly restore homeostasis by clearing the bacterial invader, healing the mucosal barrier, and down-regulating the inflammatory response once the offending stimulus has been eliminated. In contrast, a genetically susceptible host fails to appropriately inhibit the inflammatory process, which can become self-perpetuating due to continued uptake of luminal bacterial products. Genetic susceptibility factors include defective mucosal barrier function, defective/incomplete bacterial clearance, and ineffective ability to down-regulate inflammation. Commensal bacteria provide the constant antigenic and adjuvant stimuli that perpetuate chronic intestinal inflammation in susceptible hosts, but induce homeostatic signals in normal hosts. Thus, we hypothesize that the inflammatory bowel diseases are the result of an overly aggressive cell-mediated immune response to a relatively small subset of commensal bacteria in a genetically susceptible host. Dominant microbial antigens may be unique for each individual. Genetic susceptibility is determined by defective genes that encode barrier function, bacterial clearance, and immunoregulation. The onset or activation of disease is triggered by environmental factors that injure the epithelial barrier and initiate inflammatory responses. These triggering factors include
NSAIDs or self-limited bacterial, viral, or parasitic infections. A more thorough understanding of the mechanisms of induction and perpetuation of intestinal inflammation should provide the means to selectively inhibit these responses and restore mucosal homeostasis. Based on our results, we predict that appropriate therapy will need to be individualized to each patient based on their genetic susceptibility, their enteric bacterial profiles, and their T-cell and serologic responses to these commensal bacteria.
Acknowledgments
These studies were supported by NIH grants, DK 40249 and DK 53347, as well as the Crohn’s and Colitis Foundation of America and the Broad Research Foundation. The author thanks Susie May for her excellent secretarial assistance.
HOW DO CHANGES IN MICROECOLOGY AFFECT THE HUMAN HOST?
María G. Domínguez-Bello5
In a living world that has been microbial since its origins, all other forms of life have evolved in interaction with microorganisms. Therefore not only pathogens but also microbial mutualists must have shaped animals’ immunity. Self-recognition and recognition of indigenous microbes is key to the function of the immune system, which should act specifically against pathogenic invaders. This implies that there has to be some degree of resistance of indigenous microbes to host innate and adaptive immunity.
Germs have had a great impact on human perception of microbes, and it is recently that indigenous microbes are being recognized to have an important physiological function. The perception of microbes as entities capable of causing disease has triggered a cultural and technological antimicrobial response. The effect of hygiene practices and use of broad-spectrum antimicrobials is nonspecific. Modern practices have become arms of microbial mass destruction, to which, of course, many infectious disease agents have succumbed. Yet, as infectious diseases disappear in modern societies, autoimmune disorders and obesity are increasing.
This paper discusses some aspects of the interaction between animals and their microbiomes and points out the importance of recognizing that prokaryotes and eukaryotes naturally colonizing human surfaces and cavities have shaped our evolution. They are likely to play a role in promoting health and protecting from disease.
Beneficial Microbes: The Rumen as an Example of True Mutualism
Microbes interact with all other forms of life. Animals have coevolved with bacteria, responding to their selective pressures and also driving bacterial evolution. In certain animals, some microbes became so closely associated with their hosts, that together they establish a functional unit, an organ, vital for both microbes and hosts. The rumen is one of these examples of “true mutualism.” Indigestible plant cell walls and dietary toxins have exerted the evolutionary pressure for the appearance of symbiotic foregut fermentative digestion. Microbes contribute with digestive enzymes the host animal lacks, expanding the animal’s nutritional niche by fermenting plant structural carbohydrates and other dietary components into short-chain fatty acids (Figure 3-15), the most important energy source for the animal (Van Soest, 1994).
Because dietary protein is also fermented, the main amino acids source for a ruminant on a natural diet comes from the microbial biomass itself. Because bacterial cell walls—enclosing proteins—are also indigestible, foregut fermenters have multiplied the lysozyme gene and recruited the enzyme to act as a digestive enzyme (Dobson et al., 1984). Rather than feeding on plants, ruminants and other foregut fermenters really feed on the microbes they farm in the foregut.
Foregut fermentative digestion evolved convergently in mammals—ruminants, howler monkeys, sloth—and in at least one bird, the hoatzin, that ferments in the crop and also harbors a complex microbiota (Domínguez-Bello et al., 1993; Grajal et al., 1989).
The rumen ecosystem is populated by a high diversity of microorganisms, including bacteria, archaea, fungi, protozoa, (Hungate, 1972; Nelson et al., 2003; Ogimoto and Imai, 1981; Tajima et al., 2001a,b; Van Soest, 1994) and viruses (Klieve and Bauchop, 1988; Klieve and Swain, 1993; Klieve et al., 1989; Nemcova et al., 1993). Ribosomal DNA sequence data has recently revealed that gastrointestinal bacterial communities in cattle appear to be dominated by Firmicutes, particularly those related to the genus Clostridium (Nelson et al., 2003; Tajima et al., 2001a; Whitford et al., 1998). Novel rumen Archaea, not associated with known rumen methanogens, have also been recently discovered (Tajima et al., 2001b).
Culturable anaerobic bacteria from the rumen have been intensively studied over the past 40 years (Hungate, 1966; Ogimoto and Imai, 1981). Much work on this subject was published between 1960 and 1980. In those days, there were crowded scientific meetings about the rumen—such as the Chicago Conference on Rumen Function (now Conference on Gastrointestinal Function) started in 1951. Rumen studies have been seminal to the understanding of digestive processes in other animals and in humans. Unfortunately, since research on animal nutrition has become a low funding priority, scientists who previously worked on rumen microbiology have now moved to other fields. The rumen represents a case of true microbial mutualism and should be viewed as a model to further the understanding of the function of human colonic fermentation.
Changes in the Human Gastrointestinal Microbiome: Indigenous Microbes in Indigenous Peoples
Bacteria are important in the bowel function and for the correct function of the gut immune system (Cash and Hooper, 2005; Hooper et al., 2001, 2003; Stappenbeck et al., 2002). As a consequence, disrupting intestinal host-bacterial relationships may lead to immune disorders. Diversity and population structure of intestinal bacteria is dynamic and complex. They are influenced by niche resources, diet, antimicrobials, competing sympatric species, and mucosal immunity. Innate and adaptive immunity are important to reach a healthy equilibrium of the microbial ecosystem. Paneth cells in the small intestine secrete microbicidal peptides (lysozyme, Ang4), and Peyer’s patch dendritic cells present microbial antigens to lymphocytes (Figure 3-16).
Host immunity and indigenous microbes may protect against the assault of primary pathogens through what is known as colonization resistance, while host immunity acts specifically against invaders. Resistance of indigenous microbes to innate immunity is still a poorly understood phenomenon, but some rumen bacteria can develop resistance to cow gastric lysozyme (Domínguez-Bello et al., 2004). Also, Bacteroides thetaiotaomicron is resistant to the antimicrobial peptide Ang4 whose secretion is induced by the intestinal presence of the bacterium (Hooper et al., 2003).
When we think of intestinal microbes, we still think of bacteria. Recent molecular methods have unveiled an unexpected diversity of 500 bacterial species, with 30 to 40 of them accounting for 99 percent of the total intestinal population (Harmsen et al., 2002) and a great number of novel sequences (Eckburg et al., 2005). We know little about the ecological role of Archaea in the human intestine (Bäckhed et al., 2005), and nothing about the role of viruses, except that phages are the most abundant entity, with a total of 1,200 viral genotypes in the intestinal community (Breitbart and Rohwer, 2005; Edwards and Rohwer, 2005). Parasites and phages surely modulate bacterial populations in the intestine, but what are their effects on our immunity and health? (Figure 3-16).
Quantitative and qualitative changes in our indigenous bacteria brought by our modern life are unknown, but the disappearance of H. pylori from the stomach of people in modern societies (Pérez-Pérez et al., 2002) might be a marker for other bacteria as well. The degree of alterations of modern human microbiomes can only be studied by comparison with more natural human microbial ecosystems in primitive human societies.
Lack of intestinal parasites is perhaps the most noticeable example of microbiome alteration in modern humans. Science currently regards intestinal parasites as harmful (Dreyfuss et al., 2001). Of course we know the diseases they can produce, but we do not understand why most colonized persons remain asymptomatic. The fact that we call “parasites” these intestinal protozoa and helminths that have evolved in the human intestine, reflects our own prejudices.
Preliminary results from our studies in Venezuelan Amerindian villages (Dominguez-Bello et al., 2005) show some unexpected associations between gastrointestinal microbes and health. Living in areas of malaria risk, people in these villages do not suffer from food shortage—they fish from the river and eat seasonal fruits—but morbidity and mortality due to infant diarrhea is high. All but one of 72 asymptomatic children and adults of these communities carried parasites. Most people had Ascaris lumbricoides, Trichiura trichuris, and Entamoeba sp in their intestines, and multiparasitism had no effect on body mass index (P = .593). Other studies have also found high prevalence of asymptomatic intestinal parasitism. In Ecuador, 93 percent of pregnant women participating in a study carried at least one species of intestinal parasite, and 88 percent had Entamoeba histolytica (Dreyfuss et al., 2001). The possibility that some degree of parasitism might be beneficial for humans cannot be ruled out. Perhaps the optimal degree of parasitism varies according to local conditions. For example, anemia-inducing parasites might be beneficial in regions where malaria is endemic (Domínguez-Bello and Blaser, 2005; Kucik et al., 2004; Murray et al., 1978; Nacher et al., 2003; Oppenheimer, 2001).
In addition to parasites, most adults in our study had H. pylori (82 percent of adults and half the children). Interestingly, H. pylori-colonized children had higher cell mass and better nutritional status than H. pylori-negative children (Dominguez-Bello et al., 2005); (P < .05; subjects were 43 H. pylori positive and 29 H. pylori negative). The lack of detrimental effect of microbes on nutritional status supports the hypothesis of a mutualistic nature of the relation between some intestinal “parasites” and us. Which ones are our beneficial microbes? Which ones should children carry to optimize health? Which ones threaten to become opportunistic pathogens and when?
Microbial-Related Diseases and Modernity
Evolution of microbiomes and their protective function in concert with host immunity has taken nature a long evolutionary time. The “intelligent” use of antimicrobials by man, outside the pace of evolution, lacks selectivity. Although the fight against infectious diseases has increased human well-being and life expectancy, nature’s trade-off imposes a cost to this success: new diseases as a consequence of microbiome alterations. When microbes face a lethal force, they either die or compensate with changes conferring survival that are selected for. The wide use of antibiotics has resulted in increased resistance among indigenous and pathogenic bacteria. In the last 50 years, tetracycline has been used broadly as an antibiotic for the treatment of bacterial and protozoal infections in humans, as a growth promoter in animals, and as an immunosuppressant. A variety of tetracycline resistance genes are present in the oral (Pantoja et al., 2005; Villedieu et al., 2003) and intestinal (Salyers et al., 2004) microflora of healthy adults. Compounds other than antibiotics could also exert positive selection on antibiotic re-
sistance genes. Salicylate modulates multiple antibiotic resistance via mar genes, influencing bacterial multidrug resistance. In addition, copper selects for bacterial antibiotic resistance in agricultural soils (Berg et al., 2005).
According to the hygiene hypothesis, our antimicrobial behavior is linked to the appearance of new diseases. As societies develop and adopt Western lifestyles, the risk of some emergent and reemergent diseases, autoimmune diseases, and obesity increases (Figure 3-17). Developing countries still fall in the descendent region of the bimodal curve in Figure 3-17, suffering from primary pathogens, but perhaps keeping their normal microbiota less altered than modern societies. What are the actual changes in human microbiota as we get modern? What are their effects on immunity and health? Do we have mutualistic relations with parasites, fungi, or viruses? The gastrointestinal microbiomes of people living in primitive societies can probably help in unveiling what we have lost and what we can still gain in terms of our microbiomes.
An integration of concepts of host-microbe interactions is presented in the model of Figure 3-18. The newborn baby leaves a germ-free uterus and the external environment provides an initial colonization that leads to others in a successional way. In the mammalian colon, breast milk containing special oligosaccharides—inulin-type fructans—bypass the small intestine and enrich for bifidobacteria and lactobacilli (Forchielli and Walker, 2005). The nature of this fermentation dictates proliferation of the host tissue cells and of the microbial
FIGURE 3-17 Evolution of human societies and their diseases.
SOURCE: Domínguez-Bello (2005).
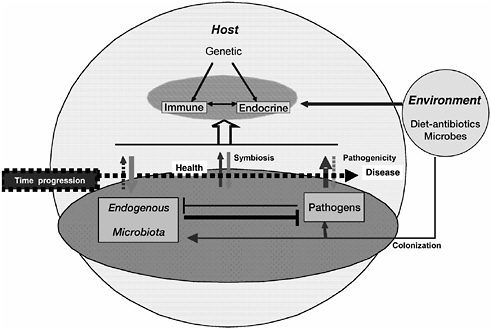
FIGURE 3-18 Host-microbial interactions shape health and disease.
SOURCE: Dominguez-Bello (2005).
community in the colon. Signals emitted in the “crosstalk” between microbes and host shape the course of colonization.
The immune signals from the newborn are strong because of maternal immunity, but the actual protective function of the gut develops by microbial stimulation. As time progresses, there is a “balanced” signaling between host and indigenous microbes. Time of acquisition of microbes and a particular succession of colonization may be very important to reach a healthy equilibrium.
Host responses to microbes, either immune (adaptive, innate) or endocrine (leptin, ghrelin), are influenced by genetic factors, but as the human microbiota is reduced in its diversity, it may contribute to changes in endocrine responses that might be involved in higher obesity risk. Although without an apparent effect on blood levels, eradication of H. pylori increases ghrelin and reduces leptin expression in the stomach (Azuma et al., 2001; Nwokolo et al., 2003). The fact that H. pylori-positive children had better nutritional status than noncolonized children in the Brazilian state of Amazonas (Dominguez-Bello et al., 2005) also supports this hypothesis. It has also been hypothesized that exposure to microbes—both exogenous pathogens and endogenous biota—might be critical environmental determinants of the expression of human height (Beard and Blaser, 2002).
Finally, in our model of Figure 3-18, endogenous microbiota exert coloniza-
tion resistance, but when pathogens break the barrier, they can disturb the ecological equilibrium. Ecologic disturbances can lead to monopolization of the niche by a primary pathogen or by indigenous microbes that become opportunistic pathogens. As the host grows old, the impairment of the immune response leads to disturbance of this dynamic equilibrium and to disease.
Adequate and complete identification of the indigenous human microbiome is the first step in elucidating its role in the physiology of our colonized organs and in the maintenance of health.
REFERENCES
Akopyants NS, Clifton SW, Kersulyte D, Crabtree JE, Youree BE, Reece CA, Bukanov NO, Drazek ES, Roe BA, Berg DE. 1998. Analyses of the cag pathogenicity island of Helicobacter pylori. Molecular Microbiology 28(1):37–53.
Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300(5624):1430–1434.
Araki A, Kanai T, Ishikura T, Makita S, Uraushihara K, Iiyama R, Totsuka T, Takeda K, Akira S, Watanabe M. 2005. MyD88-deficient mice develop severe intestinal inflammation in dextran sodium sulfate colitis. Journal of Gastroenterology 40(1):16–23.
Aras RA, TakataT, Ando T, Van der Ende A, Blaser NJ. 2001. Regulation of the HpyII restriction-modification system of Helicobacter pylori by gene deletion and horizontal reconstitution. Molecular Microbiology 42(2):369–382.
Aras RA, Small AJ, Ando T, Blaser MJ. 2002. Helicobacter pylori interstrain restriction-modification diversity prevents genome subversion by chromosomal DNA from competing strains. Nucleic Acids Research 30(24):5391–5397.
Aras RA, Kang J, Tschumi A, Harasaki Y, Blaser MJ. 2003a. Extensive repetitive DNA facilitates prokaryotic genome plasticity. Proceedings of the National Academy of Science USA 100(23):13579–13584.
Aras RA, Lee Y, Kim S-K, Israel D, Peek RM, Blaser MJ. 2003b. Natural variation in populations of persistently colonizing bacteria affect human host cell phenotype. Journal of Infectious Diseases 188(4):486–96.
Axelsson LG, Midtvedt T, Bylund-Fellenius AC. 1996. The role of intestinal bacteria, bacterial translocation and endotoxin in dextran sodium sulphate-induced colitis in the mouse. Microbial Ecology in Health and Disease 9:225–37
Azuma T, Suto H, Ito Y, Ohtani M, Dojo M, Kuriyama M, Kato T. 2001. Gastric leptin and helicobacter pylori infection. Gut 49(3):324–329.
Azuma T, Yamakawa A, Yamazaki S, Fukuta K, Ohtani M, Ito Y, Dojo M, Yamazaki Y, Kuriyama, M. 2002. Correlation between variation of the 3’ region of the cagA gene in Helicobacter pylori and disease outcome in Japan. Journal of Infectious Diseases 186(11):1621–1630.
Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. 2005. Host-bacterial mutualism in the human intestine. Science 307(5717):1915–1920.
Beard, AS, Blaser MJ. 2002. The ecology of height: The effect of microbial transmission on human height. Perspectives in Biology and Medicine 45(4):475–498.
Berg, JA, Tom-Petersen A, Nybroe O. 2005. Copper amendment of agricultural soil selects for bacterial antibiotic resistance in the field. Letters in Applied Microbiology 40(2):146–151.
Blaser MJ. 1990. Helicobacter pylori and the pathogenesis of gastroduodenal inflammation. Journal of Infectious Diseases 161(4):626–633.
Blaser MJ. 1992. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 102(2):720–727.
Blaser MJ. 1997. Ecology of Helicobacter pylori in the human stomach. Journal of Clinical Investigation 100(4):759–762.
Blaser MJ. 1998. Helicobacters are indigenous to the human stomach: Duodenal ulceration is due to changes in gastric microecology in the modern era. Gut 43(5):721–727.
Blaser MJ. 1999. The changing relationships of Helicobacter pylori and humans: Implications for health and disease. Journal of Infectious Diseases 179(6):1523–1530.
Blaser MJ. 2005 (March 16). Session I: Host-Pathogen Interactions: Defining the Concepts of Pathogenicity, Virulence, Colonization, Commensalism, and Symbiosis. Presentation at the Forum on Microbial Threats Workshop Ending the War Metaphor: The Changing Agenda for Unraveling the Host-Microbe Relationship, Washington, D.C., Institute of Medicine, Forum on Microbial Threats.
Blaser MJ, Atherton JC. 2004. Helicobacter pylori persistence: Biology and disease. Journal of Clinical Investigation 113(3):321–333.
Blaser MJ, Kirschner D. 1999. Dynamics of Helicobacter pylori colonization in relation to the host response. Proceedings of the National Academy of Sciences USA 96(15):8359–8364.
Blaser MJ, Chyou PH, Nomura A. 1995a. Age at establishment of Helicobacter pylori infection and gastric carcinoma, gastric ulcer, and duodenal ulcer risk. Cancer Research 55(3):562–565.
Blaser MJ, Pérez-Pérez GI, Kleanthous H, Cover TL, Peek RM, Chyou PH, Stemmermann GN, Nomura A. 1995b. Infection with Helicobacter pylori strains possessing cagA associated with an increased risk of developing adenocarcinoma of the stomach. Cancer Research 55(10):2111–2115.
Breitbart M, Rohwer F. 2005. Here a virus, there a virus, everywhere the same virus? Trends in Microbiology 13(6):278–284.
Calam J. 1995. Helicobacter pylori, acid and gastrin. European Journal of Gastroenterology and Hepatology 7:310–317.
Cash HL, Hooper LV. 2005. Commensal bacteria shape intestinal immune system development. American Society of Microbiology News 71(2):77–83.
Censini S, Lange C, Xiang ZY, Crabtree JE, Ghiara P, Borodovsky M, Rappuoli R, Covacci A. 1996. Cag, a pathogenicity island of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proceedings of the National Academy of Sciences USA 93(25):14648–14653.
Chen LW, Egan L, Li ZW, Greten FR, Kagnoff MF, Karin M. 2003. The two faces of IKK and NF-kappaB inhibition: Prevention of systemic inflammation but increased local injury following intestinal ischemia-reperfusion. Nature Medicine 9(5):575–581.
Chow WH, Blaser MJ, Blot WJ, Gammon MD, Vaughan TL, Risch HA, Pérez-Pérez GI, Schoenberg JB, Stanford JL, Rotterdam H, West AB, Fraumeni JF. 1998. An inverse relation between cagA+ strains of Helicobacter pylori infection and risk of esophageal and gastric cardia adenocarcinoma. Cancer Research 58(4):588–590.
Cockburn TA. 1971. Infectious diseases in ancient populations. Current Anthropology 12:45–62.
Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. 1975. A model for gastric cancer epidemiology. Lancet 2(7924):58–60.
Covacci A, Censini S, Bugnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N, Rappuoli. 1993. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proceedings of the National Academy of Sciences USA 90(12):5791–5795.
Cover TL, Dooley CP, Blaser MJ. 1990. Characterization and human serologic response to proteins in Helicobacter pylori broth culture supernatants with vacuolizing cytotoxin activity. Infection and Immunity 58(3):603–610.
Crabtree JE, Taylor JD, Wyatt JI, Heatley RV, Shallcross TM, Tompkins DS, Rathbone BJ. 1991. Mucosal IgA recognition of Helicobacter pylori 120 kDa protein, peptic ulceration, and gastric pathology. Lancet 338(8763):332–335.
de Martel C, Llosa AE, Farr SM, Friedman GD, Vogelman JH, Orentreich N, Corley DA, Parsonnet J. 2005. Helicobacter pylori infection and the risk of development of esophageal adenocarcinoma. Journal of Infectious Diseases 191(5):761–767.
Devesa SS, Blot WJ, Fraumeni JK Jr. 1998. Changing patterns in the incidence of esophageal and gastric carcinoma in the United States. Cancer 83(10):2049–2053.
Dieleman LA, Ridwan BU, Tennyson GS, Beagley KW, Bucy RP, Elson CO. 1994. Dextran sulfate sodium-induced colitis occurs in severe combined immunodeficient mice. Gastroenterology 107(6):1643–1652.
Dieleman LA, Goerres M, Arends A, Sprengers D, Torrice C, Hoentjen F, Grenther WB, Sartor RB. 2003. Lactobacillus GG prevents recurrence of colitis in HLA-B27 transgenic rats after antibiotic treatment. Gut 52(3):370–376.
Dobrindt U, Hochhut B, Hentschel U, Hacker J. 2004. Genomic islands in pathogenic and environmental microorganisms. National Review of Microbiology 2(5):414–424.
Dobson, DE, Prager EM, Wilson AC. 1984. Stomach lysozymes of ruminants. I. Distribution and catalytic properties. Journal of Biological Chemistry 259(18):11607–11616.
Dominguez-Bello MG, Blaser MJ. 2005. Are iron-scavenging parasites protective against malaria? Journal of Infectious Diseases 191(4):646.
Dominguez-Bello MG, Ruiz MC, Michelangeli F. 1993. Evolutionary significance of foregut fermentation in the hoatzin (Opisthocomus hoazin; Aves, Opisthocomidae). Journal of Comparative Physiology B 163:594–601.
Dominguez-Bello MG, Pacheco MA, Ruiz MC, Michelangeli F, Leippe M, de Pedro MA. 2004. Resistance of rumen bacteria murein to bovine gastric lysozyme. BioMed Central Ecology 4:7.
Dominguez-Bello MG, Marini GE, Maldonado AL, Hidalgo G, Cabras S, Buffa R, Marini E, Floris G, Racugno W, Pericchi LR, Castellanos ME, Blaser MJ, Gröschl M. 2005. No Evidence of Detrimental Effect of Helicobacter pylori and Intestinal Parasites on the Nutritional Status of Amerindians in Venezuela. ASM Conference on Beneficial Microbes, Lake Tahoe, Nevada.
Dreyfuss ML, Msamanga GI, Spiegelman D, Hunter DJ, Urassa EJ, Hertzmark E, Fawzi WW. 2001. Determinants of low birth weight among HIV-infected pregnant women in Tanzania. American Journal of Clinical Nutrition 74(6):814–826.
Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. 2005. Diversity of the human intestinal microbial flora. Science 308(5728):1635–1638.
Edwards RA, Rohwer F. 2005. Viral metagenomics. Nature Reviews Microbiology 3(6):504–510.
Egan LJ, Eckmann L, Greten FR, Chae S, Li ZW, Myhre GM, Robine S, Karin M, Kagnoff MF. 2004. IkappaB-kinasebeta-dependent NF-kappaB activation provides radioprotection to the intestinal epithelium. Proceedings of the National Academy of Sciences USA 101(8):2452–2457.
El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF Jr, Rabkin CS. 2000. Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 412(6842):99.
Falkow S. 1998. Who speaks for the microbes? Emerging Infectious Diseases 4(3):495–497.
Falkow S. 2005 (March 17). Session III:Understanding the Dynamic Relationships of Host-Microbe Interactions. Presentation at the Forum on Microbial Threats Workshop Ending the War Metaphor: The Changing for Unraveling the Host-Microbe Relationship, Washington, D.C., Institute of Medicine, Forum on Microbial Threats.
Falush D, Kraft C, Taylor NS, Correa P, Fox JG, Achtman M, Suerbaum S. 2001. Recombination and mutation during long-term gastric colonization by Helicobacter pylori: Estimates of clock rates, recombination size, and minimal age. Proceedings of the National Academy of Sciences USA 98(26):15056–15061.
Falush D, Wirth T, Linz B, Pritchard JK, Stephens M, Kidd M, Blaser MJ, Graham DY, Vacher S, Perez-Perez GI, Yamaoka Y, Negraud F, Otto K, Reichard U, Katzowitsch E, Wang X, Achtman M, Suerbaum S. 2003. Traces of human migration in Helicobacter pylori populations. Science 299(5629):1582–1585.
Forchielli ML, Walker WA. 2005. The role of gut-associated lymphoid tissues and mucosal defence. British Journal of Nutrition 93(Suppl 1):S41–S48.
Ghose C, Perez-Perez GI, Dominguez-Bello MG, Pride DT, Bravi CM, Blaser MJ. 2002. East Asian genotypes of Helicobacter pylori: Strains in Amerindians provide evidence for its ancient human carriage. Proceedings of the National Academy of Sciences USA 99(23):15107–15111.
Ghose C, Perez-Perez GI, van Doorn LJ, Dominguez-Bello MG, Blaser MJ. 2005. High frequency of gastric colonization with multiple Helicobacter pylori strains in Venezuelan subjects. Journal of Clinical Microbiology 43(6):2635–2641.
Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ. 2003. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. Journal of Biological Chemistry 278(11):8869–8872.
Goodman KJ, Correa P. 2000. Transmission of Helicobacter pylori among siblings. Lancet 355(9201): 358–362.
Goodman KJ, Correa P, Tengana Aux HJ, Ramirez H, DeLany JP, Guerrero Pepinosa O, Lopez Quinones M, Collazos Parra T. 1996. Helicobacter pylori infection in the Colombian Andes: A population-based study of transmission pathways. American Journal of Epidemiology 144(3): 290–299.
Govoni G, Gros P. 1998. Macrophage NRAMP1 and its role in resistance to microbial infections. Inflammation Research 47(7):277–284.
Grajal A, Strahl S, Parra R, Dominguez MG, Neher A. 1989. Foregut fermentation in the hoatzin, a Neotropical leaf-eating bird. Science 245(4923):1236–1238.
Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, Kagnoff MF, Karin M. 2004. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118(3):285–296.
Halfvarson J, Bodin L, Tysk C, Lindberg E, Jarnerot G. 2003. Inflammatory bowel disease in a Swedish twin cohort: A long-term follow-up of concordance and clinical characteristics. Gastroenterology 124(7):1767–1773.
Harford WV, Barnett C, Lee E, Perez-Perez G, Blaser MJ, Peterson WL. 2000. Acute gastritis with hypochlorhydria: Report of 35 cases with long-term follow-up. Gut 47(4):467–472.
Harmsen, HJ, Raangs GC, He T, Degener JE, Welling GW. 2002. Extensive set of 16S rRNA-based probes for detection of bacteria in human feces. Applied Environmental Microbiology 68(6): 2982–2990.
Hatakeyama M. 2004. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nature Reviews Cancer 4(9):688–694.
Hayes W. 1968. Genetics of Bacteria and Their Viruses: Studies in Basic Genetics and Molecular Biology. 2nd ed. New York: John Wiley & Sons, Inc.
Hazell SL, Lee A, Brady L, Hennessy W. 1986. Campylobacter pyloridis and gastritis: Association with intercellular spaces and adaptation to an environment of mucus as important factors in colonization of the gastric epithelium. Journal of Infectious Diseases 153(4):658–663.
Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B, Mankertz J, Gitter AH, Burgel N, Fromm M, Zeitz M, Fuss I, Strober W, Schulzke JD. 2005. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 129(2):550–64.
Hentschel E, Brandstatter G, Dragosics B, Hirschl AM, Nemec H, Schutze K, Taufer M, Wurzer H. 1993. Effect of ranitidine and amoxicillin plus metronidazole on the eradication of Helicobacter pylori and the recurrence of duodenal ulcer. New England Journal of Medicine 328(5):308–312.
Higashi H, Tsutsumi R, Muto S, Sugiyama T, Azuma T, Asaka M, Hatakeyama M. 2001. SHP-2 tyrosine phosphatase as an intracellular target of Helicobacter pylori CagA protein. Science 295(5555):683–686.
Hisamatsu T, Suzuki M, Reinecker HC, Nadeau WJ, McCormick BA, Podolsky DK. 2003. CARD15/NOD2 functions as an anti-bacterial factor in human intestinal epithelial cells. Gastroenterology 124(4):993–1000.
Hoentjen F, Harmsen HJ, Braat H, Torrice CD, Mann BA, Sartor RB, Dieleman LA. 2003. Antibiotics with a selective aerobic or anaerobic spectrum have different therapeutic activities in various regions of the colon in interleukin-10 gene deficient mice. Gut 52(12):1721–1727.
Hooper, LV, Wong MH, Thelin A, Hansson L, Falk PG, Gordon JI. 2001. Molecular analysis of commensal host-microbial relationships in the intestine. Science 291(5505): 881–884.
Hooper LV, Stappenbeck TS, Hong CV, Gordon JI. 2003. Angiogenins: A new class of microbicidal proteins involved in innate immunity. Nature Immunology 4(3):269–273.
Horvath TL, Diano S, Sotonyi P, Heiman P, Tschöp M. 2001. Minireview: Ghrelin and the regulation of energy balance—a hypothalamic perspective. Endocrinology 142(10):4163–4169.
Howson CP, Hiyama T, Wynder EL. 1986. The decline in gastric cancer: Epidemiology of an unplanned triumph. Epidemiology Review 8:1–27.
Hugot JP, Chamaillard M, Zouali H, Lesage S, Cezard JP, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel JF, Sahbatou M, Thomas G. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411(6837):599–603.
Hungate RE. 1966. The rumen and its microbes. New York: Academic Press.
Hungate RE. 1972. Relationships between protozoa and bacteria of the alimentary tract. American Journal of Clinical Nutrition 25(12):1480–1484.
IARC (International Agency for Research on Cancer). 1994. Infection with Helicobacter pylori. IARC Monograph Evaluation of Carcinogenic Risks in Humans 61: 77–240.
Isomoto H, Nakazato M, Ueno H, Date Y, Nishi Y, Mukae H, Mizuta Y, Ohtsuru A, Yamashita S, Kohno S. 2004. Low plasma ghrelin levels in patients with Helicobacter pylori-associated gastritis. American Journal of Medicine 117(6):429–432.
Israel D, Lou A, Blaser MJ. 2000. Characteristics of Helicobacter pylori natural transformation. FEMS Microbiology Letters 186(2):275-280.
Israel D, Salama N, Krishna U, Rieger UM, Atherton JC, Falkow S, Peek RM. 2001. Helicobacter pylori genetic diversity within the gastric niche of a single human host. Proceedings of the National Academy of Sciences USA 98(25):14625–14630.
Karnes WE Jr, Samloff IM, Siurala M, Kekki M, Sipponen P, Kim SW, Walsh JH. 1991. Positive serum antibody and negative tissue staining for Helicobacter pylori in subjects with atrophic body gastritis. Gastroenterology 101(1):167–174.
Kersulyte D, Chalkauskas H, Berg DE. 1999. Emergence of recombinant strains of Helicobacter pylori during human infection. Molecular Microbiology 31(1):31–43.
Kim SC, Tonkonogy SL, Bower M, Sartor RB. 2004. Dual-association of gnotobiotic IL-10-/- mice with two nonpathogenic commensal bacterial species accelerates colitis [abstract]. Gastroenterology 126:A291.
Kim SC, Tonkonogy SL, Albright CA, Sartor RB. 2005a. Different host genetic backgrounds determine disease phenotypes induced by selective bacterial colonization [abstract]. Gastroenterology 128:A512.
Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB. 2005b. Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128(4):891–906.
Kirschner DE, Blaser MJ. 1995. The dynamics of Helicobacter pylori infection of the human stomach. Journal of Theoretical Biology 176(2):281–290.
Klein PD, Graham DY, Gaillour A, Opekun AR, Smith EO. 1991. Water source as risk factor for Helicobacter pylori infection in Peruvian children. Gastrointestinal Physiology Working Group. Lancet 337(8756):1503–1506.
Klieve AV, Bauchop T. 1988. Morphological diversity of ruminal bacteriophages from sheep and cattle. Applied Environmental Microbiology 54(6):1637–1641.
Klieve AV, Swain RA. 1993. Estimation of ruminal bacteriophage numbers by pulsed-field gel electrophoresis and laser densitometry. Applied Environmental Microbiology 59:2299–2303.
Klieve AV, Hudman JF, Bauchop T. 1989. Inducible bacteriophages from ruminal bacteria. Applied Environmental Microbiology 55:1630–1634.
Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nunez G, Flavell RA. 2005. Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307(5710): 731–734.
Kucik CJ, Martin GL, Sortor BV. 2004. Common intestinal parasites. American Family Physician 69(5):1161–1168.
Kuipers EJ, Uyterlinde AM, Peña AS, Roosendaal R, Pals G, Nelis GF, Festen HP, Meuwissen SG. 1995a. Long-term sequelae of Helicobacter pylori gastritis. Lancet 345(8964):1525–1528.
Kuipers EJ, Pérez-Pérez GI, Meuwissen SGM, Blaser MJ. 1995b. Helicobacter pylori and atrophic gastritis: Importance of the cagA status. Journal of the National Cancer Institute 87(23):1777–1780.
Kuipers EJ, Israel DA, Kusters JG, Gerrits MM, Weel J, van der Ende A, van der Hulst RWM, Wirth H-P, Hôôk-Nikanne J, Thompson SA, Blaser MJ. 2000. Quasispecies development of Helicobacter pylori observed in paired isolates obtained years apart in the same host. Journal of Infectious Diseases 181(1):273–282.
Lagergren J, Bergström R, Lindgren A, Nyrén O. 1999. Symptomatic gastroesophageal reflux as a risk factor for esophageal adenocarcinoma. New England Journal of Medicine 340(11): 825–831.
Lala S, Ogura Y, Osborne C, Hor SY, Bromfield A, Davies S, Ogunbiyi O, Nunez G, Keshav S. 2003. Crohn’s disease and the NOD2 gene: A role for Paneth cells. Gastroenterology 125(1):47–57.
Lederberg J. 2000. Infectious history. Science 288(5464):287–293.
Lee HM, Wang G, Englander EW, Kojima M, Greeley Jr. GH. 2002. Ghrelin, a new gastrointestinal endocrine peptide that stimulates insulin secretion: Enteric distribution, ontogeny, influence of endocrine, and dietary manipulations. Endocrinology 143(1):185–190.
Loffeld RJLF, Werdmuller BFM, Kusters JG, Blaser MJ, Pérez-Pérez GI, Kuipers EJ. 2000. Colonization with cagA-positive H. pylori strains inversely associated with reflux oesophagitis and Barrett’s oesophagitis. Digestion 62(2-3):95–99.
Lu X, Francois F, Yang L, Zhou M, Bini E, Blaser MJ, Pei Z. 2005 (June). Alteration of the bacterial biota in reflux esophagitis. Paper presented at: 105th General Meeting of the American Society of Microbiology; Atlanta, GA.
MacDonald TT, Gordon JN. 2005. Bacterial regulation of intestinal immune responses. Gastroenterology Clinic of North America 34(3):401-412.
Machado JC, Figueiredo C, Canedo P, Pharoah P, Carvalho R, Nabais S, Castro Alves C, Campos ML, Van Doorn LJ, Caldas C, Seruca R, Carneiro F, Sobrinho-Simoes M. 2003. A pro-inflammatory genetic profile increases the risk for chronic atrophic gastritis and gastric carcinoma. Gastroenterology 125(2):364–371.
Mackowiak PA. 1982. The normal microbial flora. New England Journal of Medicine 307(2):83–93.
Mann BA, Kim SC, Sartor RB. 2003. Selective induction of experimental colitis by monoassociation of HLA-B27 transgenic rats with various enteric Bacteroides species [abstract]. Gastroenterology 124:A322.
Matson CA, Reid DF, Cannon TA, Ritter RC. 2000. Cholecystokinin and leptin act synergistically to reduce body weight. American Journal of Physiology, Regulatory, Integrative and Comparative Physiology 278(4):R882–R890.
McNeill WH. 1976. Plagues and Peoples. Garden City, NY: Anchor Books.
Moss SF, Calam J. 1993. Acid secretion and sensitivity to gastrin in patients with duodenal ulcer: Effect of eradication of Helicobacter pylori. Gut 34(7):888–892.
Moss SF, Legon S, Bishop AE, Polak JM, Calam J. 1992. Effect of Helicobacter pylori on gastric somatostatin in duodenal ulcer disease. Lancet 340(8825):930–932.
Murray J, Murray A, Murray M, Murray C. 1978. The biologicalsuppression of malaria: An ecological and nutritional interrelationship of a host and two parasites. American Journal of Clinical Nutrition 31(8):1363–1366.
Nacher M, McGready R, Stepniewska K, Cho T. Looareesuwan S, White NJ, Nosten F. 2003. Haematinic treatment of anaemia increases the risk of Plasmodium vivax malaria in pregnancy. Transactions of the Royal Society of Tropical Medicine and Hygiene 97(3):273–276.
Naumann M, Wessler S, Bartsch C, Wieland B, Covacci A, Haas R, Meyer TF. 1999. Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. Journal of Biological Chemistry 274(4):31655–31662.
Nelson KE, Zinder SH, Hance I, Burr P, Odongo D, Wasawo D, Odenyo A, Bishop R. 2003. Phylogenetic analysis of the microbial populations in the wild herbivore gastrointestinal tract: Insights into an unexplored niche. Environmental Microbiology 5(11):1212–1220.
Nemcova R, Styriak I, Stachova M, Kmet V. 1993. Isolation and partial characterization of three rumen Lactobacillus plantarum bacteriophages. New Microbiology 16(2):177–180.
Newman B, Siminovitch KA. 2005. Recent advances in the genetics of inflammatory bowel disease. Current Opinions in Gastroenterology 21(4):401–407.
Nomura AM, Stemmermann GN, Chyou P-H, Pérez-Pérez GI, Blaser MJ. 1994. Helicobacter pylori infection and the risk for duodenal and gastric ulceration. Annals of Internal Medicine 120(12): 977–981.
Nomura AM, Perez-Perez GI, Lee J, Stemmermann G, Blaser MJ. 2002a. Relationship between H. pylori cagA status and risk of peptic ulcer disease. American Journal of Epidemiology 155(11): 1054–1059.
Nomura AM, Lee J, Stemmermann G, Nomura RY, Perez-Perez GI, Blaser MJ. 2002b. Helicobacter pylori cagA seropositivity and gastric carcinoma risk in a Japanese American population. Journal of Infectious Diseases 186(8):1138–1144.
Nwokolo CU, Freshwater DA, O’Hare P, Randeva HS. 2003. Plasma ghrelin following cure of Helicobacter pylori. Gut 52(5):637–640.
Occhialini A, Marais A, Urdaci M, Sierra R, Munoz N, Covacci A, Megraud F. 2001. Composition and gene expression of the cag pathogenicity island in Helicobacter pylori strains isolated from gastric carcinoma and gastritis patients in Coast Rica. Infection and Immunity 69(3):1902–1908.
Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287(5457):1497–1500.
Ogimoto K, Imai S. 1981. Atlas of Rumen Microbiology. Tokyo, Japan: Scientific Society Press.
Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar JP, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nunez G, Cho JH. 2001. A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411(6837):603–606.
Oliveira AM, Queiroz DM, Rocha GA, Mendes EN. 1994. Seroprevalence of Helicobacter pylori infection of children of low socioeconomic level in Belo Horizonte, Brazil. American Journal of Gastroenterology 89(12):2201–2204.
Oppenheimer SJ. 2001. Iron and its relation to immunity and infectious disease. Journal of Nutrition 131(2S-2):616S-633S.
Pantoja, I, Mojica M, Vargas-Pinto G, Scott K, Patterson A, Flint H, Blaser M, Dominguez-Bello MG. 2005. Tetracycline Resistance Genes in Bolivian Amerindians with Low Antibiotic Exposure. San Francisco, CA: Infectious Diseases Society of America.
Parsonnet J. 1995. The incidence of Helicobacter pylori infection. Alimentary Pharmacology and Therapeutics 9(Suppl 2):45–51.
Parsonnet J, Friedman GD, Orentreich N, Vogelman H. 1997. Risk for gastric cancer in people with CagA positive or CagA negative Helicobacter pylori infection. Gut 40(3):297–301.
Peek RM, Blaser MJ. 2002. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature Reviews Cancer 2(1):28–37.
Peek RM Jr, Peek RM, Vaezi MF, Falkow S, Goldblum JR, Pérez-Pérez GI, Richter JE, Blaser MJ. 1999. The role of Helicobacter pylori cagA+ strains and specific host immune responses on the development of premalignant and malignant lesions of the gastric cardia. International Journal of Cancer 82:520–524.
Pei Z, Bini EJ, Yang L, Zhou M, Francois F, Blaser MJ. 2004. Bacterial biota in the human distal esophagus. Proceedings of the National Academy of Sciences USA 101(12):4250–4255.
Pei Z, Yang L, Peek RM, Levine SM, Pride DT, Blaser MJ. 2005. Bacterial biota in reflux esophagitis and Barrett’s esophagus. World Journal of Gastroenterology In press.
Peltekova VD, Wintle RF, Rubin LA, Amos CI, Huang Q, Gu X, Newman B, Van Oene M, Cescon D, Greenberg G, Griffiths AM, St George-Hyslop PH, Siminovitch KA. 2004. Functional variants of OCTN cation transporter genes are associated with Crohn disease. Nature Genetics 36(5):471–475.
Perez-Perez GI, Salomaa A, Kosunen TU, Daverman B, Rautelin H, Aromaa A, Knekt P, Blaser MJ. 2002. Evidence that cagA+Helicobacter pylori strains are disappearing more rapidly than cagA-strains. Gut 50(3):295–298.
Perez-Perez GI, Sack RB, Reid R, Santosham M, Croll J, Blaser MJ. 2003. Transient and persistent Helicobacter pylori colonization in Native American children. Journal of Clinical Microbiology 41(6):2401–2407.
Pohl H, Gilbert H. 2005. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. Journal of the National Cancer Institute 97(2):142–146.
Queiroz DM, Guerra JB, Rocha GA, Rocha AM, Santos A, De Oliveira AG, Cabral MM, Nogueira AM, De Oliveira CA. 2004. IL1B and IL1RN polymorphic genes and Helicobacter pylori cagA strains decrease the risk of reflux esophagitis. Gastroenterology 127(1):73–79.
Rakoff-Nahoum S, Paglino J, Eslami-Varzaneh F, Edberg S, Medzhitov R. 2004. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118(2): 229–241.
Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE Jr, Balish E, Taurog JD, Hammer RE, Wilson KH, Sartor RB. 1996. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. Journal of Clinical Investigation 98(4):945-953.
Rath HC, Wilson KH, Sartor RB. 1999. Differential induction of colitis and gastritis in HLA-B27 transgenic rats selectively colonized with Bacteroides vulgatus and Escherichia coli. Infection and Immunity 67(6):2969-2974.
Rath HC, Schultz M, Freitag R, Dieleman LA, Li F, Linde HJ, Scholmerich J, Sartor RB. 2001. Different subsets of enteric bacteria induce and perpetuate experimental colitis in rats and mice. Infection and Immunity 69(4):2277–2285.
Rauws EA, Langenberg W, Houthoff HJ, Zanen HC, Tytgat GN. 1998. Campylobacter pyloridis-associated chronic active antral gastritis: A prospective study of its prevalence and the effects of antibacterial and antiulcer treatment. Gastroenterology 94(1):33–40.
Rehnberg-Laiho L, Rautelin H, Koskela P, Sarna S, Pukkala E, Aromaa A, Knekt P, Kosunen TU. 2001. Decreasing prevalence of helicobacter antibodies in Finland, with reference to the decreasing incidence of gastric cancer. Epidemiology and Infection 126(1):37–42.
Rosebury T. 1962. Microorganisms Indigenous to Man. New York: McGraw Hill.
Rothenbacher D, Blaser MJ, Bode G, Brenner H. 2000. An inverse relationship between gastric colonization by Helicobacter pylori and diarrheal illnesses in children: Results of a population-based cross-sectional study. Journal of Infectious Diseases 182(5):1446–1449.
Salyers A, Gupta A, Wang Y. 2004. Human intestinal bacteria as reservoirs for antibiotic resistance genes. Trends in Microbiology 12(9):412–416.
Sartor RB. 2004. Microbial influences in inflammatory bowel disease: Role in pathogenesis and clinical implications. In: Sartor RB, Sandborn WJ, eds. Kirsner’s Inflammatory Bowel Diseases. Philadelphia, PA: Elsevier Publishers; Pp. 138–162.
Sartor RB. 2005 (March 16). Session I: Host-Pathogen Interactions: Defining the Concepts of Pathogenicity, Virulence, Colonization, Commensalism, and Symbiosis. Presentation at the Forum on Microbial Threats Workshop Ending the War Metaphor: The Changing Agenda for Unraveling the Host-Microbe Relationship, Washington, D.C., Institute of Medicine, Forum on Microbial Threats.
Sartor RB, Hoentjen F. 2005. Proinflammatory cytokines and signaling pathways in intestinal innate immune cells. In: Mestecky J, Lamm ME, Strober W, Bienenstock J, McGhee JR, Mayer L, eds. Mucosal Immunology. London: Elsevier Academic Press; Pp. 681–701.
Savage DC. Microbial ecology of the gastrointestinal tract. 1977. Annual Review of Microbiology 31:107–133.
Schreiber S, Rosenstiel P, Albrecht M, Hampe J, Krawczak M. 2005. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nature Reviews Genetics 6(5):376–388.
Segal ED, Cha J, Lo J, Falkow S, Tompkins LS. 1999. Altered states: Involvement of phosphorylated CagA in the induction of host cellular growth changes by Helicobacter pylori. Proceedings of the National Academy of Sciences USA 96(25):14559–14564.
Segal S, Hill AV. 2003. Genetic susceptibility to infectious disease. Trends in Microbiology 11(9): 445–448.
Sellon RK, Tonkonogy S, Schultz M, Dieleman LA, Grenther W, Balish E, Rennick DM, Sartor RB. 1998. Resident enteric bacteria are necessary for development of spontaneous colitis and immune system activation in interleukin-10-deficient mice. Infection and Immunity 66(11): 5224–5231.
Smythies LE, Sellers M, Clements RH, Mosteller-Barnum M, Meng G, Benjamin WH, Orenstein JM, Smith PD. 2005 Human intestinal macrophages display profound inflammatory anergy despite avid phagocytic and bacteriocidal activity. Journal of Clinical Investigation 115(1):66–75.
Sobhani I, Buyse M, Goiot H, Weber N, Laigneau JP, Henin D, Soul JC, Bado A. 2002. Vagal stimulation rapidly increases leptin secretion in human stomach. Gastroenterology 122(2): 259–263.
Stappenbeck TS, Hooper LV, Gordon JI. 2002. Developmental regulation of intestinal angiogenesis by indigenous microbes via Paneth cells. Proceedings of the National Academy of Sciences USA 99(24):15451–15455.
Strober W, Fuss I, Boirivant M, Kitani A. 2004. Insights into the mechanism of oral tolerance derived from the study of models of mucosal inflammation. Annual of the New York Academy of Sciences 1029:115–131.
Suerbaum S, Smith JM, Bapumia K, Morelli G, Smith NH, Kunstmann E, Dyrek I, Achtman M. 1998. Free recombination with Helicobacter pylori. Proceedings of the National Academy of Sciences USA 95(21):12619–12624.
Swidsinski A, Ladhoff A, Pernthaler A, Swidsinski S, Loening-Baucke V, Ortner M, Weber J, Hoffmann U, Schreiber S, Dietel M, Lochs H. 2002. Mucosal flora in inflammatory bowel disease. Gastroenterology 122(1):44–54.
Tajima K, Aminov RI, Nagamine T, Ogata K, Nakamura M, Matsui H. 2001a. Rumen bacterial diversity as determined by sequence analysis of 16S rDNA libraries. FEMS Microbiology Ecology 29:159–169.
Tajima K, Nagamine T, Matsui H, Nakamura M, Aminov RI. 2001b. Phylogenetic analysis of archaeal 16S rRNA libraries from the rumen suggests the existence of a novel group of archaea not associated with known methanogens. FEMS Microbiology Letters 200(1):67–72.
Tham KT, Peek RM, Atherton JC, Cover TL, Perez-Perez GI, Shyr Y, Blaser MJ. 2001. Helicobacter pylori genotypes, host factors, and gastric mucosal histopathology in peptic ulcer disease. Human Pathology 32(3):264–273.
Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388(6642):539–547.
Tummuru MKR, Cover TL, Blaser MJ. 1993. Cloning and expression of a high molecular weight major antigen of Helicobacter pylori: Evidence of linkage to cytotoxin production. Infection and Immunity 61(5):1799–1809.
Tummuru MKR, Sharma SA, Blaser MJ. 1995. Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Molecular Microbiology 18(5):867–876.
Vaezi MF, Falk GW, Peek RM, Vicari JJ, Goldblum JR, Perez-Perez GI, Rice TW, Blaser MJ, Richter JE. 2000. cagA-positive strains of Helicobacter pylori may protect against Barrett’s esophagus. American Journal of Gastroenterology 95(9):2206–2211.
Van Soest PJ. 1994. Nutritional Ecology of the Ruminant. Ithaca, NY: Comstock.
Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Memet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL. 2004. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nature Immunology 5(11):1166–1174.
Vicari JJ, Peek RM, Falk GW, Goldblum JR, Easley KA, Schnell J, Pérez-Pérez GI, Halter SA, Rice TW, Blaser MJ, Richter JE. 1998. The seroprevalence of cagA-positive Helicobacter pylori strains in the spectrum of gastroesophageal reflux disease. Gastroenterology 115(1):50–57.
Villedieu A, Diaz-Torres ML, Hunt N, McNab R, Spratt DA,Wilson M, Mullany P. 2003. Prevalence of tetracycline resistance genes in oral bacteria. Antimicrobial Agents in Chemotherapy 47(3): 878–882.
Warburton-Timmsa VJ, Charlettd A, Valorib RM, Uffc JS, Shepherdc NA, Barrb H, McNultya CAM. 2001. The significance of cagA+Helicobacter pylori in reflux oesophagitis. Gut 49(3):341–346.
Warren JR, Marshall BJ. 1983. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1(8336):1273–1275.
Watanabe T, Kitani A, Murray PJ, Strober W. 2004. NOD2 is a negative regulator of Toll-like receptor 2-mediated T helper type 1 responses. Nature Immunology 5(8):800–808.
Wehkamp J, Harder J, Weichenthal M, Schwab M, Schaffeler E, Schlee M, Herrlinger KR, Stallmach A, Noack F, Fritz P, Schroder JM, Bevins CL, Fellermann K, Stange EF. 2004. NOD2 (CARD15) mutations in Crohn’s disease are associated with diminished mucosal alpha-defensin expression. Gut 53(11):1658–1664.
Whitford MF, Forster RJ, Beard CE, Gong J, Teather RM. 1998. Phylogenetic analysis of rumen bacteria by comparative sequence analysis of cloned 16S rRNA genes. Anaerobe 4:153–163.
Wirth HP, Yang M, Peek RM, Hook-Nikanne J, Fried M, Blaser MJ. 1999. Phenotypic diversity in Lewis expression of Helicobacter pylori isolates from the same host. Journal of Laboratory and Clinical Medicine 133(5):488–500.
Wu AH, Crabtree JE, Bernstein L, Hawtin P, Cockburn M, Tseng C, Forman D. 2003. Role of Helicobacter pylori cagA+ strains and risk of adenocarcinoma of the stomach and esophagus. International Journal of Cancer 103(6):815–821.
Yamaji Y, Mitsushima T, Ikuma H, Okamoto M, Yoshida H, Kawabe T, Shiratori Y, Saito K, Yokouchi K, Omata M. 2001. Inverse background of Helicobacter pylori anti-cancer: Analysis of 5732 Japanese subjects. Gut 49(3):335–340.
Yamaoka Y, Kodama T, Kashima K, Graham DY, Sepulveda AR. 1998. Variants of the 3’ region of the cagA gene in Helicobacter pylori isolates from patients with different H. pylori-associated diseases. Journal of Clinical Microbiology 36(8):2258–2263.
Ye W, Held M, Lagergren J, Engstrand L, Blot WJ, McLaughlin JK, Nyrén O. 2004. Helicobacter pylori infection and gastric atrophy: Risk of adenocarcinoma and squamous-cell carcinoma of the esophagus and adenocarcinoma of the gastric cardia. Journal of the National Cancer Institute 96(5):388–396.

























































