
Summary
Dioxins are a class of chemicals, and the most toxic of these compounds is 2,3,7,8-tetrachlorodibenzo-p-dioxin (commonly referred to as TCDD or dioxin). There are many forms of dioxins and “dioxin-like compounds” (DLCs) that share most, if not all, of the toxic potential of TCDD, although nearly all are considerably less potent. Included in the list of DLCs are chlorinated forms of dibenzofurans and certain polychlorinated biphenyls (PCBs).
Combustion, metal processing, chemical manufacturing and processing, and other sources emit TCDD, other dioxins, and DLCs into the environment. Unlike PCBs, TCDD and other dioxins have never been intentionally produced. TCDD, other dioxins, and DLCs persist and bioaccumulate in the environment, which means that they break down slowly and build up through the food chain. Human exposure to TCDD, other dioxins, and DLCs occurs primarily from eating foods, such as beef, dairy products, fish, shellfish, and pork. In recent years, efforts to reduce the amount of TCDD, other dioxins, and DLCs in the environment have resulted in reductions in measured concentrations in the environment and in human blood.
TCDD, other dioxins, and DLCs share a common mode of action in producing toxic effects in humans and animals. They bind to a specific receptor, called the aromatic hydrocarbon receptor or Ah receptor; such binding is a necessary, but not sufficient, step toward producing adverse health effects.
A few industrial accidents and occupational exposures to substantial amounts of TCDD, other dioxins, and DLCs have provided opportunities
to assess the toxicity of these compounds to humans. Several episodes of high-level human exposure to TCDD have been found to cause a specific type of persistent, potentially disfiguring skin lesion called chloracne. In 2004, the media widely publicized the suspected intentional poisoning of Viktor Yushchenko with TCDD after he developed chloracne during the Ukraine presidential campaign. In contrast to the undisputed high-dose effects of chloracne, the potential adverse effects of TCDD, other dioxins, and DLCs in humans after long-term, low-level environmental exposures remain controversial. The major controversies include how to classify the potential of these compounds to cause cancer in humans (as either “carcinogenic to humans” or “likely to be carcinogenic to humans”), how to estimate the potential health risks at very low doses typical of actual population exposures, and how to assess the toxicity of each of the compounds and various mixtures of them in the environment.
TCDD, other dioxins, and DLCs have been regulated extensively worldwide. In the early 1980s, the U.S. Environmental Protection Agency (EPA) and other organizations, such as the World Health Organization (WHO), began collecting and evaluating scientific information about the sources, fate, and effects of the compounds. In 1985, EPA produced an initial assessment of the human health risks from environmental exposure to TCDD. Later, as new scientific information became available, EPA reassessed the human health risks in an open process involving participation of numerous scientists external to the agency, a series of public meetings, and peer review.
An Interagency Working Group (IWG) made up of representatives of seven federal agencies was established in 2000 to coordinate federal strategies for risk management of TCDD, other dioxins, and DLCs. Members of the IWG, EPA’s Science Advisory Board, and the public commented on earlier drafts of EPA’s dioxin risk assessment, and after further revisions, EPA released the 2003 draft document titled Exposure and Human Health Reassessment of Tetrachlorodibenzo-p-Dioxin (TCDD) and Related Compounds (referred to as the Reassessment). The IWG recommended further review of the new document, and in 2004, EPA asked the National Research Council (NRC) to convene an expert committee to review independently EPA’s 2003 draft Reassessment and to determine whether EPA’s risk estimates are scientifically robust and whether there is clear delineation of all substantial uncertainties and variabilities (Box S-1).
This report presents the committee’s conclusions and recommendations. In general, the committee recommends that EPA substantially augment its Reassessment to improve the transparency about assumptions used to estimate risk and how these assumptions affect estimates. The committee also recommends that EPA re-estimate the risks using several assumptions and communicate the uncertainty in these estimates to the public.
|
BOX S-1 Statement of Task The National Academies’ National Research Council will convene an expert committee that will review EPA’s 2003 draft reassessment of the risks of dioxins and dioxin-like compounds to assess whether EPA’s risk estimates are scientifically robust and whether there is a clear delineation of all substantial uncertainties and variability. To the extent possible, the review will focus on EPA’s modeling assumptions, including those associated with the dose-response curve and points of departure; dose ranges and associated likelihood estimates for identified human health outcomes; EPA’s quantitative uncertainty analysis; EPA’s selection of studies as a basis for its assessments; and gaps in scientific knowledge. The study will also address the following aspects of the EPA reassessment: (1) the scientific evidence for classifying dioxin as a human carcinogen; and (2) the validity of the non-threshold linear dose-response model and the cancer slope factor calculated by EPA through the use of this model. The committee will also provide scientific judgment regarding the usefulness of toxicity equivalence factors (TEFs) in the risk assessment of complex mixtures of dioxins and the uncertainties associated with the use of TEFs. The committee will also review the uncertainty associated with the reassessment’s approach regarding the analysis of food sampling and human dietary intake data and, therefore, human exposures, taking into consideration the Institute of Medicine’s report Dioxin and Dioxin-Like Compounds in the Food Supply: Strategies to Decrease Exposure. The committee will focus particularly on the risk characterization section of EPA’s reassessment report and will endeavor to make the uncertainties in such risk assessments more fully understood by decision makers. The committee will review the breadth of the uncertainty and variability associated with risk assessment decisions and numerical choices—for example, modeling assumptions, including those associated with the dose-response curve and points of departure. The committee will also review quantitative uncertainty analyses, as feasible and appropriate. The committee will identify gaps in scientific knowledge that are critical to understanding dioxin reassessment. |
CARCINOGENIC CLASSIFICATION
In 1985, EPA classified TCDD as a “probable human carcinogen” based on the data available at the time, but the latest Reassessment (2003) stated that TCDD was better characterized as “carcinogenic to humans.” EPA and the International Agency for Research on Cancer (IARC), an arm of WHO, have established criteria for qualitatively classifying chemicals into various carcinogenic categories based on the weight of scientific evidence from animal, human epidemiological, and mechanism or mode-of-action studies. In 1997, an expert panel convened by IARC concluded that the weight of scientific evidence for dioxin carcinogenicity in humans supported its classification as a Class 1 carcinogen—“carcinogenic to humans.”
In 2001, the U.S. National Toxicology Program (NTP) upgraded its classification of dioxin to “known to be a human carcinogen.”
After reviewing EPA’s 2003 Reassessment and other scientific information and in light of EPA’s recently revised 2005 Guidelines for Carcinogen Risk Assessment (cancer guidelines), the committee concludes that the classification of TCDD as “carcinogenic to humans”—a designation suggesting the greatest degree of certainty about carcinogenicity—versus “likely to be carcinogenic to humans”—the next highest designation—is somewhat subjective and depends largely on the definition and interpretation of the criteria used for classification. The true weight of evidence lies on a continuum, with no obvious point or “bright line” that readily distinguishes those two categories.
Referring to the specific definitions in EPA’s 2005 cancer guidelines for qualitative classification of chemical carcinogens, the NRC committee was split on whether the evidence met all the criteria necessary for classification of TCDD as “carcinogenic to humans,” although the committee unanimously agreed on a classification of at least “likely to be carcinogenic to humans.” The committee concludes that the weight of epidemiological evidence supporting classification of TCDD as a human carcinogen is not “strong.” The committee points out, however, that the human data available from occupational studies show a modest positive association between relatively high concentrations of TCDD in the body and increased mortality from all cancers. Animal studies and mechanistic data provide additional support for classifying TCDD as a human carcinogen.
The committee concludes that the distinction between those two qualitative categories of cancer risk classification depends more on semantics than on science and that the public health implications of the two terms appeared identical, and for these reasons the committee did not focus much attention on the issue of classification. To the extent that EPA can be consistent with regulatory requirements, the committee recommends that EPA focus its energies and resources on more carefully quantifying risks and uncertainties for TCDD, other dioxins, and DLCs rather than on whether its carcinogenicity is probable or proven. Because the 2005 cancer guidelines’ definition of “carcinogenic to humans” has changed since EPA completed its 2003 Reassessment, the committee recommends that EPA reevaluate its conclusion that TCDD satisfies the criteria for designation as either “carcinogenic to humans” or “likely to be a human carcinogen” based on the criteria set out in EPA’s 2005 cancer guidelines.
The committee agrees with EPA in classifying dioxins, other than TCDD, and DLCs as “likely to be carcinogenic to humans.” However, because mixtures of DLCs may also contain dioxins, including TCDD, EPA should reconsider its classification of such mixtures as “likely to be carcinogenic to humans” if it continues to classify dioxin as “carcinogenic to humans.”
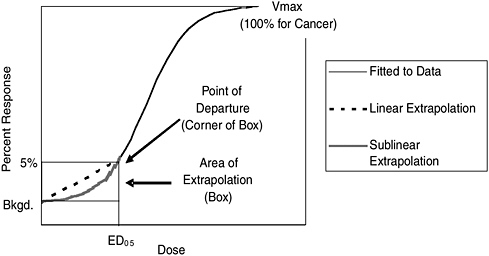
FIGURE S-1 Conceptual illustration of the effect of the selection of the point of departure and of the mathematical model used to extrapolate below the point of departure on the risk estimate. Note that the 5% response rate is not drawn to scale. If it were, the area of extrapolation box would be much smaller.
ESTIMATING CANCER RISK
Because nearly all data (both human epidemiological studies and experimental animal bioassays) relevant to cancer risk are for doses much higher than those to which the general human population is typically exposed, analysts must extrapolate below the doses observed when estimating risks. This extrapolation depends on first fitting a dose-response curve to the observed data from a given study and choosing a “point-of-departure” (POD) dose, which corresponds to the lowest dose associated with adverse effects within the range of the data from the experiment or study. The POD dose is an incremental “effect” observed; for example, analysts would call a POD corresponding to a 5% increase in effects (above no exposure) a 5% effective dose or an ED05.
Estimating risks below the POD may require extrapolating down to background levels of exposure. See Figure S-1 for a conceptual illustration of a dose-response extrapolation to background levels using the 5% response rate and ED05 as the POD. This extrapolation must be based on assumptions about how TCDD, other dioxins, and DLCs might cause cancer. Thus, the selection of the type of mathematical model used to extrapolate below the POD is a critical decision in the cancer risk assessment process. In the 2003 Reassessment, EPA chose to extrapolate below the POD with a “linear” model, which assumes that the biological response
increases proportionally with the level of exposure starting at a dose of zero. Risk estimates based on this approach are generally higher than those based on alternative “nonlinear” assumptions, where the biological response does not vary proportionally with the dose. However, EPA took the position that scientific data were inadequate to rule out its default linear assumption.
Selection of the POD also is an important choice in cancer risk assessment modeling because it determines the range of extrapolation below the observed data range. For example, more extrapolation below the POD is necessary when using a POD equal to the ED10 than when using a POD equal to the ED01. However, using an ED01 requires more data because the analyst must be able to detect a 1% increase in effects instead of a larger increase (e.g., a 5% increase for an Ed05).
After reviewing EPA’s 2003 Reassessment and additional scientific data published since completion of the Reassessment, the committee unanimously agreed that the current weight of scientific evidence on the carcinogenicity of dioxin is adequate to justify the use of nonlinear methods consistent with a receptor-mediated response to extrapolate below the POD. The committee points out that data from NTP released after EPA generated the 2003 Reassessment provide the most extensive information collected to date about TCDD carcinogenicity in test animals, and the committee found the NTP results to be compelling. The committee concludes that EPA should reevaluate how it models the dose-response relationships for TCDD, other dioxins, and DLCs. Specifically, the committee determined that the scientific evidence is consistent with receptor-mediated responses and favors the use of a nonlinear model over the default linear assumption to extrapolate below the POD for dioxin-related cancer risk. The committee recognizes that a linear response at doses below the POD cannot be entirely excluded, especially if background exposures are not orders of magnitude below the POD and additivity of risk from other types of chemicals is considered.
Because the committee concludes that the data support the hypothesis that the dose-response relationship for dioxin and cancer is sublinear, it recommends that EPA include a nonlinear model for cancer risk estimates but also use the current linear models for comparative purposes. EPA should then describe the scientific strengths and weaknesses of each approach to inform risk managers about the importance of these assumptions. The committee recognizes that additional evidence about dioxin carcinogenicity will continue to develop and concludes that EPA should proceed with completing its quantitative cancer risk assessment and include the recent NTP data and appropriate nonlinear dose-response models.
ESTIMATING NONCANCER RISK
To characterize the risks of adverse health effects other than cancer at very low doses, EPA typically identifies a dose called the reference dose (RfD) below which it anticipates no adverse effects from exposure, even among sensitive members of the population. To estimate the RfD, EPA usually starts with a benchmark dose (BMD), which is the level of exposure in an epidemiological study or an animal experiment that produces a certain specified level of response. For example, the BMD might be defined as the level of exposure at which 5% of exposed animals or people exhibit a specific type of adverse effect. EPA then calculates the RfD by dividing the BMD by a series of uncertainty factors intended to take into account several sources of uncertainty. These sources of uncertainty include extrapolation from animals to humans (allowing for a more sensitive response in humans than that in the test animals), extrapolation within the population (allowing for more sensitive members of the human population), and database sufficiency considerations (allowing for the possibility that more data might reveal more sensitive effects).
EPA did not estimate an RfD for TCDD, other dioxins, and DLCs in the Reassessment. However, the committee noted that defining an RfD would provide useful guidance to risk managers to help them (1) assess potential health risks in that portion of the population with intakes above the RfD, (2) assess risks to population groups, such as those with occupational exposures, and (3) estimate the risk contributions of the major food sources and other environmental sources for those individuals with high intakes. Alternatively, EPA could undertake risk characterization for different adverse effects by comparing noncancer dose-response data to relevant human exposure data in the calculation of margins of exposure (MOEs),1 as was done in the Reassessment. Such MOEs, accompanied by a description of associated uncertainties, could provide risk managers with information that would help to inform their decisions. Although EPA concluded that calculating RfDs would not provide useful information, the committee concludes that the information might be useful if EPA also considered that the use of body burden (estimate of the total amount of chemical in the body at steady state for a defined rate of exposure) as a dose metric would already take into account some of the uncertainty factors that EPA would typically use to adjust the BMD or POD in estimating an RfD. Estimates of background exposures in the United States also appear to have continued to decline, in part due to enhanced analytical detection.
|
1 |
EPA defines MOE as the lowest “ED10 or other point of departure divided by the actual or projected environmental exposure of interest” (http://www.epa.gov/iris/gloss8.htm#m). |
The committee concludes that EPA did not adequately justify the use of the 1% response level (the ED01) as the POD for analyzing epidemiological or animal bioassay data for both cancer and noncancer effects. The committee recommends that EPA more explicitly address the importance of the selection of the POD and its impact on risk estimates by calculating risk estimates using alternative assumptions (e.g., the ED05).
The committee commends EPA’s extensive dose-response modeling efforts of a large number of data sets, particularly those for noncancer effects, but remains concerned about selection of the final model for computing the POD. It is critical that the model used for determining a POD fit the data well, especially at the lower end of the observed responses. Whenever feasible, mechanistic and statistical information should be used to estimate the shape of the dose-response curve at lower doses. At a minimum, EPA should use rigorous statistical methods to assess model fit, and to control and reduce the uncertainty of the POD caused by a poorly fitted model. The overall quality of the study design is also a critical element in deciding which data sets to use for quantitative modeling.
UNCERTAINTY AND VARIABILITY IN RISK ESTIMATES
Risk assessors must make many choices as they develop models to characterize risks. Some of the initial choices are selecting appropriate data sets for low-dose extrapolation, selecting appropriate dose-response models, selecting critical end points, and selecting an appropriate POD (e.g., ED01 versus ED05). Risk estimates routinely reflect numerous sources of both uncertainty (which describes the range of plausible risk estimates arising because of limitations in knowledge) and variability (which describes the range of risks arising because of true differences—for example, genetic and age differences among members of the population). Failure to fully characterize uncertainty and variability can convey a false sense of precision in the conclusions of the risk assessment. EPA should include a detailed discussion of both uncertainty and variability.
Overall, the committee concludes that EPA addressed many sources of uncertainty and variability qualitatively, but it did not adequately quantify either the uncertainty or the variability of many. In the case of its cancer risk estimates, EPA should provide quantitative estimates corresponding to (1) central, upper-bound, and lower-bound estimates of the POD; (2) the use of different plausible POD values; (3) different plausible mathematical functions fit to the observed epidemiological data; and (4) different assumptions for estimating historical exposures among subjects in the epidemiological studies. In the case of the noncancer risk estimates, EPA should characterize the uncertainty associated with (1) fitting a dose-response relationship to the available data and (2) selecting a POD. If necessary, EPA
should acknowledge that the information available remains insufficient to support a meaningful point estimate.
EPA’s discussion of epidemiological studies in Part III of the Reassessment, Integrated Summary and Risk Characterization for TCDD and Related Compounds, should clearly specify inclusion criteria for those studies used as a basis to support quantitative risk estimates. The committee notes that EPA could substantially improve the transparency and management of the uncertainties and complexities of the risk assessment for TCDD, other dioxins, and DLCs by creating an ongoing process for clearly identifying and updating the key assumptions that support the quantitative risk assessment.
ESTIMATING TOXICITY OF DLCS AND MIXTURES
Many DLCs and dioxins, other than TCDD, present in the environment are capable of producing toxicological effects similar or identical to those of TCDD. Substantial efforts have been aimed at simplifying estimation of risk for these compounds and for mixtures of them. EPA and international public health organizations have tended to take the approach of assigning each compound (dioxins, other than TCDD, and DLCs) a toxic equivalency factor (TEF), which represents a scaling factor for estimating the toxicity of the compound relative to TCDD. For example, a substance with a TEF of 0.1 is estimated to be 10% as toxic as dioxin per unit mass. Estimation of TEFs is a critically important part of the risk assessment of environmental mixtures of TCDD, other dioxins, and DLCs, because any environmental sample typically contains a dozen or more similar substances, but often very little TCDD. TCDD, other dioxins, and DLCs break down at different rates in the environment and have different elimination rates in humans. Thus, although analysts may reasonably estimate the relative potency value for a given compound based on toxicity tests, the compound’s contribution to total risk in an environmental (or biological) sample may change over a period of many years. This change may occur because the relative concentration in a sample may change with time, even though the potency remains constant, and the estimated risk in a given sample depends on both potency and concentration. Because these mixtures may contain little or no TCDD but relatively large amounts of low-potency dioxins and/ or DLCs, TEFs are a critical factor in determining the mixture’s overall estimated toxicity. Analysts refer to the aggregate weighting by TEF of a mixture as the mixture’s toxic equivalent quotient (TEQ).
The recent NTP studies on TCDD and several other dioxins and DLCs provide additional evidence in support of the TEF approach. Uncertainty about the validity of the approach led the NTP to specifically test the TEF value for one particular PCB (126) in its analyses, and the results showed
excellent agreement between the predicted TEF for PCB 126 and the value observed in the NTP experiment.
Overall, even given the inherent uncertainties, the committee agrees that the TEF method is reasonable, scientifically justifiable, and widely accepted for the estimation of the relative toxic potency of TCDD, other dioxins, and DLCs. The TEF approach has also been used in other contexts. WHO’s International Programme on Chemical Safety used the approach in assessing the risks of different polycyclic aromatic hydrocarbons (PAHs) relative to benzo(a)pyrene as an indicator PAH.
The committee concludes that the recent NTP results, released after EPA completed its 2003 Reassessment, provide important additional support for the TEF approach. However, EPA should acknowledge the need for better uncertainty analysis of the TEF values and should, as a follow-up to the Reassessment, establish a task force to begin to address this uncertainty by developing “consensus probability density functions” for dioxins and DLCs. The committee recommends that EPA clearly address TEF uncertainties in the Reassessment.
SCALING DATA FROM ANIMAL STUDIES
For risk assessments that rely on experimental animal data, determining the most appropriate way to scale the data from the animal model (usually rats and mice) to humans is another important risk assessment choice. Numerous options for choosing dose metrics exist, and they can yield results different from the traditional daily dose metric based on per unit of body weight. For highly persistent chemicals like TCDD, other dioxins, and DLCs, substantial differences in the rates of elimination from the body will result in very different amounts of chemical accumulated in the body over time, even with the same daily dose rate expressed in body weight or body surface area units. In the 2003 Reassessment, EPA used an estimate of the total amount of chemical in the body at steady state for a defined rate of exposure, called the body burden, as the dose metric to adjust for differences in body weight (or surface area) and in elimination rates.
The committee agrees with EPA’s conclusion that use of body burden as the dose metric appears to be the most reasonable and pragmatic approach for dioxin risk assessment, but EPA should address important uncertainties quantitatively in more detail when possible. One such uncertainty, not quantitatively addressed in EPA’s 2003 Reassessment, relates to species differences in body fat expressed as a percentage of total body weight. Differences in body fat content have a potentially large impact on dioxin concentrations present in nonfatty tissues, including such organs as the liver.
Large errors may also arise from trying to estimate the overall body burden TEQs for humans based on intake TEFs from rats. The errors result
from uncertainties in the differences in how long TCDD, other dioxins, and DLCs persist in humans and rodents and uncertainties about how these compounds concentrate in tissues.
The committee recommends that EPA’s Reassessment use basic physiologically based pharmacokinetic (PBPK) models to estimate the differences between humans and rodents in the relationship between total body burden at steady state, as calculated from the intake, half-life, bioavailability, and tissue concentrations, and use the results to modify the estimated human equivalent intakes. The committee also recommends that EPA provide a clear evaluation of the impact of using body burden as the dose metric, relative to other possible options such as intake, on the final risk estimates.
HUMAN EXPOSURE TO TCDD, OTHER DIOXINS, AND DLCS
Estimating human exposure levels, including those representative of background levels (e.g., typical dietary intake levels) and levels resulting from specific exposure scenarios (e.g., accidental, occupational, and highly exposed communities), is a critical component of any chemical risk assessment. The extensive environmental persistence of TCDD, other dioxins, and DLCs and their global environmental distribution create many possible sources and routes of exposure to these compounds, and determining typical background rates of exposure is difficult. EPA’s 2003 Reassessment addresses exposure to TCDD, other dioxins, and DLCs in terms of sources, environmental fate, environmental media concentrations, food concentrations, background exposures, and potentially highly exposed populations.
To assess total dioxin and DLC emissions, EPA used a “bottom-up” approach in which it attempted to identify all source categories and then estimated the emissions for each category. However, a “top-down” approach that attempts to account for measured levels and considers the emission sources required to account for those levels would provide useful additional information. Such alternative approaches may give rise to significantly different estimates of the historical levels of dioxin and DLC emissions. Both approaches come with uncertainties, and EPA could benefit substantially from using the approaches simultaneously to set plausible bounds on historical trends and current levels in emissions.
The committee also recommends that EPA more explicitly define its procedures for addressing analytical measurements that fall below the limit of detection in environmental and exposure media samples. Consideration of the detection limits is important in assessing background exposure estimates. Typically, samples that contain small or no amounts of dioxin (“nondetects”) are given a value of 50% of the lowest level measurable by the instrument (the detection limit). For example, if the detection limit was 1 part per billion (ppb), a sample that contained 0.1 ppb would be assigned
a value of 0.5 ppb (half of the detection limit), or 5 times greater than its actual value. If the detection limit decreased to 0.05 ppb, the actual value of 0.1 ppb would be reported. In addition, as analytical detection limits improve, the estimates of contaminants in background environmental samples become more accurate as nondetect samples become fewer and the range of uncertainty is narrowed.
Although beyond the scope of the review of the EPA Reassessment, the committee notes that it would be useful for EPA to set up a compound-specific, active database of typical concentrations for the range of TCDD, other dioxins, and DLCs present in dietary and other environmental sources. This database should undergo regular updates to capture new data as they appear in the peer-reviewed literature. Such a database should include clear requirements of data quality and traceability (chemical analysis, representative and targeted sampling, representative of consumer exposure, presentation of data, and handling and presentation of “nondetects”). Chapter 4 provides several additional recommendations about the exposure assessment section of the 2003 Reassessment.
IMMUNOTOXICITY OF TCDD, OTHER DIOXINS, AND DLCS
TCDD, other dioxins, and DLCs have well-known effects on the immune systems of experimental animals. Chemically induced alterations in immune function could result in various adverse health outcomes because the immune system plays a critical role in fighting off infections, killing cancer cells at early stages, and implementing numerous other health-protective functions.
In light of the large database showing that TCDD, other dioxins, and DLCs produce immunotoxic responses in laboratory animal studies, combined with sparse human data, the committee agrees with EPA’s conclusion that these compounds are potential human immunotoxicants.
However, EPA’s conclusion that dioxins, other than TCDD, and DLCs are immunotoxic at “some dose level” is inadequate. At a minimum, EPA should add a section or paragraph that discusses the immunotoxicology of these compounds in the context of current Ah receptor biology. EPA should also include some discussion about the implications of using genetically homogeneous inbred mice to characterize immunotoxicological risk in the genetically variable human population.
REPRODUCTIVE AND DEVELOPMENTAL TOXICITY OF TCDD, OTHER DIOXINS, AND DLCS
Reproduction and embryonic and fetal development are sensitive end points from rodent exposure to TCDD, other dioxins, and DLCs. Although
the fetal rodent consistently appears to be more susceptible to adverse effects of these compounds than the adult rodent, comparable human data do not exist, and the susceptibility of humans to these end points is less well determined.
EPA’s 2003 Reassessment comprehensively covers developmental and reproductive toxicity of TCDD, other dioxins, and DLCs in several models. One rodent model included TCDD administration during pregnancy and thus tested the disruption of development of the pups and their reproductive function later in life. The 2003 Reassessment presented a comprehensive overview of the pregnancy model, but it did not provide an adequate discussion of the doses used in the studies or the relationships of animal studies to human reproductive and developmental toxicity. The committee recommends that EPA more thoroughly address how the effective doses used in the animal pregnancy models relate to human reproductive and developmental toxicity and risk information, including TEFs and TEQs. The 2003 Reassessment also did not provide an adequate discussion of other models (e.g., effects of TCDD on ovulation in adult rats).
OTHER TOXIC END POINTS
Although TCDD, other dioxins, and DLCs have received wide recognition for their potential to cause cancer, birth defects, reproductive disorders, immunotoxicity, and chloracne, animal and human studies have demonstrated other potential toxic end points, including liver disease, thyroid dysfunction, lipid disorders, neurotoxicity, cardiovascular disease, and metabolic disorders, such as diabetes.
The committee agrees that EPA has in general adequately addressed the available data on the likelihood that exposure to TCDD, other dioxins, and DLCs is a significant risk factor for other toxic end points. EPA cautiously stated its overall conclusions about noncancer risks due to TCDD, other dioxins, and DLCs exposures and acknowledged the uncertainty of suspected relationships. Nonetheless, the committee notes that EPA did not uniformly address the limitations of individual human studies. Similarly, EPA did not discuss the broad 95% confidence intervals accompanying some reported statistically significant effects in the context of the uncertainty (and, perhaps, individual variability) that these broad confidence limits imply. Conversely, the 2003 Reassessment highlights statistically non-significant effects in some cases, suggesting an implied potential for unobserved detrimental effects without a supporting presentation of a firm evidence base. The committee recommends that EPA establish formal principles and mechanisms for evidence-based classification and systematic statistical review, including meta-analysis when possible, for available human, clinical, and noncancer end-point data.
New studies of the effects of dioxin on the developing vascular system suggest a potentially sensitive target for TCDD, other dioxins, and DLCs. The committee recommends that EPA identify this area as an important data gap in the understanding of the potential adverse effects of these compounds.
EPA’S OVERALL APPROACH TO RISK CHARACTERIZATION
Risk characterization is the culminating step in risk assessment. It should attempt to pull together all the relevant scientific information on toxicity and exposure for a coherent, quantitative understanding of potential health risks and on the uncertainties that surround the estimates of risk. Ideally, the risk characterization component of a risk assessment provides risk managers with a user-friendly synopsis of the scientific basis that underpins an agent’s potential impact on public health under defined exposure conditions and scenarios.
As discussed previously, selection of the default linear extrapolation approach for carcinogenicity emerged as one of the most critical decisions in the 2003 Reassessment. The committee concludes that EPA did not support its decision adequately to rely solely on this default linear model and recommends that EPA add a scientifically rigorous evaluation of a nonlinear model that is consistent with receptor-mediated responses and uses the recent NTP cancer bioassay studies. The committee determined that the available data support the use of a nonlinear model, which is consistent with receptor-mediated responses and a potential threshold, with subsequent calculations and interpretation of MOEs. EPA’s sole use of the default assumption of linearity and selection of the ED01 as the only POD to quantify cancer risk does not provide an adequate quantitative characterization of the overall range of uncertainty associated with the final estimates of cancer risk.
Because EPA decided not to derive an RfD, its traditional noncancer metric, or any other alternative for noncancer effects, the 2003 Reassessment does not provide important detailed risk characterization information about noncancer risks. Typically, when EPA estimates an RfD, the risk characterization will include (1) estimates of the proportion of the population with intakes above the RfD; (2) detailed assessment of population groups, such as those with occupational exposures; and (3) contributions of the major food sources and other environmental sources for those individuals with high intakes. If a nonlinear model consistent with a threshold were used for cancer risk assessment, these same types of risk characterization details could also be provided for cancer risk. The lack of such a focus in the risk characterization section of the 2003 Reassessment results in a risk
characterization that is difficult to follow and does not provide clear guidance with respect to noncancer end points.
The committee recommends that EPA revise its risk characterization chapter to clearly describe the following:
-
The effects seen at the lowest body burdens that are the primary focus for any risk assessment—the “critical effects.”
-
The modeling strategy used for each noncancer effect modeled, paying particular attention to the critical effects, and the selection of a point of comparison based on the biological significance of the effect; if the ED01 is retained, then the biological significance of the response should be defined and the precision of the estimate given.
-
The precision and uncertainties associated with the body burden estimates for the critical effects at the point of comparison, including the use of total body burden rather than modeling steady-state concentrations for the relevant tissue.
-
The committee encourages EPA to calculate RfDs as part of its effort to develop appropriate margins of exposure for different end points and risk scenarios, including the proportions of the general population and of any identified groups that might be at increased risk (See Table A-1 in the Reassessment, Part III Appendix, for the different effects; appropriate exposure information would need to be generated.) Interpretation of the calculated values should take into consideration the uncertainties in the POD values and intake estimates.
-
Consideration of individuals in susceptible life stages or groups (e.g., children, women of childbearing age, and nursing infants) who might require estimation of a separate MOE using specific exposure data.
-
Distributions that provide clear insights about the uncertainty in the risk assessments, along with discussion of the key contributors to the uncertainty.
The committee recommends that EPA substantially revise the risk characterization section of Part III of the Reassessment to include a more comprehensive risk characterization and discussion of the uncertainties surrounding key assumptions and variables.
CONCLUDING REMARKS
The committee appreciates the dedication and hard work that went into the creation of the Reassessment and commends EPA for its detailed evaluation of an extremely large volume of scientific literature (particularly Parts I and II of the Reassessment). This NRC report focuses its review on
Part III of the Reassessment and offers its recommendations with the intention of helping to guide EPA in its efforts to make and implement environmental policies that adequately protect human health and the environment from the potential adverse effects of TCDD, other dioxins, and DLCs. The committee recognizes that it will require a substantial amount of effort for EPA to incorporate all the changes recommended in this report. Nevertheless, the committee encourages EPA to finalize the current Reassessment as quickly, efficiently, and concisely as possible after addressing the major recommendations in this report. The committee notes that new advances in the understanding of TCDD, other dioxins, and DLCs could require reevaluation of key assumptions in EPA’s risk assessment document. The committee recommends that EPA routinely monitor new scientific information related to TCDD, other dioxins, and DLCs, with the understanding that future revisions may be required to maintain a risk assessment based on the current state-of-the-art science. However, the committee also recognizes that stability in regulatory policy is important to the regulated community and therefore suggests that EPA establish criteria for identifying when compelling new information would warrant science-based revisions in its risk assessment. The committee finds that the recent dose-response data released by the NTP after submission of the Reassessment are good examples of new and compelling information that warrants consideration in a revised risk assessment.
COMMITTEE’S KEY FINDINGS
The committee identified three areas that require substantial improvement in describing the scientific basis for EPA’s dioxin risk assessment to support a scientifically robust risk characterization:
-
Justification of approaches to dose-response modeling for cancer and noncancer end points.
-
Transparency and clarity in selection of key data sets for analysis.
-
Transparency, thoroughness, and clarity in quantitative uncertainty analysis.
The following points represent Summary recommendations to address the key concerns:
-
EPA should compare cancer risks by using nonlinear models consistent with a receptor-mediated mechanism of action and by using epidemiological data and the new NTP animal bioassay data. The comparison should include upper and lower bounds, as well as central estimates of
-
risk. EPA should clearly communicate this information as part of its risk characterization.
-
EPA should identify the most important data sets to be used for quantitative risk assessment for each of the four key end points (cancer, immunotoxicity, reproductive effects, and developmental effects). EPA should specify inclusion criteria for the studies (animal and human) used for derivation of the benchmark dose (BMD) for different noncancer effects and potentially for the development of RfD values and discuss the strengths and limitations of those key studies; describe and define (quantitatively to the extent possible) the variability and uncertainty for key assumptions used for each key end-point-specific risk assessment (choices of data set, POD, model, and dose metric); incorporate probabilistic models to the extent possible to represent the range of plausible values; and assess goodness-of-fit of dose-response models for data sets and provide both upper and lower bounds on central estimates for all statistical estimates. When quantitation is not possible, EPA should clearly state it and explain what would be required to achieve quantitation.
-
When selecting a BMD as a POD, EPA should provide justification for selecting a response level (e.g., at the 10%, 5%, or 1% level). The effects of this choice on the final risk assessment values should be illustrated by comparing point estimates and lower bounds derived from selected PODs.
-
EPA should continue to use body burden as the preferred dose metric but should also consider physiologically based pharmacokinetic modeling as a means to adjust for differences in body fat composition and for other differences between rodents and humans.

















