
4
Liver Toxicity and Cancer
This chapter reviews information on the effects of trichloroethylene and its principal metabolites (trichloroacetic acid, dichloroacetic acid, and chloral hydrate) on the liver, particularly information generated since the U.S. Environmental Protection Agency (EPA) released its draft health risk assessment (EPA 2001b). Trichloroethylene metabolism occurs primarily in the liver and is critical to understanding its toxicity and carcinogenicity. Background information on trichloroethylene metabolism is provided in Appendix C. In this chapter, hepatotoxicity and liver cancer are discussed separately, although they are not necessarily independent end points. A review of current knowledge on the proposed modes of action for trichloroethylene-induced liver cancer (peroxisome proliferator-activated receptor agonism, genotoxicity and mutagenicity) and their relevance to humans is provided.
HEPATOTOXICITY
Animal Studies
It is well documented that trichloroethylene produces hepatotoxicity in experimental animals and humans (ATSDR 1997a; EPA 2001b). Table 4-1 provides the details of some recent studies, and selected findings are discussed below.
Rodents exposed to high doses of trichloroethylene or some of its metabolites develop hepatocellular necrosis. Different studies have localized the injury to midzonal, periportal, or centrilobular hepatocytes (Buben
TABLE 4-1 Hepatotoxicity of Trichloroethylene and Metabolites in Animal Stud
|
Species (Sex) |
Doses/Concentrations |
Duration of Exposure |
Route/Vehicle |
|
Swiss-Cox mice (males) |
TCE, 0-3,200 mg/kg/day |
6 wk |
Gavage/corn oil |
|
Sprague-Dawley rats (males) |
TCE, 250, 500, 1,250, 2,500 mg/kg |
Single dose |
i.p./corn oil |
|
Sprague-Dawley rats (males) |
TCE, 16 and 64 mg/kg, |
Single dose |
i.v. or via portal vein cannula/ vegetable oil |
|
Autoimmune prone MRL−/−mice (female) |
TCE, 0, 0.1, 0.5, 2.5 mg/mL |
4 and 32 wk |
Drinking water |
|
Autoimmune prone MRL−/−mice (female) |
TCE, 0 or 2.5 mg/mL, in presence of diallyl sulfide (CYP2E1 inhibitor) |
4 wk |
TCE in drinking water; diallyl sulfide via osmotic pump |
|
Autoimmune prone MRL−/−mice (female) |
TCA or chloral, 0, 0.1, 0.9 mg/mL |
4 wk |
Drinking water |
|
Wistar rats (male) |
TCE, 376 ppm, 4 hr/day, 5 days/wk |
8, 12, 24 wk |
Inhalation |
|
B6C3F1 mice (female) |
chloral hydrate, 25, 50, and 100 mg/kg, 5 days/wk |
3, 6, 12 mo, 24 mo |
Gavage/in water |
|
Swiss-Webster mice (male) |
DCVC, 15, 30, 75 mg/kg |
Single dose |
i.p./in water |
|
B6CF1 mice (male) |
chloral hydrate, 1 g/L; DCA, 0.5 g/L |
104 wk |
Drinking water |
|
B6C3F1 mice (male) |
DCA, 0.5 or 5 g/L |
0, 5, 15, 20, 30 days |
Drinking water |
|
B6C3F1 mice (male and female) |
DCA or TCA, 1 or 2 g/L |
52 wk |
Drinking water |
|
B6C3F1 mice (male and female) |
DCA or TCA, 1 or 2 g/L |
52 wk |
Drinking water |
|
B6C3F1 mice (male) |
DCA, 0.1 to 3 g/L |
1, 2 and 8 wk |
Drinking water |
|
Features of Hepatotoxicity |
Reference |
|
Increases in serum glutamic pyruvic transaminase evident only with the two highest doses; histologic findings: swollen hepatocytes and minimal evidence of necrosis. |
Buben and O’Flaherty 1985 |
|
Midzonal injury that spreads to centrilobular regions with the highest dose. |
Soni et al. 1998, 1999 |
|
Periportal necrosis with portal vein administration. No necrosis with i.v. administration. This is reflective of solvent-related injury of TCE to portal areas. |
Lee et al. 2000 |
|
CD4+ T-cell activation; mononuclear infiltration in portal regions consistent with autoimmune hepatitis; slight but significant increase of serum ALT. |
Griffin et al. 2000a |
|
Blockade of TCE protein adduct formation; reversal of CD4+ T-cell-mediated autoimmunity by TCE. |
Griffin et al. 2000b |
|
CD4+ T-cell activation by these metabolites as shown for TCE. |
Blossom et al. 2004 |
|
This dose (1/25 LC50) resulted in hepatomegaly and fatty infiltration; fatty changes with marked necrosis were more evident at 12 and 24 wk. No elevation in serum liver transaminases or mortality was detected. |
Kumar et al. 2001a |
|
No evidence of dose-dependent elevation in serum liver transaminases, only AST elevated at the 50-mg/kg dose. |
NTP 2002b |
|
Transient elevation in liver transaminases at the highest dose; no histologic evidence of liver injury. |
Vaidya et al. 2003a |
|
Increased liver weight and hepatocellular necrosis with both metabolites. |
Daniel et al. 1992 |
|
Dose- and time-dependent liver enlargement; morphologic evidence of focal necrosis and apoptotic bodies. |
Carter et al. 1995 |
|
Enlarged livers, significant glycogen accumulation, focal areas of necrosis seen with DCA. No focal necrosis with TCA, modest hyperthrophy and glycogen accumulation. |
Bull et al. 1990 |
|
Hepatocytes from DCA-treated mice contained large amounts of glycogen evenly distributed throughout the liver; less glycogen accumulation with TCA treatment, which was more prominent in periportal regions. |
Bull et al. 1990 |
|
Dose-dependent liver glycogen accumulation associated with decreased glycogen synthase activity. No effect in glycogen phosphorylase or glucose-6-phosphatase (enzymes involved glycogen metabolism). |
Kato-Weinstein et al. 1998 |
|
Species (Sex) |
Doses/Concentrations |
Duration of Exposure |
Route/Vehicle |
|
B6C3F1 mice (male) |
DCA, 0.1 to 2 g/L |
2-10 wk |
Drinking water |
|
Fresh hepatocytes in culture from B6C3F1 mice (male) |
DCA, 10-500 μM |
16-hr incubation |
N/A |
|
B6C3F1 mice (female) |
DCA, 3.2 g/L, with or without L-methionine at 4 or 8 g/kg |
8 and 44 wk |
DCA in drinking water; L-methionine in diet |
|
Fisher 344 rats (male) and B6C3F1 mice (female) |
dibromoacetic acid, 0, 1, 2 g/kg |
2, 4, 7, 28 days |
Drinking water |
|
Sprague-Dawley rats (males) |
TCE, 0.01, 0.1, 1, 5, and 10 mmol/kg |
Daily dosing for 3 days |
i.p./corn oil |
|
Fresh hepatocytes in culture from Sprague-Dawley rats (males) |
Cells dosed by vapor phase in 25-mL flasks; exposure: TCE, 0, 0.5, 1, 2, 4 μL per flask |
20-min exposure |
N/A |
|
Fresh hepatocytes in culture from Sprague-Dawley rats (males) |
Cells dosed by vapor phase in 25-mL flasks; exposure: 1,1,1-trichloroethane, 0, 2, 5, 10 μL per flask (concentration range: 230-1,000 μM) |
20-min exposure |
N/A |
|
ABBREVIATIONS: ALT, alanine aminotransferase; AST, aspartate aminotransferase; DCA, dichloroacetic acid; DCVC, S-1,2-dichlorovinyl-L-cysteine; i.p., intraperitoneal; i.v., intravenous; LC50, concentration lethal to 50% of test animals; N/A, not applicable; ppm, parts per million; TC, taurocholate; TCA, trichloroacetic acid; TCE, trichloroethylene. |
|||
and O’Flaherty 1985; Soni et al. 1998, 1999; Lee et al. 2000). This lack of consistency in location of injury might reflect the routes of administration, doses, strain, or species of rodents used in the different studies. For example, Soni et al. (1999) conducted dose-response studies with trichloroethylene (250-2,500 mg/kg) to investigate the time course of liver injury and compensatory hepatocyte regeneration. Hepatocellular necrosis was evident after 24 hours at all doses. Injury was detected in midzonal areas of the liver lobule with no evidence of necrosis in hepatocytes adjacent to the central vein (centrilobular hepatocytes). This study also showed that the dose of trichloroethylene can influence the location of injury. At a trichloroethylene
|
Features of Hepatotoxicity |
Reference |
|
Significant reduction in serum insulin levels, insulin receptor expression, and protein kinase B. Increases in liver glycogen preceded these changes. |
Lingohr et al. 2001 |
|
Dose- and time-dependent glycogen accumulation. Presence or absence of insulin in culture media did not affect this DCA effect. However, glycogen accumulation is dependent on phosphatidylinositol 3-kinase activity. |
Lingohr et al. 2002 |
|
L-Methionine prevented liver DNA hypomethylation completely, while blocking only 25% of glycogen accumulation produced by DCA. |
Pereira et al. 2004 |
|
Dose- and time-dependent hypomethylation of liver DNA in both species. Significant increases in liver glycogen in both species, although longer exposure is required in rats for this effect. |
Tao et al. 2004a |
|
Increases in total and some individual serum bile acids (TC was most sensitive). No elevation in transaminases (except for ALT at the highest dose) or morphologic evidence of injury. Separate inhalation studies showed similar elevation in serum TC levels. |
Wang and Stacey 1990 |
|
Dose-dependent inhibition of bile acid (TC) uptake into hepatocytes. Time-dependent inhibition of bile acid accumulation. No significant intracellular enzyme or potassium leakage. No changes in bile acid efflux from hepatocytes with TCE exposure. |
Bai and Stacey 1993 |
|
No ALT, lactate dehydrogenase or potassium leakage at any dose level. Concentration-dependent inhibition of TC, ouabain, and 2-aminoisobutyric acid uptake. Decrease ATP levels and activity of ATP-dependent membrane ATPases. No overt morphologic changes in TCE-exposed hepatocytes. |
Kukongviriyapan et al. 1990 |
dose of 2,500 mg/kg, centrilobular injury was clearly evident after 24 hours. High doses of trichloroethylene administered into the portal vein caused periportal liver injury via a direct solvent action rather than a mechanism dependent on activation by the enzyme cytochrome P-450 (CYP-450) (Lee et al. 2000).
Reynolds and Moslen (1977) proposed that reactive intermediates of trichloroethylene generated by CYP-450 bind covalently to cellular components, resulting in cell necrosis. More recent evidence from mouse studies suggests that an autoimmune response might play a role in trichloroethylene-mediated liver disease (Griffin et al. 2000a). Administration of
trichloroethylene at concentrations of 0 to 2.5 mg/mL in drinking water to autoimmune-prone MRL+/+ mice for 34 weeks resulted in an inflammatory response in the liver. Metabolic activation of trichloroethylene by CYP2E1 was demonstrated to be an obligatory step in the development of autoimmune hepatitis in the mice. The metabolites trichloroacetic acid and chloral hydrate also have the potential to induce autoimmunity in the same autoimmune-prone mice (Blossom et al. 2004).
Recent studies investigated the hepatotoxicity produced by trichloroethylene in rats exposed via inhalation at 376 parts per million for 8, 12, or 24 weeks. Liver enlargement with necrotic cells and fatty infiltration was more prominent in rats in the 12- and 24-week treatment groups. The authors also detected elevated markers of lysosomal disruption. They recorded no mortality in any of the treatment groups (Kumar et al. 2001a).
Human Studies
Table 4-2 presents findings from human studies of hepatotoxicity. There is some evidence that occupational exposure to trichloroethylene results in several forms of noncancer liver disease such as hepatic necrosis, fatty liver, and cirrhosis. It is well established that acute occupational exposure to trichloroethylene does not produce liver injury, whereas chronic exposure does. Case reports have linked occupational exposure to trichloroethylene with Stevens-Johnson syndrome (erythema multiforme major) of abrupt onset (Phoon et al. 1984). All these cases demonstrated liver involvement ranging from mild jaundice to fatal liver failure. Another case report documented that repeated exposure to trichloroethylene in the work setting resulted in chronic cirrhosis and portal hypertension (Thiele et al. 1982). The most recent report of trichloroethylene hepatotoxicity associated with occupational exposure comes from a watch manufacturing plant in Thailand, where two female workers developed generalized skin lesions, fever, and hepatitis. One case resulted in fatal hepatic damage 2 weeks after the onset of symptoms. Both workers cleaned watch metal straps by dipping them in containers that contained trichloroethylene (Pantucharoensri et al. 2004).
These case reports support data from animal studies indicating that an autoimmune response might be important in trichloroethylene-induced hepatitis. Genetic and environmental factors that influence xenobiotic metabolizing enzymes can favor the formation of trichloroethylene metabolites capable of triggering an immune response against the liver.
Contribution of Metabolites to Hepatotoxicity
Chloral hydrate, a metabolic intermediate of trichloroethylene, has been reported to contribute to the hepatotoxic potential of this solvent. In a 2-
TABLE 4-2 Hepatotoxicity of Trichloroethylene and Metabolites in Human Studies
|
Subjects |
Doses/Concentrations |
Duration of Exposure |
Route/Vehicle |
Features of Hepatotoxicity |
Reference |
|
Five case reports (males and females) |
TCE, 50-912 mg/m3 |
3-5 wk |
Inhalation of vapors in workplace |
Stevens-Johnson syndrome (erythema multiforme), jaundice, hepatomegaly, and hepatic encephalopathy. Other solvents besides TCE might be involved. |
Phoon et al. 1984 |
|
Two case reports (females) |
TCE, 15-45 ppm |
4-5 wk |
Inhalation of vapors in workplace |
Stevens-Johnson syndrome, generalized skin eruptions, and hepatitis with no jaundice (case 1). Fulminant hepatitis (case 2). |
Pantucharoensri et al. 2004 |
|
Cross-sectional study (148 workers) and a 2-yr follow- up study (13 workers) |
Low, moderate,and high TCE exposure based on concentrations of total trichloro compounds detected in urine |
Duration of employment: 0.1 to 36.6 yr; average: 7 yr |
Ambient air; occupational |
Increases in high-density lipoprotein cholesterol in the absence of elevation in plasma liver transaminases, indicating that low level exposure to TCE affects cholesterol metabolism without causing hepatocellular necrosis. Alcohol intake is an influential factor in the cross-sectional study. |
Nagaya et al. 1993 |
|
Human workers (21 men, 1 woman) |
Regular exposures of TCE at <5 ppm; peak exposures for 2 workers at >250 ppm |
Mean duration of employment for TCE-exposed workers: 7 yr |
Ambient air; occupational |
Highly significant increases in individual and total serum bile acids in the exposed group (controlled for age and alcohol intake). No abnormalities in liver function tests. No relationship between plasma bile acid and cholesterol was detected. |
Driscoll et al. 1992 |
|
Human workers |
TCE, 8.9 ± 3.1 ppm |
Mean duration ofexposure: 3.4 yr |
Ambient air; occupational |
Elevation in total serum bile acids and some individual bile acids; normal hepatobiliary function tests. |
Neghab et al. 1997 |
|
ABBREVIATION: TCE, trichloroethylene. |
|||||
year National Toxicology Program (NTP 2002b) study, male B6C3F1 mice given chloral hydrate by gavage at 25, 50, or 100 mg/kg showed no significant changes in three serum liver transaminases, except for a significant increase in aspartate aminotransferase activity in the 50-mg/kg group.
Lash et al. (1995) investigated the toxicity of trichloroethylene and its metabolites with freshly isolated rat hepatocytes in culture. The studies showed that exposure to only S-(1,2-dichlorovinyl)-L-cysteine resulted in hepatocellular damage. None of the other metabolites (CYP450 dependant or reduced glutathione dependent) or trichloroethylene produced hepatocellular injury. The metabolites tested included trichloroacetic acid, dichloroacetic acid, chloral hydrate, trichloroethanol, oxalic acid, and S-1,2-dichlorovinyl-L-glutathione. Despite this lack of cytotoxicity, trichloroethylene and its metabolites produced mitochondrial dysfunction. This in vitro study showed that S-1,2-dichlorovinyl-L-cysteine is the only trichloroethylene-derived compound cytotoxic to rat hepatocytes in culture.
In contrast, in vivo studies showed that S-1,2-dichlorovinyl-L-cysteine has a very low hepatotoxic potential. Acute toxicity studies investigating the nephrotoxicity of S-1,2-dichlorovinyl-L-cysteine in male Swiss-Webster mice showed transient elevations in serum liver transaminases 12 hours after administration of the highest dose tested (75 mg/kg). This dose resulted in significant lethality due to nephrotoxicity at later times. No liver histopathology was detected at any of the doses or time points examined (Vaidya et al. 2003a).
Other studies investigating the epigenetic mechanisms of dichloroaceticacid-induced carcinogenesis revealed morphologic evidence of liver injury that includes loss of cell membrane integrity, focal areas of cell debris, and appearance of apoptotic bodies in B6C3F1 mice undergoing short-term exposure to dichloroacetic acid at 0.5 or 5 g/L for up to 30 days (Carter et al. 1995). Hepatocellular necrosis was also detected in male and female Swiss-Webster mice receiving dichloroacetic acid in their drinking water at 300, 1,000, or 2,000 mg/L for up to 14 days. This was accompanied by a marked increase in liver weights. No such changes were seen with trichloroacetic acid under the same dosing regimen (Bull et al. 1990; Sanchez and Bull 1990). Exposure of male B6C3F1 mice to dichloroacetic acid (0.5 g/L) or chloral hydrate (1 g/L) via drinking water resulted in hepatocellular necrosis after 104 weeks of exposure (Daniel et al. 1992).
Changes in Liver Glycogen Status
Exposure to trichloroethylene produces effects in the liver other than hepatocellular injury. Treating mice with dichloroacetic acid results in marked dose-dependent accumulation of liver glycogen (Bull et al. 1990;
Kato-Weinstein et al. 1998). This dose-response relationship parallels that for the development of hepatocellular carcinomas. Furthermore, patients with glycogen storage disorders have a greater propensity for developing liver tumors (Labrune et al. 1997). These observations have prompted investigators to study in depth the relationship between increased liver glycogen storage and carcinogenesis.
Studies have been carried out to assess the effect of dichloroacetic acid treatment on insulin secretion, insulin receptor expression, and activity and expression of protein kinases controlled by insulin receptor signaling due to the role of these gene products in glycogen synthesis and homeostasis (Lingohr et al. 2001). Mice treated with dichloroacetic acid in drinking water at 0.1 to 2.0 g/L for 2-10 weeks showed a significant reduction in expression of the insulin receptor in the liver. As early as 2 weeks after the initiation of dichloroacetic acid treatment, insulin concentrations were significantly reduced. Dichloroacetic acid similarly reduced the expression of protein kinase B (an insulin-sensitive enzyme involved in glycogen homeostasis). Because dichloroacetic-acid-induced glycogen accumulation precedes down-regulation of the insulin receptor and insulin-dependent signaling pathways, these changes in gene expression for insulin and related genes are considered to be compensatory responses to changes in glycogen homeostasis.
Lingohr et al. (2002) investigated whether the changes in glycogen accumulation brought about by dichloroacetic acid were insulin dependent. Freshly isolated mouse hepatocytes exposed to dichloroacetic acid accumulated more glycogen than control hepatocytes. This response was dose dependent. Omitting insulin from the culture media did not prevent the enhanced accumulation of glycogen produced by dichloroacetic acid. By contrast, dichloroacetic-acid-induced glycogen deposition was fully blocked by inhibition of the enzyme phosphatidylinositol 3-kinase. Phosphatidylinositol 3-kinase participates in the signal transduction pathway leading to glycogen synthesis initiated by activation of the insulin receptor. This suggests that dichloroacetic-acid-induced glycogen accumulation involves a phosphatidylinositol 3-kinase-dependent pathway downstream of the insulin receptor (Lingohr et al. 2002). In addition to regulating glycogen synthesis, phosphatidylinositol 3-kinase has been implicated in the proliferative and antiapoptotic effects of peroxisome proliferators (Mounho and Thrall 1999), which further establishes an association between abnormal liver glycogen status and the carcinogenic effect of trichloroethylene and its metabolites. Other halogenated solvents such as bromochloroacetate and dibromoacetate induce glycogen accumulation in the liver to a similar degree as dichloroacetic acid (Kato-Weinstein et al. 2001).
Another potential link between aberrant liver glycogen homeostasis and the carcinogenicity of trichloroethylene is provided by the effect of
this chemical and its metabolites on DNA methylation status. Exposure to dichloroacetic acid results in hypomethylation of protooncogenes and other genes involved in cell growth, such as insulin-like growth factor II in mouse liver (Tao et al. 2000a, 2004b; Ge et al. 2001). This response has been causally linked to the development of hepatocellular adenomas and carcinomas induced by these chemicals.
Dibromoacetic acid, a related haloacetic acid, similarly induced hepatic DNA hypomethylation in female mice and male rats receiving 1 or 2 g/L in their drinking water for up to 28 days. Specifically, hypomethylation of c-myc and insulin-like growth factor II was detected along with increases in mRNA expression of both genes in mouse liver. Only c-myc mRNA expression was increased in rat liver. Glycogen accumulation and induction of markers of peroxisome proliferation were also observed in mice and rats receiving dibromoacetic acid (Tao et al. 2004a). These results further support the relationship between glycogen accumulation and liver tumors induced by trichloroethylene and its metabolites, making it more uncertain whether trichloroethylene-induced glycogen accumulation can be considered a non-cancer liver effect or an early event in carcinogenesis. More recently, Pereira et al. (2004) showed that methionine treatment not only prevents the DNA hypomethylation induced by dichloroacetic acid and related solvents, but it also prevents liver tumor formation in mice. Interestingly, methionine did not prevent glycogen accumulation completely. Dichloroacetic-acid-induced glycogen storage was reduced by only 25% with methionine treatment. This new information suggests dissociation between glycogen accumulation and the carcinogenic effects of dichloroacetic acid. Clearly, more studies are needed to clarify the relationship between altered glycogen storage and liver cancer in response to trichloroethylene.
Elevation of Serum Bile Acids
Occupational exposure to trichloroethylene has been reported to increase serum bile acid and cholesterol concentrations. Chronic occupational exposure to low concentrations of trichloroethylene appears to alter cholesterol metabolism in the absence of noticeable hepatocellular damage, as evidenced by lack of increase in serum liver transaminases (Nagaya et al. 1993). Similarly, serum concentrations of total and individual bile acids were significantly elevated in a group of workers exposed to trichloroethylene (Driscoll et al. 1992; Neghab et al. 1997). Because no association was observed between elevated plasma bile acids and conventional markers of liver injury, it was concluded that this perturbation in bile acid homeostasis could be indicative of early changes in liver function independent of hepatocellular damage.
Similar alterations in bile acid status have been observed in experimental animals exposed to trichloroethylene and its metabolites. Exposure to trichloroethylene via inhalation or intraperitoneal injection resulted in elevation of serum bile acid concentrations at doses that did not produce changes in markers of liver function, such as serum liver transaminases and bilirubin concentrations (Wang and Stacey 1990). To determine whether this increase in serum bile acids after low exposure to trichloroethylene is indicative of early liver dysfunction, the mechanism(s) responsible for these changes was investigated by examining the effect of trichloroethylene on bile acid transport in freshly isolated rat hepatocytes in culture (Bai and Stacey 1993). The uptake of the bile acids taurocholic and cholic acids into rat hepatocytes was inhibited in a dose-dependent manner by trichloroethylene at concentrations up to 1.84 mM. These concentrations did not result in significant leakage of transaminases or intracellular potassium into the culture media. Trichloroethylene inhibition of bile acids uptake was determined to be noncompetitive. These results indicate that the increase in serum bile acids produced by trichloroethylene occurs through interference with uptake into hepatocytes. The same study also determined that trichloroethylene does not affect the efflux of bile acids from hepatocytes. These observations clearly show that alterations in transport processes for bile acids (primarily by inhibiting uptake) occur in the absence of pathologic evidence of liver dysfunction.
The fact that inhibition of bile acid uptake by trichloroethylene was determined to be noncompetitive strongly argues against direct competition between trichloroethylene and bile acids, such as taurocholic and cholic acids, for the main basolateral carrier in hepatocytes for bile acids, the sodium taurocholate transporter polypeptide. This effect on bile acids is not unique for trichloroethylene, because occupational exposure to a mixture of other organic solvents including toluene, xylene, acetone, n-butanol, and ethylacetate similarly increases serum bile acids (Franco et al. 1986). These results also argue against competition for common transport as it is very unlikely that the uptake of all these solvents into hepatocytes requires the same transport process. Because of their high lipid solubility, these solvents can readily partition into the plasma membrane of hepatocytes by diffusion, which does not require transport protein function.
A decrease in hepatic bile acid uptake by trichloroethylene can be the result of changes in or disruption of plasma membrane lipids and changes in membrane fluidity. Changes in the fluidity of the plasma membrane lipid bilayer are known to affect the function of uptake transporters and other transmembrane proteins. Again, this is supported not only by the noncompetitive nature of the inhibition of the uptake of bile acids by trichloroethylene but also by its reversibility with time. In contrast, the lack of changes
in bile acid efflux in trichloroethylene-exposed rat hepatocytes does not support the idea that changes in membrane fluidity contribute to overall reduction in plasma membrane transport protein function, because changes in membrane fluidity should also affect the function of plasma membrane efflux transporters.
Earlier studies also demonstrated that 1,1,1-trichloroethane, a solvent similar to trichloroethylene, decreases ATP concentrations and inhibits the activity of plasma membrane ATPases in cultured rat hepatocytes in a dose-dependent manner. The only ultrastructural alteration detected in 1,1,1-trichloroethane-exposed hepatocytes was loss of membrane microvilli that was not associated with cell death (Kukongviriyapan et al. 1990). This reduction in ATP concentration could lead to impairment of energy-dependent transport processes across the plasma membrane of hepatocytes, thus providing a possible mechanistic explanation for the reduction in bile acid uptake into the liver of experimental animals. On the other hand, the main efflux transporter for bile acids in hepatocytes, known as the bile-acid-exporting pump, is also an ATP-dependent carrier. A reduction in cellular ATP quantities by 1,1,1-trichloroethane should also affect the efflux of bile acids from hepatocytes via the bile-acid-exporting pump if a reduction in cellular ATP status is a critical factor in the abnormal handling of bile acids with 1,1,1-trichloroethane exposure. However, this is not the case. Additional studies are required to better define the mechanisms by which trichloroethylene affects the vectorial transport of bile acids across hepatocytes.
The interference of 1,1,1-trichloroethane with the influx of chemicals into hepatocytes is not limited to bile acids because 1,1,1-trichloroethane also inhibited the uptake of ouabain and 2-aminoisobutyric acid (Kukong-viriyapan et al. 1990). These two compounds are known to enter hepatocytes via an energy-dependent transport-mediated process. It is worth noting that the transport of cadmium chloride and 3-O-methyl-D-glucose does not change. Furthermore, occupational exposure to a mixture of organic solvents including toluene, xylene, acetone, n-butylacetate, n-butanol, and ethylacetate also results in elevation of mean serum bile acid concentrations in the absence of changes in biochemical markers of liver injury (Franco et al. 1986). Exposure to toluene by itself produces the same response (Neghab and Stacey 1997).
LIVER CANCER
This section provides an overview of the evidence on liver cancer caused by trichloroethylene from animal and human studies. Tables detailing dose-response data from animal studies are included to provide a context for assessing risks from environmental exposures and to conduct physiologi-
cally based pharmacokinetic modeling. The potential modes of action of carcinogenesis for trichloroethylene and its metabolites are then discussed.
Animal Studies
Trichloroethylene
Trichloroethylene induction of hepatic tumors in rodents was extensively reviewed by Bull et al. (2002). Gavage administration of trichloroethylene in corn oil has been shown to produce hepatic cancer in B6C3F1 mice (NCI 1976; NTP 1990). One study (NCI 1976) used technical grade trichloroethylene (1,000 and 2,000 mg/kg for males, 700 and 1,400 mg/kg for females) that contained 0.09% epichlorohydrin, a known rodent carcinogen. In another study (NTP 1990), a single dose of epichlorohydrin-free trichloroethylene was administered by gavage in corn oil to male and female B6C3F1 mice at 1,000 mg/kg/day. Significant increases in the incidence of hepatocellular cancer were found in the mice.
Both pure amine-based-stabilized trichloroethylene and technical grade trichloroethylene with stabilizers (epichlorohydrin and 1,2-expoxybutane) were tested at 1.8 or 2.4 g/kg in corn oil in Swiss ICR/HA mice (Henschler et al. 1984). Neither compound produced liver tumors in the mice given daily doses for 18 months.
Stabilizer-free trichloroethylene was administered to male and female F344/N rats at 500 and 1,000 mg/kg for 103 weeks (NTP 1990). Reduced survival was observed in male rats and a dose-related increase in hepatic cytomegaly occurred in both sexes. However, no hepatic adenomas or carcinomas were reported. Exposure to trichloroethylene likewise did not produce significant hepatic tumors in four other strains of rat of both sexes administered trichloroethylene by gavage in corn oil for 103 weeks at doses from 125 to 2,000 mg/kg (NTP 1988).
Trichloroacetic Acid
Trichloroacetic acid is a peroxisome proliferator and a species-specific carcinogen. As shown in Table 4-3, it induces hepatocellular carcinomas when administered in drinking water to male and female B6C3F1 mice (a susceptible mouse strain). Dose-related increases in the incidence of malignant tumors and precancerous lesions have been observed with concentrations in drinking water up to 5 g/L. Significant increases in benign hyperplastic nodules and adenomas were found at concentrations in drinking water as low as 0.35 g/L. However, trichloroacetic acid has not induced significant hepatic tumors in male F344 rats under similar treatment conditions.
TABLE 4-3 Hepatocarcinogenic Effects of Trichloroacetic Acid in Drinking Water Studies with Mice and Rats
|
Species (sex) |
Concentration (g/L) |
Duration (wk) |
Combined Hyperplastic Nodule and Hepatocellular Adenoma |
Hepatocellular Carcinoma |
Reference |
||
|
Incidence |
Tumor/n (multiplicity) |
Incidence |
Tumor/n (multiplicity) |
||||
|
B6C3F1 mice (M) |
0 |
61 |
2/22 |
0.09 |
0/22 |
0 |
Herren-Freund, mice (M) |
|
|
5 |
61 |
8/22 |
0.5 |
7/22 |
0.5 |
|
|
B6C3F1 mice (M) |
0 |
52 |
1/35 |
0.03 |
0/35 |
0 |
Bull et al. 1990 |
|
|
1 |
52 |
5/11 |
0.45 |
2/11 |
0.18 |
|
|
|
2 |
52 |
15/24 |
1.04 |
4/24 |
0.17 |
|
|
|
2 |
37 |
2/11 |
0.18 |
3/11 |
0.27 |
|
|
B6C3F 1 mice (M) |
0 |
60-95 |
Not reported |
Not reported |
6.7-10% |
0.07-0.15 |
DeAngelo et al. 1991 |
|
|
0.05 |
60 |
Not reported |
Not reported |
22% |
0.31 |
|
|
|
0.5 |
60 |
Not reported |
Not reported |
38% |
0.55 |
|
|
|
4.5 |
95 |
Not reported |
Not reported |
87% |
2.2 |
|
|
|
5 |
60 |
Not reported |
Not reported |
55% |
0.97 |
|
|
B6C3F 1 mice (F) |
0 |
52 |
1/40 |
0.03 |
0/40 |
0 |
Pereira 1996 |
|
|
0.35 |
52 |
6/40 |
0.15 |
0/40 |
0 |
|
|
|
1.2 |
52 |
3/19 |
0.16 |
0/19 |
0 |
|
|
|
3.5 |
52 |
2/20 |
0.10 |
5/20 |
0.25 |
|
|
|
0 |
81 |
2/90 |
0.02 |
2/90 |
0.02 |
|
|
|
0.35 |
81 |
14/53 |
0 |
0/53 |
0 |
|
|
|
1.2 |
81 |
12/27 |
0 |
5/27 |
0 |
|
|
|
3.5 |
81 |
18/18 |
1.0 |
5/18 |
0.28 |
|
|
F344 rats (M) |
0 |
104 |
2/23 |
0.087 |
0/23 |
0 |
Daniel et al 1993 |
|
|
0.05 |
104 |
2/24 |
0.083 |
0/24 |
0 |
|
|
|
0.5 |
104 |
5/20 |
0.25 |
0/20 |
0 |
|
|
|
5 |
104 |
1/22 |
0.045 |
1/22 |
0.045 |
|
|
F344/N rats (M) |
0 |
104 |
0 |
0 |
0 |
0 |
DeAngelo et al. 1997 |
|
|
0.05 |
104 |
0 |
0 |
0 |
0 |
|
|
|
0.5 |
104 |
0 |
0 |
0 |
0 |
|
|
|
5 |
104 |
0 |
0 |
0 |
0 |
|
A large amount of trichloroacetic acid is formed in susceptible mouse strains after exposure to trichloroethylene (Green and Prout 1985), whereas only a minor portion is found in unresponsive strains of mice (Dekant et al. 1984, 1986a). Saturation of oxidative metabolism of trichloroethylene in rats results in trichloroacetic acid insufficient to induce peroxisome proliferation (Green 1990). Humans, like rats, may exhibit lower rates of oxidation and higher rates of conjugation than do mice.
The carcinogenic potential of peroxisome proliferators, such as trichloroacetic acid, in rodents might be associated with the ability of these agents to increase the rate of hepatocellular proliferation, resulting in hepatocellular hyperplasia and hepatomegaly (Marsman et al. 1988; Popp et al. 1994; Gonzalez et al. 1998). This mode of action is discussed later in this chapter.
Dichloroacetic Acid
Dichloroacetic acid, which is metabolized much more rapidly than trichloroacetic acid, is an effective inducer of hepatic tumors in mice and rats (see Table 4-4). It is a major metabolite of trichloroethylene in B6C3F1 mice but is below the limit of detection in similarly dosed Sprague-Dawley rats (Larson and Bull 1992a). Hepatoproliferative lesions increased sharply in male B6C3F1 mice when drinking water concentration increased from 1 to 2 g/L (Bull et al. 1990). Observations of greatly enlarged livers and marked cytomegaly in dichloroacetic-acid-treated mice led to the conclusion that tumorigenesis might depend largely on stimulation of cell division secondary to hepatotoxic damage. A follow-up histologic study to determine the dose relatedness of premalignant hepatic lesions was conducted (Carter et al. 2003) in liver tissues from male B6C3F1 mice treated in a study by DeAngelo et al. (1999). End points measured were altered hepatic foci, large foci of cellular alternations, adenomas, and carcinomas. Altered hepatic foci, large foci of cellular alterations, and adenomas demonstrated neoplastic progression with time; however, independent of dose and length of exposure, signs of toxicity were also observed in noninvolved liver. The authors interpreted these results as indicating that dichloroacetic acid behaves as a nongenotoxic carcinogen at doses below which genotoxicity has been observed.
In male F344 rats, dichloroacetic acid induced observable signs of toxicity in the nervous system, liver, and myocardium; however, treatment-related neoplastic lesions were observed only in the liver (DeAngelo et al. 1996). The low concentration of dichloroacetic acid in rats exposed to trichloroethylene may explain their relative insensitivity to hepatocarcinogenic effects. Dichloroacetic acid inhibits its own metabolism in the dose range 0.5 to 1 g/L in water; thus, sharp inconsistencies in blood concentrations are seen (ranging from <1 μM to 300-500 μM) (Kato-Weinstein et al. 1998) likely
TABLE 4-4 Hepatocarcinogenic Effects of Dichloroacetic Acid in Drinking Water Studies with Mice and Rats
|
Species (sex) |
Concentration (g/L) |
Duration (wk) |
Combined Hyperplastic Nodule and Hepatocellular Adenoma |
Hepatocellular Carcinoma |
Reference |
||
|
Incidence |
Tumor/n (multiplicity) |
Incidence |
Tumor/n (multiplicity) |
||||
|
B6C3F1 (M) |
0 |
61 |
|
|
|
|
Herren-Freund et al. 1987 |
|
|
5 |
61 |
25/26 |
4.6 |
21/26 |
1.7 |
|
|
B6C3F 1 (M) |
1 |
52 |
2/11 |
0.3 |
NR |
NR |
Bull et al. 1990 |
|
|
2 |
52 |
23/24 |
3.6 |
5/24 |
0.25 |
|
|
|
2 |
37 |
7/11 |
2.2 |
0/11 |
0 |
|
|
B6C3F 1 (M) |
0 |
60 |
0/10 |
0 |
8/12 |
1.7 |
DeAngelo et al. 1999 |
|
|
0.5 |
60 |
|
|
25/30 |
2.2 |
|
|
|
3.5 |
60 |
12/12 |
2.3 |
|
|
|
|
|
5 |
60 |
27/30 |
2.3 |
|
|
|
|
|
0 |
75 |
2/28 |
0.07 |
|
|
|
|
|
0.05 |
75 |
4/29 |
0.31 |
|
|
|
|
|
0.5 |
75 |
3/27 |
0.11 |
|
|
|
|
|
0 |
104 |
1/20 |
0.05 |
2/20 |
0.1 |
Daniel et al. 1992 |
|
|
0.5 |
104 |
12/24 |
0.5 |
15/24 |
0.63 |
|
|
B6C3F 1 (F) |
0 |
52 |
1/40 |
0.03 |
0/40 |
0 |
Pereira 1996 |
|
|
0.28 |
52 |
0/40 |
0 |
0/40 |
0 |
|
|
|
0.93 |
52 |
3/20 |
0.20 |
0/20 |
0 |
|
|
|
2.8 |
52 |
7/20 |
0.45 |
1/20 |
0.1 |
|
|
|
0 |
81 |
2/90 |
0.02 |
2/90 |
0.02 |
|
|
|
0.28 |
81 |
3/50 |
0.06 |
0/50 |
0 |
|
|
|
0.93 |
81 |
7/28 |
0.32 |
1/28 |
0.04 |
|
|
|
2.8 |
81 |
16/19 |
5.6 |
5/19 |
0.37 |
|
|
B6C3F1 (M) |
0 |
100 |
14/50 |
0.25 |
5/50 |
0.28 |
DeAngelo et al. 1999 |
|
|
0.05 |
100 |
11/33 |
0.5 |
NR |
NR |
|
|
|
0.5 |
100 |
11/24 |
0.32 |
5/24 |
0.68 |
|
|
|
1 |
100 |
23/32 |
0.8 |
16/32 |
1.29 |
|
|
|
2 |
100 |
13/14 |
0.85 |
6/14 |
2.47 |
|
|
|
3.5 |
100 |
8/8 |
0.64 |
4/8 |
2.9 |
|
|
F344 (M) |
0 |
60 |
0/7 |
0 |
0/7 |
0 |
DeAngelo et al. 1996 |
|
|
0.05 |
60 |
0/7 |
0 |
0/7 |
0 |
|
|
|
0.5 |
60 |
0/7 |
0 |
0/7 |
0 |
|
|
|
2.4 |
60 |
26/27 |
0.96 |
1/27 |
0.04 |
|
|
|
0 |
104 |
1/23 |
0.04 |
0/23 |
0 |
|
|
|
0.05 |
104 |
0/26 |
0 |
0/26 |
0 |
|
|
|
0.5 |
104 |
9/29 |
0.31 |
2/29 |
0.1 |
|
|
|
2.4 |
104 |
Not done |
Not done |
Not done |
Not done |
|
to lead to some variation in tumor yield in the mouse and other findings studied in this dose range (Bull et al. 2002).
Chloral Hydrate
Chloral hydrate produces hepatic tumors in male B6C3F1 mice but not in female B6C3F1 mice or in F344 male rats (Table 4-5). A single dose of chloral hydrate at 10 mg/kg administered by intragastric intubation to mice at 15 days of age increased the number of tumors between 48 and 92 wk based on the appearance of three adenomas and three carcinomas among eight animals (Rijhsinghani et al. 1986). Chloral hydrate administered to male B6C3F1 mice for 2 years in drinking water at an average dose of 166 mg/kg/day resulted in a 71% incidence of hepatic tumors (combined adenomas and carcinomas) (Daniel et al. 1992).
In another study, male B6C3F1 mice and male F344/N rats were given chloral hydrate in drinking water for 2 years (George et al. 2000). Time-weighted mean daily doses in rats were 7.4, 37.4, and 162.6 mg/kg/day. No increase in prevalence (percentage of animals with a tumor) or multiplicity (tumors/animal) of hepatocellular tumors was seen in male rats. Time-weighted mean daily doses in mice were 13.5, 65.0, and 146.6 mg/kg/day. Water consumption, survival, and body and organ weights were not altered from control values in any of the chloral hydrate treatment groups for either species. It was concluded that chloral hydrate induced hepatocellular neoplasia in the mouse, with a significant increase in the prevalence of hepatoadenoma and multiplicity at all doses tested and a significant increase in hepatocellular carcinomas in the high-dose group.
A 2-year NTP study in female B6C3F1 mice exposed to chloral hydrate administered in water by gavage was negative for induction of hepatic tumors up to a dose of 100 mg/kg (NTP 2002a). However, a 2-year NTP study in male B6C3F1 mice at the same doses found some evidence of carcinogenic activity based on increased incidences of hepatocellular adenoma or carcinoma (combined) in mice fed ad libitum and increased incidences of hepatocellular carcinoma in dietary-controlled mice (NTP 2002a,b; Leakey et al. 2003a). Dietary-controlled mice received variably restricted feed allocations to maintain their body weight on a predetermined “idealized” weight curve predictive of a terminal background liver tumor incidence of 15% to 20%. A statistically significant dose response to chloral hydrate was observed in the dietary-controlled animals (terminally adjusted liver tumor incidence 23.4%, 23.9%, 29.7%, and 38.6% for the four dose groups) but not the test groups fed ad libitum (terminally adjusted liver tumor incidence 33.4%, 52.6%, 50.6%, and 46.2%) (Leakey et al. 2003b). Dietary control was deemed to improve survival and decrease interassay variation.
Overall, chloral hydrate appears to be a species-, strain-, and sex-spe-
TABLE 4-5 Hepatocarcinogenic Effects of Chloral Hydrate in Mice
|
Species (sex) |
Dose (mg/kg) |
Exposure Route/Vehicle |
Duration (wk) |
Combined Hyperplastic Nodule and Hepatocellular Adenoma |
Hepatocellular Carcinoma |
Reference |
||
|
Incidence |
Tumor/n (multiplicity) |
Incidence |
Tumor/n (multiplicity) |
|||||
|
C57BL × C3HF1 single dose to neonates |
0 |
Oral/drinking water |
92 |
0/19 |
0 |
2/19 |
0.11 |
Rijhsinghani et al. 1986 |
|
|
5 |
92 |
2/9 |
0.22 |
1/9 |
0.11 |
||
|
|
10 |
92 |
3/8 |
0.38 |
3/8 |
0.38 |
||
|
B6C3F1 |
0 |
Oral/drinking water |
104 |
1/20 |
0.05 |
2/20 |
0.10 |
Daniel et al. 1992 |
|
|
166 |
104 |
8/24 |
0.33 |
11/24 |
0.46 |
||
|
B6C3F1 (M) |
0 |
Oral/drinking water |
104 |
21.4% |
0.21 |
54.8% |
0.74 |
George et al. 2000 |
|
|
13.5 |
104 |
43.5% |
0.65 |
54.3% |
0.72 |
||
|
|
65.0 |
104 |
51.3% |
0.95 |
59.0% |
1.03 |
||
|
|
146.6 |
104 |
50.0% |
0.72 |
84.4% |
0.72 |
||
|
B6C3F1 (F) |
0 |
Oral gavage/distilled water |
104 |
0/37 |
0 |
0 |
0 |
NTP 2002a |
|
|
25 |
104 |
0/48 |
0 |
0 |
0 |
||
|
|
50 |
104 |
0/43 |
0 |
0 |
0 |
||
|
|
100 |
104 |
0/36 |
0 |
0 |
0 |
||
|
B6C3F1 (M) fed ad libitum |
0 |
Oral gavage/distilled water |
104 |
12/48 |
— |
4/48 |
— |
NTP 2002b |
|
|
25 |
104 |
19/48 |
|
10/48 |
|
||
|
|
50 |
104 |
17/47 |
|
10/47 |
|
||
|
|
100 |
104 |
17/48 |
|
7/48 |
|
||
|
B6C3F1 (M) fed ad libitum |
0 |
Oral gavage/ distilled water |
104 |
— |
— |
16/48a |
— |
NTP 2002b |
|
|
25 |
104 |
|
|
25/48a |
|
||
|
|
50 |
104 |
|
|
23/47a |
|
||
|
|
100 |
104 |
|
|
22/48a |
|
||
|
B6C3F1 (M) controlled dietary intake |
0 |
Oral gavage/ distilled water |
104 |
9/48 |
— |
2/48 |
— |
NTP 2002b; Leakey et al. 2003a |
|
|
25 |
104 |
7/48 |
|
5/48 |
|
||
|
|
50 |
104 |
10/48 |
|
4/48 |
|
||
|
|
100 |
104 |
10/48 |
|
8/48 |
|
||
|
aAdenoma or carcinoma. |
||||||||
cific weak carcinogen. Furthermore, because two of the metabolites of chloral hydrate are trichloroacetic acid and dichloroacetic acid, it is difficult to assess the direct contribution of chloral hydrate to the specific carcinogenic effects observed solely in male B6C3F1 mice.
Collective Assessment of Animal Data
Trichloroacetic acid and chloral hydrate appear to be capable of inducing liver tumors only in mice, but dichloroacetic acid also induces liver tumors in rats. The blood concentration of trichloroacetic acid required to induce liver cancer in mice approaches the millimolar range; trichloroacetic acid is a peroxisome proliferator in the same dose range that induces liver cancer. The concentration of dichloroacetic acid associated with liver cancer is in the submicromolar range (Kalkuhl et al. 1998). The weak carcinogenicity of chloral hydrate is largely due to its metabolic conversion to trichloroacetic acid or dichloroacetic acid.
Altered gene expression in specific genes involved in the functional categories of cell growth, tissue remodeling, apoptosis, cancer progression, and xenobiotic metabolism has been observed in mouse liver after administration of a tumorigenic dose of dichloroacetic acid at 2 g/L of drinking water for 4 weeks (Thai et al. 2003). Dichloroacetic acid produces tumors in mice that display immunoreactivity to a c-Jun antibody, whereas trichloroethylene-induced tumors do not show this antibody reactivity (Stauber and Bull 1997). More recent work, in which trichloroacetic acid and dichloroacetic acid were given to mice alone and in various dose combinations, showed that dichloroacetic acid and trichloroacetic acid produced some tumors that were c-Jun+, and many that were c-Jun−; the number with a mixed phenotype increased with the relative dose of dichloroacetic acid (Bull et al. 2002). Mutation frequency of the H-ras protooncogene in mouse tumors induced by trichloroacetic acid alone was significantly different from that observed in trichloroethylene-induced tumors (0.44 versus 0.21), but that observed with dichloroacetic-acid-induced tumors (0.33) was not significantly different from that observed with trichloroethylene. No significant difference was observed in mutation spectra of tumors produced by the three compounds. Thus, dichloroacetic acid appears to produce liver tumors with a different phenotype than those produced as a result of trichloroacetic acid exposure. Dichloroacetic acid also induces markedly enlarged livers associated with cytomegaly (Bull 2000).
Both trichloroacetic acid and dichloroacetic acid are effective as rodent liver carcinogens at doses that do not produce cytotoxicity. Trichloroacetic acid produces liver tumors in mice with a phenotype common to peroxisome proliferators, whereas dichloroacetic acid increases the growth of liver tumors and produces tumors with a phenotype distinct in several aspects
from those produced by trichloroacetic acid. An initiation-promotion study of each compound alone or in pairwise combinations of trichloroacetic acid, dichloroacetic acid, and carbon tetrachloride was conducted in male B6C3F1 mice (Bull et al. 2004). Carbon tetrachloride was chosen for its ability to promote growth of liver tumors through cytotoxicity, producing a reparative hyperplasia growth stimulus. Thus, trichloroacetic acid, dichloroacetic acid, and carbon tetrachloride were hypothesized to have individually different modes of action as promoters. In general, interactions between carbon tetrachloride and trichloroacetic acid were seen to be additive and likely acting via different mechanisms whereas interactions between carbon tetrachloride and dichloroacetic acid were generally less than additive with a consistent dose-dependent decrease in the growth rate of tumors promoted by carbon tetrachloride. Dichloroacetic acid appears to exert an inhibitory effect on the growth of trichloroacetic-acid-promoted tumors. Thus, interactions were inhibitory or additive, but there appeared to be no evidence of synergy.
Differences between mice and rats in the development of hepatocellular adenoma and carcinoma from trichloroethylene and its metabolites is consistent with other nonmutagenic compounds and is not particularly useful for determination of mechanism of action or extrapolation to humans. This is due to controversy surrounding the validity of results in mouse liver resulting from the large number of nonmutagens that induce such tumors and the high and variable spontaneous tumor rates in some strains. Gold and Sloane (1995) examined the Carcinogenic Potency Database where 174 chemicals were evaluated as liver carcinogens in rats and mice. More mutagens than nonmutagens have been identified as liver carcinogens in each species (in mice 84 mutagens and 70 nonmutagens; in rats 75 mutagens and 32 nonmutagens). Their analysis indicated a species difference in the predominance of liver cancer in mice compared to rats. Among chemicals with positive results in the mouse, 55% (84/154) of mutagens compared to 71% (70/99) of nonmutagens induce liver tumors, while the proportions among positive chemicals in the rat are 39% (75/194) and 33% (32/98). Thus, while the proportion of rat carcinogens that are positive in the liver is similar for mutagens and nonmutagens, a higher proportion of nonmutagenic mouse carcinogens are positive in the liver than mutagenic carcinogens. This finding in mice reflects that chlorinated compounds (composed solely of chlorine, carbon, hydrogen, and, optionally, oxygen) are frequently positive in the mouse liver and are not mutagenic. Excluding the chlorinated compounds, results in mice are similar for mutagenic and nonmutagenic carcinogens; 56% (79/142) of mutagens and 59% (40/68) of nonmutagens are mouse liver carcinogens. In the Carcinogenic Potency Database, 261 rodent carcinogens have been tested in both rats and mice, and 82 (31%) induce tumors in only one target site of one species. The mouse liver is the
most common single-site, single-species target organ for both mutagens (12 chemicals) and nonmutagens (19 chemicals). Many of the nonmutagens in this group are chlorinated compounds. Thus, the species difference in the potency of trichloroethylene and its metabolites to induce liver tumors must be put in context with this historical data.
Human Studies
Because it was not possible for the committee to provide a comprehensive evaluation of the epidemiologic evidence on trichloroethylene and different cancers, it borrowed a previously compiled summary of the epidemiologic evidence on liver cancer from the Institute of Medicine (IOM 2003) to give some perspective on the evidence for liver cancer (see Table 4-6). The list was updated with one study published since the IOM report. Some common limitations of the studies that were reviewed include a relatively small number of cases of liver cancer and a lack of control for potential confounding by risk factors such as alcohol consumption and hepatitis B (see methodology and exposure information on some of the specific studies reviewed in Chapter 3, Tables 3-4 and 3-6). Another issue is that some studies reported findings for primary liver cancer, and others reported findings for biliary and primary liver cancer combined. This could result in misclassification of the outcome if these two cancer sites (liver and biliary) are etiologically distinct with respect to the effects of trichloroethylene exposure. In addition, only large cohort studies would have adequate statistical power to estimate excess risks and exposure-response relationships, as the incidence of liver cancer in the United States is low; the age-adjusted rate of cancer of the liver and intrahepatic bile duct is 6 per 100,000 people (SEER 2005). The American Cancer Society (2006) estimates that approximately 18,500 people will be diagnosed liver and intrahepatic bile duct cancer in 2006.
Cohort Studies
Excess incidence of liver cancer was observed in most cohort studies that specifically examined exposures to trichloroethylene (Axelson et al. 1994; Antilla et al. 1995; Hansen et al. 2001; Morgan and Cassady 2002; Raaschou-Nielsen et al. 2003). These findings were generally based on a small number of incident cases and thus were statistically unstable. Only one study reported a statistically significant excess of liver cancer incidence for the entire cohort (Raaschou-Nielsen et al. 2003). An excess among women (relative risk [RR] = 2.8; 95% confidence interval [CI] = 1.1, 5.8), but not among males (RR = 1.1, 95% CI = 0.7, 1.6), was reported in this study. Liver cancer incidence among females was significantly increased among women with 1 to 4.9 years of exposure (RR = 4.1, 95%CI = 1.1, 10.5). The inci-
TABLE 4-6 Selected Epidemiologic Data on Liver Cancer or Hepatobiliary Cancers and Exposure to Trichloroethylene
|
Reference |
Study Population |
Exposed Cases |
Estimated Relative Risk (95% CI) |
|
Cohort Studies—Incidence |
|||
|
Raaschou-Nielsen et al. 2003 |
Workers in Denmark |
|
|
|
Males |
|
|
|
|
All TCE-exposed workers |
27a |
1.1 (0.7-1.6) |
|
|
<1 yr employed |
9 |
1.3 (0.6-2.5) |
|
|
1-4.9 yr employed |
9 |
1.0 (0.5-1.9) |
|
|
≥5 yr employed |
9 |
1.1 (0.5-2.1) |
|
|
Females |
|
|
|
|
All TCE-exposed workers |
7 |
2.8 (1.1-5.8) |
|
|
<1 year employed |
2 |
2.8 (0.3-10.0) |
|
|
1-4.9 yr employed |
4 |
4.1 (1.1-10.5) |
|
|
≥5 yr employed |
1 |
1.3 (0.0-7.1) |
|
|
Morgan and Cassady 2002 |
Redlands, CA, community exposed to TCE in drinking water |
28a |
1.29 (99% CI 0.74-2.05) |
|
Hansen et al. 2001 |
Biologically monitored Danish workers |
|
|
|
Males |
5b |
2.6 (0.8-6.0) |
|
|
|
Females |
0 |
— |
|
Blair et al. 1998 |
Aircraft-maintenance workers in Utah |
|
|
|
Males |
|
|
|
|
No exposure |
1a |
0.8 (0.1-12.0) |
|
|
<5 unit-yr |
2 |
1.2 (0.1-13.8) |
|
|
5-25 unit-yr |
1 |
1.0 (0.1-16.0) |
|
|
>25 unit-yr |
3 |
2.6 (0.3-25.0) |
|
|
Antilla et al. 1995 |
Biologically monitored workers in Finland |
|
|
|
Entire period since first measurement |
5a |
2.27 (0.74-5.29) |
|
|
0-9 yr |
0 |
— (0.0-6.59) |
|
|
10-19 yr |
2 |
1.74 (0.21-6.29) |
|
|
≥20 yr |
3 |
6.07 (1.25-17.7) |
|
|
Mean personal U-TCA level |
|
|
|
|
<100 μmol/L |
2 |
1.64 (0.20-5.92) |
|
|
100+ μmol/L |
2 |
2.74 (0.33-9.88) |
|
|
Axelson et al. 1994 |
Biologically monitored Swedish workers |
4a |
1.41 (0.38-3) |
|
Cohort Studies—Mortality |
|||
|
Chang et al. 2003 |
Electronics-manufacturing workers in Taiwanc |
|
|
|
|
Males |
0 |
0.00 (NA) |
|
|
Females |
0 |
0.00 (NA) |
|
Reference |
Study Population |
Exposed Cases |
Estimated Relative Risk (95% CI) |
|
Boice et al. 1999 |
Aircraft manufacturing workers in California |
|
|
|
|
All exposed factory workers employed at least 1 yr since 1960 with routine exposure |
4b |
0.54 (0.15-1.38) |
|
|
Duration of potential exposure (routine or intermittent) |
|
|
|
|
<1 year exposed |
4a |
0.53 (0.18-1.60) |
|
|
1-4 yr exposed |
3 |
0.52 (0.15-1.79) |
|
|
≥5 yr exposedo |
6 |
0.94 (0.36-2.46) |
|
Ritz 1999 |
White male U.S. uranium processing workers |
|
|
|
|
TCE, cutting fluids, or kerosene TCE, light exposure |
8a |
1.66 (0.71-3.26) |
|
|
>2 yr, no latency |
3 |
0.93 (0.19-4.53) |
|
|
>2 yr, 15-yr latency |
3 |
1.16 (0.24-5.60) |
|
|
>5 yr, no latency |
3 |
1.90 (0.35-10.3) |
|
|
>5 yr, 15-yr latency |
3 |
2.86 (0.48-17.3) |
|
|
TCE, moderate exposure |
|
|
|
|
>2 yr, no latency |
1 |
4.97 (0.48-51.1) |
|
|
>2 yr, 15-yr latency |
1 |
5.53 (0.54-56.9) |
|
|
>5 yr, no latency |
1 |
8.82 (0.79-98.6) |
|
|
>5 yr, 15-yr latency |
1 |
12.1 (1.03-144) |
|
Blair et al. 1998 |
Aircraft maintenance workers in Utah Primary liver cancer for all TCE exposed |
4 |
1.7 (0.2-16.2) |
|
|
Liver and biliary cancer by cumulative TCE exposure |
|
|
|
|
Males |
|
|
|
|
No exposure |
3 |
0.5 (0.1-2.4) |
|
|
<5 unit-yr |
6 |
1.1 (0.3-4.1) |
|
|
5-25 yr |
3 |
0.9 (0.2-4.3) |
|
|
>25 unit-yr |
3 |
0.7 (0.2-3.2) |
|
|
Females |
|
|
|
|
No exposure |
3 |
4.2 (0.7-25.0) |
|
|
<5 unit-yr |
1 |
1.6 (0.2-18.2) |
|
|
5-25 unit-yr |
0 |
— |
|
|
>25 unit-yr |
2 |
2.3 (0.3-16.7) |
|
Morgan et al. 1998 |
Aerospace workers in Arizona TCE-exposed subcohort: |
6b |
0.98 (0.36-2.13) |
|
|
Low cumulative exposure |
3 |
1.32 (0.27-3.85) |
|
|
High cumulative exposure |
3 |
0.78 (0.16-2.28) |
|
|
Peak and cumulative exposurec: |
|
|
|
|
Peak: medium and high versus low and no exposure |
3a |
0.98 (0.29-3.35) |
|
|
Cumulative (low) |
3 |
2.12 (0.59-7.66) |
|
|
Cumulative (high) |
3 |
1.19 (0.34-4.16) |
dence of liver cancer was not significantly elevated in the highest exposure group (RR = 1.3, 95%CI = 0.0,7.1); however, there was only one case and less than one case expected in this group and, thus, the findings were highly unstable for this group. Evidence for an exposure-response relationship between the incidence of liver cancer and trichloroethylene exposure was also observed in the study by Antilla et al. (1995), who reported a statistically significant excess of liver cancer incidence in their highest duration of exposure category (>20 years; RR = 6.07, 95% CI = 1.25, 17.7).
Findings from the cohort studies that reported findings for mortality were mixed, with one study reporting no difference (Garabrant et al. 1988), three studies reporting a deficit (Greenland et al. 1994; Morgan et al. 1998; Boice et al. 1999), and two studies reporting an excess (Blair et al. 1998; Ritz 1999) in deaths from liver cancer. One study (Ritz 1999) found evidence of an exposure-response relationship; mortality from liver cancer was found to increase with degree (light versus moderate) and duration of exposure and time since first exposure (>15 years). A statistically significant excess of liver cancer (RR = 12.1) was reported among workers with moderate exposure, greater than 5 years of exposure, and at least 15 years since the first exposure; this finding was based on only one case and thus was not statistically stable (95% CI = 1.03, 144).
Case-Control Studies
A strength of the case-control studies is that they can have greater statistical power than cohort studies for evaluating rare outcomes such as liver cancer, but the power also depends on the prevalence of the exposure of interest, which is often low in general populations. A frequent weakness of population-based case-control studies is their inability to reliably document and estimate workplace exposures. IOM (2003) identified four case-control studies of liver cancer and exposure to organic solvents in general (Stemhagen et al. 1983; Hardell et al. 1984; Hernberg et al. 1988; Heinemann et al. 2000). One study has been published since then in which exposure to trichloroethylene was investigated. Lee et al. (2003) conducted a population-based case-control study in a Taiwanese village downstream from an electronic factory that contaminated community wells with trichloroethylene, tetrachloroethylene, and 1,1-dichloroethylene. Trichloroethylene concentrations in the well water were reported to be an order of magnitude higher than those for tetrachloroethylene and 1,1-dichloroethylene. This study reported increased mortality odds ratios among males for all cancer and for liver cancer for the periods after 10 years of latency—namely, 1980-1989 and 1990-1997. The adjusted mortality odds ratios for liver cancer in males was 2.6 (95% CI = 1.2, 5.5), with a significant linear trend for the period effect. This study did not address potential confounding related to hepatitis viral infection status, a risk factor for liver cancer, or potential misclassification due to the inclusion of secondary liver cancer among the case series.
Mode of Action
A number of modes of action have been proposed for the carcinogenic action of trichloroethylene and its metabolites in the liver. This section reviews the available evidence on genotoxicity, mutagenicity, and activation of the nuclear receptor peroxisome proliferator-activated receptor α (PPARα) and their relevance to humans. Although these modes of action are discussed separately, it is likely that multiple modes of action are involved in the carcinogenic process.
Mutagenicity and Genotoxicity
Mutagenicity refers to the ability of a chemical to induce heritable mutations (damage that can pass to daughter somatic cells), whereas genotoxicity is a broader term that includes mutational end points, cytogenetic analysis, and primary DNA damage.
Most mutagenicity assays for trichloroacetic acid, dichloroacetic acid,
and chloral hydrate are negative. Dichloroacetic acid and trichloroacetic acid do not consistently induce DNA damage in the livers of mice treated with hepatotoxic doses (IARC 1995a). The weight of evidence on the mutagenicity of chloral hydrate, dichloroacetic acid, and trichloroacetic acid indicates that a chemically induced mutation is unlikely to be a key event in the induction of tumors (Moore and Harrington-Brock 2000). In general, these chemicals require very high doses to elicit positive results, principally in in vitro tests. For example, chloral hydrate was positive in approximately 10 in vitro genotoxicity studies; however, in vivo results were mixed. Moreover, the potency of chloral hydrate in these studies was very low. Dichloroacetic acid has been the most extensively studied and has shown positive results in the standard Ames test protocol and in vitro mouse lymphoma assay; it was shown to induce very small increases in mouse bone marrow micronuclei and to increase DNA strand breaks in mouse and rat liver cells in vivo. However, DNA damage assays do not prove that a chemical can cause mutational damage. The collective evidence indicates that dichloroacetic acid is likely mutagenic but very weakly so. Trichloroacetic acid is the least mutagenic of the three metabolites, being negative in the Salmonella test and only weakly positive in the mouse lymphoma assay. It is unlikely that trichloroacetic acid would contribute to tumor formation through a mutational mechanism.
Neonatal B6C3F1 mice were administered chloral hydrate, trichloroacetic acid, and trichloroethylene by intraperitoneal injection at 8 and 15 days of age (Von Turgeln et al. 2002). At 12 months, only male mice treated with the positive control compounds had significant induction of liver tumors. Additional male mice were treated as above and livers were excised 24 and 48 hours and 7 days after the final dose. At 24 and 48 hours, mice treated with chloral hydrate or trichloroacetic acid showed significantly higher 8-oxo-2′deoxyguanosine formation, indicating increased endogenous DNA adduct formation through lipid peroxidation or oxidative stress; the authors concluded that neonatal B6C3F1 male mice are not sensitive to chloral hydrate or trichloroacetic acid as liver carcinogens. DNA and insulin-like growth factor II were demonstrated to be hypomethylated in mouse liver tumors in an initiation-promotion experiment (Tao et al. 2004b). DNA in both dichloroacetic-acid- and trichloroacetic-acid-promoted tumors was shown to be hypomethylated. Specific genes involved in several functional categories, including cell growth, tissue remodeling, apoptosis, cancer progression, and xenobiotic metabolism, were shown to have altered gene expression in dichloroacetic-acid-induced mouse liver tumors (Thai et al. 2003). Overall, the evidence indicates that none of the three metabolites under consideration here is likely to act principally by a mutational or genotoxic mechanism as liver carcinogens.
Peroxisome Proliferator-Activated Receptor Agonism
Peroxisome proliferators are a class of compounds that when fed to laboratory animals result in liver cancer (see Appendix E for detail and perspective). A key mode of action in this carcinogenic process is activation of the nuclear receptor PPARα. The human relevance of PPAR agonism is a subject of debate in the scientific community that has resulted in at least two important working groups and subsequent publications (Klaunig et al. 2003; IAS 2005). Several review articles have detailed the role of peroxisome proliferators and PPARs in the carcinogenic process (Green 1992, 1995; Green and Wahli 1994; Cattley et al. 1995; James et al. 1998; Gelman et al. 1999; Vanden Heuvel 1999a,b; Bull 2000; Corton et al. 2000a,b; Yeldandi et al. 2000; Melnick 2001; Guan 2002; Youssef and Badr 2002, 2005; Yu et al. 2003; Zhao and Jiang 2003; Kennedy et al. 2004; Lai 2004; Bosgra et al. 2005; Corton and Lapinskas 2005; O’Brien et al. 2005). Trichloroethylene, trichloroacetic acid, and dichloroacetic acid are considered peroxisome proliferators and they induce morphologic and biochemical changes that typify this class of chemicals. Chloral hydrate is considered either a very weak or a nonperoxisome proliferator. Thus, at least in terms of trichloroethylene, trichloroacetic acid, and dichloroacetic acid, the PPARα agonism (i.e., peroxisome proliferation) mode of action is a viable possibility which will be examined in more detail herein. The general applicability of PPARα agonism to human health is discussed in Appendix E.
Peroxisome Proliferators and Liver Cancer
The proposed mode of action for peroxisome proliferators is depicted in Figure 4-1, which shows several key events that ultimately result in rodent liver tumors. First, peroxisome proliferators activate PPARα, which regulates the transcription of genes involved in peroxisome proliferation, cell cycle and apoptosis, and lipid metabolism. Alterations in growth regulatory genes lead to perturbations in cell proliferation and apoptosis. Suppression of apoptosis coupled with stimulation of cell proliferation allows DNA-damaged cells to persist and proliferate, giving rise to preneoplastic foci and ultimately to tumors via further clonal expansion. Peroxisome proliferation per se is considered to be evidence of PPARα activation but it might or might not be related to the tumor formation. However, peroxisome proliferation could lead to oxidative stress, which could contribute to the mode of action by causing indirect DNA damage or by contributing to the stimulation of cell proliferation. As with other tumor promoters, PPARα ligands also inhibit gap junction intercellular communication, an event associated with increased cell proliferation. Several peroxisome proliferators stimulate non-parenchymal hepatic Kupffer cells and these resident macrophages affect
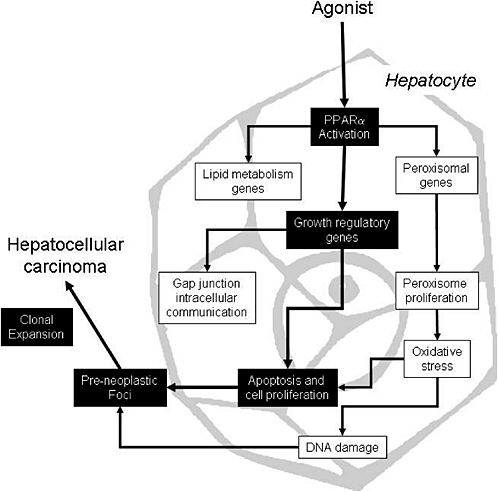
FIGURE 4-1 Proposed mode of action for liver tumor formation by peroxisome proliferators. Events causally related to adenoma or carcinoma formation are shown in black boxes; associated events are in white boxes. Source: Adapted from Klaunig et al. 2003.
the cell proliferation; this event is not depicted in Figure 4-1 because the importance of this event is controversial and no studies have been performed in this regard with trichloroethylene. The weight of evidence for the causally related events is discussed elsewhere (Klaunig et al. 2003; IAS 2005).
A minimal set of data elements to support a convincing demonstration that rodent liver tumors have arisen as a result of a PPARα mode of action would consist of PPARα agonism combined with light- or electron-microscopic evidence for peroxisome proliferation. Alternatively, evidence for PPARα agonism (in a receptor assay) combined with increases in liver weight and one (induction of acyl CoA oxidase) or more (e.g., induction of CYP4A) of the specific in vivo markers of peroxisome proliferation would suffice. Demonstration that liver growth was accompanied by at least tran-
siently increased rates of replicative DNA synthesis or decreased apoptosis also would significantly strengthen the case. The most convincing information showing that a particular compound induces liver cancer by PPARα mode of action would be through the use of the null mouse model. If the compound induces tumors in the wildtype, but not the PPARα-/- mouse, then this mode of action would be verified. Short of this information, the minimal criteria listed above (PPARα agonism with accompanying altered proliferation and growth characteristics) would be considered highly supportive. The absence of liver tumors in PPARα-null mice would definitively demonstrate the role of PPARα. Whether trichloroethylene and its metabolites meet this minimal data set is discussed below.
Trichloroethylene
The PPARα mode of action relative to trichloroethylene is summarized in Table 4-7; many of these effects might be attributable to trichloroethylene metabolites—in particular, trichloroacetic acid and dichloroacetic acid. Trichloroethylene activates mouse and human PPARα, albeit at high concentrations (1 mM), and can regulate known target genes for PPARα (Maloney and Waxman 1999; Nakajima et al. 2000; Laughter et al. 2004). Recent studies in PPARα null mice have shown that several events depend on this protein, including regulation of peroxisomal enzymes, cell proliferation, and perhaps some cell cycle regulatory genes (Klaunig et al. 1991; Stauber and Bull 1997; Tao et al. 1999, 2000b; Laughter et al. 2004). To the committee’s knowledge, a long-term bioassay in this mouse model system has not been performed. An important characteristic of all tumor promoters is their ability to selectively enhance survival of a particular phenotype of foci that ultimately gains a growth advantage. The work of Stauber and Bull (1997) and Bull et al. (2002) examining oncogene expression and mutations as well as that Tao et al. (1999, 2000a,b) examining DNA methylation have provided substantial information on the tumor phenotypes in trichloroacetic-acid- and dichloroacetic-acid-treated rodents, although the data for trichloroethylene are much less extensive. However, trichloroethylene causes tumors that are mixed for c-jun expression but consistently contain codon 61 mutations in c-Ha-ras. Interestingly, the tumor phenotypes of trichloroethylene-, trichloroacetic-acid, and dichloroaceticacid-induced tumors are not identical.
The species difference in tumor-promoting effects between rats and mice can be examined relative to the PPARα mode of action. The species difference in sensitivity to palmitoyl CoA oxidation activity was studied in F344 rats after treatment with trichloroethylene, tetrachloroethylene, and trichloroacetic acid (Goldsworthy and Popp 1987), and in Osborne-Mendel and Alderly Park rats and B6C3F1 and Alderley Park mice treated with
TABLE 4-7 Trichloroethylene and PPARα Mode of Action
|
Event |
Comments |
References |
|
Causal Events |
|
|
|
PPARα activation |
Human and mouse PPARα activated in transient transfection reporter assays. Studies from PPARα null mice show that the effects on cell proliferation and peroxisome proliferator target genes are PPARα dependent. |
Maloney and Waxman 1999; Nakajima et al. 2000; Laughter et al. 2004 |
|
Regulation of growth regulatory genes |
Increased c-jun and c-myc mRNA levels in nontumor tissue. Several potential growth regulatory target genes examined using microarrays showing a PPARα-dependent response. |
Tao et al. 1999, 2000b; Laughter et al. 2004 |
|
Cell proliferation or apoptosis |
Although there is no or little increase in hepatocyte labeling index in rats, mice exposed to trichloroethylene have higher rates of cell proliferation. This event is PPARα dependent. Trichloroethylene inhibited intercellular communication in mouse hepatocytes and not in rat hepatocytes. |
Klaunig et al. 1989; 1991; Stauber and Bull 1997; Laughter et al. 2004 |
|
Clonal expansion |
Tumors that arise from trichloroethylene treatment are basophilic with a relatively consistent mutational spectrum (c-Ha-ras codon 61). |
Bull et al. 2002 |
|
Associative Events |
||
|
Peroxisome proliferation and regulation of lipid metabolism genes |
Increased peroxisomes and peroxisomal enzymes are seen in mice but much less in rats. Increases in CN-insensitive palmitoyl CoA oxidation is induced by trichloroethylene but to a lesser extent than fibrates. ACO and CYP4A induction is PPARα dependent. |
Goldsworthy and Popp 1987; NTP 1988; Nakajima et al. 2000; Laughter et al. 2004 |
|
Oxidative stress |
Increased thiobarbituric acid reactive substances and decreases in reduced glutathione in mouse liver. |
Watanabe and Fukui 2000 |
trichloroethylene (Elcombe et al. 1985). The data indicate that rats treated with trichloroethylene (or tetrachloroethylene) do not show increases in peroxisomal enzyme activities, whereas rats treated with trichloroacetic acid have significant increases in peroxisomal enzyme activity. On the other hand, mice responded to treatment with trichloroethylene, exhibiting increases in peroxisomal volume density and induction of peroxisomal enzyme activities, catalase, and palmitoyl CoA oxidation. The accepted explanation for this species difference in sensitivity is a difference in metabolism. Trichloroethylene is metabolized by cytochrome P-450s and other noncytochrome P-450 oxidative enzymes to trichloroacetic acid and dichloroacetic acid. Goldsworthy and Popp (1987) clearly demonstrated that trichloroacetic
acid was a peroxisome proliferator in rats. Yet rats metabolize trichloroethylene more slowly than mice and metabolism appears to be saturable (Green and Prout 1985; Prout et al. 1985; Green et al. 1997a). Thus, the lack of tumorigenesis in rats compared with mice can be explained by a difference in metabolism of the active metabolite.
Trichloroacetic Acid
Trichloroacetic acid is most often cited as the hepatocarcinogenic metabolite of trichloroethylene. Thus, much of the same evidence for a PPARα mode of action for trichloroethylene could be provided for trichloroacetic acid. However, as outlined in Table 4-8, there are some differences in the strength of the data relative to trichloroethylene. There have been no studies showing the PPARα dependence on cell proliferation induced by trichloroethylene, although hypertrophy was not seen in PPARα null mice. A more extensive characterization of trichloroacetic-acid-induced tumors is available that clearly shows clonal expansion occurs as a result of treatment with this chemical (greater evidence than provided by trichloroethylene).
Dichloroacetic Acid
Although the initial events in the PPARα mode of action associated with dichloroacetic acid seem similar to those of trichloroethylene and trichloroacetic acid, there are some intriguing differences (see Table 4-9). For example, although dichloroacetic acid can activate PPARα (perhaps slightly less than trichloroacetic acid does) and cause peroxisome proliferation, the liver hypertrophy is not PPARα dependent. Also, the clonal expansion of preneoplastic foci by dichloroacetic acid is quite different with different phenotypic and genetic markers than by trichloroacetic acid and trichloroethylene.
Chloral Hydrate
Although chloral hydrate is used medically as a sedative or hypnotic and as a rubefacient in topical preparations, it has not been studied extensively in terms of the PPARα mode of action (see Table 4-10). The most extensive study was performed by the National Toxicology Program (NTP 2002b). Groups of male mice received chloral hydrate in distilled water by gavage at doses of 25, 50, or 100 mg/kg 5 days per week for 104 to 105 weeks. Each dose group was divided into two dietary groups of mice. The mice fed ad libitum had free access to feed, and the diet-controlled mice received feed in measured daily amounts calculated to maintain body weight on a previously computed idealized body weight curve. Chloral hydrate did not significantly induce either lauric acid 4-hydroxylase activity or CYP4A immunoreactive
TABLE 4-8 Trichloroacetic Acid and PPARα Mode of Action
|
Event |
Comments |
References |
|
Causal Events |
|
|
|
PPARα activation |
Mouse PPARα activated in transient transfection reporter assays with mixed results regarding human PPARα. Studies from PPARα null mice show that effects on peroxisome proliferator target genes are PPARα dependent. |
Maloney and Waxman 1999; Walgren et al. 2000a,b; Laughter et al. 2004 |
|
Regulation of growth regulatory genes |
Has not been studied. |
|
|
Cell proliferation or apoptosis |
Increased labeling index and dose-dependent increases in cell proliferation were observed. Hypertrophy was noted in the PPARα wild-type but not PPARα null mice. Trichloroacetic acid inhibited intercellular communication in mouse hepatocytes but not in rate hepatocytes. |
Stauber and Bull 1997; Klaunig et al. 1989; Ge et al. 2001; Laughter et al. 2004 |
|
Clonal expansion |
Increased clonal expansion of tumors that resemble those seen by peroxisome proliferators (and unlike DCA). Basophilic foci lacking GST-pi. See spectrum of mutations in K- and H-ras. Expansion not due to cytotoxicity. DNA hypomethylation is seen in TCA-induced tumors in the promoters of c-myc, IGF-II, and c-jun. |
Ferreira-Gonzalez et al. 1995; Pereira 1996; Pereira and Phelps 1996; Pereira et al. 1997, 2001; Tao et al. 1996, 2004b; Latendresse and Pereira 1997; Bull et al. 2004 |
|
Associative Events |
|
|
|
Peroxisome proliferation and regulation of lipid metabolism genes |
Increases in peroxisomal enzymes are seen in mouse and to a lesser extent in rat liver. CYP4A induction is PPARα dependent. |
Goldsworthy and Popp 1987; Odum et al. 1988; Walgren et al. 2004 |
|
Oxidative stress |
Has not been studied. |
|
|
ABBREVIATIONS: DCA, dichloroacetic acid; GST, glutathione S-transferase; IGF-II, insulin-like growth factor II; TCA, trichloroacetic acid. |
||
protein in any of the dosed groups of mice fed ad libitum. However, the high dose significantly induced both lauric acid 4-hydroxylase activity and CYP4A immunoreactive protein in the diet-controlled mice. Moreover, the induction-response profile of CYP4A was similar to the increase in the incidence of liver neoplasms at 2 years in the diet-controlled mice, with the major effect occurring in the 100-mg/kg group.
TABLE 4-9 Dichloroacetic Acid and PPARα Mode of Action
|
Event |
Comments |
References |
|
Causal Events |
|
|
|
PPARα activation |
Mouse PPARα activated in transient transfection reporter assays with mixed results regarding human PPARα. Studies from PPARα null mice show that the effects on peroxisome proliferator target genes are PPARα dependent. |
Maloney and Waxman 1999; Walgren et al. 2000a,b; Laughter et al. 2004 |
|
Regulation of growth regulatory genes |
Has not been studied. |
|
|
Cell proliferation or apoptosis |
Increased labeling index and dose-dependent increases in cell proliferation were observed. Hypertrophy was noted in the PPARα wild-type and PPARα null mice. |
Stauber and Bull 1997; DeAngelo et al. 1999; Walgren 2000a,b; Ge et al. 2001; Laughter et al. 2004 |
|
Clonal expansion |
Increased clonal expansion of tumors that do not resemble those seen by peroxisome proliferators (and unlike TCA and trichloroethylene). Eosinophilic foci positive for GST-pi, TGF-α, c-jun, and c-myc and negative for c-fos. DNA hypomethylation is seen in DCA-induced tumors in the promoters of c-myc and IGF-II. This hypomethylation and tumor formation can be reversed by methionine (although peroxisome proliferation may not be). Reversal of hypomethylation can be reversed after removal of DCA (unlike TCA). Loss of heterozygosity is observed in DCA-induced tumors. |
Anna et al. 1994; Ferreira-Gonzalez et al. 1995; Pereira 1996; Pereira and Phelps 1996; Pereira et al. 1997, 2004; Tao et al. 1996, 2004b; Latendresse and Pereira 1997; Miller et al. 2000; Carter et al. 2003 |
|
Associative Events |
|
|
|
Peroxisome proliferation and regulation of lipid metabolism genes |
Increases in peroxisomal enzymes are seen in mouse and to a lesser extent in rat liver. CYP4A induction is PPARα dependent. |
Everhart et al. 1998; DeAngelo et al. 1999; Pereira et al. 2004; Walgren et al. 2004 |
|
ABBREVIATIONS: DCA, dichloroacetic acid; GST, glutathione S-transferase; IGF-II, insulin-like growth factor II; TCA, trichloroacetic acid; TGF-α, transforming growth factor type α. |
||
Weight of Evidence
Table 4-11 summarizes the committee’s evaluation of the weight of evidence for a PPARα mode of action in rodents for trichloroethylene and its metabolites. The assessment is based on the general framework described
TABLE 4-10 Chloral Hydrate and PPARα Mode of Action
|
Event |
Comments |
References |
|
Causal Events |
|
|
|
PPARα activation |
Has not been studied. |
|
|
Regulation of growth regulatory genes |
Has not been studied. |
|
|
Cell proliferation or apoptosis |
Although hepatocelluar carcinoma was observed in mice and not rats, there was no increase in cell proliferation in either species. Chloral hydrate had no effect on hepatocyte intercellular communication in either rat or mouse cells. |
Klaunig et al. 1989; George et al. 2000 |
|
Clonal expansion |
Has not been studied. |
|
|
Associative Events |
|
|
|
Peroxisome proliferation and regulation of lipid metabolism genes |
Although hepatocellular carcinoma was observed in mice and not rats, there was no increase in palmitoyl CoA oxidation in either species. In diet-controlled mice, peroxisome proliferation and an increase in CYP4A protein and enzyme activity were seen. |
George et al. 2000; NTP 2002b |
|
Oxidative stress |
Has not been studied. |
|
by Cohen et al. (2003) and illustrated for PPARα agonists by Klaunig et al. (2003). Briefly, strong weight of evidence is defined as several studies that support the mode of action, and weak weight of evidence is defined by having a single study from a single laboratory or a significant amount of contradiction in the literature.
Dose-Response Relationships
The dose-response relationships for the key events in the PPARα mode of action are shown in the Table 4-12. Note the results from the comparison of wild-type with PPARα null mice that show the role of this receptor in the toxicity of trichloroethylene or its metabolites (discussed earlier in this chapter). PPARα activation per se is reserved for transactivation assays, as that is the most direct and definitive way to examine nuclear receptor agonism.
TABLE 4-11 Strength of the Weight of Evidence for PPARα Mode of Action for Trichloroethylene and Its Metabolites
|
Chemical |
Weight of Evidencea |
Comments |
|
TCE |
Strong |
TCE activates PPARα at high concentrations and regulates a variety of target genes. Studies with PPARα null mice show that most responses are dependent on this receptor, including peroxisome proliferation, cell proliferation, and target gene expression. More evidence could be provided by a long-term bioassay in this model system. |
|
TCA |
Strong |
TCA activates PPARα at high concentrations. Less extensive characterization of PPARα dependence on cell proliferation is provided than is known for TCE. However, the evidence of clonal expansion and phenotypic characteristics of tumors is strong and shows similarity to peroxisome proliferators. |
|
DCA |
Strong |
DCA activates PPARα at high concentrations. As is the case with TCA, PPARα dependence of DCA’s effects on cell proliferation is less than for trichloroethylene. Increasingly, it appears that DCA-induced clonal expansion is dissimilar to that of TCA and TCE. The reason for this discrepancy is not known but may require examination of tumors from PPARα null mice. Also, liver weight changes (and presumably cell proliferation) are not dependent on PPARα, indicating a potential for other modes of action that would be different than that of TCA. |
|
Chloral hydrate |
Weak |
There is no evidence of PPARα activation by chloral hydrate aside from it being a weak peroxisome proliferator. |
|
aA strong weight of evidence is defined as evidence from several studies which support the mode of action, while a weak weight of evidence is defined as having a single study from a single laboratory or a significant amount of contradiction in the literature (Klaunig et al. 2003). ABBREVIATIONS: DCA, dichloroacetic acid; TCA, trichloroacetic acid; TCE, trichloroethylene. |
||
FINDINGS AND RECOMMEDATIONS
Hepatotoxicity
The existing data clearly demonstrate that trichloroethylene produces hepatotoxicity in experimental animals and humans that is dependent on generation of reactive intermediates by CYP-450 in the liver. Besides its hepatotoxic potential, trichloroethylene and its metabolites produce liver effects categorized as independent of hepatotoxicity. These effects include elevations in plasma bile acids and accumulation of liver glycogen in the absence of subclinical evidence of liver dysfunction. The absence of liver dysfunction in rodents has been documented in studies using serum markers
TABLE 4-12 PPARα Mode-of-Action Dose-Response Relationships
|
Effect |
Dose/Concentrationa |
Route |
Vehicle |
Duration |
Gender |
Species/Strain |
Reference |
|
Causal Events |
|
|
|
|
|
|
|
|
PPARα Activation |
|
|
|
|
|
|
|
|
|
TCA, 1.0 mM; 5 mM |
In vitro |
DMSO |
24 hr |
N/A |
Cos-1 cells transfected with human and mouse PPARα |
Maloney and Waxman 1999 |
|
|
DCA, 1.0 mM, 5 mM |
In vitro |
DMSO |
24 hr |
N/A |
Cos-1 cells transfectedwith human and mouse PPARα |
Maloney and Waxman 1999 |
|
|
TCA, 4 mM (DCA at 4 mM, no effect) |
In vitro |
Unknown |
24 hr |
N/A |
HL8.5 cells transfected with mouse PPARα |
Walgren et al. 2000b |
|
Regulation of Growth Regulatory Genes |
|||||||
|
c-jun, c-myc |
TCE, 1,000 mg/kg |
Oral gavage, 5 days/wk |
Corn oil |
33 days |
Female |
B6C3F1 mice |
Tao et al. 1999 |
|
c-jun, c-myc (in tumors) |
DCA, 20 mmol/L |
Drinking water |
Water |
46 wk |
Female |
B6C3F1 mice |
Tao et al. 1999 |
|
c-jun, c-myc (in tumors) |
TCA, 20 mmol/L |
Drinking water |
Water |
46 wk |
Female |
B6C3F1 mice |
Tao et al. 1999 |
|
c-jun, c-myc |
TCE, 1,000 mg/kg |
Oral gavage |
Corn oil |
5 days |
Female |
B6C3F1 mice |
Tao et al. 2000b |
|
c-jun, c-myc |
TCA, 500 mg/kg |
Oral gavage |
Water |
5 days |
Female |
B6C3F1 mice |
Tao et al. 2000b |
|
Effect |
Dose/Concentrationa |
Route |
Vehicle |
Duration |
Gender |
Species/Strain |
Reference |
|
c-jun, c-myc |
DCA, 500 mg/kg |
Oral gavage |
Water |
5 days |
Female |
B6C3F1 mice |
Tao et al. 2000b |
|
Growth regulatory genes via microarray |
TCE, 1,500 mg/kg |
Oral gavage |
Methyl cellulose (0.1%) |
3 days |
Male |
SV129 wild-type and PRARα null mice |
Laughter et al. 2004 |
|
Cell Proliferation or Apoptosis |
|||||||
|
BrdU labeling |
TCE, 500 and 1,000 mg/kg/day (not observed in PPARAα null) |
Oral gavage |
Methyl cellulose (0.1%) |
3 wk |
Male |
SV129 wild-type and PPARAα null mice |
Laughter et al. 2004 |
|
[3H]Thymidine incorporation |
TCE, 500 mg/kg |
Oral gavage |
Corn oil |
7, 14 days |
Male |
B6C3F1 mice |
Klaunig et al. 1991 |
|
[3H]Thymidine incorporation |
TCE, no effect |
Oral gavage |
Corn oil |
3, 7, 14 days |
Female |
B6C3F1 mice |
Klaunig et al. 1991 |
|
[3H]Thymidine incorporation |
TCE, no effect |
Oral gavage |
Corn oil |
3, 7, 14 days |
Male |
F344 rat |
Klaunig et al. 1991 |
|
[3H]Thymidine incorporation |
TCE, no effect |
Oral gavage |
Corn oil |
3, 7, 14 days |
Male |
F344 rat |
Klaunig et al. 1991 |
|
BrdU labeling (not within tumors) |
DCA, 2 g/L |
Drinking Water |
Water |
14 days |
Male |
B6C3F1 mice |
Stauber and Bull 1997 |
|
BrdU labeling (not within tumors) |
TCA, 2 g/L |
Drinking water |
Water |
14, 28 days |
Male |
B6C3F1 mice |
Stauber and Bull 1997 |
|
[3H]Thymidine incorporation |
TCE, 250-2,500 mg/kg |
Intraperitoneal |
Corn Oil |
24 hr (500, 1,250) 36 hr (250-1,250) 48 hr (all doses) 72 hr (250, 1,250, 2,500) 96 hr (none) |
Male |
Sprague-Dawley rats |
Soni et al. 1998 |
|
Anchorage-independent growth |
TCA, DCA, 0.5-2 mM |
In vitro |
Media |
10-25 days |
Male |
Hepatocytes from B6C3F1 mice |
Stauber et al. 1998 |
|
[3H]Thymidine incorporation |
TCA, DCA, 0.1-1 mM. Varied based on individual |
In vitro |
Media |
72 hr |
Male and Female |
Human primary cultures |
Walgren et al. 2000a |
|
PCNA labeling |
TCA, DCA, 500 mg/kg |
Oral gavage |
Saline |
72-96 hr |
Female |
B6C3F1 mice |
Ge et al. 2001 |
|
Clonal Expansionb |
|||||||
|
Associative Events |
|||||||
|
Peroxisome Proliferation and Regulation of Lipid Metabolism Genes |
|||||||
|
CYP 4a12 mRNA |
TCE, 1,500 mg/kg/day (not observed in PPARAα null) |
Oral gavage |
Methyl cellulose (0.1%) |
3 days |
Male |
SV129 wild-type and PPARAα null mice |
Laughter et al. 2004 |
|
CYP4A, ACO protein |
TCE, 125, 500, 1,000 mg/kg/day (not observed in PPARAα null) |
Oral gavage |
Methyl cellulose (0.1%) |
3 wk |
Male |
SV129 wild-type and PPARAα null mice |
Laughter et al. 2004 |
|
Palmitoyl CoA oxidase |
TCE, 1,500 mg/kg/day (not observed in PPARAα null) |
Oral gavage |
Methyl cellulose (0.1%) |
3 wk |
Male |
SV129 wild-type and PPARα null mice |
Laughter et al. 2004 |
|
Effect |
Dose/Concentrationa |
Route |
Vehicle |
Duration |
Gender |
Species/Strain |
Reference |
|
CYP4A, ACO protein |
TCA, 1.0, 2.0 M (not observed in PPARα null) |
Drinking water |
Water |
1 wk |
Male |
SV129 wild-type and PPARα null mice |
Laughter et al. 2004 |
|
Palmitoyl CoA oxidase |
TCA, 2 M (not observed in PPARα null) |
Drinking water |
Water |
3 wk |
Male |
SV129 wild-type and PPARα null mice |
Laughter et al. 2004 |
|
CYP4A protein |
DCA, 2 M (not observed in PPARα null) |
Drinking water |
Water |
3 wk |
Male |
SV129 wild-type and PPARα null mice |
Laughter et al. 2004 |
|
Palmitoyl CoA oxidase |
TCA, DCA, 600 mg/kg |
In vitro |
Media |
24 hr |
Male and Female |
B6C3F1 homogenate and primary culture. No effects seen with LEH rat or human cells/cultures mouse liver |
Walgren et al. 2000a |
|
Laural CoA oxidase |
DCA, 3.2 g/L Unaffected by methionine |
Drinking water |
Water |
8 and 44 wk |
Female |
B6C3F1 mice |
Pereira et al. 2004 |
|
Lauric acid oxidase; CYP4A protein |
CH, 100 mg/kg |
Gavage (5 days/week) |
Water |
104 wk |
Male |
B6C3F1/Nctr mice |
Leakey et al. 2003a |
|
Palmitoyl CoA oxidase activity |
TCA, 5 g/L MCA, no effect |
Drinking water |
Water |
15-104 wk |
Male |
F344 rats |
DeAngelo et al. 1997 |
|
Palmitoyl CoA oxidase activity |
Perc, 200, 400 ppm |
Inhalation |
Air |
28 days |
Male and Female |
B6CF1 mice F344 rats |
Odum et al. 1988 |
of liver injury and is further supported by histopathologic examinations that showed no ultrastructural changes. However, the absence of liver dysfunction in humans has been based entirely on measures of serum markers of liver injury (e.g., plasma transaminases, bilirubin concentrations). Therefore, the possibility that humans might have discrete ultrastructural changes in the liver that can affect bile acid homeostasis cannot be ruled out. There are some mechanistic data addressing the nature of the elevation in bile acids in plasma but the precise mode of action remains unknown.
Elevation of plasma bile acids could result in their accumulation in other tissues, which could conceivably have detrimental effects on those organs. In addition to their lipid-solubilizing effect, bile acids are also signaling molecules that regulate gene transcription. The farnesoid X receptor functions as a bile acid nuclear receptor which regulates transcription of multiple genes responsible for maintaining cholesterol and bile acid homeostasis. Thus, accumulation of bile acids might contribute to the adverse effects of trichloroethylene exposure in organs other than the liver through a detergent effect or altered cellular signaling. However, it is not clear whether or not this is a significant effect.
The human relevance of liver glycogen accumulation observed in rodents exposed to dichloroacetic acid remains unclear. There are no studies documenting this effect in humans. Furthermore, all research on glycogen accumulation has been carried out using dichloroacetic acid, and not trichloroethylene. In light of this, it is not known whether exposure to trichloroethylene at environmentally relevant concentrations results in glycogen accumulation in rodents or humans.
Investigators have been able to dissociate the glycogen deposition effect from the peroxisome proliferation produced by trichloroethylene and its metabolites because dichloroacetic acid, which produces significant liver enlargement and no peroxisome proliferation, induces a marked accumulation of liver glycogen. In contrast, exposure to trichloroacetic acid produces only modest glycogen accumulation while stimulating considerable peroxisome proliferation.
Data from studies with autoimmune-prone mice also suggest that trichloroethylene and its metabolites are capable of triggering an immunemediated reaction against the liver. This observation is highly relevant to humans because there are multiple case reports of workers exposed to trichloroethylene with Stevens-Johnson syndrome who developed generalized skin reactions often accompanied by hepatitis of acute onset.
In summary, data generated since EPA (2001b) released its draft health risk assessment have not significantly advanced understanding of whether some of the noncancer liver effects of trichloroethylene and its metabolites are independent of early ultrastructural and discrete pathologic changes.
Also, the relationship of these effects to hepatocarcinogenesis remains unclear.
Liver Cancer
Data on trichloroethylene indicate that relatively high doses are needed to induce liver cancer, even in susceptible strains of mice. The three major metabolites of trichloroethylene—trichloroacetic acid, dichloroacetic acid, and chloral hydrate—can contribute to liver cancer in mice. None of the three is directly mutagenic or genotoxic as the principal mode of action. Trichloroacetic acid and dichloroacetic acid have been shown to promote liver cancer in classic initiation-promotion experimental protocols. The concentrations of trichloroacetic acid in blood required to induce liver cancer approach the millimolar range, whereas dichloroacetic acid concentrations in blood associated with carcinogenesis are in the submicromolar range. The carcinogenic activity of chloral hydrate is largely dependent on its conversion to trichloroacetic acid and dichloroacetic acid. Dichloroacetic acid and trichloroacetic acid adequately account for the hepatocarcinogenic responses to trichloroethylene.
There is sufficient weight of evidence to conclude that the mode of action of trichloroacetic acid as a rodent liver carcinogen is principally as a liver peroxisome proliferator in a specific strain of mouse, B6C3F1. This strain also has a particularly high background incidence of liver tumors. Moreover, F344 rats in which peroxisome proliferation is not induced do not show induction of liver cancer at the same doses at which B6C3F1 mice do. Altered cellular metabolism leading to transient changes in cell proliferation and cell regulation is related to induction of peroxisome proliferation in rodents.
Dichloroacetic acid produces liver tumors with a different phenotype than trichloroacetic acid. Its tumorigenic effects are closely associated with differential effects on cell replication rates in tumors, normal hepatocytes, and suppression of apoptosis. There is sufficient weight of evidence to conclude that the mode of action of dichloroacetic acid at high doses in rodents includes hepatomegaly and marked cytomegaly, which are closely associated with its activity as a differential promoter with effects on increased cell replication rates in tumors and normal hepatocytes and suppression of apoptosis. High-dose treatments alter activities of key enzymes in metabolism and cell growth. Dichloroacetic acid induces liver tumorigenesis in both mice and rats by this mode of action. However, dichloroacetic acid is a minor metabolite of trichloroethylene and whether it is formed in humans has not been clearly established.
The mode of action of chloral hydrate as a weak rodent liver carcinogen is dominated by induction of peroxisome proliferation activity in male
B6C3F1 mice. Female mice and rats are resistant to the carcinogenic effects of chloral hydrate. Because the metabolites of chloral hydrate are trichloroacetic acid and dichloroacetic acid, the contribution to liver tumor induction of the specific modes of action of each of these metabolites is also likely; however, an overall lack of potency for chloral hydrate in the carcinogenic response is notable.
Induction of peroxisome proliferation in human liver is not a prominent feature; therefore, this key event related to trichloroacetic acid liver carcinogenesis is not likely to occur in humans. The promotional activity of dichloroacetic acid includes a significant effect on cellular metabolism, differentiation function, and proliferation that encompass a mitogenic mode of action. Repeated exposure to dichloroacetic acid results in an inhibition of both mitosis and apoptosis and eventual formation of focal eosinophilic hyperplastic lesions. Assuming that the underlying mode of action for dichloroacetic acid as a liver carcinogen in rodents is promotional events affecting and culminating in mitogenesis, genotypic species differences between mice (one transforming growth factor type-β growth factor receptor allele) and humans with two functional copies of the gene suggest that humans would be phenotypically much less susceptible to liver carcinogenesis from agents that demonstrate a mitogenic mode of action (Andersen et al. 1995).
The weak carcinogenic activity of chloral hydrate in the liver of male B6C3F1 mice (with no liver cancer induction in female mice and rats) combined with lower rates of oxidation and higher rates of conjugation in humans compared with mice indicates that the mode of action in mice is not likely to be relevant to humans.
Exposure to trichloroethylene at concentrations relevant to the general public is not likely to induce liver cancer in humans. However, it is possible that much higher exposures to trichloroethylene, such as in certain high-risk occupations or in heavily contaminated locales, could result in increased risks of liver toxicity and cancer. In addition, the existence of sensitive populations due to genetics, disease, or life stage cannot be discounted.
The epidemiologic evidence for an association between liver cancer and trichloroethylene exposure is inconclusive. Excess liver cancer incidence was observed in most of the cohort studies that examined this outcome. However, cohort studies of mortality and population-based case-control studies yielded mixed results. Of particular interest is a recent case-control study that found an association between liver cancer mortality and trichloroethylene concentrations in well water in a community that was downstream from a Taiwanese factory. Although this study suffers from several methodologic weaknesses, it is the first to show an association between environmental exposures to trichloroethylene and liver cancer mortality.
Recommendations:
-
Additional laboratory studies are needed to establish the significance of increased bile acids in relation to the hepatotoxic potential of trichloroethylene, as well as in relation to other systemic effects. Such studies will help clarify whether elevation of serum bile acids is an early indicator of changes in liver function or is a marker of exposure to trichloroethylene (or other halogenated solvents which induce this effect).
-
More research is also needed to assess whether increases in serum bile acids in humans exposed to trichloroethylene are independent of discrete pathologic changes in the liver. Because histopathologic assessment would be difficult to perform in human subjects, new, highly sensitive, and noninvasive toxicologic parameters are needed to clarify the toxicologic importance of these effects of trichloroethylene.
-
Additional studies of the effects of trichloroethylene on glycogen accumulation, perhaps using cultured human hepatocytes, should shed some light on the significance of this effect and its relevance to humans.
-
More research is needed to determine whether an autoimmune response might play a role in trichloroethylene-mediated liver disease. Adducts formed between metabolites of trichloroethylene and liver proteins can result in the formation of neoantigens. These neoantigens can lead to antibody-dependent hepatocellular injury. The same process has been reported with chemicals such as halothane, which is well known to produce immune-mediated hepatotoxicity. Studies similar to those carried out with halothane could be instrumental in elucidating whether autoimmunity is a causal factor in the hepatotoxicity of trichloroethylene.
-
Studies are needed to determine the metabolic pathway and yield for forming dichloroacetic acid from trichloroethylene either via trichloroacetic acid or via other pathway(s). If dichloroacetic acid is found to be a metabolite of concern, additional studies may be needed to understand its role in the toxicities associated with trichloroethylene.













































